De Novo Assembly and Analysis of Tartary Buckwheat (Fagopyrum tataricum Garetn.) Transcriptome Discloses Key Regulators Involved in Salt-Stress Response

Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Materials and Growth Conditions
2.2. Physiological Analysis
2.3. RNA Extraction and cDNA Library Preparation
2.4. Illumina Sequencing and Sequences Assembly
2.5. Coding Sequence Prediction, Functional Annotation and Classification of Unigenes
2.6. Identification of Transcription Factor Families
2.7. Expression Analysis
2.8. Identification of Simple Sequence Repeats (SSRs)
2.9. Reverse Transcription and Real-Time PCR
2.10. Statistical Analysis
3. Results
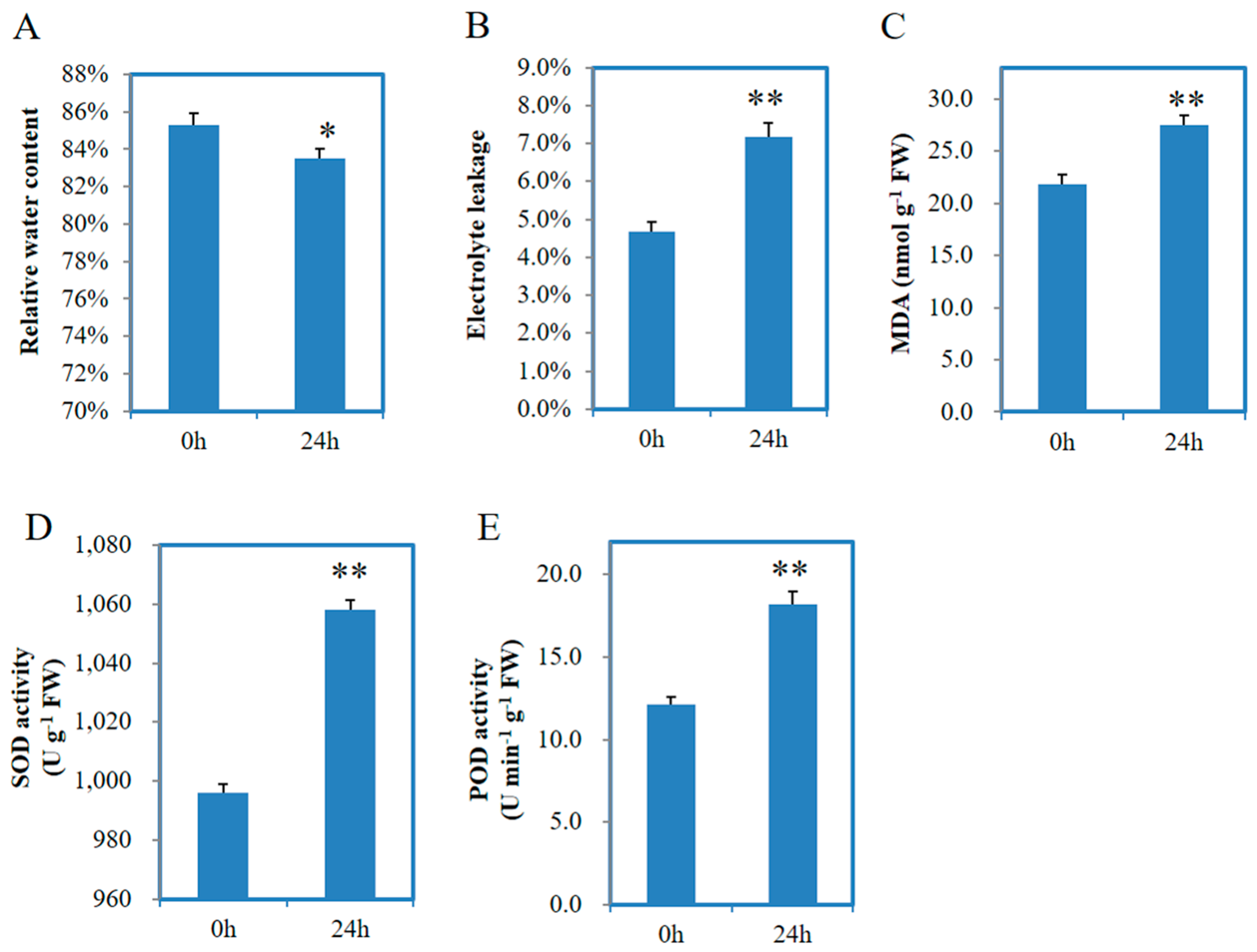
3.1. Physiological Analysis
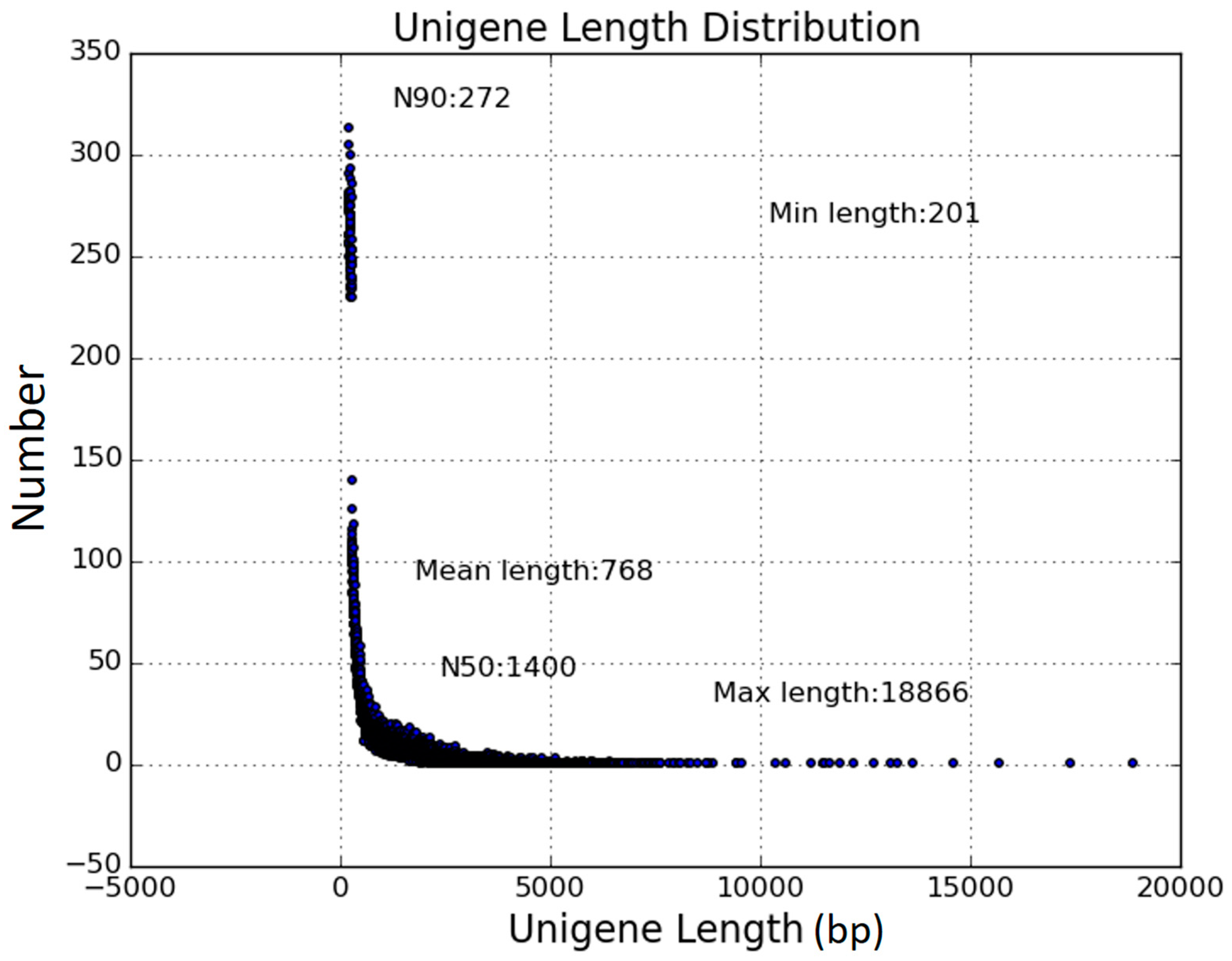
3.2. RNA-Seq and De Novo Assembly of the F. tataricum Transcriptome
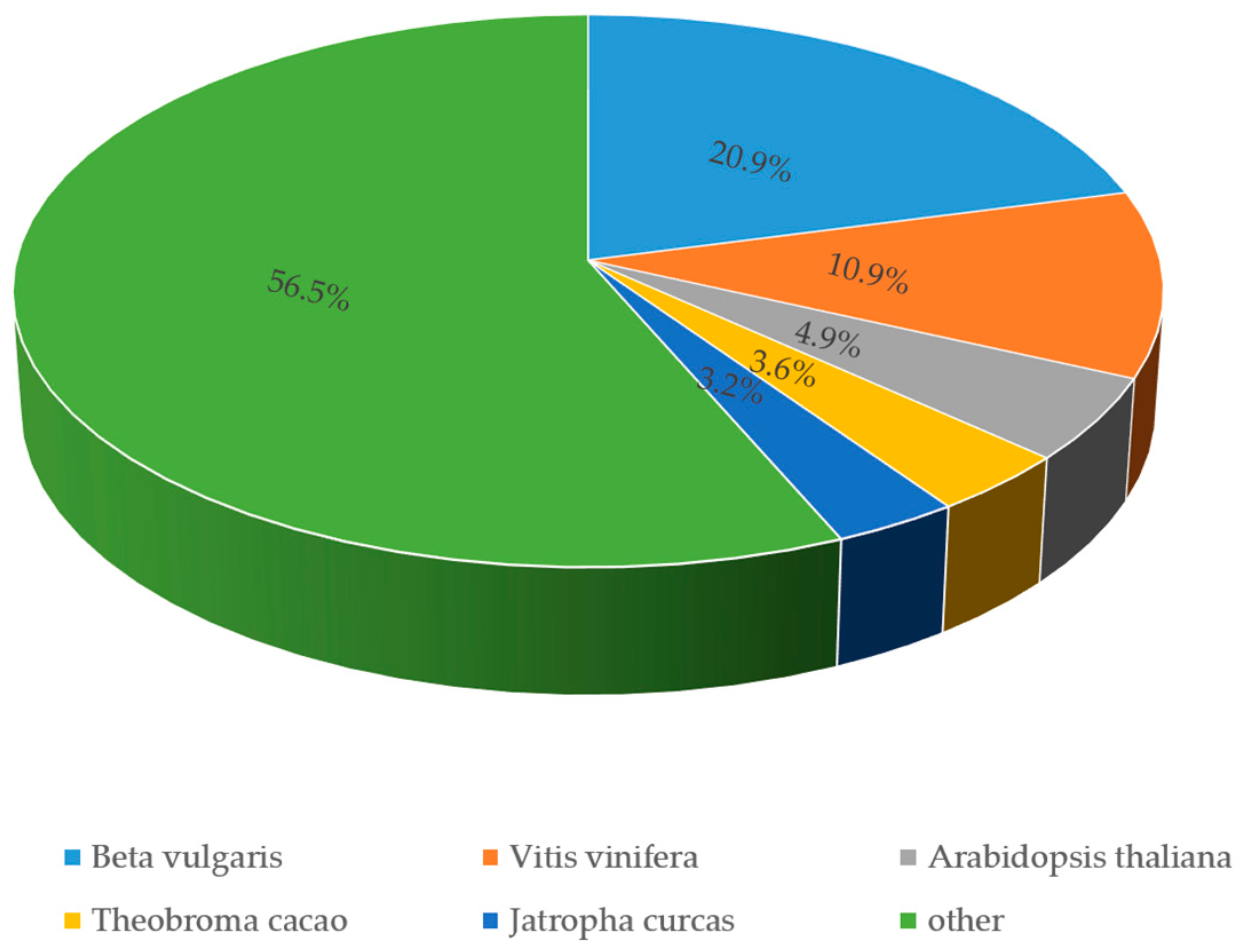
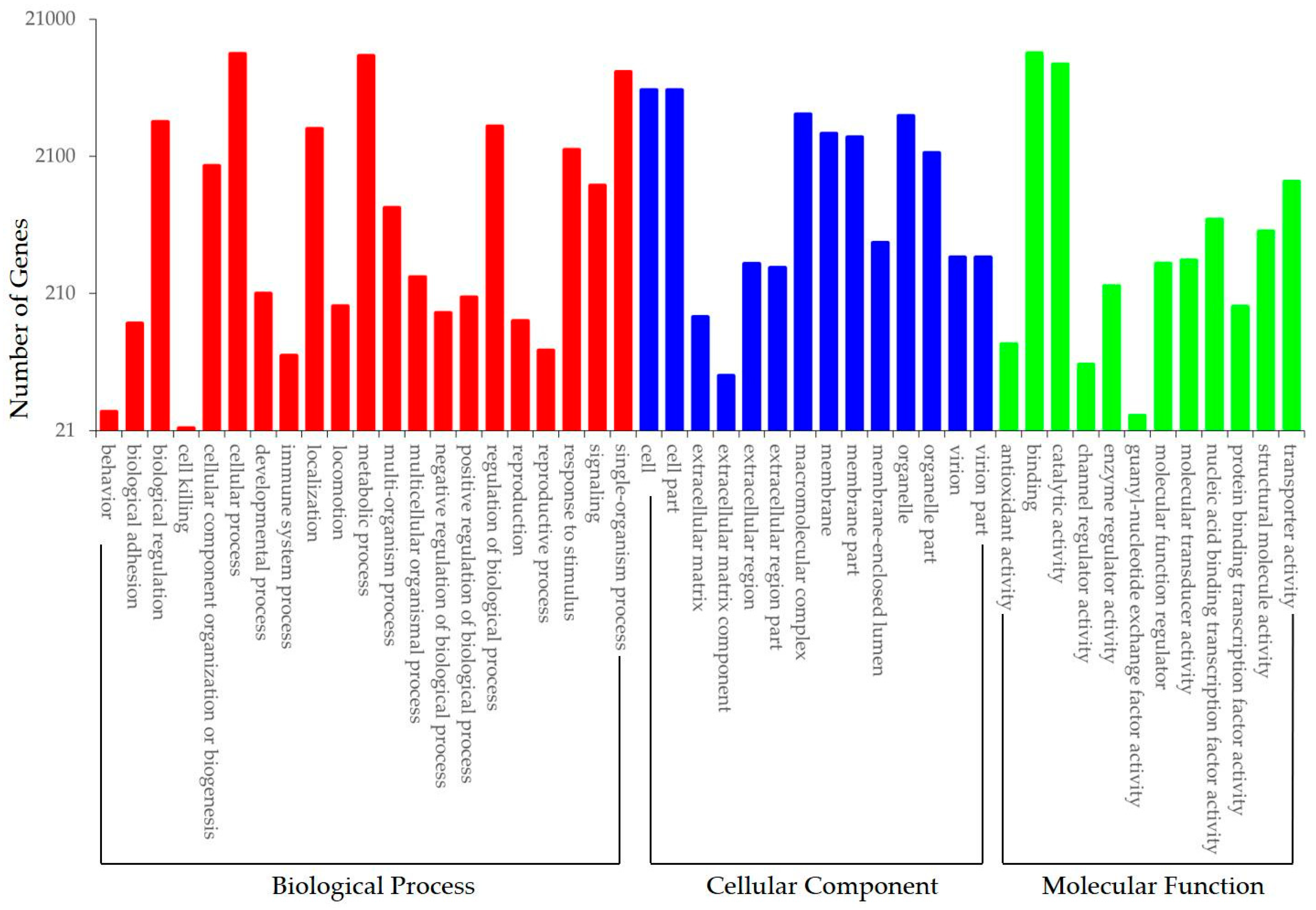
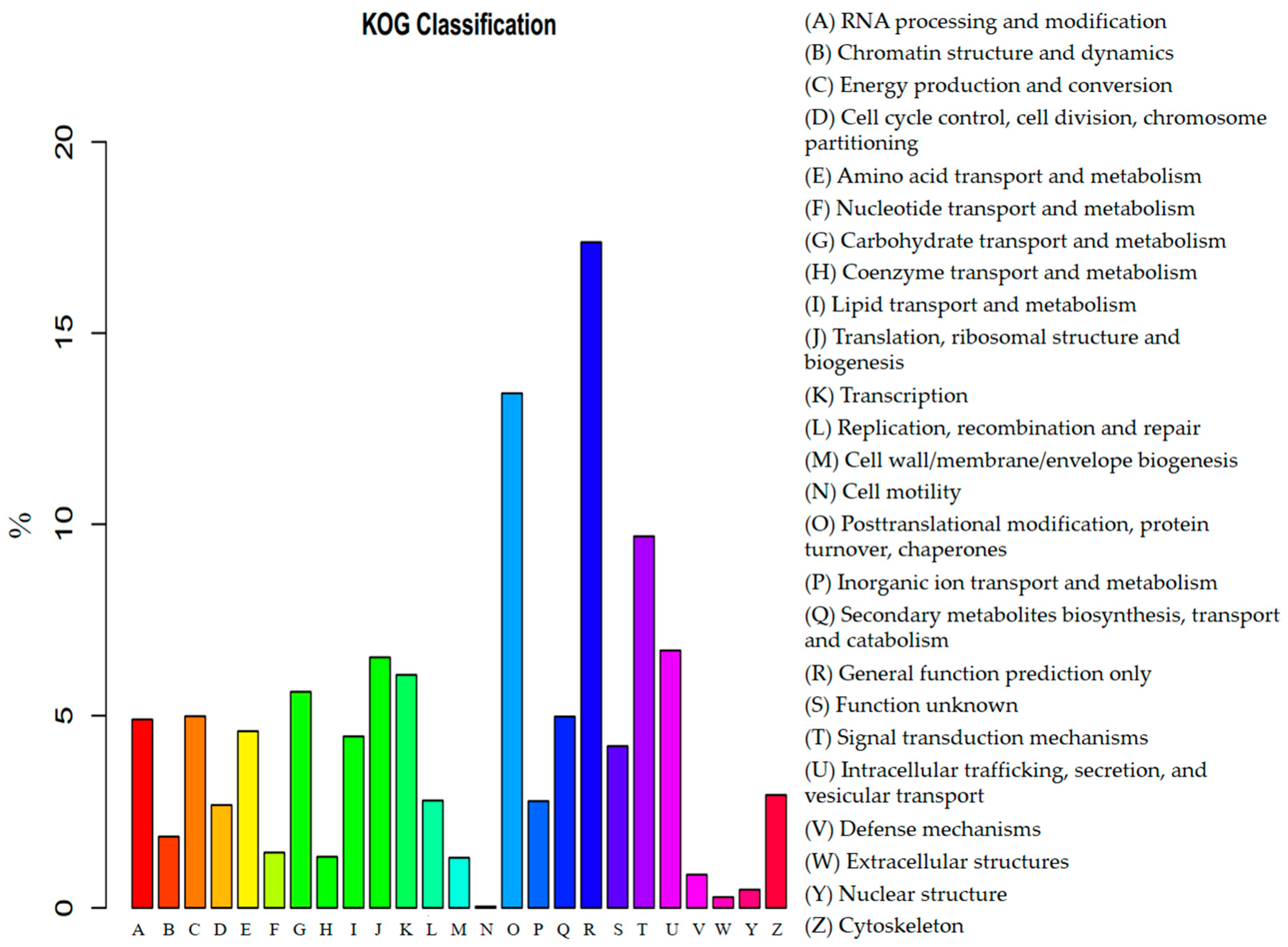
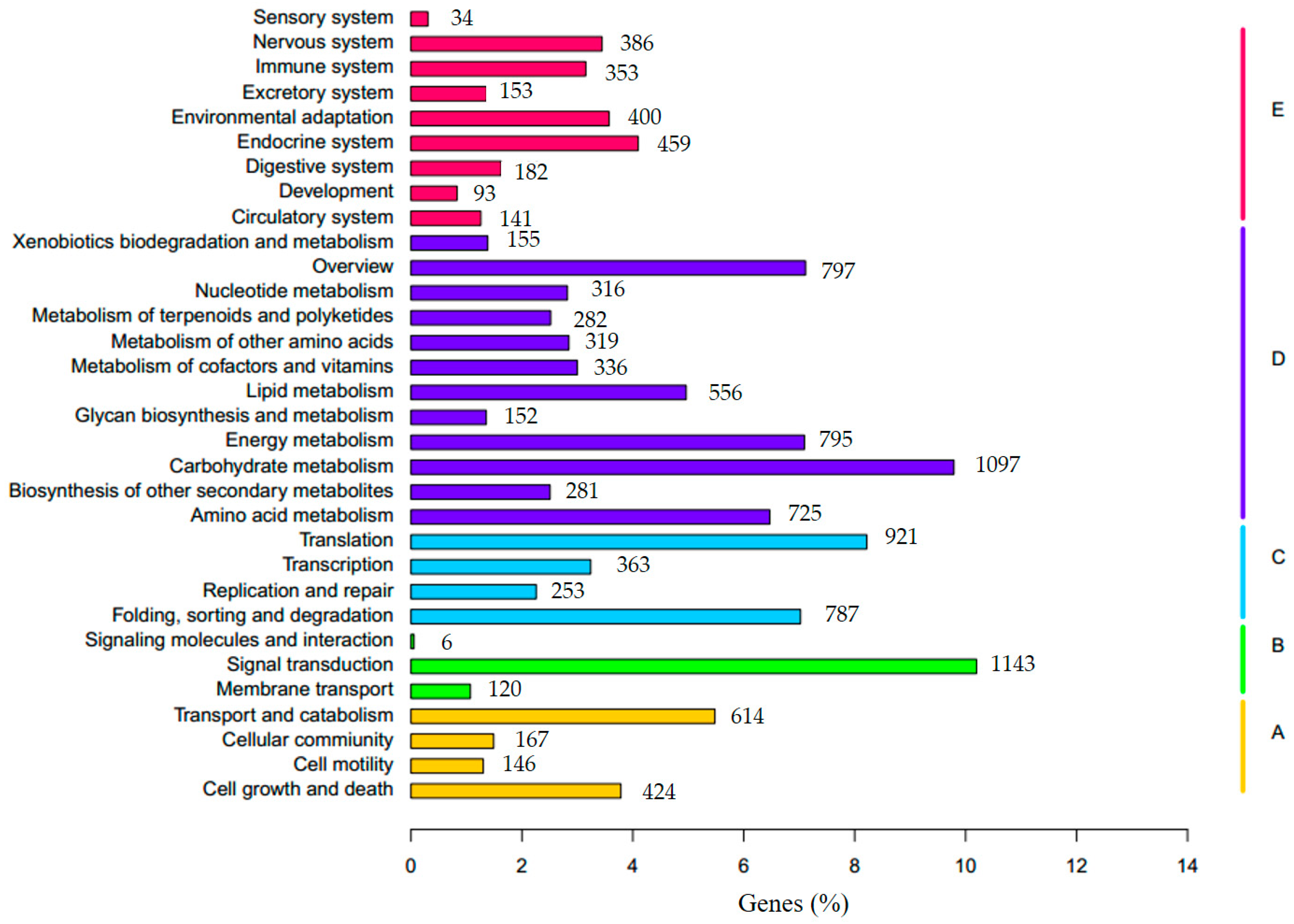
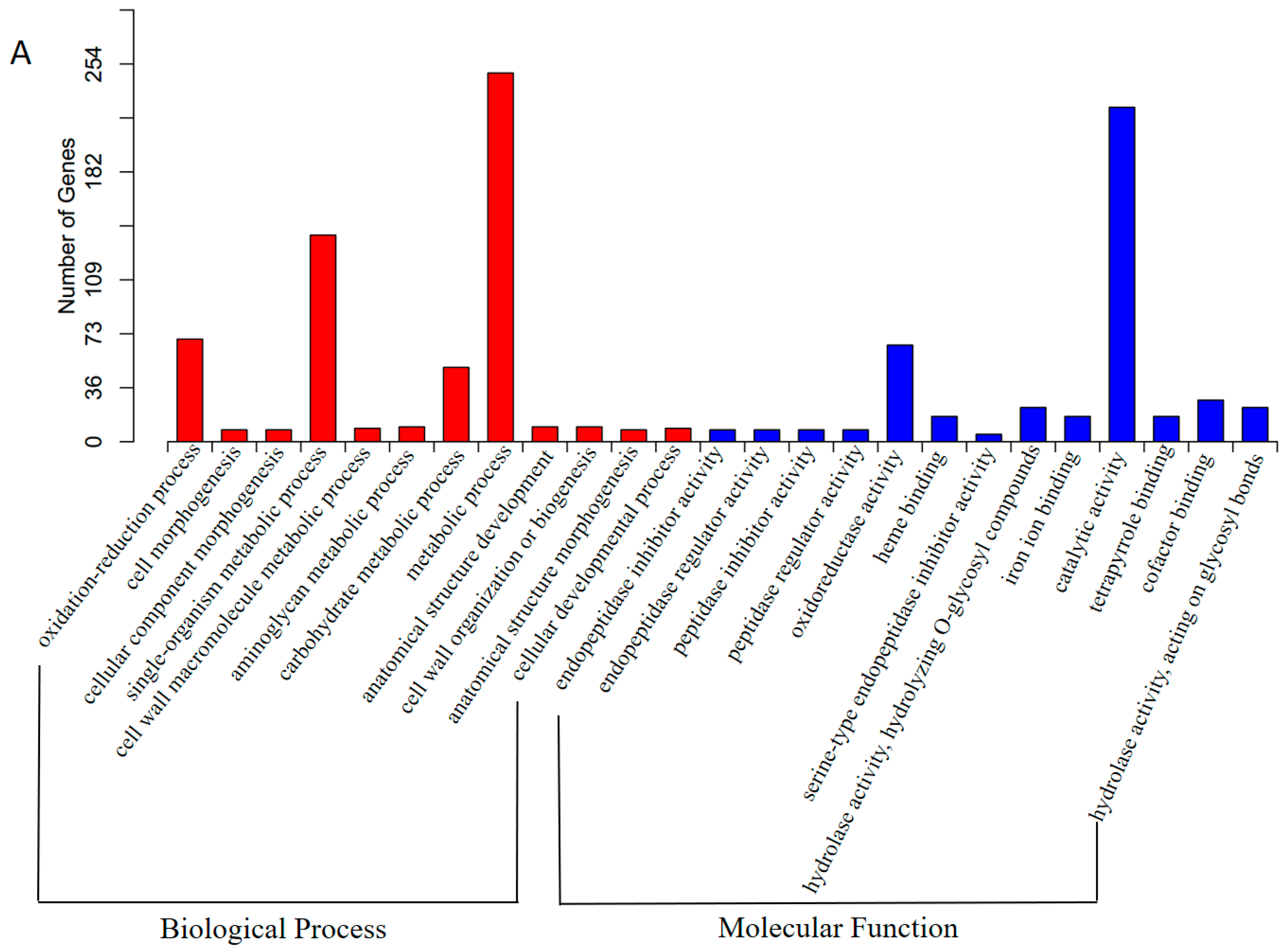
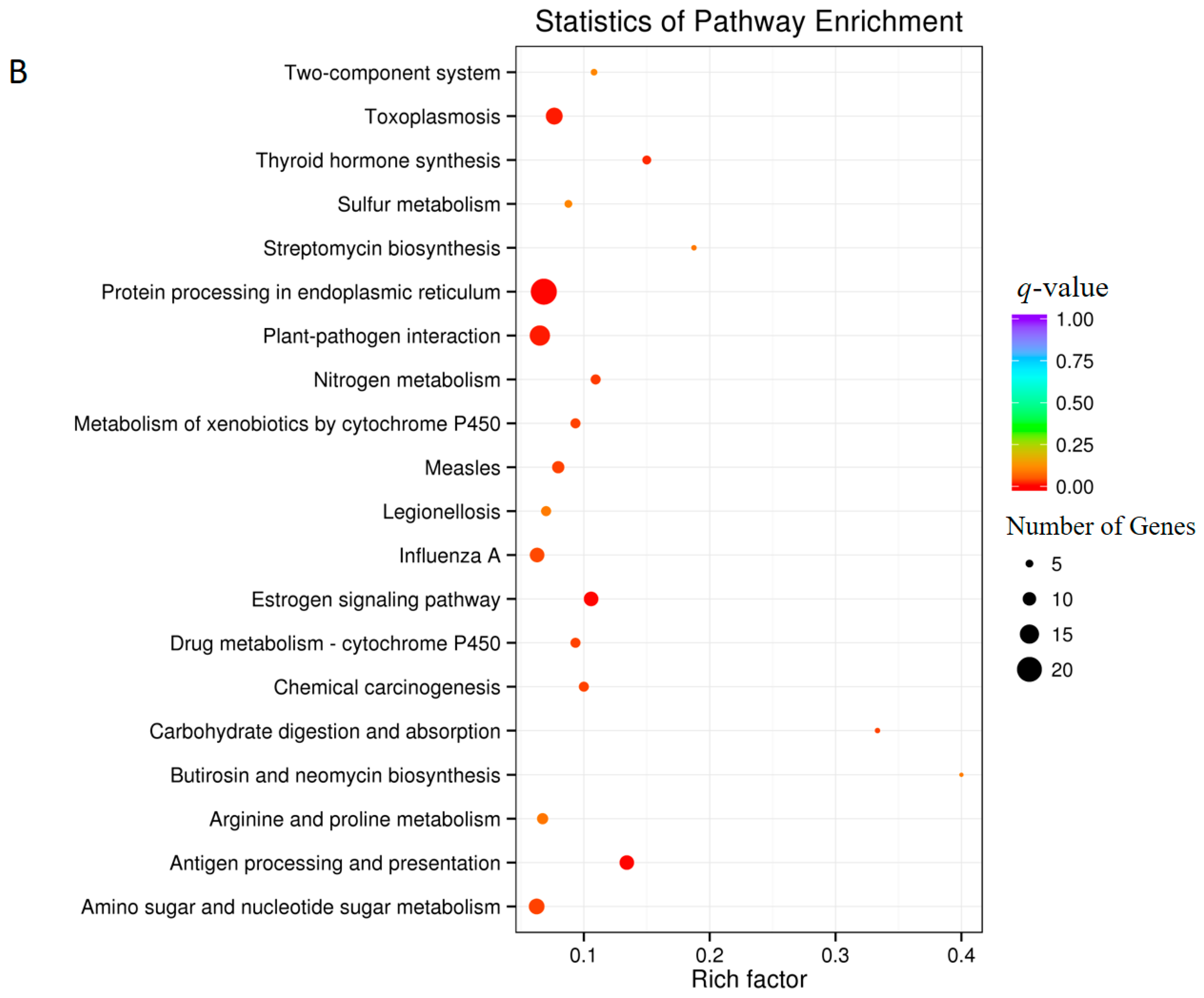
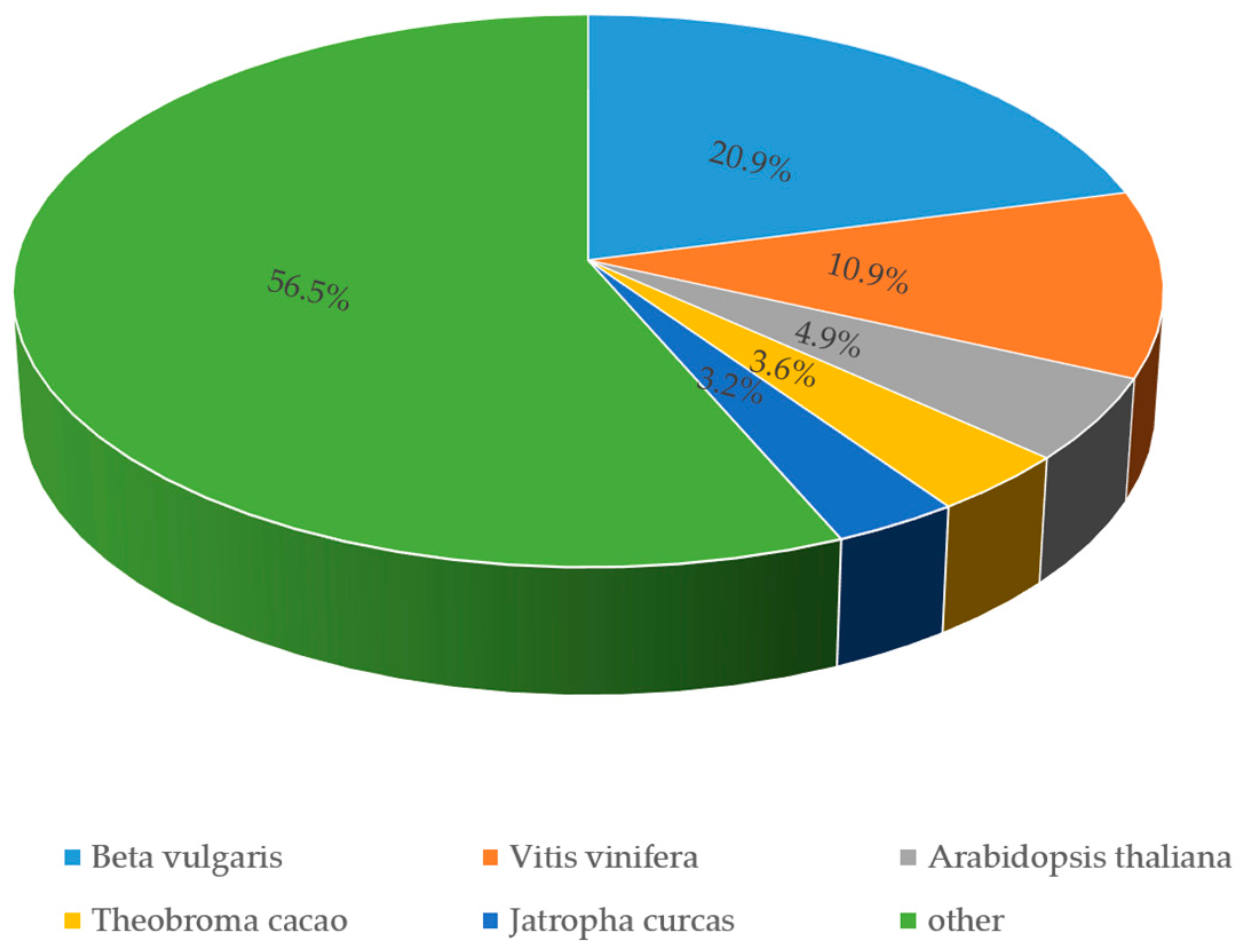
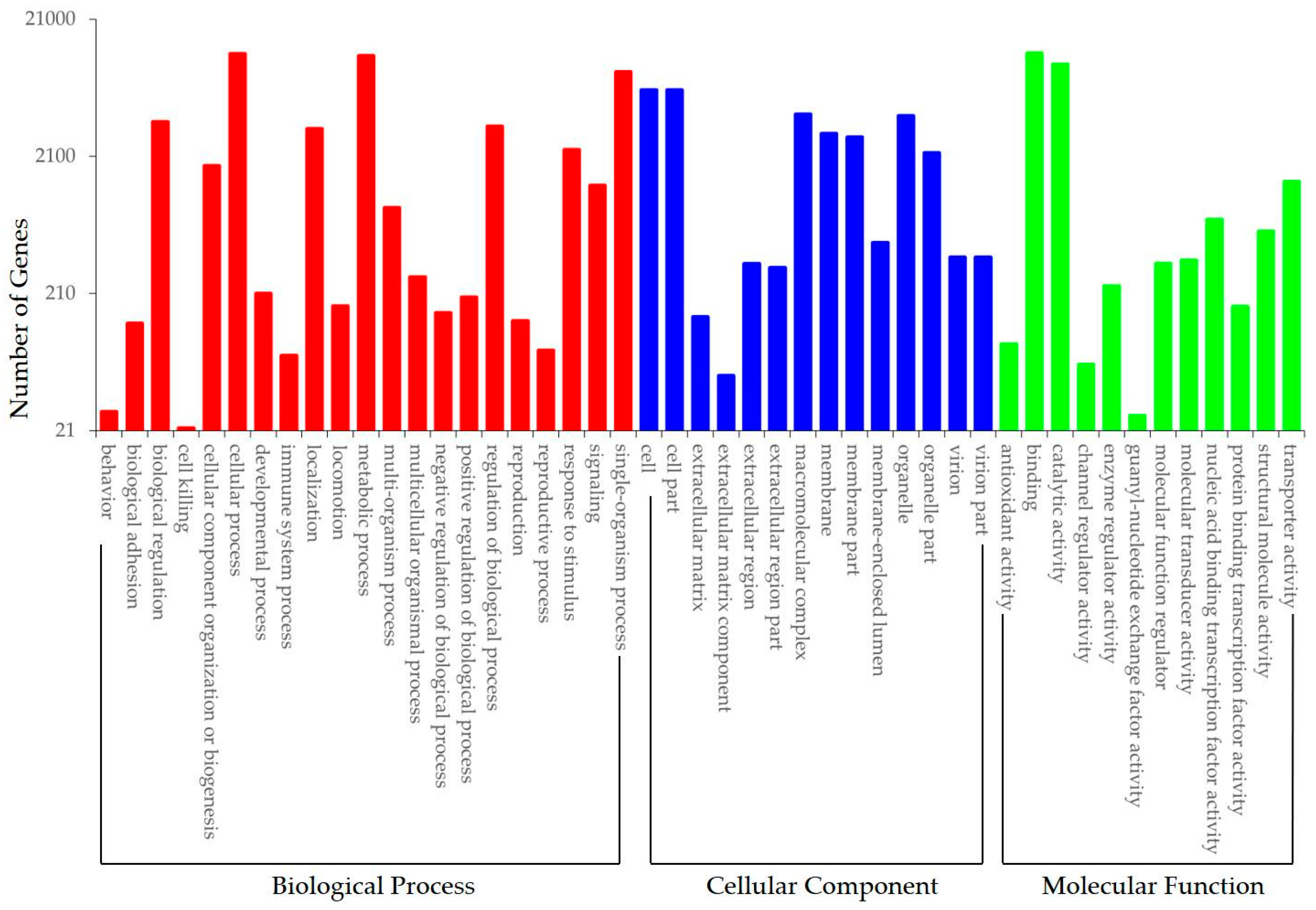
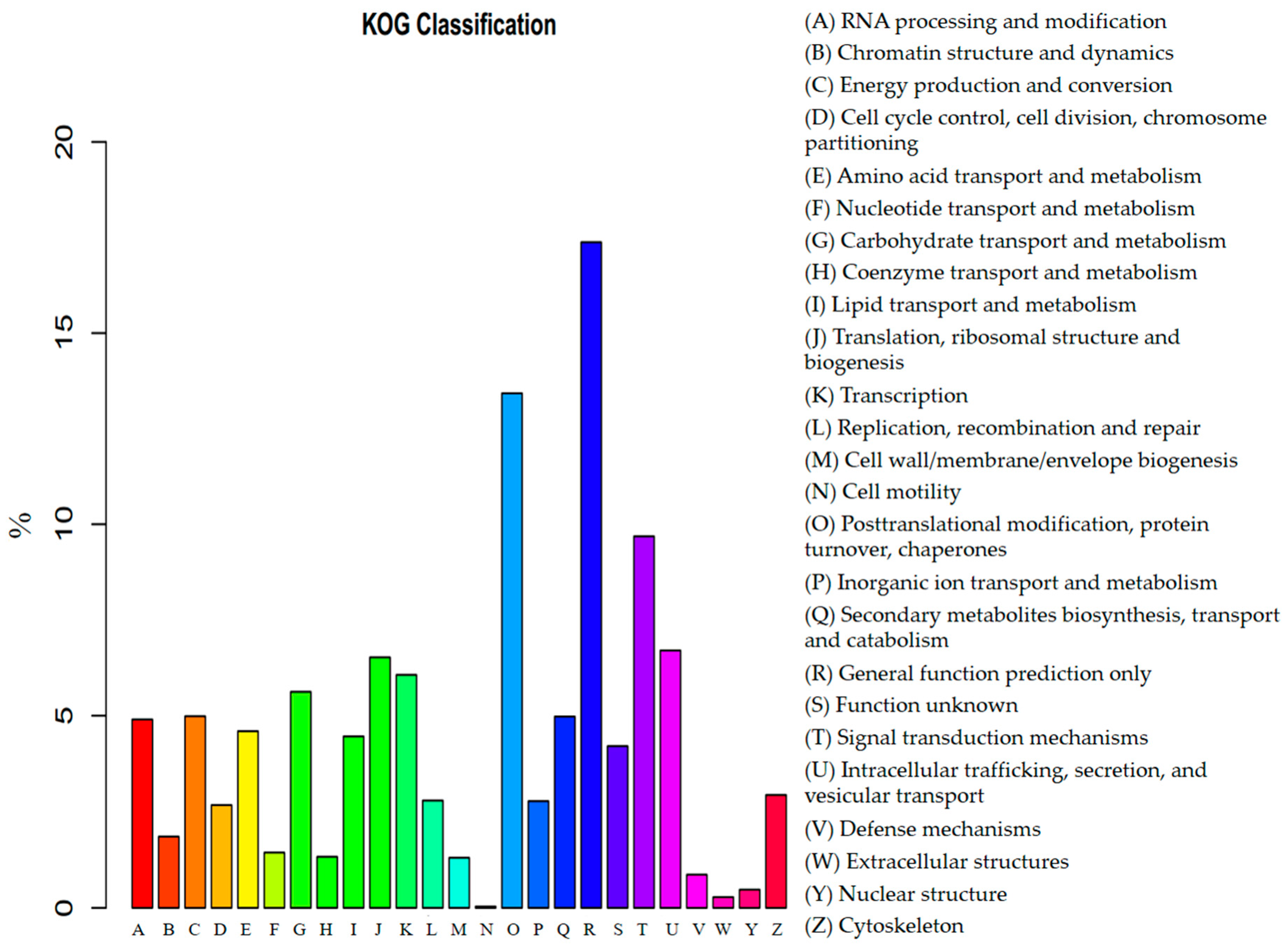
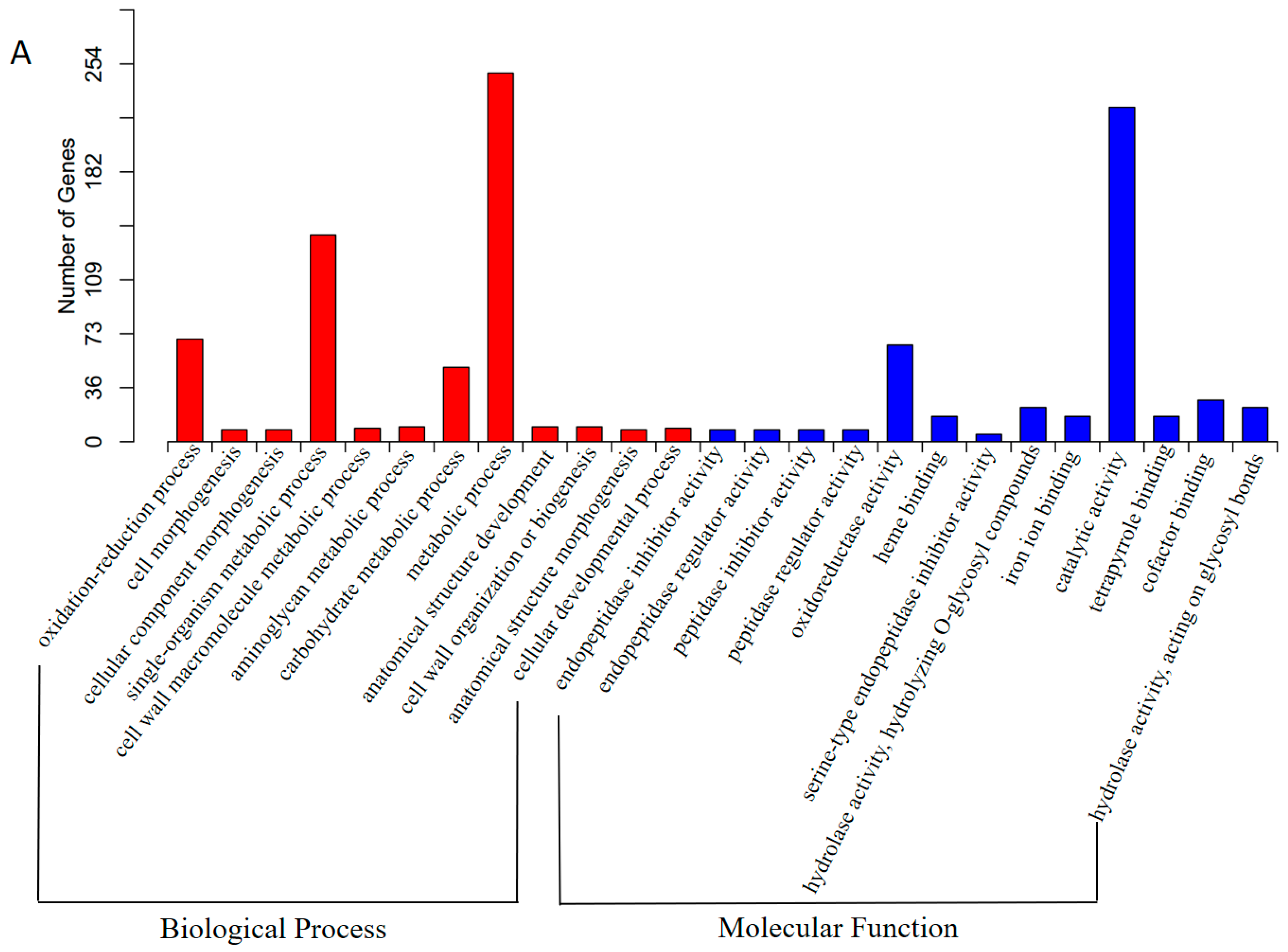
3.3. Functional Annotation and Classification of Transcriptome Sequences
3.4. Identification of Transcription Factor Families
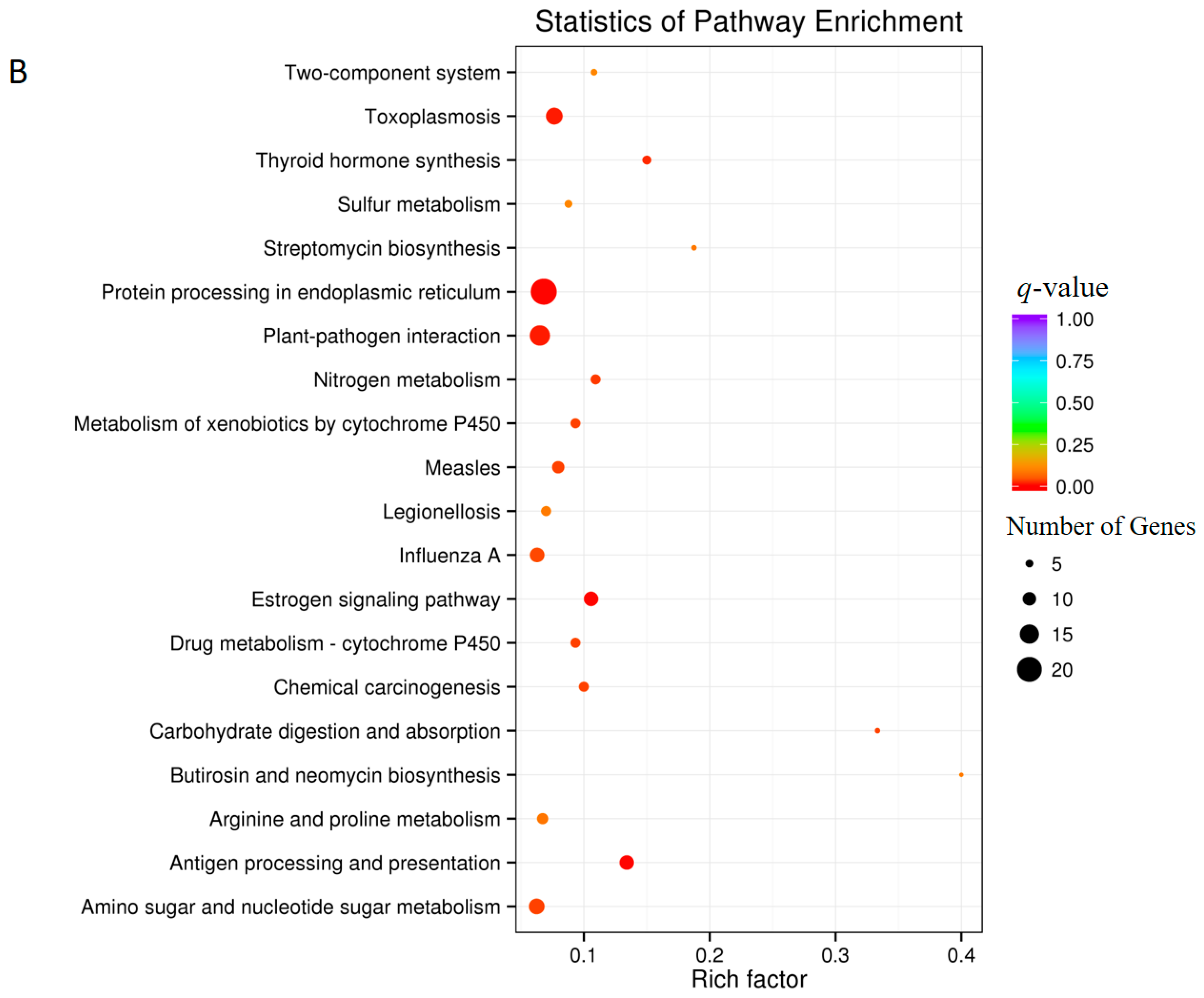
3.5. Analysis of Differentially Expressed Genes
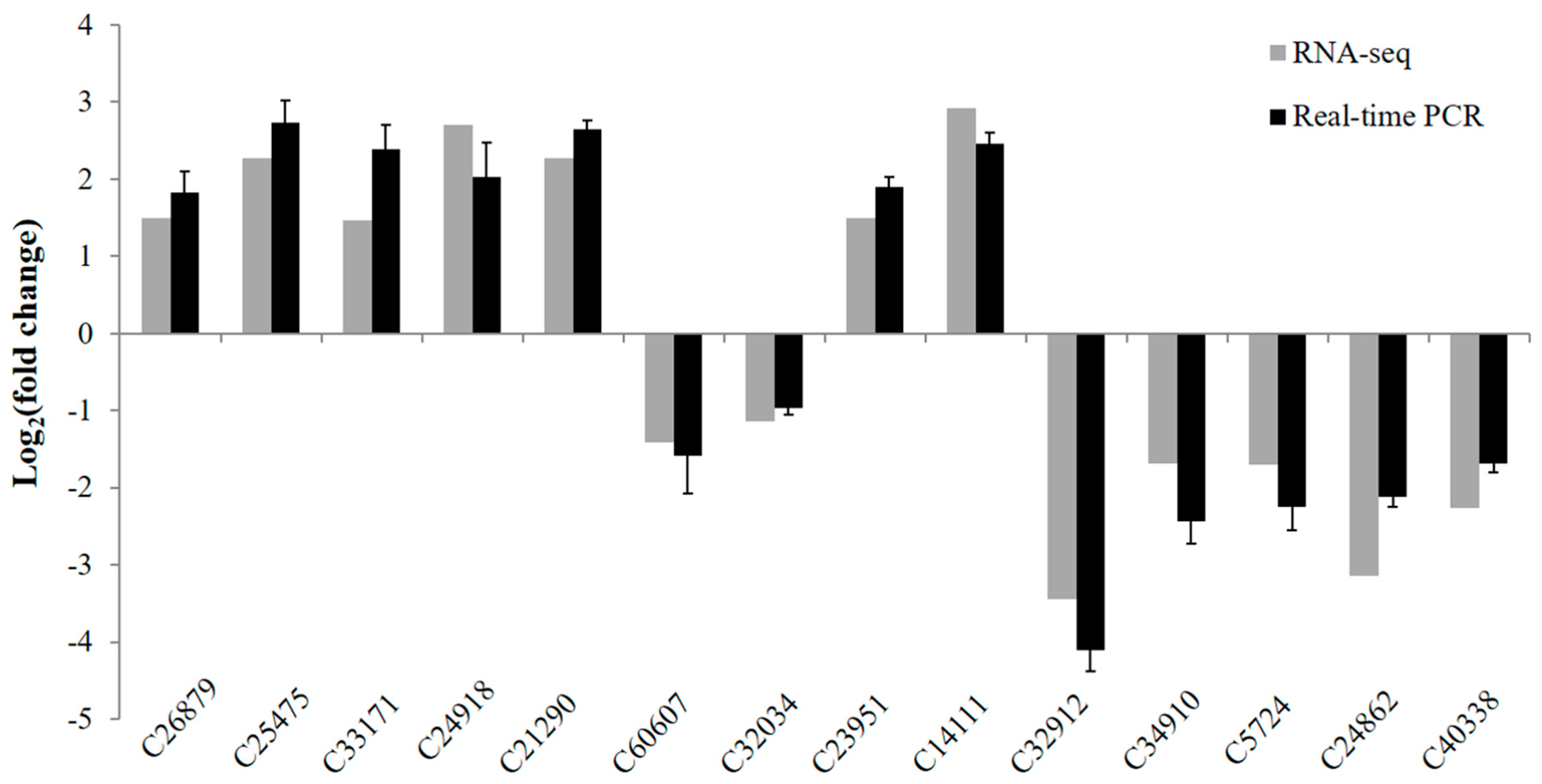
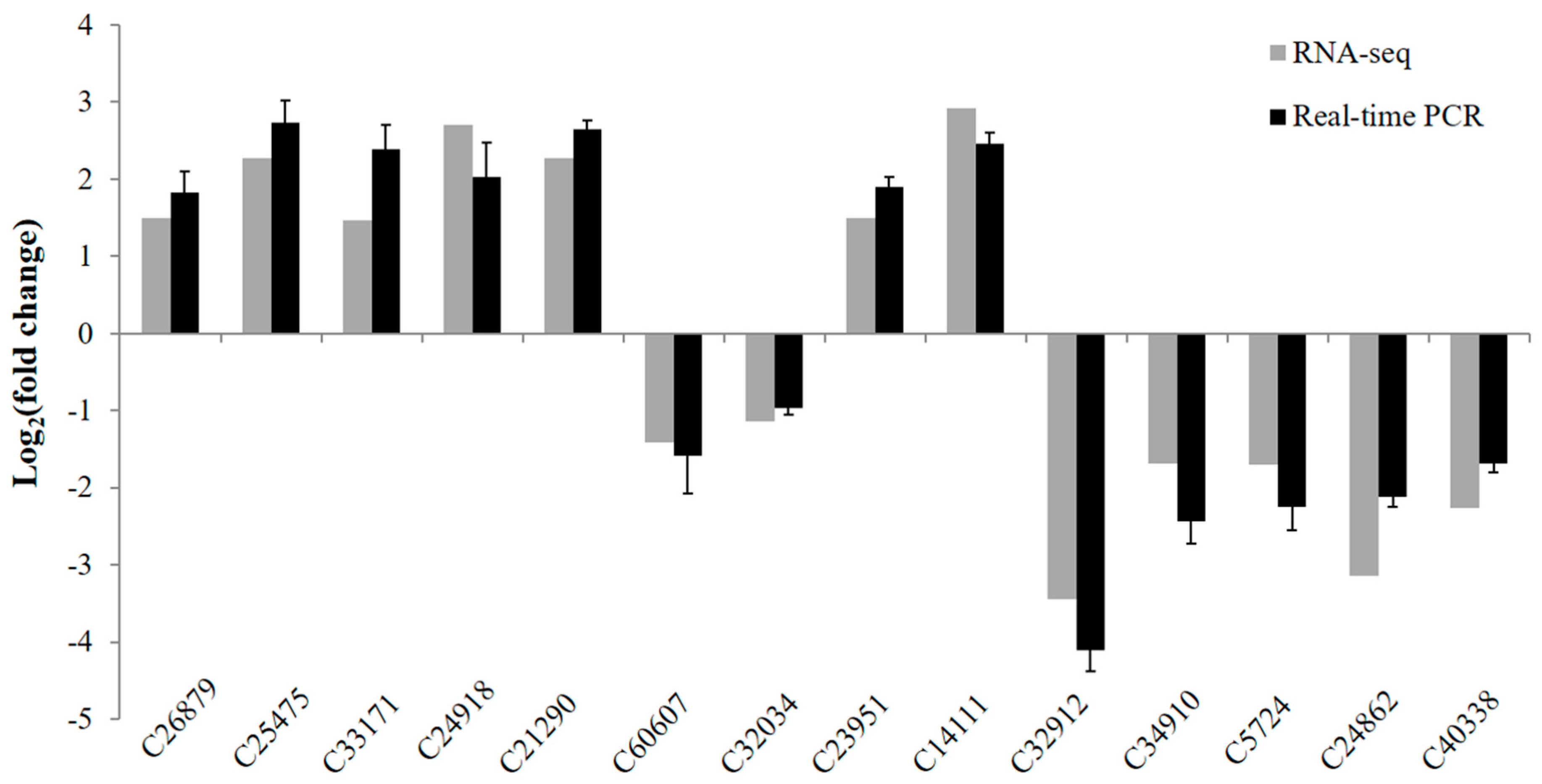
3.6. Validation of Differentially Expressed Genes by Real-Time PCR
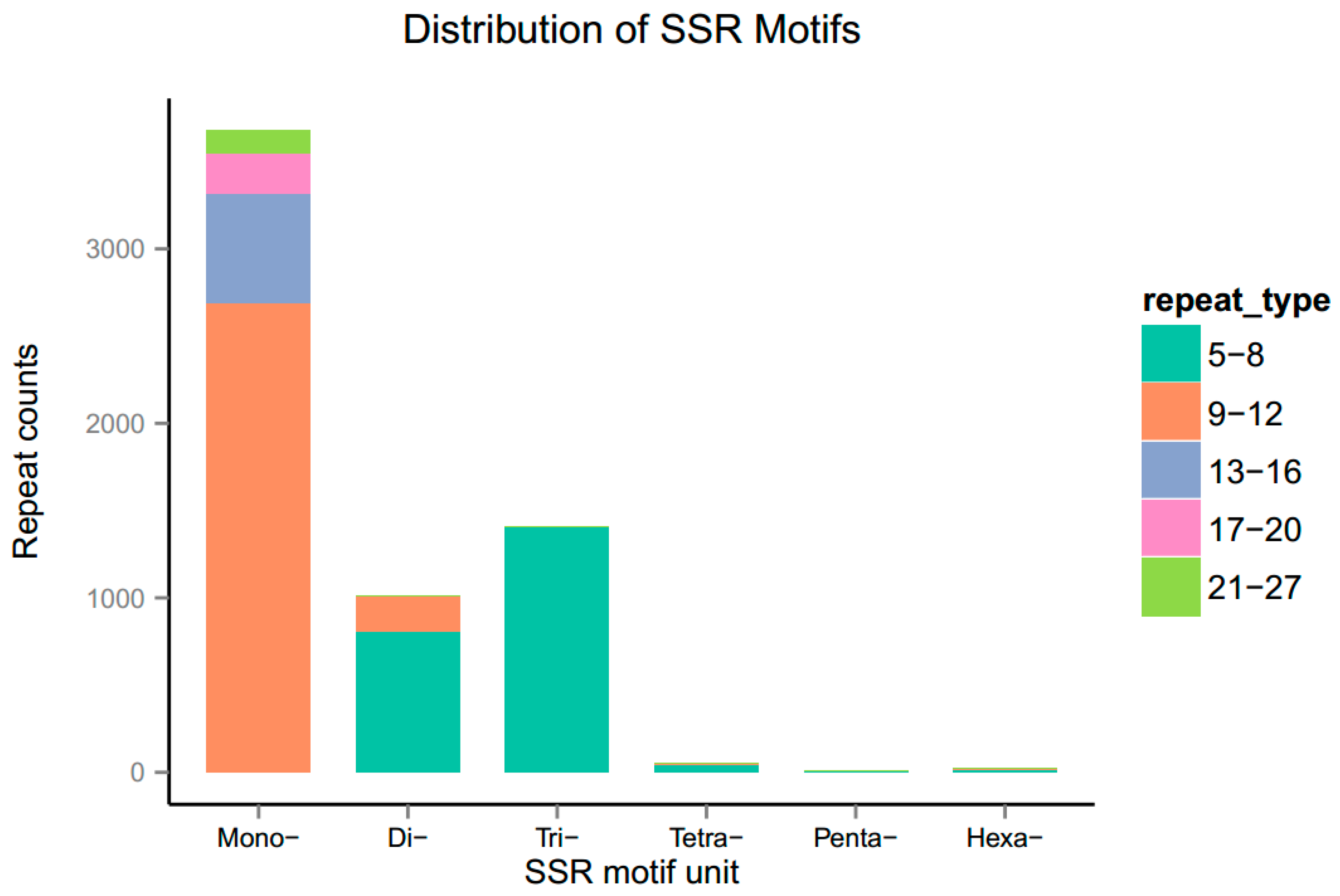
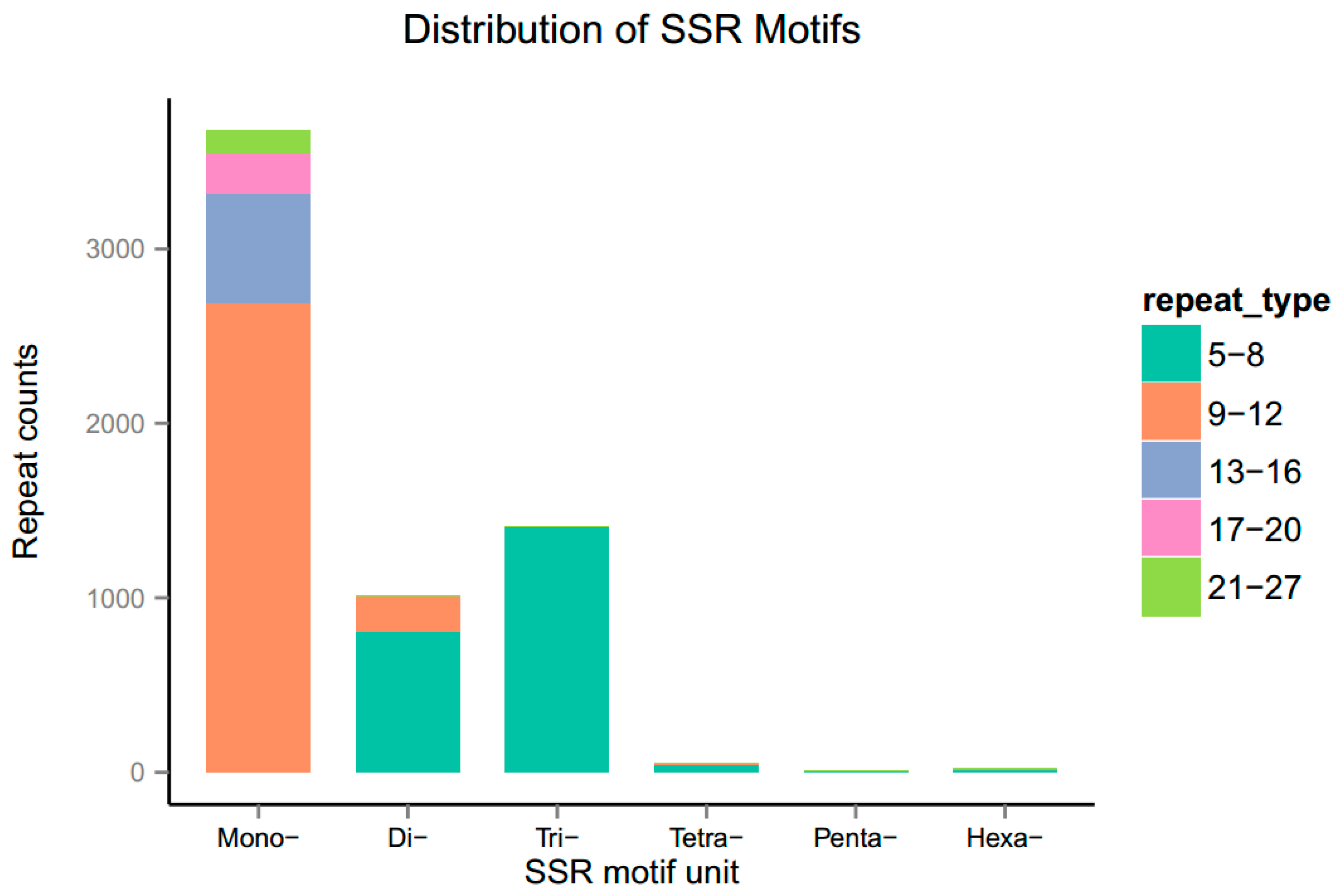
3.7. Identification of Simple Sequence Repeats
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, P.M.; Bressan, R.A.; Zhu, J.K.; Bohnert, H.J. Plant Cellular and Molecular Responses to High Salinity. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 463–499. [Google Scholar] [CrossRef] [PubMed]
- Weinl, S.; Kudla, J. The CBL-CIPK Ca2+-decoding signaling network: function and perspectives. New Phytol. 2009, 184, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Cabello, J.V.; Lodeyro, A.F.; Zurbriggen, M.D. Novel perspectives for the engineering of abiotic stress tolerance in plants. Curr. Opin. Biotechnol. 2014, 26, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed]
- Ciftci-Yilmaz, S.; Morsy, M.R.; Song, L.; Coutu, A.; Krizek, B.A.; Lewis, M.W.; Warren, D.; Cushman, J.; Connolly, E.L.; Mittler, R. The EAR-motif of the Cys2/His2-type zinc finger protein Zat7 plays a key role in the defense response of Arabidopsis to salinity stress. J. Biol. Chem. 2007, 282, 9260–9268. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xu, Y.; Ma, Q.; Xu, W.; Wang, T.; Xue, Y.; Chong, K. Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic Arabidopsis. Plant Physiol. 2007, 143, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Y.; Tian, A.G.; Zou, H.F.; Xie, Z.M.; Lei, G.; Huang, J.; Wang, C.M.; Wang, H.W.; Zhang, J.S.; Chen, S.Y. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol. J. 2008, 6, 486–503. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.J.; Wei, W.; Song, Q.X.; Chen, H.W.; Zhang, Y.Q.; Wang, F.; Zou, H.F.; Lei, G.; Tian, A.G.; Zhang, W.K.; et al. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant. J. 2011, 68, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.F.; Wei, W.; Zhou, Q.Y.; Tian, A.G.; Hao, Y.J.; Zhang, W.K.; Ma, B.; Lin, Q.; Zhang, Z.B.; Zhang, J.S.; Chen, S.Y. Wheat WRKY genes TaWRKY2 and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell. Environ. 2012, 35, 1156–1170. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Li, J.; Liu, P.; Duan, J.; Zhao, Y.; Guo, X.; Li, Y.; Zhang, H.; Ali, J.; Li, Z. Overexpression of OsMYB48–1, a novel MYB-related transcription factor, enhances drought and salinity tolerance in rice. PLoS ONE 2014, 9, e92913. [Google Scholar] [CrossRef] [PubMed]
- Bedre, R.; Mangu, V.R.; Srivastava, S.; Sanchez, L.E.; Baisakh, N. Transcriptome analysis of smooth cordgrass (Spartina alterniflora Loisel), a monocot halophyte, reveals candidate genes involved in its adaptation to salinity. BMC Genomics 2016, 17, 657. [Google Scholar] [CrossRef] [PubMed]
- Goyal, E.; Amit, S.K.; Singh, R.S.; Mahato, A.K.; Chand, S.; Kanika, K. Transcriptome profiling of the salt-stress response in Triticum aestivum cv. Kharchia Local. Sci. Rep. 2016, 6, 27752. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.B.; Cassin, A.; Keeble-Gagnere, G.; Doblin, M.S.; Bacic, A.; Roessner, U. De novo transcriptome assembly and analysis of differentially expressed genes of two barley genotypes reveal root-zone-specific responses to salt exposure. Sci. Rep. 2016, 6, 31558. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Dong, D.; Yang, Q.; Zhu, D. Salt-Responsive Transcriptome Profiling of Suaeda glauca via RNA Sequencing. PLoS ONE 2016, 11, e0150504. [Google Scholar] [CrossRef] [PubMed]
- Shankar, R.; Bhattacharjee, A.; Jain, M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016, 6, 23719. [Google Scholar] [CrossRef] [PubMed]
- Mallikarjuna, G.; Mallikarjuna, K.; Reddy, M.K.; Kaul, T. Expression of OsDREB2A transcription factor confers enhanced dehydration and salt stress tolerance in rice (Oryza sativa L.). Biotechnol. Lett. 2011, 33, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lu, G.; Hao, Y.; Guo, H.; Guo, Y.; Zhao, J.; Cheng, H. ABP9, a maize bZIP transcription factor, enhances tolerance to salt and drought in transgenic cotton. Planta 2017, 246, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhu, J.K. The broad roles of CBF genes: From development to abiotic stress. Plant Signal. Behav. 2016, 11, e1215794. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Ramanathan, V.; Nallathambi, J.; Duraialagaraja, S.; Muthurajan, R. Over-expression of a NAC 67 transcription factor from finger millet (Eleusine coracana L.) confers tolerance against salinity and drought stress in rice. BMC Biotechnol. 2016, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Shi, T.L.; Zhang, Z.W. Development of microsatellite markers from Tartary buckwheat. Biotechnol. Lett. 2007, 29, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kobayashi, K.; Yoshida, Y.; Ohsawa, R. Genetic analyses of agronomic traits in Tartary buckwheat (Fagopyrum tataricum (L.) Gaertn.). Breed. Sci. 2012, 62, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.H.; Zhang, B.; Xu, Z.Q. Salt tolerance conferred by overexpression of Arabidopsis vacuolar Na+/H+ antiporter gene AtNHX1 in common buckwheat (Fagopyrum esculentum). Transgenic Res. 2008, 17, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, X.; Ma, B.; Gao, Q.; Du, H.; Han, Y.; Li, Y.; Cao, Y.; Qi, M.; Zhu, Y.; et al. The Tartary Buckwheat Genome Provides Insights into Rutin Biosynthesis and Abiotic Stress Tolerance. Mol. Plant 2017, 10, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Baldwin, C.M.; Hu, Q.; Liu, H.; Luo, H. Heterologous expression of Arabidopsis H+-pyrophosphatase enhances salt tolerance in transgenic creeping bentgrass (Agrostis stolonifera L.). Plant Cell Environ. 2010, 33, 272–289. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, M.; Xiong, L.; Lee, H.; Stevenson, B.; Zhu, J.K. HOS1, a genetic locus involved in cold-responsive gene expression in Arabidopsis. Plant Cell 1998, 10, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Liang, Z.; Shao, M. Changes of anti-oxidative enzymes and MDA content under soil water deficits among 10 wheat (Triticum aestivum L.) genotypes at maturation stage. Colloids Surf. B. Biointerfaces 2005, 45, 7–13. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed]
- Kuimelis, R.G.; Livak, K.J.; Mullah, B.; Andrus, A. Structural analogues of TaqMan probes for real-time quantitative PCR. Nucleic Acids Symp. Ser. 1997, 37, 255–256. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 138–148. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2^(−ΔΔ CT) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sinharoy, S.; Udvardi, M.; Zhao, P.X. PlantTFcat: An online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics 2013, 14, 321. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2008, 36, D281–D288. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-Seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-Seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-Seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zhang, X.; Gao, J.; Wang, P.; Xu, X.; Liu, Z.; Shen, S.; Feng, B. A Buckwheat (Fagopyrum esculentum) DRE-Binding Transcription Factor Gene, FeDREB1, Enhances Freezing and Drought Tolerance of Transgenic Arabidopsis. Plant Mol. Biol. Report. 2015, 33, 1510–1525. [Google Scholar] [CrossRef]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, Z.; Peng, Z.; Yu, Y.; Liao, M.; Wei, S. Development of a high-density linkage map and mapping of the three-pistil gene (Pis1) in wheat using GBS markers. BMC Genom. 2017, 18, 567. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fan, C.; Li, Y.; Zhang, J.; Sun, J.; Chen, Y.; Tian, C.; Su, X.; Lu, M.; Liang, C.; et al. Effects of drought and salt-stresses on gene expression in Caragana korshinskii seedlings revealed by RNA-seq. BMC Genom. 2016, 17, 200. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.J.; Negrao, S.; Tester, M. Salt resistant crop plants. Curr. Opin. Biotechnol. 2014, 26, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, H.; Zhu, Y.; Zou, J.; Zhao, F.J.; Huang, C.F. Genome-wide transcriptomic and phylogenetic analyses reveal distinct aluminum-tolerance mechanisms in the aluminum-accumulating species buckwheat (Fagopyrum tataricum). BMC Plant Biol. 2015, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Logacheva, M.D.; Kasianov, A.S.; Vinogradov, D.V.; Samigullin, T.H.; Gelfand, M.S.; Makeev, V.J.; Penin, A.A. De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum). BMC Genom. 2011, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Taji, T.; Seki, M.; Satou, M.; Sakurai, T.; Kobayashi, M.; Ishiyama, K.; Narusaka, Y.; Narusaka, M.; Zhu, J.K.; Shinozaki, K. Comparative genomics in salt tolerance between Arabidopsis and Arabidopsis-related halophyte salt cress using Arabidopsis microarray. Plant Physiol. 2004, 135, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Li, P.; Ma, S.; Indu Rupassara, S.; Bohnert, H.J. Salinity stress adaptation competence in the extremophile Thellungiella halophila in comparison with its relative Arabidopsis thaliana. Plant J. 2005, 44, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Do Amaral, M.N.; Arge, L.W.; Benitez, L.C.; Danielowski, R.; Silveira, S.F.; Farias Dda, R.; de Oliveira, A.C.; da Maia, L.C.; Braga, E.J. Comparative transcriptomics of rice plants under cold, iron, and salt stresses. Funct. Integr. Genom. 2016, 16, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, H.; Pitzschke, A.; Hirt, H. Emerging MAP kinase pathways in plant stress signalling. Trends Plant Sci. 2005, 10, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K.; Jaggi, M.; Raghuram, B.; Tuteja, N. Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signal Behav. 2011, 6, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Kiegerl, S.; Cardinale, F.; Siligan, C.; Gross, A.; Baudouin, E.; Liwosz, A.; Eklof, S.; Till, S.; Bogre, L.; Hirt, H.; et al. SIMKK, a mitogen-activated protein kinase (MAPK) kinase, is a specific activator of the salt stress-induced MAPK, SIMK. Plant Cell 2000, 12, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Knight, H.; Trewavas, A.J.; Knight, M.R. Calcium signalling in Arabidopsis thaliana responding to drought and salinity. Plant J. 1997, 12, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Ju, H.W.; Min, J.H.; Zhang, X.; Kim, S.H.; Yang, K.Y.; Kim, C.S. Overexpression of L-type lectin-like protein kinase 1 confers pathogen resistance and regulates salinity response in Arabidopsis thaliana. Plant Sci. 2013, 203, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhan, C.; Huang, B. Heat shock proteins in association with heat tolerance in grasses. Int. J. Proteom. 2011, 2011, 529648. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, C.; Li, Z.; Hu, Q.; Han, L.; Luo, H. AsHSP17, a creeping bentgrass small heat shock protein modulates plant photosynthesis and ABA-dependent and independent signalling to attenuate plant response to abiotic stress. Plant Cell Environ. 2016, 39, 1320–1337. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Huebner, M.; Weber, A.P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genom. 2007, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Perlin, M.H.; Andrews, J.; Toh, S.S. Essential letters in the fungal alphabet: ABC and MFS transporters and their roles in survival and pathogenicity. Adv. Genet. 2014, 85, 201–253. [Google Scholar] [PubMed]
- Kim, D.Y.; Jin, J.Y.; Alejandro, S.; Martinoia, E.; Lee, Y. Overexpression of AtABCG36 improves drought and salt stress resistance in Arabidopsis. Physiol. Plant 2010, 139, 170–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.; Lam, H.M. A putative λ class glutathione S-transferase enhances plant survival under salinity stress. Plant Cell Physiol. 2014, 55, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Le Martret, B.; Poage, M.; Shiel, K.; Nugent, G.D.; Dix, P.J. Tobacco chloroplast transformants expressing genes encoding dehydroascorbate reductase, glutathione reductase, and glutathione-S-transferase, exhibit altered anti-oxidant metabolism and improved abiotic stress tolerance. Plant Biotechnol. J. 2011, 9, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, Y.; Rao, J.; Wang, G.; Li, H.; Ge, F.; Chen, C. Overexpression of the glutathione S-transferase gene from Pyrus pyrifolia fruit improves tolerance to abiotic stress in transgenic tobacco plants. Mol. Biol. 2013, 47, 591–601. [Google Scholar] [CrossRef]
- Gou, X.; Yin, H.; He, K.; Du, J.; Yi, J.; Xu, S.; Lin, H.; Clouse, S.D.; Li, J. Genetic evidence for an indispensable role of somatic embryogenesis receptor kinases in brassinosteroid signaling. PLoS Genet. 2012, 8, e1002452. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.Y.; Zhang, L.Y.; Gampala, S.S.; Zhu, S.W.; Song, W.Y.; Chong, K.; Wang, Z.Y. Functions of OsBZR1 and 14–3-3 proteins in brassinosteroid signaling in rice. Proc. Natl. Acad. Sci. USA 2007, 104, 13839–13844. [Google Scholar] [CrossRef] [PubMed]
- Deinlein, U.; Stephan, A.B.; Horie, T.; Luo, W.; Xu, G.; Schroeder, J.I. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Ye, R.; Gao, L.; Zhang, L.; Wang, R.; Mao, T.; Zheng, Y.; Li, D.; Lin, Y. Characterization and Ectopic Expression of CoWRI1, an AP2/EREBP Domain-Containing Transcription Factor from Coconut (Cocos nucifera L.) Endosperm, Changes the Seeds Oil Content in Transgenic Arabidopsis thaliana and Rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Kim, Y.S.; Baek, K.H.; Jung, H.; Ha, S.H.; Do Choi, Y.; Kim, M.; Reuzeau, C.; Kim, J.K. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Kim, Y.; Song, L.; Coutu, J.; Coutu, A.; Ciftci-Yilmaz, S.; Lee, H.; Stevenson, B.; Zhu, J.K. Gain- and loss-of-function mutations in Zat10 enhance the tolerance of plants to abiotic stress. FEBS Lett. 2006, 580, 6537–6542. [Google Scholar] [CrossRef] [PubMed]
- Nozue, K.; Covington, M.F.; Duek, P.D.; Lorrain, S.; Fankhauser, C.; Harmer, S.L.; Maloof, J.N. Rhythmic growth explained by coincidence between internal and external cues. Nature 2007, 448, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Harmer, S.L. The circadian system in higher plants. Annu. Rev. Plant Biol. 2009, 60, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Gehan, M.A.; Greenham, K.; Mockler, T.C.; McClung, C.R. Transcriptional networks-crops, clocks, and abiotic stress. Curr. Opin. Plant Bio.l 2015, 24, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Ali, Z.; Park, H.J.; Park, S.J.; Cha, J.Y.; Perez-Hormaeche, J.; Quintero, F.J.; Shin, G.; Kim, M.R.; Qiang, Z.; et al. Release of SOS2 kinase from sequestration with GIGANTEA determines salt tolerance in Arabidopsis. Nat. Commun. 2013, 4, 1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yue, W.; Wang, M.; Qiu, W.; Zhou, L.; Shou, H. Mutation of OsGIGANTEA Leads to Enhanced Tolerance to Polyethylene Glycol-Generated Osmotic Stress in Rice. Front. Plant Sci. 2016, 7, 465. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | CQ_0h | CQ_Na24h |
| Raw reads number | 23,828,556 | 29,351,455 |
| Clean reads | 23,816,967 | 29,340,188 |
| Base number (Gb) | 7.14 | 8.80 |
| GC content (%) | 48.76% | 46.70% |
| Q20 percentage | 97.85% | 98.03% |
| Unigenes | ||
| Number of unigenes | 57,921 | |
| Min length of unigenes (bp) | 201 | |
| Mean length of unigenes (bp) | 768 | |
| Max length of unigenes (bp) | 18,866 | |
| N50 of unigenes (bp) | 1,400 | |
| Total Nucleotides of unigenes (bp) | 44,508,371 | |
| Number of Unigenes | % | |
|---|---|---|
| Annotated in Genebank NR | 30,500 | 52.65 |
| Annotated in Genebank NT | 21,064 | 36.36 |
| Annotated in KEGG | 11,206 | 19.34 |
| Annotated in SwissProt | 23,445 | 40.47 |
| Annotated in Pfam | 20,859 | 36.01 |
| Annotated in GO | 20,834 | 35.96 |
| Annotated in KOG | 11,791 | 20.35 |
| Annotated in all databases | 4813 | 8.3 |
| Annotated in at least one database | 36,688 | 63.34 |
| Not annotated in all database | 21,233 | 36.66 |
| Total Unigenes | 57,921 | 100 |
| Gene_ID | CQ_Na24_Readcount | CQ_0_Readcount | log2.Fold_Change | p−Value | q−Value | Annotation |
|---|---|---|---|---|---|---|
| MAPK | ||||||
| c33984_g1 | 149.2283698 | 47.67405994 | 1.6462 | 1.10E−14 | 1.77E−12 | Mitogen-activated protein kinase 9-like (Nelumbo nucifera) |
| c34545_g1 | 59.3142507 | 5.238326231 | 3.5012 | 1.99E−13 | 2.75E−11 | Mitogen-activated protein kinase 2-like (Citrus sinensis) |
| Serine−threonine protein kinase | ||||||
| c1037_g1 | 36.96338405 | 1.587371585 | 4.5414 | 3.94E−10 | 3.99E−08 | probable serine-threonine protein kinase (Beta vulgaris subsp. vulgaris) |
| c25455_g1 | 32.83888317 | 1.957758288 | 4.0681 | 9.25E−09 | 7.77E−07 | serine-threonine protein kinase (Ricinus communis) |
| c33021_g2 | 18.93342307 | 1.005335337 | 4.2352 | 1.04E−05 | 0.0005562 | serine-threonine protein kinase (Ricinus communis) |
| c25455_g1 | 32.83888317 | 1.957758288 | 4.0681 | 9.25E−09 | 7.77E−07 | serine-threonine protein kinase (Ricinus communis) |
| c22689_g1 | 45.015981 | 15.55624154 | 1.5329 | 5.30E−05 | 0.0024396 | serine/threonine protein kinase (Eucalyptus grandis) |
| CDPKs | ||||||
| c38839_g1 | 35.23502178 | 8.042682698 | 2.1313 | 9.55E−06 | 0.00051234 | Calcium-dependent protein kinase 19 (Theobroma cacao) |
| c31052_g1 | 232.8033275 | 105.0294992 | 1.1483 | 8.13E−14 | 1.17E−11 | Calcium-dependent protein kinase 26 isoform X1 (Vitis vinifera) |
| c60566_g1 | 41.9127851 | 14.39216904 | 1.5421 | 8.99E−05 | 0.0039211 | Calcium-dependent protein kinase 11 (Beta vulgaris subsp. vulgaris) |
| CIPKs | ||||||
| c49233_g1 | 703.207759 | 280.4356467 | 1.3263 | 3.54E−47 | 2.54E−44 | CBL-interacting serine/threonine-protein kinase 4-like (Beta vulgaris subsp. vulgaris) |
| LPK | ||||||
| c10866_g1 | 25.21837679 | 2.751444081 | 3.1962 | 3.82E−06 | 0.00022042 | kinase, putative (Ricinus communis) |
| PP2C | ||||||
| c38748_g1 | 173.1111939 | 63.54777579 | 1.4458 | 2.75E−14 | 4.10E−12 | protein phosphatase 2C 6 (Morus notabilis) |
| PTPase | ||||||
| c60922_g1 | 38.06325095 | 6.137836796 | 2.6326 | 2.23E−07 | 1.57E−05 | tyrosine-protein phosphatase (Beta vulgaris subsp. vulgaris) |
| HSPs | ||||||
| c10992_g1 | 595.8528933 | 171.2244817 | 1.7991 | 2.80E−60 | 2.80E−57 | heat shock protein 70 (Ageratina adenophora) |
| c22339_g1 | 128.9593941 | 50.9017155 | 1.3411 | 4.77E−10 | 4.76E−08 | heat shock protein, putative (Ricinus communis) |
| c26417_g3 | 280.7410263 | 129.0003975 | 1.1219 | 7.62E−16 | 1.36E−13 | Heat shock cognate 70 kDa protein (Glycine soja) |
| c29031_g1 | 123.4207786 | 52.065788 | 1.2452 | 7.83E−09 | 6.64E−07 | hypothetical protein B456_013G139100 (Gossypium raimondii) |
| c29386_g1 | 1492.912194 | 346.1528304 | 2.1086 | 1.92E−180 | 1.06E−176 | heat shock protein 83 (Beta vulgaris subsp. vulgaris) |
| c29386_g2 | 864.9274743 | 402.3986969 | 1.104 | 2.04E−44 | 1.37E−41 | heat shock protein 90-2 (Populus euphratica) |
| c30033_g1 | 222.2909568 | 78.20450677 | 1.5071 | 6.84E−19 | 1.60E−16 | heat shock protein 70 (Gossypium hirsutum) |
| c31857_g1 | 196.9940181 | 78.25741915 | 1.3319 | 1.90E−14 | 2.88E−12 | class I heat shock protein-like (Nelumbo nucifera) |
| c32499_g1 | 674.21841 | 200.4321188 | 1.7501 | 1.27E−65 | 1.50E−62 | heat shock protein 2 (Sesamum indicum) |
| c32766_g1 | 93.5279675 | 46.50998745 | 1.0079 | 1.78E−05 | 0.00091511 | heat shock 70 kDa protein 14-like (Vitis vinifera) |
| c35678_g1 | 57.62516939 | 7.354821678 | 2.9699 | 1.37E−11 | 1.59E−09 | class II heat shock protein-like (Populus euphratica) |
| c38902_g1 | 275.9880301 | 87.94038582 | 1.65 | 6.68E−26 | 2.40E−23 | class I heat shock protein (Vitis vinifera) |
| c54547_g1 | 852.1611621 | 267.4721121 | 1.6717 | 1.06E−77 | 1.40E−74 | heat shock protein (Solanum lycopersicum) |
| ABC Transporter | ||||||
| c39646_g1 | 110.6151854 | 44.97552825 | 1.2983 | 1.77E−08 | 1.44E−06 | ABC transporter B family member 25 (Vitis vinifera) |
| c30623_g2 | 110.5759045 | 22.06446503 | 2.3252 | 1.56E−16 | 2.92E−14 | ABC transporter B family member 11-like (Beta vulgaris subsp. vulgaris) |
| c38901_g1 | 526.9933692 | 238.2115626 | 1.1455 | 3.36E−29 | 1.32E−26 | ABC transporter C family member 4 (Gossypium arboreum) |
| c33388_g1 | 55.58255943 | 10.10626576 | 2.4594 | 1.58E−09 | 1.45E−07 | ABC transporter C family member 3-like (Pyrus x bretschneideri) |
| GST | ||||||
| c39155_g1 | 250.1804389 | 40.74253735 | 2.6184 | 5.03E−40 | 2.97E−37 | glutathione S-transferase (Eucalyptus grandis) |
| c39970_g1 | 45.17310484 | 12.32858598 | 1.8735 | 3.69E−06 | 0.00021425 | glutathione transferase (Populus trichocarpa) |
| c4684_g2 | 107.5119895 | 14.60381858 | 2.8801 | 8.78E−20 | 2.30E−17 | glutathione S-transferase (Eucalyptus grandis) |
| c59876_g1 | 188.4307686 | 61.59001751 | 1.6133 | 1.09E−17 | 2.21E−15 | glutathione S-transferase (Citrus sinensis) |
| c60552_g1 | 79.93675509 | 7.19608452 | 3.4736 | 1.79E−17 | 3.55E−15 | glutathione S-transferase U17-like (Citrus sinensis) |
| SERK1 | ||||||
| c25693_g1 | 21.29028072 | 0.634948634 | 5.0674 | 1.37E−06 | 8.58E−05 | somatic embryogenesis receptor kinase 1-like (Zea mays) |
| 14-3-3 | ||||||
| c39300_g1 | 82.49001753 | 39.10225338 | 1.077 | 2.27E−05 | 0.0011344 | 14-3-3 protein, putative (Ricinus communis) |
| AP2/EREBP | ||||||
| c21290_g1 | 39.55592746 | 8.201419857 | 2.2699 | 1.13E−06 | 7.13E−05 | ethylene−responsive transcription factor (Sesamum indicum) |
| c33171_g2 | 52.40080161 | 18.94263425 | 1.468 | 2.32E−05 | 0.0011555 | AP2-like ethylene−responsive transcription factor AIL5 (Eucalyptus grandis) |
| c44576_g1 | 67.79893822 | 28.67851331 | 1.2413 | 1.97E−05 | 0.00099714 | ethylene-responsive transcription factor ABR1-like (Populus euphratica) |
| c50089_g1 | 84.41478461 | 14.23343188 | 2.5682 | 2.66E−14 | 3.99E−12 | putative ethylene response factor 5 (Vitis aestivalis) |
| MYB | ||||||
| c23951_g1 | 55.8182452 | 19.84214482 | 1.4922 | 9.98E−06 | 0.00053404 | transcription factor MYB108 (Beta vulgaris subsp. vulgaris) |
| NAC | ||||||
| c14111_g2 | 46.90146711 | 6.190749182 | 2.9214 | 1.42E−09 | 1.32E−07 | NAC domain-containing protein (Boehmeria nivea) |
| c15464_g1 | 48.47270554 | 0.846598179 | 5.8394 | 2.27E−13 | 3.13E−11 | NAC transcription factor (Fagus sylvatica) |
| c24596_g2 | 89.3249047 | 35.76877305 | 1.3204 | 2.99E−07 | 2.07E−05 | NAC domain-containing protein 2-like (Tarenaya hassleriana) |
| c26879_g1 | 85.8288992 | 30.53044682 | 1.4912 | 4.36E−08 | 3.41E−06 | NAC domain-containing protein 78 (Vitis vinifera) |
| c44171_g1 | 65.59920442 | 29.63093626 | 1.1466 | 7.51E−05 | 0.0033343 | NAC domain protein (Medicago truncatula) |
| c44256_g1 | 232.5040066 | 87.56999912 | 1.4087 | 4.75E−18 | 1.01E−15 | NAC transcription factor (Fagus sylvatica) |
| c60319_g1 | 54.52197349 | 8.99510565 | 2.5996 | 7.35E−10 | 7.10E−08 | NAC domain-containing protein (Boehmeria nivea) |
| c8889_g1 | 97.37750165 | 31.27122023 | 1.6388 | 4.90E−10 | 4.87E−08 | NAC domain-containing protein 1 (Salvia miltiorrhiza) |
| WRKY | ||||||
| c10876_g1 | 37.67044134 | 2.222320219 | 4.0833 | 7.35E−10 | 7.10E−08 | WRKY transcription factor 31 (Nicotiana sylvestris) |
| c23849_g1 | 215.1811029 | 69.89726214 | 1.6222 | 4.05E−20 | 1.10E−17 | WRKY protein (Rheum australe) |
| c25475_g1 | 64.77430424 | 13.3868337 | 2.2746 | 4.47E−10 | 4.47E−08 | WRKY4 transcription factor (Vitis aestivalis) |
| c28962_g2 | 179.0819 | 55.55800548 | 1.6886 | 7.33E−18 | 1.52E−15 | WRKY transcription factor 33 (Glycine max) |
| c45063_g1 | 30.24633976 | 3.439305101 | 3.1366 | 5.17E−07 | 3.43E−05 | WRKY transcription factor 75 (Nelumbo nucifera) |
| c49337_g1 | 37.04194597 | 4.762114756 | 2.9595 | 6.23E−08 | 4.76E−06 | WRKY transcription factor 70-like (Citrus sinensis) |
| c49928_g1 | 18.1085229 | 0.105824772 | 7.4188 | 1.57E−05 | 0.00081139 | WRKY transcription factor 73 (Gossypium hirsutum) |
| ZAT | ||||||
| c38942_g1 | 96.94541108 | 21.85281549 | 2.1494 | 1.57E−13 | 2.22E−11 | zinc finger protein ZAT10 (Beta vulgaris subsp. vulgaris) |
| c51237_g1 | 19.79760421 | 0.423299089 | 5.5475 | 2.80E−06 | 0.00016544 | zinc finger protein ZAT11-like (Solanum tuberosum) |
| c61192_g1 | 52.32223969 | 6.08492441 | 3.1041 | 4.95E−11 | 5.54E−09 | zinc finger protein ZAT10 (Populus euphratica) |
| GIGANTEA | ||||||
| c32034_g1 | 202.2969478 | 445.575204 | −1.1392 | 1.05E−19 | 2.73E−17 | protein GIGANTEA (Beta vulgaris subsp. vulgaris) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Bai, X.; Zhao, W.; Xiang, D.; Wan, Y.; Yan, J.; Zou, L.; Zhao, G. De Novo Assembly and Analysis of Tartary Buckwheat (Fagopyrum tataricum Garetn.) Transcriptome Discloses Key Regulators Involved in Salt-Stress Response. Genes 2017, 8, 255. https://doi.org/10.3390/genes8100255
Wu Q, Bai X, Zhao W, Xiang D, Wan Y, Yan J, Zou L, Zhao G. De Novo Assembly and Analysis of Tartary Buckwheat (Fagopyrum tataricum Garetn.) Transcriptome Discloses Key Regulators Involved in Salt-Stress Response. Genes. 2017; 8(10):255. https://doi.org/10.3390/genes8100255
Chicago/Turabian StyleWu, Qi, Xue Bai, Wei Zhao, Dabing Xiang, Yan Wan, Jun Yan, Liang Zou, and Gang Zhao. 2017. "De Novo Assembly and Analysis of Tartary Buckwheat (Fagopyrum tataricum Garetn.) Transcriptome Discloses Key Regulators Involved in Salt-Stress Response" Genes 8, no. 10: 255. https://doi.org/10.3390/genes8100255