
Achieving HIV-1 Control through RNA-Directed Gene Regulation


 ,
,
Abstract
:1. Introduction
2. The Latent Reservoir
3. HIV Cure: Sterilising or Functional?
4. RNA Silencing Pathways
5. RNA Therapeutics Targeting HIV by PTGS
6. RNA Therapeutics Targeting HIV by TGS
7. RNA Therapeutics Targeting HIV by CRISPR/Cas9
8. HIV Gene Therapy
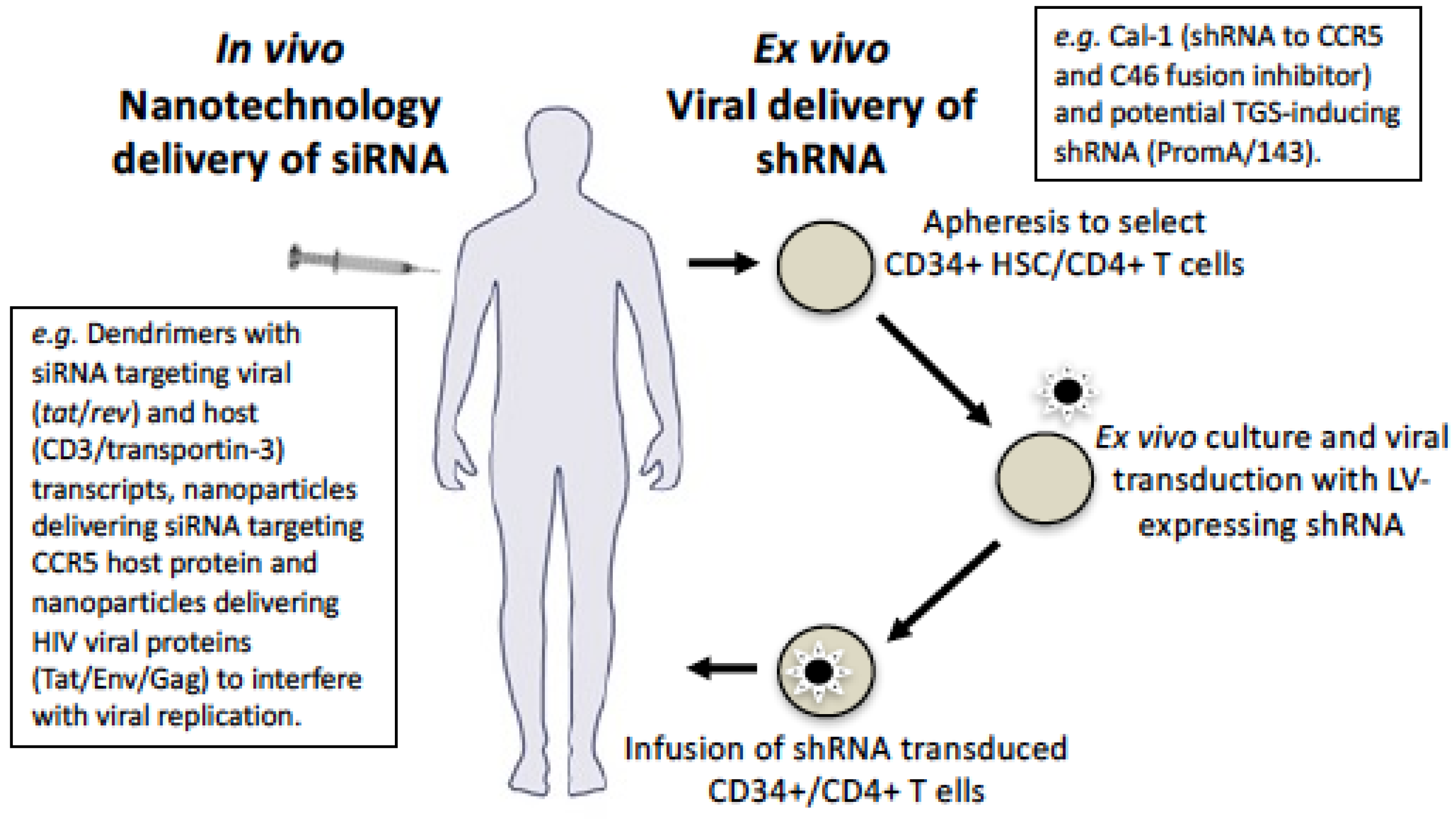
9. Delivery Using Viral Vectors
10. Delivery Using Nanotechnology
11. Future Perspectives
Acknowledgements
Conflicts of Interest
References
- Fang, C.T.; Chang, Y.Y.; Hsu, H.M.; Twu, S.J.; Chen, K.T.; Lin, C.C.; Huang, L.Y.; Chen, M.Y.; Hwang, J.S.; Wang, J.D.; et al. Life Expectancy of Patients with Newly-Diagnosed HIV Infection in the Era of Highly Active Antiretroviral Therapy. QJM 2007. [Google Scholar] [CrossRef] [PubMed]
- Guideline on when to start antiretroviral therapy and on pre-exposure prophylaxis for HIV. Avaliable online: http://apps.who.int/iris/bitstream/10665/186275/1/9789241509565_eng.pdf (accessed on 11 August 2016).
- Group, I.S.S.; Lundgren, J.D.; Babiker, A.G.; Gordin, F.; Emery, S.; Grund, B.; Sharma, S.; Avihingsanon, A.; Cooper, D.A.; Fatkenheuer, G.; et al. Initiation of Antiretroviral Therapy in Early Asymptomatic HIV Infection. N. Engl. J. Med. 2015, 373, 795–807. [Google Scholar]
- Group, T.A.S.; Danel, C.; Moh, R.; Gabillard, D.; Badje, A.; Le Carrou, J.; Ouassa, T.; Ouattara, E.; Anzian, A.; Ntakpe, J.B.; et al. A Trial of Early Antiretrovirals and Isoniazid Preventive Therapy in Africa. N. Engl. J. Med. 2015, 373, 808–822. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Chan, S.W.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Justement, J.S.; Moir, S.; Hallahan, C.W.; Maenza, J.; Mullins, J.I.; Collier, A.C.; Corey, L.; Fauci, A.S. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: Implications for eradication of virus. J. Infect. Dis. 2007. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Hey-Cunningham, W.J.; Murray, J.M.; Natarajan, V.; Amin, J.; Moore, C.L.; Emery, S.; Cooper, D.A.; Zaunders, J.; Kelleher, A.D.; Koelsch, K.K.; et al. Early antiretroviral therapy with raltegravir generates sustained reductions in HIV reservoirs but not lower T-cell activation levels. AIDS 2015, 29, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Koelsch, K.K.; Boesecke, C.; Mcbride, K.; Gelgor, L.; Fahey, P.; Natarajan, V.; Baker, D.; Bloch, M.; Murray, J.M.; Zaunders, J.; et al. Impact of treatment with raltegravir during primary or chronic HIV infection on RNA decay characteristics and the HIV viral reservoir. AIDS 2011, 25, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.M.; Mcbride, K.L.; Amin, J.; Cordery, D.V.; Kelleher, A.D.; Cooper, D.A.; Koelsch, K.K. Switching virally suppressed, treatment-experienced patients to a raltegravir-containing regimen does not alter levels of HIV-1 DNA. PLoS ONE 2012, 7, e31990s. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Mcbride, K.; Boesecke, C.; Bailey, M.; Amin, J.; Suzuki, K.; Baker, D.; Zaunders, J.J.; Emery, S.; Cooper, D.A.; et al. Integrated HIV DNA accumulates prior to treatment while episomal HIV DNA records ongoing transmission afterwards. AIDS 2012, 26, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Zaunders, J.J.; Mcbride, K.L.; Xu, Y.; Bailey, M.; Suzuki, K.; Cooper, D.A.; Emery, S.; Kelleher, A.D.; Koelsch, K.K.; et al. HIV DNA subspecies persist in both activated and resting memory CD4+ T cells during ART. J. Virol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Sgarbanti, M. HIV-1 latency: An update of molecular mechanisms and therapeutic strategies. Viruses 2014, 6, 1715–1758. [Google Scholar] [CrossRef] [PubMed]
- Cillo, A.R.; Krishnan, S.; Mcmahon, D.K.; Mitsuyasu, R.T.; Para, M.F.; Mellors, J.W. Impact of Chemotherapy for HIV-1 Related Lymphoma on Residual Viremia and Cellular HIV-1 DNA in Patients on Suppressive Antiretroviral Therapy. PLoS ONE 2014, 9, e92118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O′shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Bosch, R.J.; Aga, E.; Albrecht, M.; Demeter, L.M.; Dykes, C.; Bastow, B.; Para, M.; Lai, J.; Siliciano, R.F.; et al. No evidence for decay of the latent reservoir in HIV-1-infected patients receiving intensive enfuvirtide-containing antiretroviral therapy. J. Infect. Dis. 2010, 201, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Zheng, L.; Bosch, R.J.; Chan, E.S.; Margolis, D.M.; Read, S.; Kallungal, B.; Palmer, S.; Medvik, K.; Lederman, M.M.; et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: A randomized controlled trial. PLoS Med. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, S.M.; Ribaudo, H.; Bassett, R.; Mellors, J.W.; Demeter, L.M.; Coombs, R.W.; Currier, J.; Morse, G.D.; Gerber, J.G.; Martinez, A.I.; et al. A randomized, placebo-controlled trial of abacavir intensification in HIV-1-infected adults with virologic suppression on a protease inhibitor-containing regimen. HIV Clin. Trials 2010. [Google Scholar] [CrossRef] [PubMed]
- Mcmahon, D.; Jones, J.; Wiegand, A.; Gange, S.J.; Kearney, M.; Palmer, S.; Mcnulty, S.; Metcalf, J.A.; Acosta, E.; Rehm, C.; et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin. Infect. Dis. 2010, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Shergill, A.K.; Mcquaid, K.; Gianella, S.; Lampiris, H.; Hare, C.B.; Pandori, M.; Sinclair, E.; Gunthard, H.F.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Cheema, M.; Parker, D.; Wiegand, A.; Bosch, R.J.; Coffin, J.M.; Eron, J.; Cohen, M.; Margolis, D.M. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS ONE 2010, 5, e9390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Contreras, X.; Schweneker, M.; Chen, C.S.; Mccune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Xu, Y.; Sullivan, J.; Souder, E.; Argyris, E.G.; Acheampong, E.A.; Fisher, J.; Sierra, M.; Thomson, M.M.; Najera, R.; et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J. Clin. Investig. 2005, 115, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Ellenberg, P.; Churchill, M.; Lewin, S.R. HDAC inhibitors in HIV. Immunol. Cell. Biol. 2012, 90, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Paiardini, M. Hijacking the IL-7/IL-7R system in HIV infection. J. Leukoc. Biol. 2011, 89, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, R.M.; Prins, J.M.; Roos, M.T.; Schellekens, P.T.; Ten Berge, I.J.; Yong, S.L.; Schuitemaker, H.; Eerenberg, A.J.; Jurriaans, S.; De Wolf, F.; et al. OKT3 and IL-2 treatment for purging of the latent HIV-1 reservoir in vivo results in selective long-lasting CD4+ T cell depletion. J. Clin. Immunol. 2001, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Mckernan, L.N.; Momjian, D.; Kulkosky, J. Protein Kinase C: One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv. Virol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef] [PubMed]
- Quivy, V.; Adam, E.; Collette, Y.; Demonte, D.; Chariot, A.; Vanhulle, C.; Berkhout, B.; Castellano, R.; De Launoit, Y.; Burny, A.; et al. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: Potential perspectives for the development of therapeutic strategies. J. Virol. 2002, 76, 11091–11103. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Chun, T.W.; Belay, B.W.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.; Hallahan, C.W.; Moir, S.; Wender, P.A.; et al. Effect of histone deacetylase inhibitors on HIV production in latently infected, resting CD4(+) T cells from infected individuals receiving effective antiretroviral therapy. J. Infect. Dis. 2012, 206, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Beliakova-Bethell, N.; Zhang, J.X.; Singhania, A.; Lee, V.; Terry, V.H.; Richman, D.D.; Spina, C.A.; Woelk, C.H. Suberoylanilide hydroxamic acid induces limited changes in the transcriptome of primary CD4(+) T cells. AIDS 2013, 27, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O′connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; Mcmahon, J.; Velayudham, P. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Robb, G.B.; Brown, K.M.; Khurana, J.; Rana, T.M. Specific and potent RNAi in the nucleus of human cells. Nat. Struct. Mol. Biol. 2005, 12, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol. Ther. Nucleic Acids 2015. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ahlenstiel, C.; Marks, K.; Kelleher, A.D. Promoter Targeting RNAs: Unexpected Contributors to the Control of HIV-1 Transcription. Mol. Ther. Nucleic Acids 2015. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Juelich, T.; Lim, H.; Ishida, T.; Watanebe, T.; Cooper, D.A.; Rao, S.; Kelleher, A.D. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J. Biol. Chem. 2008. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Rand, T.A.; Ginalski, K.; Grishin, N.V.; Wang, X. Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proc. Natl. Acad. Sci. USA 2004, 101, 14385–14389. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, R.; Chiang, C.M.; Corey, D.R. Regulation of mammalian transcription and splicing by Nuclear RNAi. Nucleic Acids Res. 2016, 44, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Jakymiw, A.; Lian, S.; Eystathioy, T.; Li, S.; Satoh, M.; Hamel, J.C.; Fritzler, M.J.; Chan, E.K. Disruption of GW bodies impairs mammalian RNA interference. Nat. Cell Biol. 2005, 7, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.; Moazed, D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 2004, 303, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Shimojo, H.; Hayashi, A.; Kawaguchi, R.; Ohtani, Y.; Uegaki, K.; Nishimura, Y.; Nakayama, J. Intrinsic nucleic acid-binding activity of Chp1 chromodomain is required for heterochromatic gene silencing. Mol. Cell 2012, 47, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Schalch, T.; Job, G.; Noffsinger, V.J.; Shanker, S.; Kuscu, C.; Joshua-Tor, L.; Partridge, J.F. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell 2009, 34, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Kovochich, M.; Liong, M.; Meng, H.; Kabehie, S.; George, S.; Zink, J.I.; Nel, A.E. Polyethyleneimine coating enhances the cellular uptake of mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS Nano 2009, 3, 3273–3286. [Google Scholar] [CrossRef] [PubMed]
- Schalch, T.; Job, G.; Shanker, S.; Partridge, J.F.; Joshua-Tor, L. The Chp1-Tas3 core is a multifunctional platform critical for gene silencing by RITS. Nat. Struct. Mol. Biol. 2011, 18, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Callinan, P.A.; Feinberg, A.P. The emerging science of epigenomics. Hum. Mol. Genet. 2006. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Das, K.; Quinones, O.; Shao, W.; Tuske, S.; Alvord, W.G.; Arnold, E.; Hughes, S.H. Mutations in HIV-1 reverse transcriptase affect the errors made in a single cycle of viral replication. J. Virol. 2014, 88, 7589–7601. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.P.; Boyd, M.P.; Impey, H.; Breton, L.R.; Bartlett, J.S.; Symonds, G.P.; Hutter, G. CCR5 as a natural and modulated target for inhibition of HIV. Viruses 2014. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.P.; Levin, B.R.; Zhang, J.; Sahakyan, A.; Boyer, J.; Carroll, M.V.; Colon, J.C.; Keech, N.; Rezek, V.; Bristol, G.; et al. Engineering Cellular Resistance to HIV-1 Infection In Vivo Using a Dual Therapeutic Lentiviral Vector. Mol. Ther. Nucleic Acids 2015. [Google Scholar] [CrossRef] [PubMed]
- Digiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; T Hooft, K.; Liu, Y.P.; Centlivre, M.; Von Eije, K.J.; Berkhout, B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol. Ther. 2008. [Google Scholar] [CrossRef]
- Centlivre, M.; Legrand, N.; Klamer, S.; Liu, Y.P.; Jasmijn Von Eije, K.; Bohne, M.; Rijnstra, E.S.; Weijer, K.; Blom, B.; Voermans, C.; et al. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol. Ther. Nucleic Acids 2013. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Digiusto, D.L.; Rossi, J.J. Combinatorial RNA-based gene therapy for the treatment of HIV/AIDS. Methods Opin. Biol. Ther. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Zhang, J.; Li, H.; Ouellet, D.L.; Digiusto, D.L.; Rossi, J.J. Endogenous MCM7 microRNA cluster as a novel platform to multiplex small interfering and nucleolar RNAs for combinational HIV-1 gene therapy. Hum. Gene Ther. 2012. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.W.; Haworth, K.G.; Burke, B.P.; Polacino, P.; Norman, K.K.; Adair, J.E.; Hu, S.L.; Bartlett, J.S.; Symonds, G.P.; Kiem, H.P. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol. Ther. Methods Clin. Dev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wolstein, O.; Boyd, M.; Millington, M.; Impey, H.; Boyer, J.; Howe, A.; Delebecque, F.; Cornetta, K.; Rothe, M.; Baum, C.; et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; St Vincent’s Centre for Applied Medical Research, Sydney, Australia. Cloning and sequencing of over 60 provirus has not reported any virus escape at MOIs of 0.1 to 100. 2014. [Google Scholar]
- Singh, A.; Palanichamy, J.K.; Ramalingam, P.; Kassab, M.A.; Bhagat, M.; Andrabi, R.; Luthra, K.; Sinha, S.; Chattopadhyay, P. Long-term suppression of HIV-1C virus production in human peripheral blood mononuclear cells by LTR heterochromatization with a short double-stranded RNA. J. Antimicrob. Chemother. 2014. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.M.; Ackley, A.M.; Matrone, M.A.; Morris, K.V. Characterization of an HIV-targeted transcriptional gene-silencing RNA in primary cells. Hum. Genet. Ther. 2012. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.M.; De La Cruz, J.; Morris, K.V. Mobilization-competent Lentiviral Vector-mediated Sustained Transcriptional Modulation of HIV-1 Expression. Mol. Ther. 2009. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, C.L.; Lim, H.G.; Cooper, D.A.; Ishida, T.; Kelleher, A.D.; Suzuki, K. Direct evidence of nuclear Argonaute distribution during transcriptional silencing links the actin cytoskeleton to nuclear RNAi machinery in human cells. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ishida, T.; Yamagishi, M.; Ahlenstiel, C.; Swaminathan, S.; Marks, K.; Murray, D.; Mccartney, E.M.; Beard, M.R.; Alexander, M.; et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol. 2011, 8, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Shijuuku, T.; Fukamachi, T.; Zaunders, J.; Guillemin, G.; Cooper, D.; Kelleher, A. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J. RNAi Gene Silenc. 2005, 1, 66–78. [Google Scholar] [PubMed]
- Yamagishi, M.; Ishida, T.; Miyake, A.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K.; Watanabe, T. Retroviral delivery of promoter-targeted shRNA induces long-term silencing of HIV-1 transcription. Microbes Infect. 2009. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Hattori, S.; Marks, K.; Ahlenstiel, C.; Maeda, Y.; Ishida, T.; Millington, M.; Boyd, M.; Symonds, G.; Cooper, D.A.; et al. Promoter Targeting shRNA Suppresses HIV-1 Infection In vivo through Transcriptional Gene Silencing. Mol. Ther. Nucleic Acids 2013. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 2011. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Leibman, R.S.; Riley, J.L. Engineering T Cells to Functionally Cure HIV-1 Infection. Mol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 11 August 2016).
- Bobbin, M.L.; Burnett, J.C.; Rossi, J.J. RNA interference approaches for treatment of HIV-1 infection. Genome Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Persons, D.A. Lentiviral vector gene therapy: Effective and safe? Mol. Ther. 2010. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Vink, M.A.; Westerink, J.T.; Ramirez De Arellano, E.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010. [Google Scholar] [CrossRef] [PubMed]
- Younan, P.M.; Peterson, C.W.; Polacino, P.; Kowalski, J.P.; Obenza, W.; Miller, H.W.; Milless, B.P.; Gafken, P.; Derosa, S.C.; Hu, S.L.; et al. Lentivirus-mediated Gene Transfer in Hematopoietic Stem Cells Is Impaired in SHIV-infected, ART-treated Nonhuman Primates. Mol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.N.; Wu, H.; Shankar, P. Recent advances in RNAi-based strategies for therapy and prevention of HIV-1/AIDS. Adv. Drug Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, A.; Miyata, K.; Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016, 104, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Dis. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Choung, S.; Kim, Y.J.; Kim, S.; Park, H.-O.; Choi, Y.-C. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 2006, 342, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, B.; Sipa, K. Chemical and structural diversity of siRNA molecules. Curr. Top. Med. Chem. 2006, 6, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Allerson, C.R.; Dande, P.; Vickers, T.A.; Sioufi, N.; Jarres, R.; Baker, B.F.; Swayze, E.E.; Griffey, R.H.; Bhat, B. Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J. Med. Chem. 2005, 48, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.K.; Deleavey, G.F.; Damha, M.J. Chemically modified siRNA: Tools and applications. Drug Discov. Today 2008, 13, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Du, Q.; Wahlestedt, C.; Liang, Z. RNA Interference with chemically modified siRNA. Curr. Top. Med. Chem. 2006, 6, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Muratovska, A.; Eccles, M.R. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004, 558, 63–68. [Google Scholar] [CrossRef]
- Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K. Lactosylated poly (ethylene glycol)-siRNA conjugate through acid-labile β-thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J. Am. Chem. Soc. 2005, 127, 1624–1625. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Goldberg, M.; Qin, J.; Dorkin, J.R.; Gamba-Vitalo, C.; Maier, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Manoharan, M. Development of lipidoid–siRNA formulations for systemic delivery to the liver. Mol. Ther. 2009, 17, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-C.; Mozumdar, S.; Huang, L. Lipid-based systemic delivery of siRNA. Adv. Drug Deliv. Rev. 2009, 61, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Min, D.-H.; Singh, N.; Zhu, H.; Birjiniuk, A.; Von Maltzahn, G.; Harris, T.J.; Xing, D.; Woolfenden, S.D.; Sharp, P.A. Functional delivery of siRNA in mice using dendriworms. ACS Nano 2009, 3, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Cun, D.; Jensen, L.B.; Nielsen, H.M.; Moghimi, M.; Foged, C. Polymeric nanocarriers for siRNA delivery: Challenges and future prospects. J. Biomed. Nanotechnol. 2008, 4, 258–275. [Google Scholar] [CrossRef]
- Patil, M.L.; Zhang, M.; Taratula, O.; Garbuzenko, O.B.; He, H.; Minko, T. Internally cationic polyamidoamine PAMAM-OH dendrimers for siRNA delivery: Effect of the degree of quaternization and cancer targeting. Biomacromolecules 2009, 10, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, J.; Hafdi, N.; Behr, J.-P.; Erbacher, P.; Peng, L. PAMAM dendrimers for efficient siRNA delivery and potent gene silencing. Chem. Commun. 2006, 22, 2362–2364. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Dohmen, C.; Philipp, A.; Kiener, D.; Maiwald, G.; Scheu, C.; Ogris, M.; Wagner, E. Synthesis and biological evaluation of a bioresponsive and endosomolytic siRNA-polymer conjugate. Mol. Pharm. 2009, 6, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tang, N.; Liu, X.; Liang, W.; Xu, W.; Torchilin, V.P. siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J. Control. Release 2006, 112, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Elbakry, A.; Zaky, A.; Liebl, R.; Rachel, R.; Goepferich, A.; Breunig, M. Layer-by-layer assembled gold nanoparticles for siRNA delivery. Nano Lett. 2009, 9, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.S.; Kim, C.-K.; Han, G.; Forbes, N.S.; Rotello, V.M. Efficient gene delivery vectors by tuning the surface charge density of amino acid-functionalized gold nanoparticles. ACS Nano 2008, 2, 2213–2218. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Green, J.J.; Love, K.T.; Sunshine, J.; Langer, R.; Anderson, D.G. Gold, poly (β-amino ester) nanoparticles for small interfering RNA delivery. Nano Lett. 2009, 9, 2402–2406. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Bae, K.H.; Kim, S.H.; Lee, K.R.; Park, T.G. Amine-functionalized gold nanoparticles as non-cytotoxic and efficient intracellular siRNA delivery carriers. Int. J. Pharm. 2008, 364, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xie, J.; Zhang, F.; Wang, Z.; Luo, K.; Zhu, L.; Quan, Q.; Niu, G.; Lee, S.; Ai, H. N-Alkyl-PEI-functionalized iron oxide nanoclusters for efficient siRNA delivery. Small 2011, 7, 2742–2749. [Google Scholar] [CrossRef] [PubMed]
- Medarova, Z.; Pham, W.; Farrar, C.; Petkova, V.; Moore, A. In vivo imaging of siRNA delivery and silencing in tumors. Nat. Med. 2007, 13, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Taratula, O.; Garbuzenko, O.; Savla, R.; Andrew Wang, Y.; He, H.; Minko, T. Multifunctional nanomedicine platform for cancer specific delivery of siRNA by superparamagnetic iron oxide nanoparticles-dendrimer complexes. Curr. Drug Deliv. 2011, 8, 59–69. [Google Scholar] [CrossRef]
- Yezhelyev, M.V.; Qi, L.; O′regan, R.M.; Nie, S.; Gao, X. Proton-sponge coated quantum dots for siRNA delivery and intracellular imaging. J. Am. Chem. Soc. 2008, 130, 9006–9012. [Google Scholar] [CrossRef] [PubMed]
- Mamo, T.; Moseman, E.A.; Kolishetti, N.; Salvador-Morales, C.; Shi, J.; Kuritzkes, D.R.; Langer, R.; Von Andrian, U.; Farokhzad, O.C. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine 2010, 5, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-F.; Wang, J. Delivery systems for siRNA drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Haasnoot, J.; Westerhout, E.M.; Berkhout, B. RNA interference against viruses: Strike and counterstrike. Nat. Biotechnol. 2007, 25, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.J.; June, C.H.; Kohn, D.B. Genetic therapies against HIV. Nat. Biotechnol. 2007, 25, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yun, K.-S.; Choi, C.S.; Shin, S.-H.; Ban, H.-S.; Rhim, T.; Lee, S.K.; Lee, K.Y. T cell-specific siRNA delivery using antibody-conjugated chitosan nanoparticles. Bioconj. Chem. 2012, 23, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Ramishetti, S.; Kedmi, R.; Goldsmith, M.; Leonard, F.; Sprague, A.G.; Godin, B.; Gozin, M.; Cullis, P.R.; Dykxhoorn, D.M.; Peer, D. Systemic gene silencing in primary T lymphocytes using targeted lipid nanoparticles. ACS Nano 2015, 9, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-S.; Peer, D.; Kumar, P.; Subramanya, S.; Wu, H.; Asthana, D.; Habiro, K.; Yang, Y.-G.; Manjunath, N.; Shimaoka, M. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol. Ther. 2010, 18, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Current progress in the development of RNAi-based therapeutics for HIV-1. Gene Ther. 2011, 18, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Neff, C.P.; Liu, X.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Aboellail, T.; Huang, Y.; Du, Q. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol. Ther. 2011, 19, 2228–2238. [Google Scholar] [CrossRef] [PubMed]

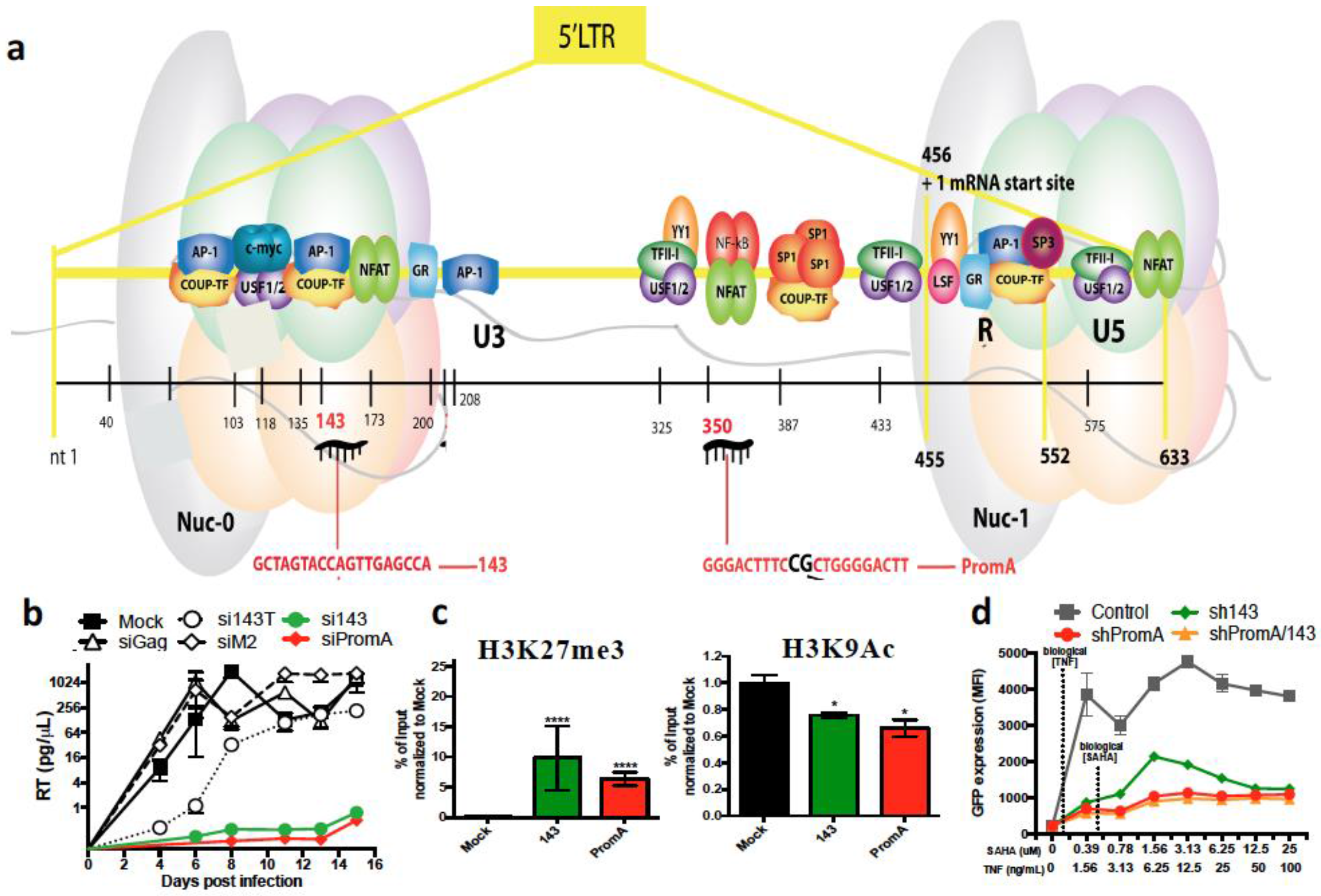
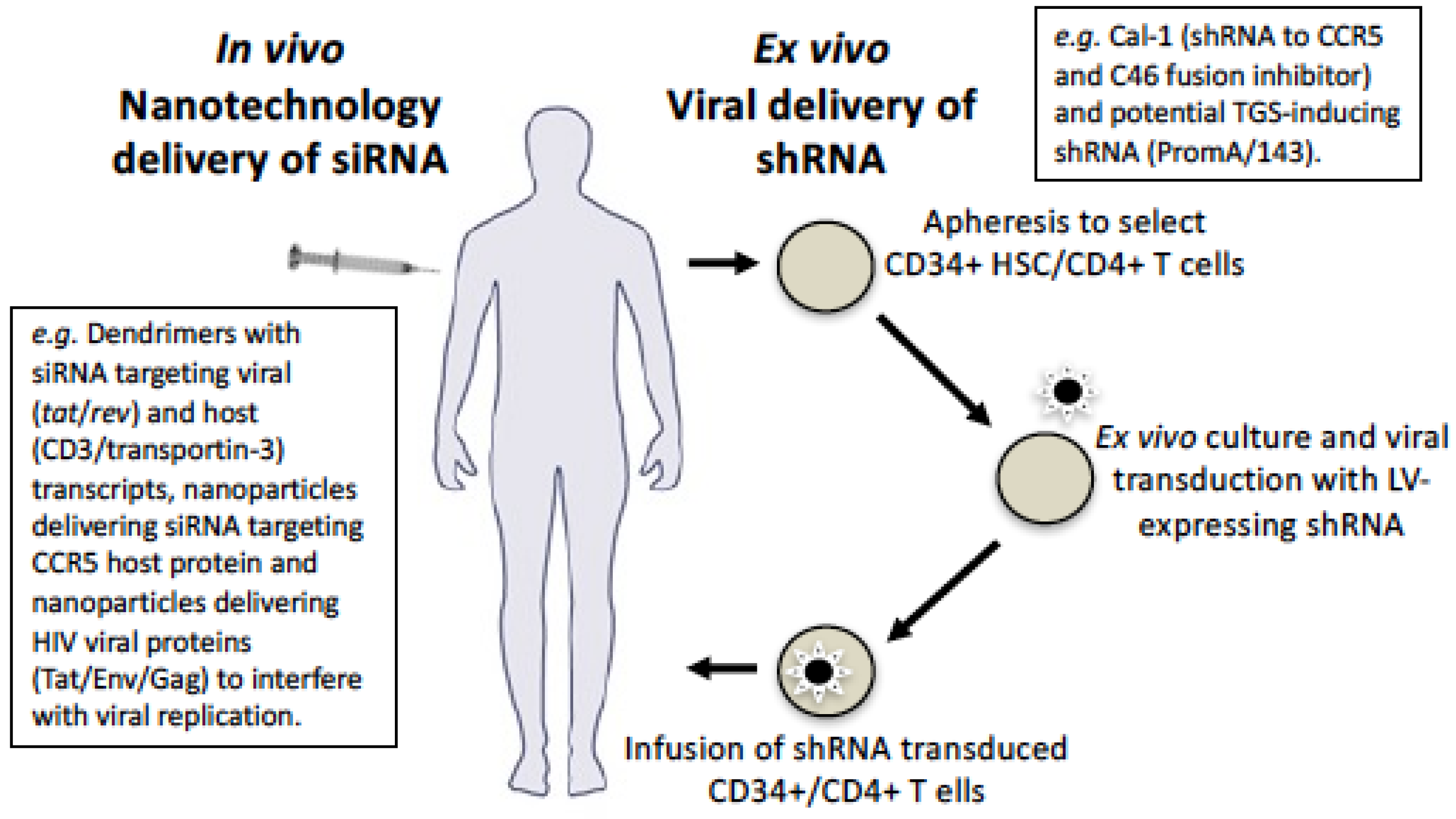

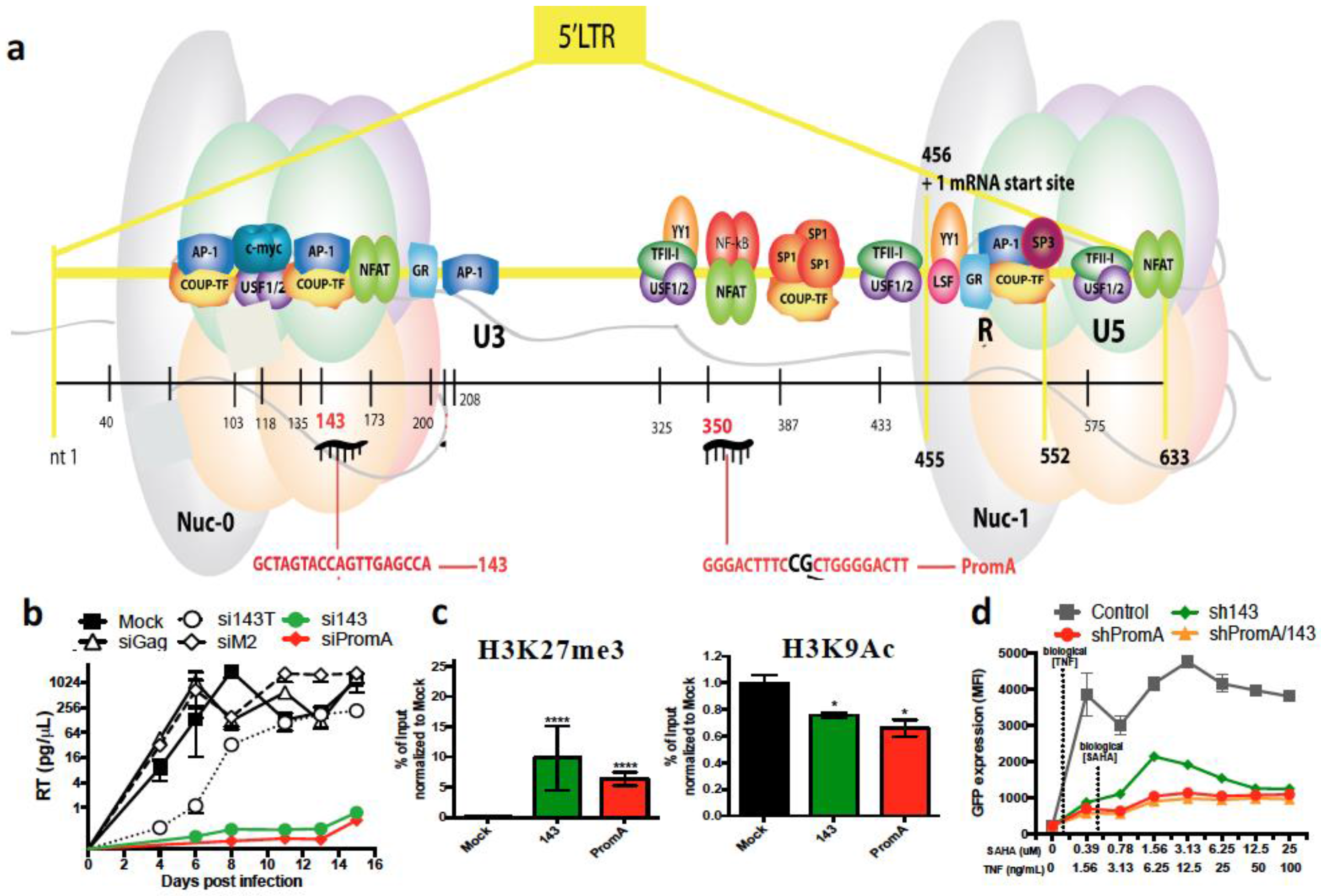
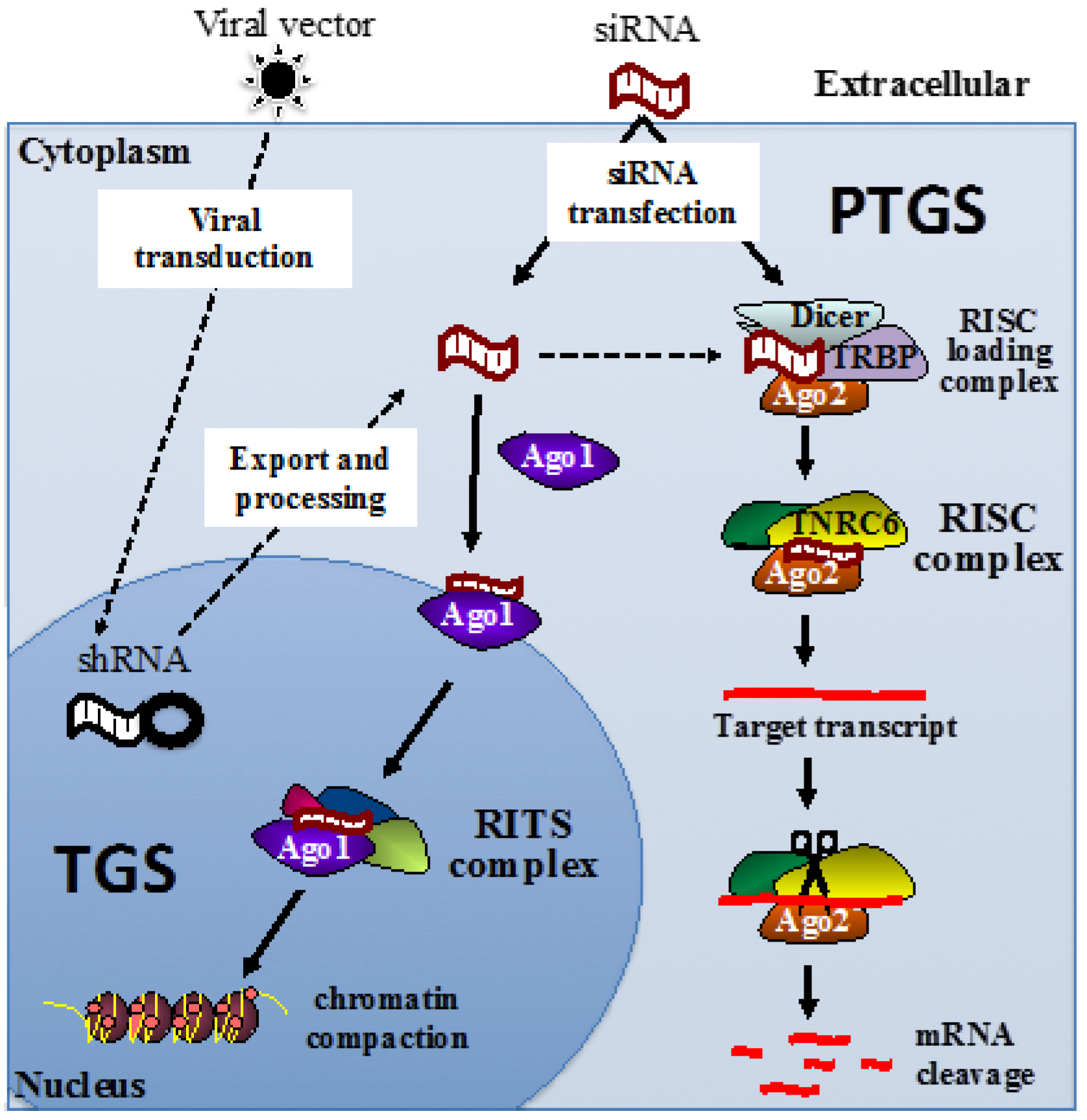{kind=link}
{kind=link}
{kind=link}
| Gene Therapy Trials | Intervention/s | Target/s | Identifier | Sponsor/Collaborator | Stage/Status |
|---|---|---|---|---|---|
| Dual Anti-HIV Gene Transfer Construct, LVsh5/C46 (Cal-1) | CCR5 shRNA C46 peptide Busulfan | Host co-receptor Viral Env | NCT01734850 | Calimmune, Inc. | Phase I/II Recruiting |
| Long Term Follow up of Delayed Adverse Events in Cal-1 Recipients | Blood tests for general health, complete blood count and Cal-1 specific analyses | NCT02390297 | Calimmune, Inc. | Recruiting by invitation | |
| Redirected MazF-CD4 Autologous T-Cells | CCR5 MazF | Host co-receptor | NCT01787994 | University of Pennsylvania | Phase I Ongoing |
| T-Cells Modified at CCR5 Gene by ZFN SB-728mR | CCR5 ZFN | CCR5 DNA | NCT02388594 | University of Pennsylvania/NIAID | Phase I Recruiting |
| SB-728mR-T After Cyclophosphamide Conditioning | CCR5 ZFN | CCR5 DNA | NCT02225665 | Sangamo Biosciences | Phase I/II Ongoing |
| Autologous T-Cells Modified at CCR5 Gene by ZFN SB-728 | CCR5 ZFN | CCR5 DNA | NCT00842634 | University of Pennsylvania/ Sangamo Biosciences | Phase I Completed |
| Redirected High Affinity Gag-Specific Autologous T Cells | WT-gag-TCR or α/6-gag-TCR | CD8 TCR | NCT00991224 | University of Pennsylvania/ Adaptimmune | Phase I Completed |
| This study was closed before any patient received T cells transduced with a high affinity A2-SL9-specific TCR [98] | |||||
| Autologous CD34+ HSCs Transduced With Anti-HIV-1 Ribozyme (OZ1) | Tat-vpr ribozyme | Tat-vpr mRNA | NCT00074997 | Janssen-Cilag Pty Ltd. | Phase II Completed |
| Long Term Follow-Up Study of OZ1 Gene Therapy | Blood tests for quantitative marking of the gene transfer product in PBMCs over time | NCT01177059 | Janssen-Cilag Pty Ltd. | Phase II Recruiting by invitation | |
| Tolerability and Therapeutic Effects of Repeated Doses of Autologous T Cells With VRX496 | VRX496 antisense RNA | Env mRNA | NCT00295477 | University of Pennsylvania/NIAID | Phase I/II Ongoing |
| Safety and Efficacy of T-Cell Genetic Immunotherapy | VRX496 antisense RNA | Env mRNA | NCT00131560 | VIRxSYS Corporation | Phase II Ongoing |
| Gene Therapy Trials | Intervention/s | Target/s | Identifier | Sponsor/Collaborator | Stage/Status |
|---|---|---|---|---|---|
| L-TR/Tat-neo in Patients With Non-Hodgkin’s Lymphoma | Tat ribozyme | Tat-rev mRNA | NCT00002221 | Ribozyome | Phase II Completed |
| M87o autologous HSCs for Patients with Malignant Diseases | C46 peptide | Viral Env | NCT00858793 | University Medical Center Hamburg-Eppendorf | Phase I/II Suspended |
| A leukaemia case was reported in patient treated with a similar vector. For safety risk recruitment was stopped. | |||||
| C46/CCR5/P140K modified autologous HSCs in patients with lymphoma | C46 peptide, CCR5 ribozyme, MGMTP140K mutant | Viral Env, CCR5 mRNA, Alkylating agent resistance | NCT02343666 | Fred Hutchinson Cancer Research Center/NCI/NHLBI | Phase I, Not yet recruiting |
| Autologous Transplantation of HSCs With LVsh5/C46 (Cal-1) for Treatment of HIV-Related Lymphoma | CCR5 shRNA, C46 peptide | Host co-receptor, Viral Env | NCT02378922 | Fred Hutchinson Cancer Research Center/NCI | Phase I Recruiting |
| rHIV7-shI-TAR-CCR5RZ-transduced HSC in patients with AIDS-related Non-Hodgkin Lymphoma | tat/rev shRNA, TAR decoy, CCR5 ribozyme, Busulfan | Viral mRNA, Viral tat proteinm, CCR5 mRNA, Transplant conditioning | NCT02337985 | City of Hope Medical Center/NCI | Pilot Recruiting |
| rHIV7-shI-TAR-CCR5RZ-transduced HSC in patients with AIDS-related non-Hodgkin’s lymphoma | tat/rev shRNA, TAR decoy, CCR5 ribozyme, Busulfan | Viral mRNA, Viral tat protein, CCR5 mRNA | NCT01961063 | City of Hope Medical Center | Pilot Recruiting |
| rHIV7-shI-TAR-CCR5RZ-transduced HSC in patients undergoing stem cell transplant for AIDS-related lymphoma | tat/rev shRNA, TAR decoy, CCR5 ribozyme, Busulfan | Viral mRNA, Viral tat protein, CCR5 mRNA, Transplant conditioning | NCT00569985 | City of Hope Medical Center/NCI | Pilot, Ongoing |
| shRNA/TRIM5alpha/TAR Decoy-transduced Autologous HSC in Patients With HIV-Related Lymphoma | CCR5 shRNA, RNF88, TAR decoy | Host co-receptor, Gag p24, Viral tat protein | NCT02797470 | AIDS Malignancy Consortium/NCI | Phase I/II |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klemm, V.; Mitchell, J.; Cortez-Jugo, C.; Cavalieri, F.; Symonds, G.; Caruso, F.; Kelleher, A.D.; Ahlenstiel, C. Achieving HIV-1 Control through RNA-Directed Gene Regulation. Genes 2016, 7, 119. https://doi.org/10.3390/genes7120119
Klemm V, Mitchell J, Cortez-Jugo C, Cavalieri F, Symonds G, Caruso F, Kelleher AD, Ahlenstiel C. Achieving HIV-1 Control through RNA-Directed Gene Regulation. Genes. 2016; 7(12):119. https://doi.org/10.3390/genes7120119
Chicago/Turabian StyleKlemm, Vera, Jye Mitchell, Christina Cortez-Jugo, Francesca Cavalieri, Geoff Symonds, Frank Caruso, Anthony Dominic Kelleher, and Chantelle Ahlenstiel. 2016. "Achieving HIV-1 Control through RNA-Directed Gene Regulation" Genes 7, no. 12: 119. https://doi.org/10.3390/genes7120119