
Transcriptomic Analysis of Long Non-Coding RNAs and Coding Genes Uncovers a Complex Regulatory Network That Is Involved in Maize Seed Development

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Isolation
2.2. Library Construction, RNA-Sequencing and Data Sets Used for the Identification of lncRNAs
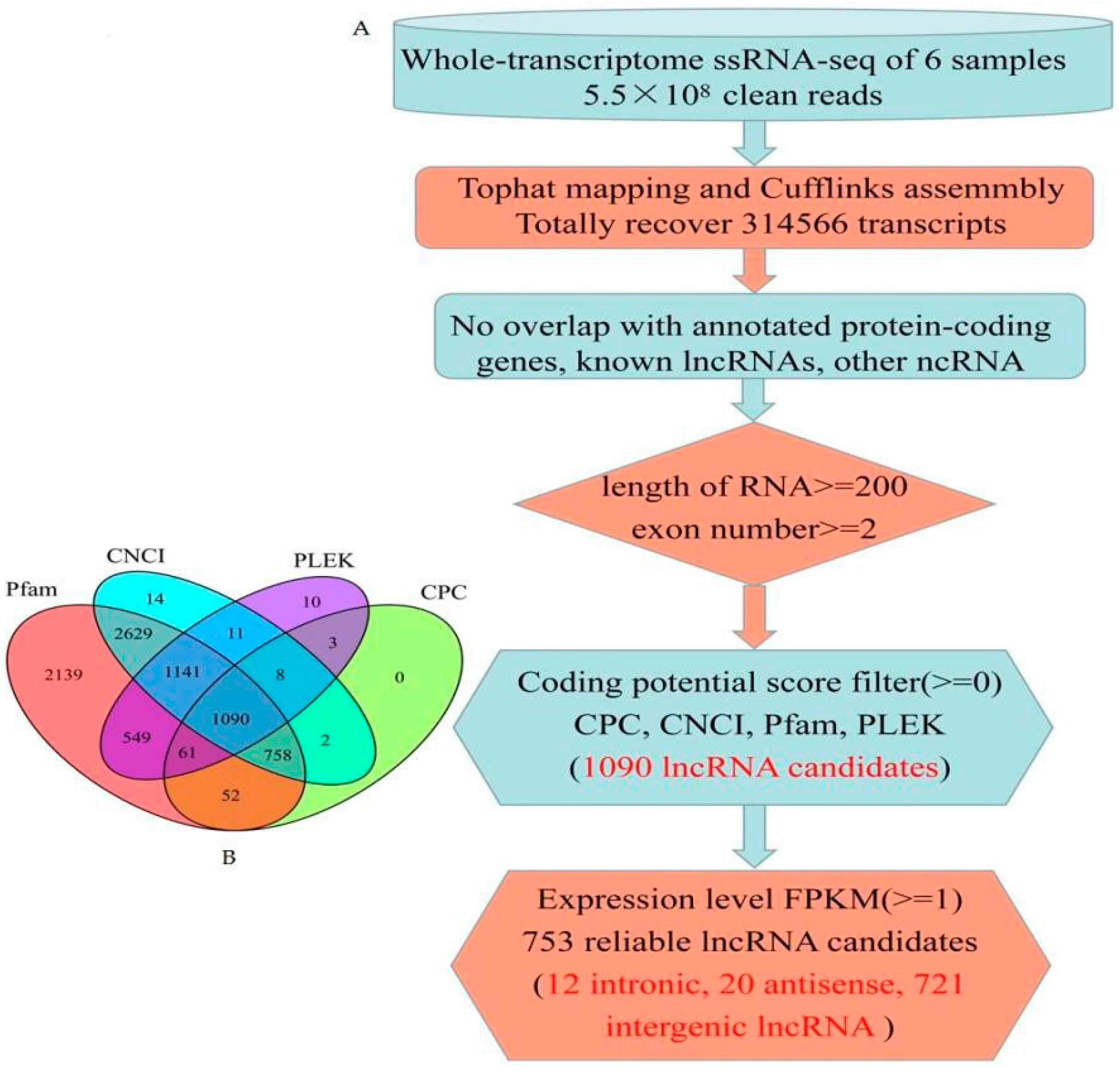
2.3. Bioinformatics Identification of Maize Seed lncRNAs
2.4. Expression Analysis
2.5. Quantitative Real Time PCR (qRT-PCR) Validation of lncRNAs
2.6. Construction and Analysis of the lncRNA-miRNA-mRNA ceRNA network
3. Results
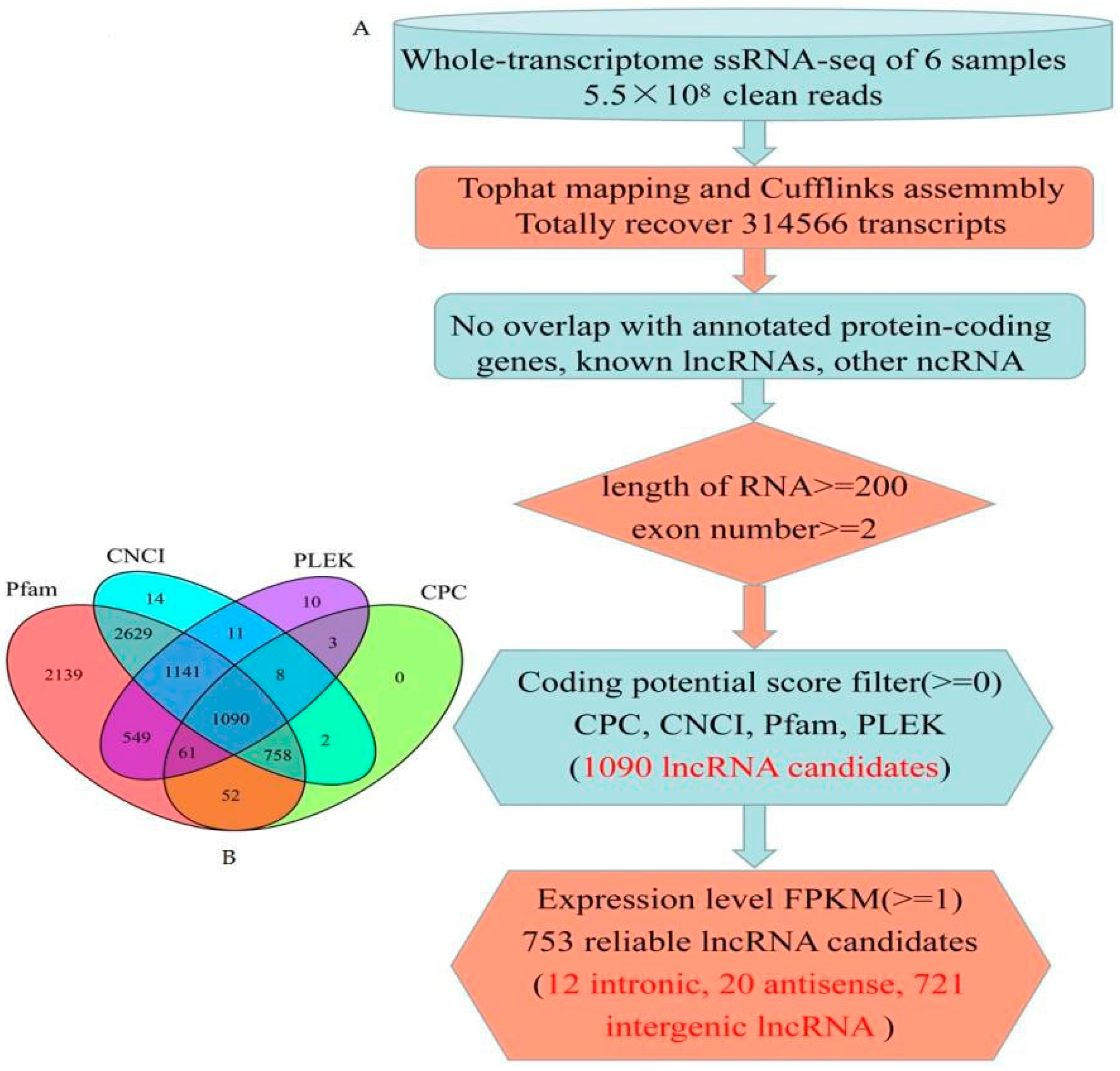
3.1. Genome-Wide Identification of lncRNAs in Maize Seed
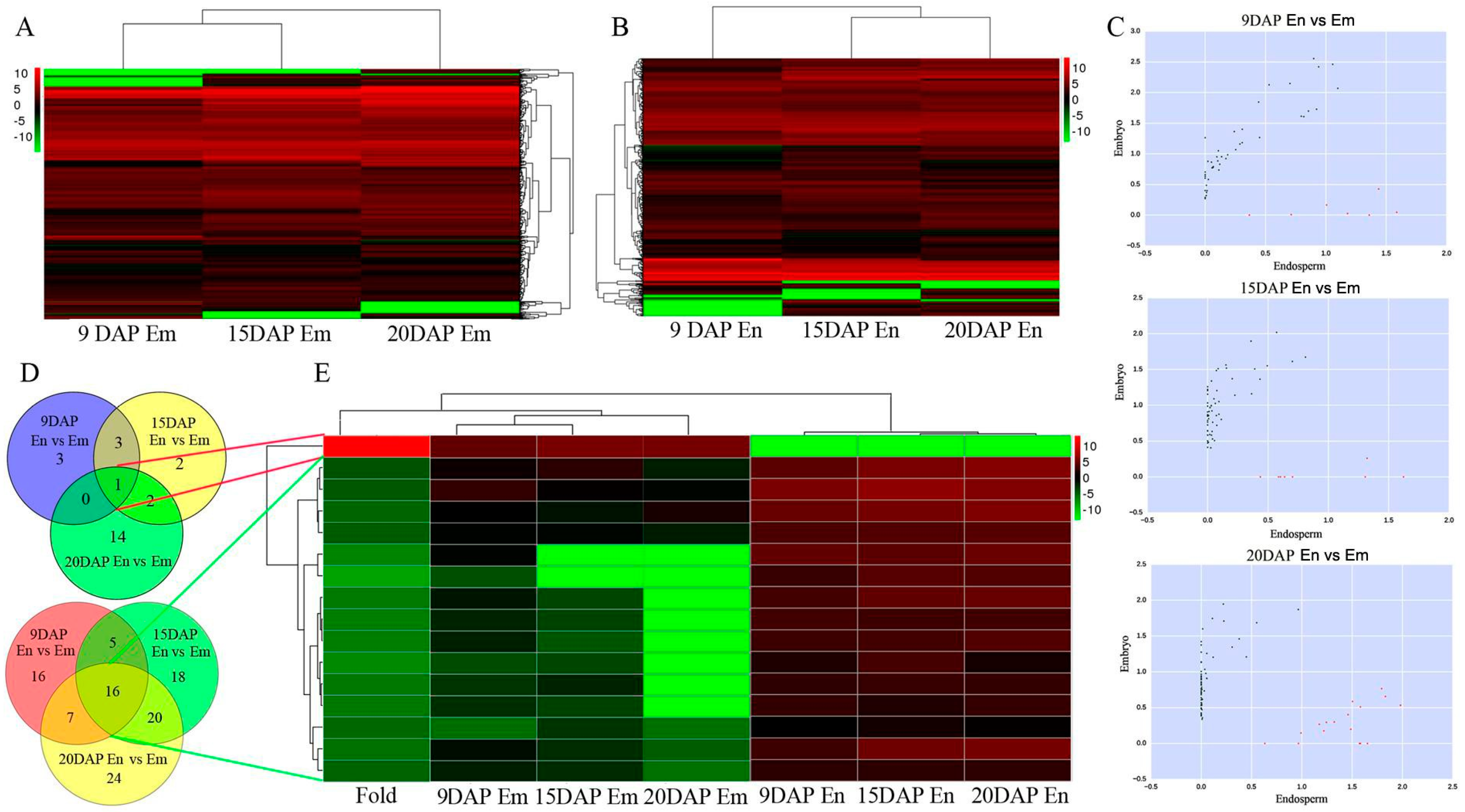
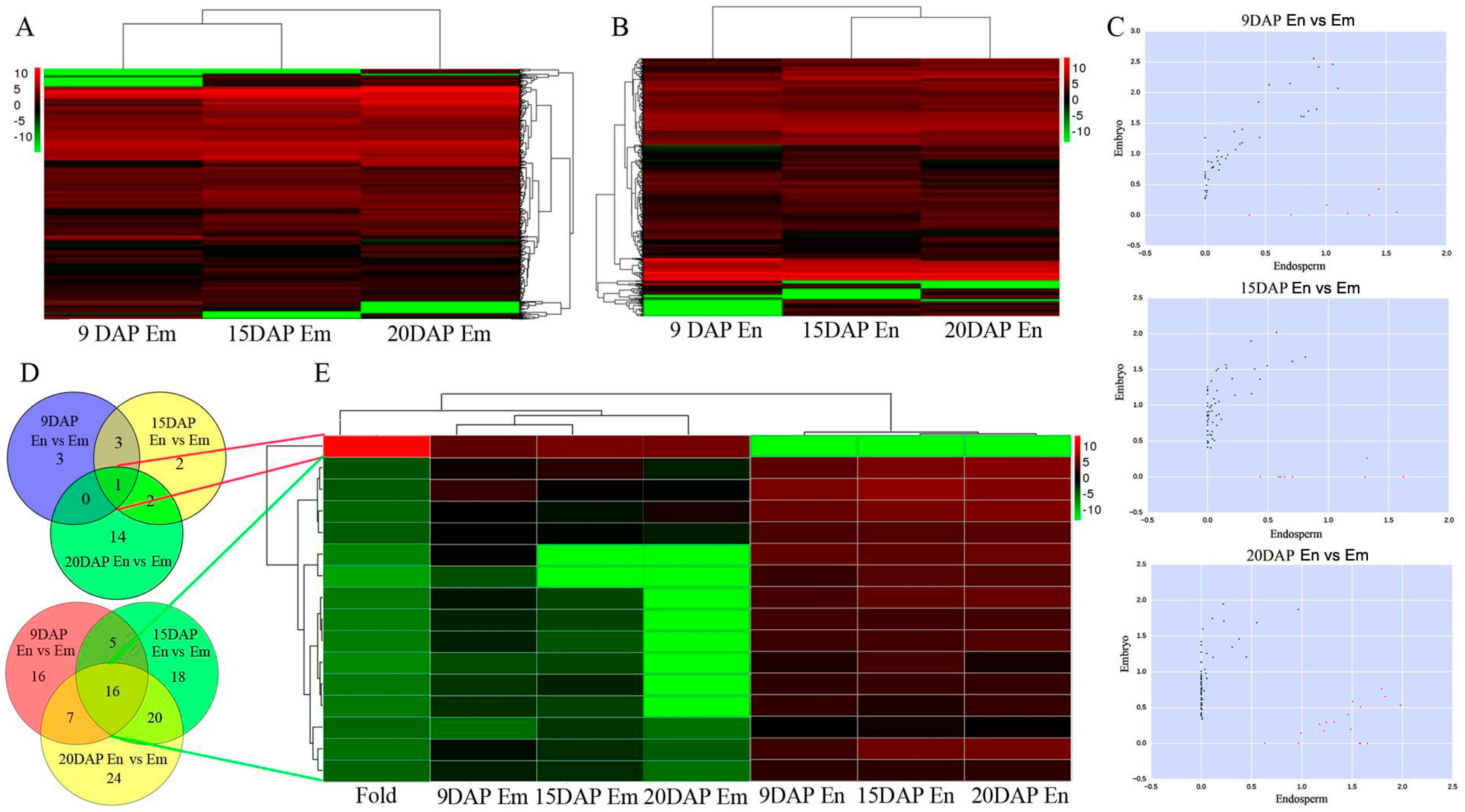
3.2. Expression Profiles of lncRNAs during Embryo and Endosperm Development
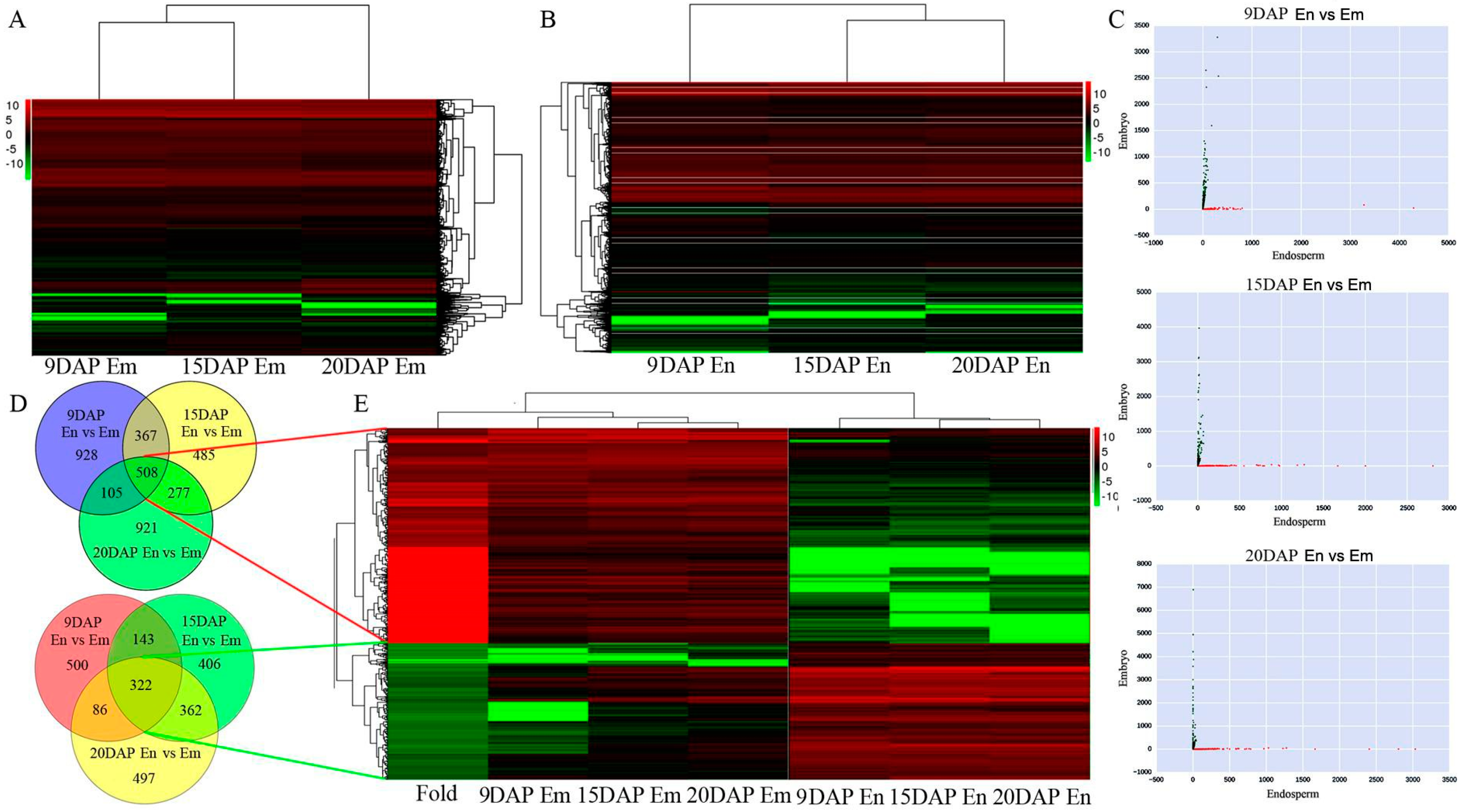
3.3. Expression Profiles of mRNAs during Embryo and Endosperm Development
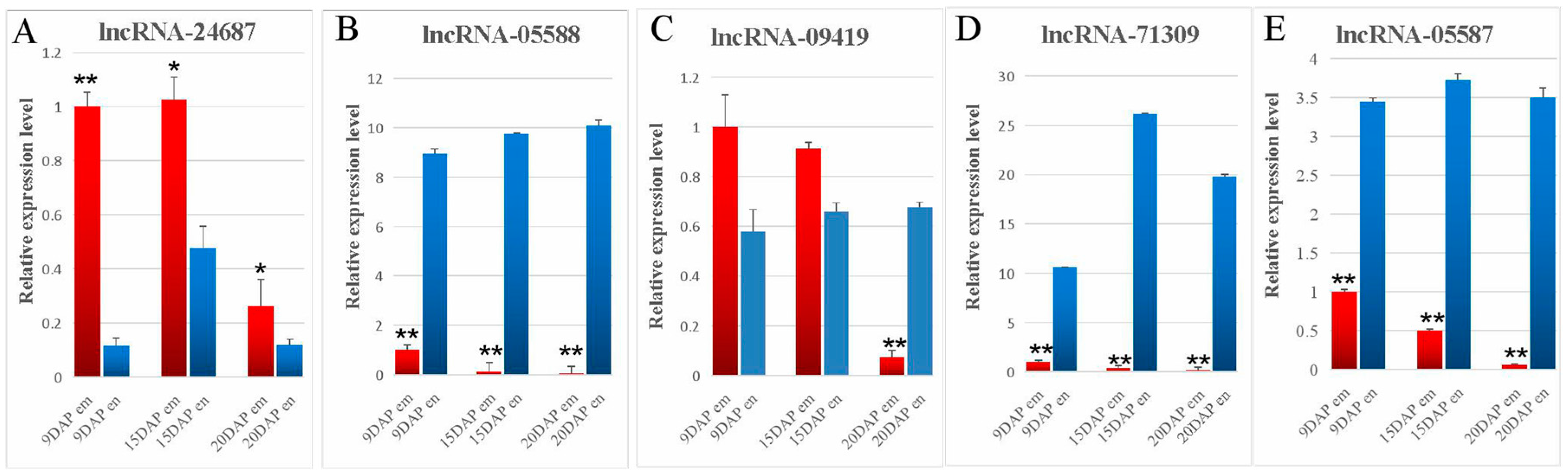
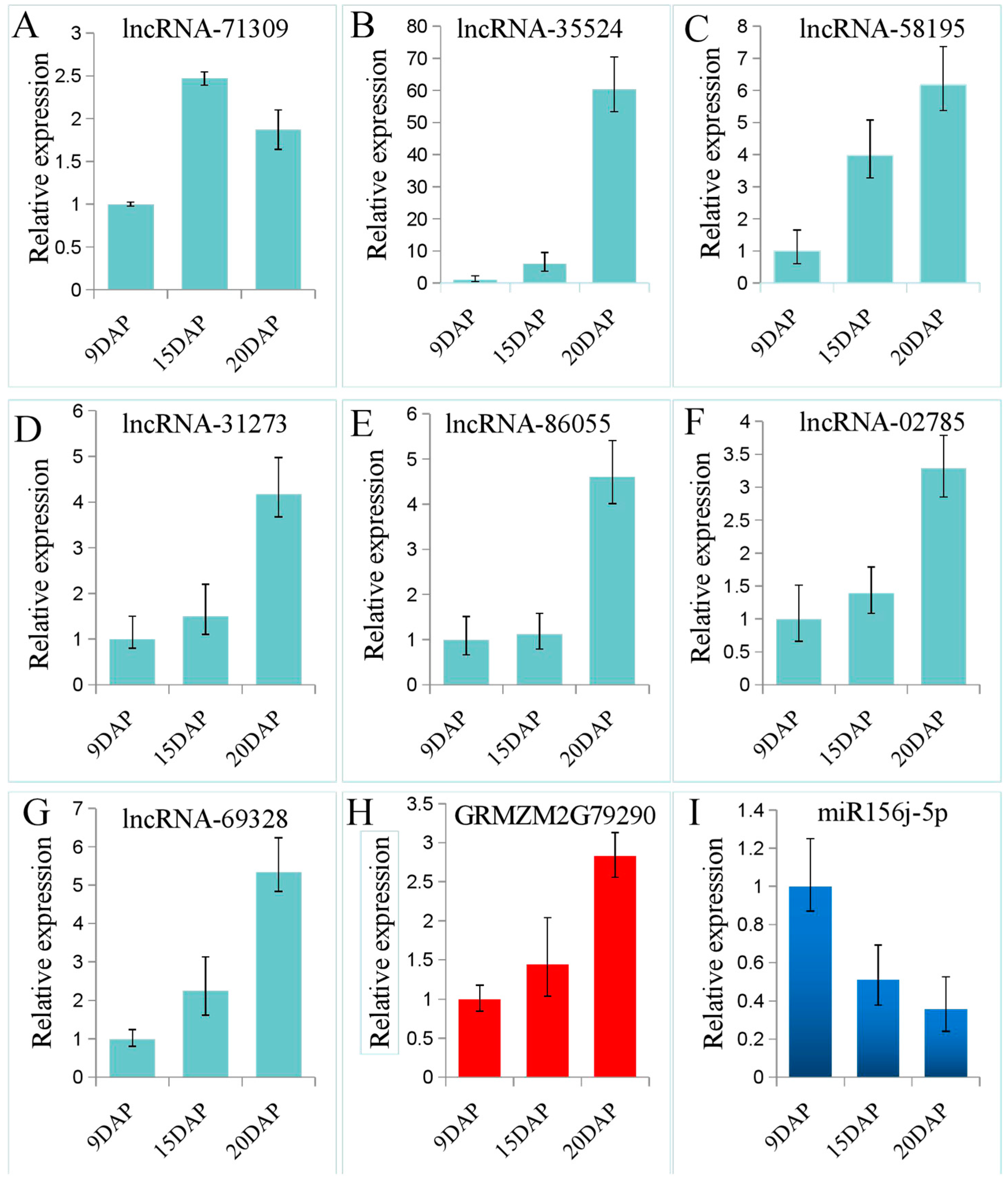
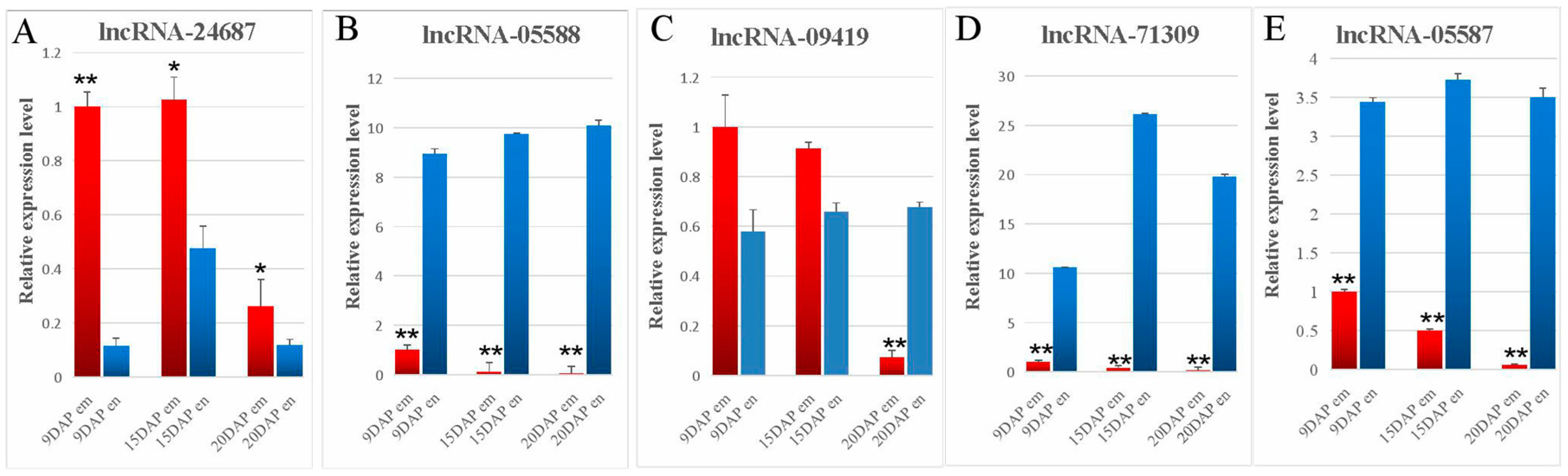
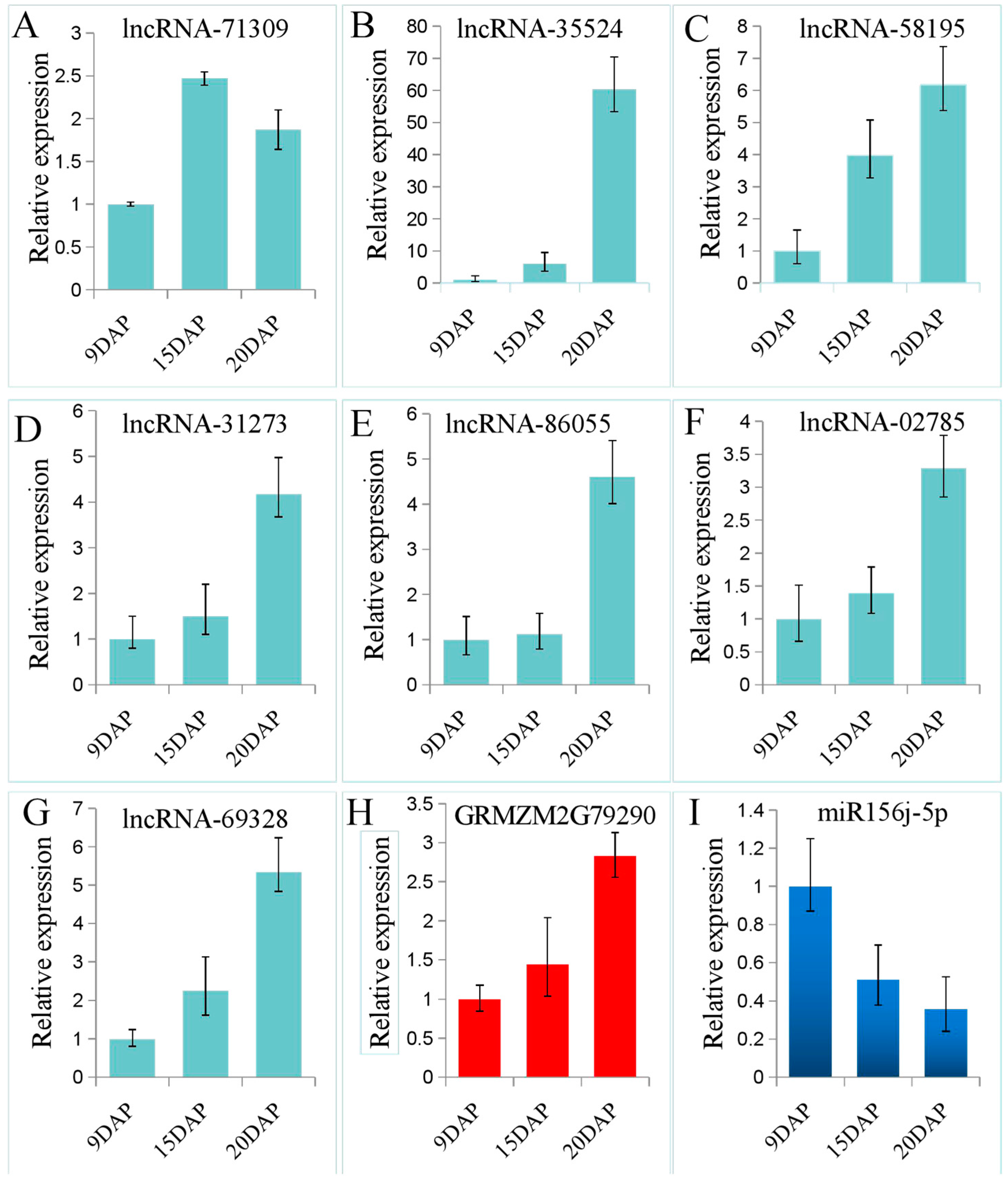
3.4. Validation of lncRNA Expression Using qRT-PCR
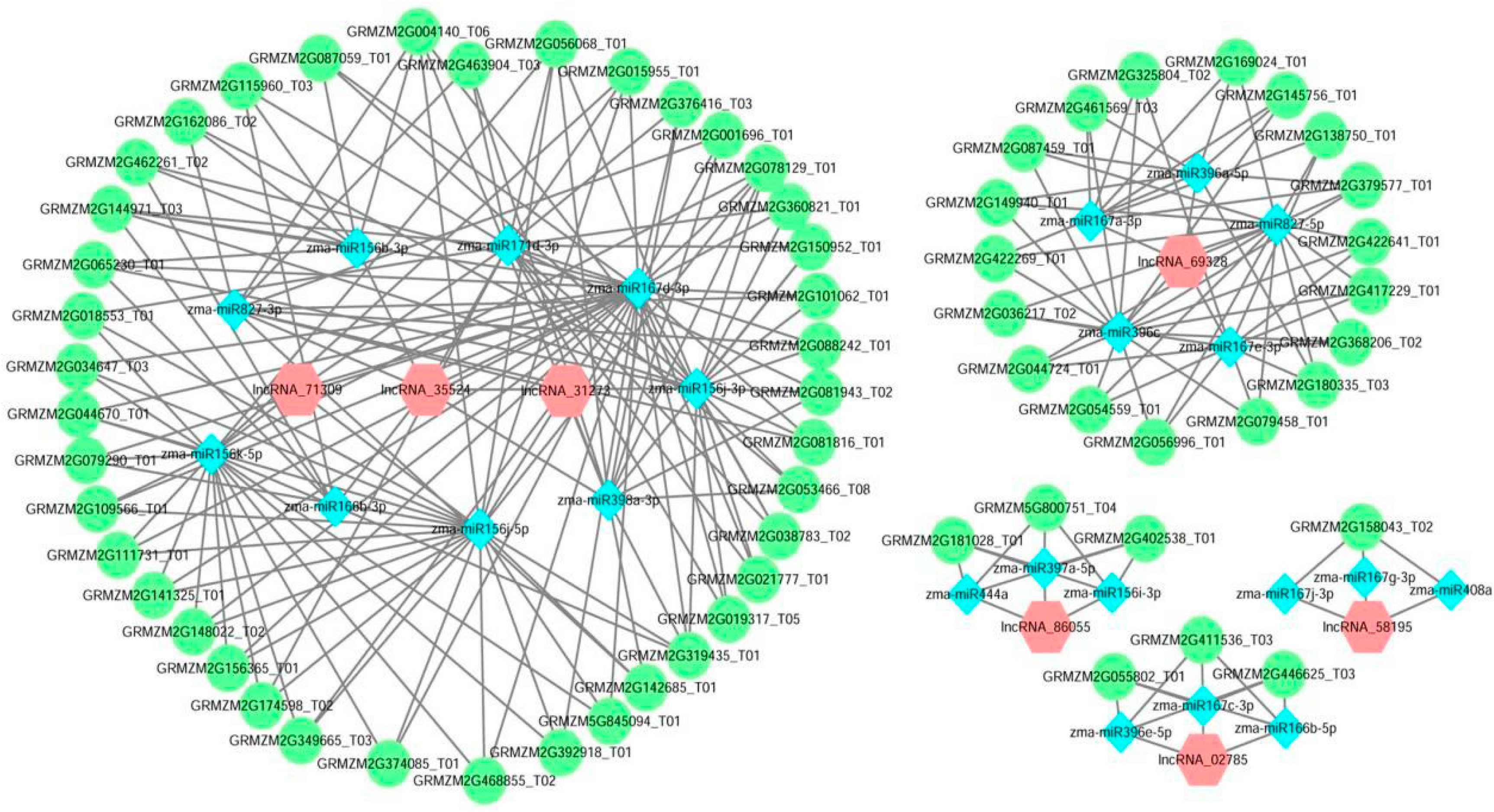
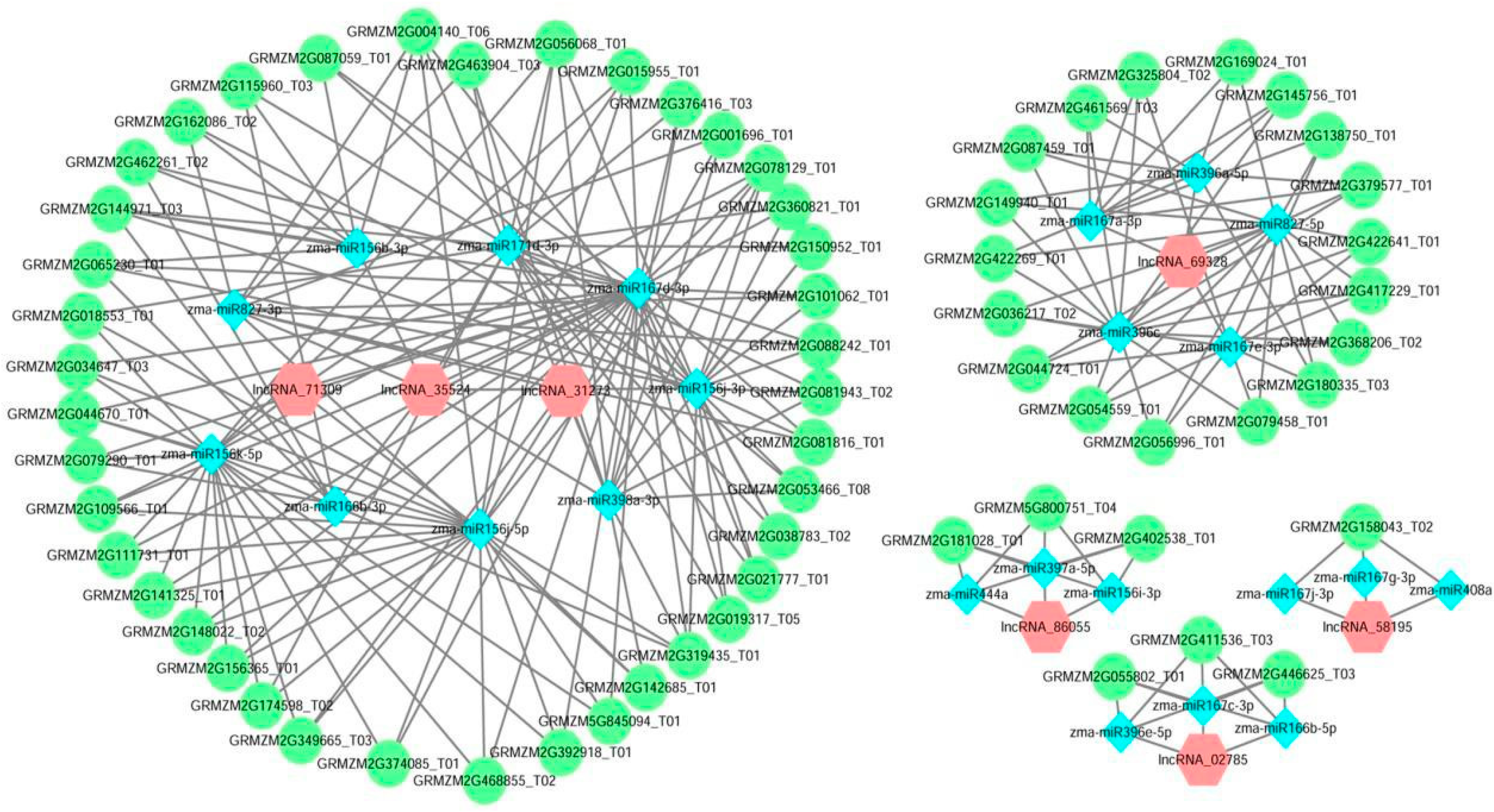
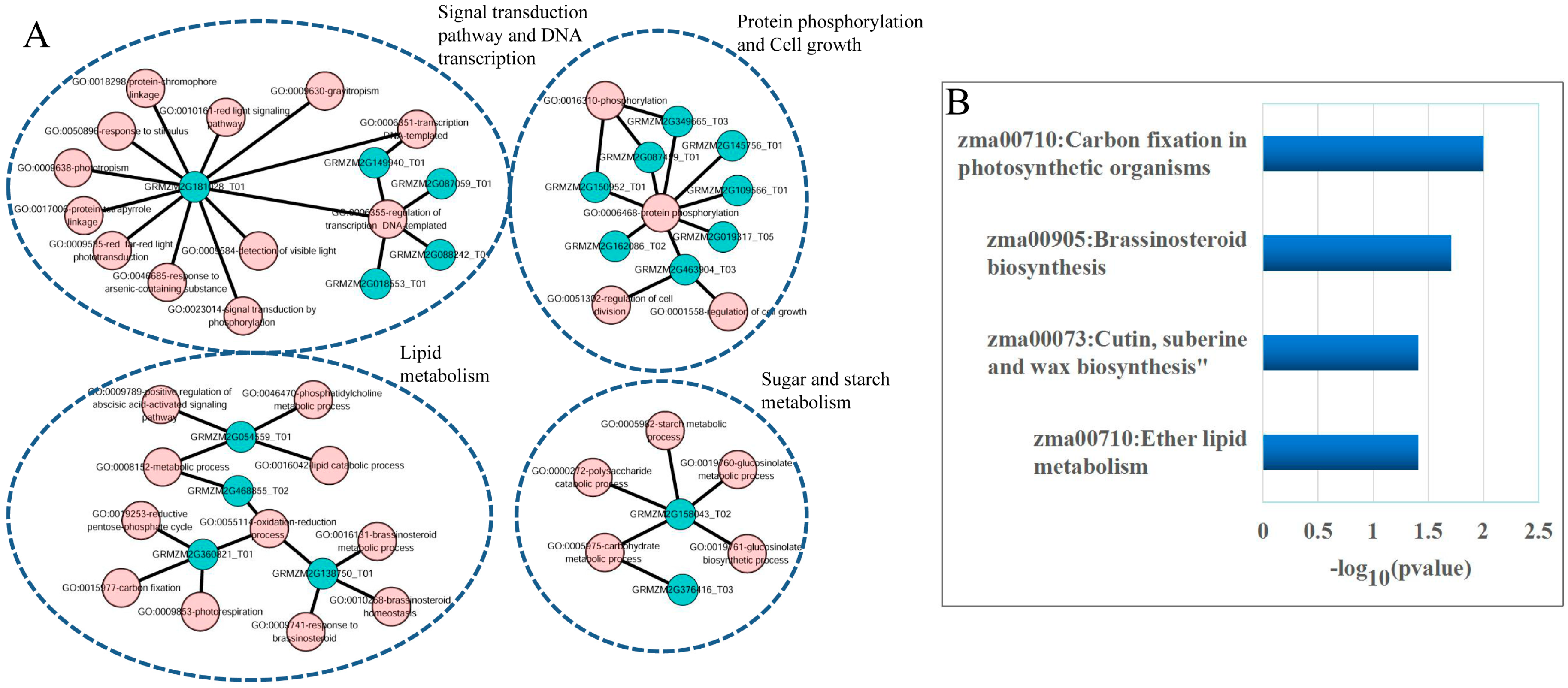
3.5. Construction and Analysis of the lncRNA-miRNA-mRNA ceRNA Network
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Faure, J.E. Double fertilization in flowering plants: Discovery, study methods and mechanisms. Comptes Rendus De Lacademie Des Sciences 2001, 324, 551–558. [Google Scholar] [CrossRef]
- Hamamura, Y.; Nagahara, S.; Higashiyama, T. Double fertilization on the move. Curr. Opin. Plant Biol. 2012, 15, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.A.; Larkins, B.A. Endosperm origin, development, and function. Plant Cell 1993, 5, 1383–1399. [Google Scholar] [CrossRef] [PubMed]
- Conover, C.A.; Ronk, M.; Lombana, F.; Powell, D.R. Structural and biological characterization of bovine insulin-like growth factor binding protein-3. Endocrinology 1990, 127, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Placette, C.; Köhler, C. Embryo and endosperm, partners in seed development. Curr. Opin. Plant Biol. 2014, 17, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, N.A. Transcriptome study outlines ontogeny of the maize shoot apical meristem. Plant Cell 2012, 24, 3169. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Chang, Y.M.; Chen, S.C.; Lu, C.H.; Wu, Y.H.; Lu, M.Y.; Chen, D.R.; Shih, A.C.; Sheue, C.R.; Huang, H.C. Anatomical and transcriptional dynamics of maize embryonic leaves during seed germination. Proc. Natl. Acad. Sci. USA 2013, 110, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.T.; Requesens, D.V.; Devaiah, S.P.; Johnson, D.; Huang, X.; Howard, J.A.; Hood, E.E. Transcriptome analysis of embryo maturation in maize. BMC Plant Biol. 2013, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, D.; Yang, R.; Logan, K.; Chen, H.; Zhang, S.; Skaggs, M.I.; Lloyd, A.; Burnett, W.J.; Laurie, J.D. Temporal patterns of gene expression in developing maize endosperm identified through transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7582–7587. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Thakare, D.; Ma, C.; Lloyd, A.; Nixon, N.M.; Arakaki, A.M.; Burnett, W.J.; Logan, K.O.; Wang, D.; Wang, X.; et al. RNA sequencing of laser-capture micro dissected compartments of the maize kernel identifies regulatory modules associated with endosperm cell differentiation. Plant Cell 2015, 27, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.C.; Lin, H. Repressing the Repressor: A lincRNA as a microRNA sponge in embryonic stem cell self-renewal. Dev. Cell 2013, 25, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Jiang, J.; Chen, X.; Kang, J.; Xiao, L.; Wu, M.; Xiong, J.; Guo, X.; Liu, H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 2013, 25, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Basak, J.; Nithin, C. Targeting non-coding RNAs in plants with the CRISPR-Cas technology is a challenge yet worth accepting. Front. Plant Sci. 2015, 6, 4640. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Cao, Z.; Yang, B.; Ding, N.; Hou, T.; Luo, F.; Kang, F.; Li, J.; Yang, X.; Jiang, H. Changing expression profiles of lncRNAs, mRNAs, circRNAs and miRNAs during osteoclasto genesis. Sci. Rep. 2016, 6, 21499. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Hao, Z.; Yan, J.; Li, G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genom. 2015, 16, 793. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, T.; Kats, L. A ceRNA hypothesis: The Rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Ghosal, S.; Das, S.; Balti, S.; Chakrabarti, J. Competing Endogenous RNA: The Key to Posttranscriptional Regulation. Sci. World J. 2014, 112, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Qi, L.; Jiang, X.; Shao, Y.; Xiao, B.; Yang, X.; Guo, J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, X.; Shi, H.; Liang, C.; Wang, Z.; Zhao, H.; Lei, Y.; Jie, S. Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget 2016, 7, 12598–12611. [Google Scholar] [CrossRef]
- Jiao, C.; Song, Z.; Chen, J.; Zhong, J.; Cai, W.; Tian, S.; Chen, S.; Yi, Y.; Xiao, Y. lncRNA-UCA1 enhances cell proliferation through functioning as a ceRNA of Sox4 in esophageal cancer. Oncol. Rep. 2016, 36, 2960. [Google Scholar] [CrossRef] [PubMed]
- NCBI SRA Database. Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 29 August 2017).
- FASTQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 August 2017).
- NGS QC TOOLKIT v2.3.3. Available online: http://59.163.192.90:8080/ngsqctoolkit/ (accessed on 29 August 2017).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Hierarchical Clustering Explorer 3.5. Available online: http://www.cs.umd.edu/hcil/hce/ (accessed on 29 August 2017).
- The MicroRNA Database. Available online: http://www.mirbase.org/cgi-bin/query.pl?terms=maize (accessed on 29 August 2017).
- Liu, K.; Yan, Z.; Li, Y.; Sun, Z. Linc2GO: A human LincRNA function annotation resource based on ceRNA hypothesis. Bioinformatics 2013, 29, 2221–2222. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ghosal, S.; Sen, R.; Chakrabarti, J. lnCeDB: Database of Human Long Noncoding RNA Acting as Competing Endogenous RNA. PLoS ONE 2014, 9, e98965. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Wang, Z.; Lu, H.; Dai, Z.; Liu, X.; Cui, L. Expression profiling of lncRNAs in C3H10T1/2 mesenchymal stem cells undergoing early osteoblast differentiation. Mol. Med. Rep. 2013, 8, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Moire, L.; Schmutz, A.; Buchala, A.; Yan, B.; Stark, R.E.; Ryser, U. Glycerol is a suberin monomer. New experimental evidence for an old hypothesis. Plant Physiol. 1999, 119, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J. L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Wang, M.B. Molecular Functions of Long Non-Coding RNAs in Plants. Genes 2012, 3, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, M.J.; Takacs, E.M. Kernel Biology. In Handbook of Maize: Its Biology, 1st ed.; Bennetzen, J.L., Hake, S.C., Eds.; Springer: New York, NY, USA, 2009; ISBN 978-0-387-79417-4. [Google Scholar]
- Lu, X.D.; Chen, D.J.; Shu, D.F.; Zhang, Z.; Wang, W.X.; Klukas, C.; Chen, L.L.; Fan, Y.L.; Chen, M.; Zhang, C.Y. The differential transcription network between embryo and endosperm in the early developing maize seed. Plant Physiol. 2013, 162, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Doll, N.M.; Depege-Fargeix, N.; Rogowsky, P.M.; Widies, T. Signaling in early maize kernel development. Mol. Plant 2017, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H. The miR156/SPL Module, a Regulatory Hub and Versatile Toolbox, Gears up Crops for Enhanced Agronomic Traits. Mol. Plant 2015, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Naser, V.; Shani, E. Auxin response under osmotic stress. Plant Mol. Biol. 2016, 91, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Peng, S.; Xian, Z.; Lin, D.; Hu, G.; Yang, L.; Ren, M.; Li, Z. Overexpression of a tomato miR171 target gene SlGRAS24 impacts multiple agronomical traits via regulating gibberellin and auxin homeostasis. Plant Biotechnol. J. 2016, 15, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, F.; Xie, K.; Zeng, X.; Cao, Y.; Zeng, J.; He, Z.; Ren, Y.; Li, W.; Deng, Q. The OsmiR396c-OsGRF4-OsGIF1 regulatory module determines grain size and yield in rice. Plant Biotechnol. J. 2016, 14, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, H.; Hamera, S.; Chen, X.; Fang, R. miR444a has multiple functions in the rice nitrate-signaling pathway. Plant J. Cell Mol. Biol. 2014, 78, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Y.; Huang, T.K.; Chiou, T.J. Nitrogen limitation adaptation, a target of microRNA827, mediates degradation of plasma membrane-localized phosphate transporters to maintain phosphate homeostasis in Arabidopsis. Plant Cell 2013, 25, 4061–4074. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNAs | stages | Upregulated | Often Upregulated | Downregulated |
|---|---|---|---|---|
| lncRNAs | 9 DAP | 7 | 1 | 44 |
| 15 DAP | 8 | 59 | ||
| 20 DAP | 17 | 67 | ||
| mRNAs | 9 DAP | 1908 | 1052 | |
| 15 DAP | 1637 | 508 | 1233 | |
| 20 DAP | 1811 | 1267 |
| ID | Genomic Location | False Discovery Rate (FDR) (9 DAP Em/En) | FDR (15 DAP Em/En) | FDR (20 DAP Em/En) |
|---|---|---|---|---|
| lncRNA_35524 | Chr1: 55243404-55249539 | 1.11 | 2.92 | 8.23 |
| 55250972-55251328 | ||||
| lncRNA_58195 | Chr6: 14305962-14306059 | 0.1 | 0.01 | 0.01 |
| 14311356-14312539 | ||||
| lncRNA_71309 | Chr7: 135515079-135515358 | 0.008 | 0/495 | 0/375 |
| 135515534-135515693 | ||||
| 135520571-135520702 | ||||
| 135520779-135521159 | ||||
| lncRNA_31273 | Chr3: 118012172-118012521 | 1.56 | 6.54 | 24 |
| 118013700-118014996 | ||||
| lncRNA_86055 | Chr1: 137900175-137900970 | 0/64.5 | 0/62.9 | 0/21.2 |
| 137901054-137901119 | ||||
| lncRNA_02785 | Chr1: 134602111-134603048 | 0.08 | 0.06 | 0.004 |
| 134603153-134603273 | ||||
| lncRNA_69328 | Chr7: 3875642-3879270 | 15.2 | 6 | 4 |
| 3879378-3879427 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, M.; Zhang, M.; Xing, L.; Li, W.; Jiang, H.; Wang, L.; Xu, M. Transcriptomic Analysis of Long Non-Coding RNAs and Coding Genes Uncovers a Complex Regulatory Network That Is Involved in Maize Seed Development. Genes 2017, 8, 274. https://doi.org/10.3390/genes8100274
Zhu M, Zhang M, Xing L, Li W, Jiang H, Wang L, Xu M. Transcriptomic Analysis of Long Non-Coding RNAs and Coding Genes Uncovers a Complex Regulatory Network That Is Involved in Maize Seed Development. Genes. 2017; 8(10):274. https://doi.org/10.3390/genes8100274
Chicago/Turabian StyleZhu, Ming, Min Zhang, Lijuan Xing, Wenzong Li, Haiyang Jiang, Lei Wang, and Miaoyun Xu. 2017. "Transcriptomic Analysis of Long Non-Coding RNAs and Coding Genes Uncovers a Complex Regulatory Network That Is Involved in Maize Seed Development" Genes 8, no. 10: 274. https://doi.org/10.3390/genes8100274