
The MXL-3/SBP-1 Axis Is Responsible for Glucose-Dependent Fat Accumulation in C. elegans

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Fat Storage Staining
2.3. Fluorescence Imaging
2.4. RNA Extraction and Quantitative RT-PCR
2.5. Lipid Extraction and Gas Chromatography/Mass Spectrometry Assays
3. Results
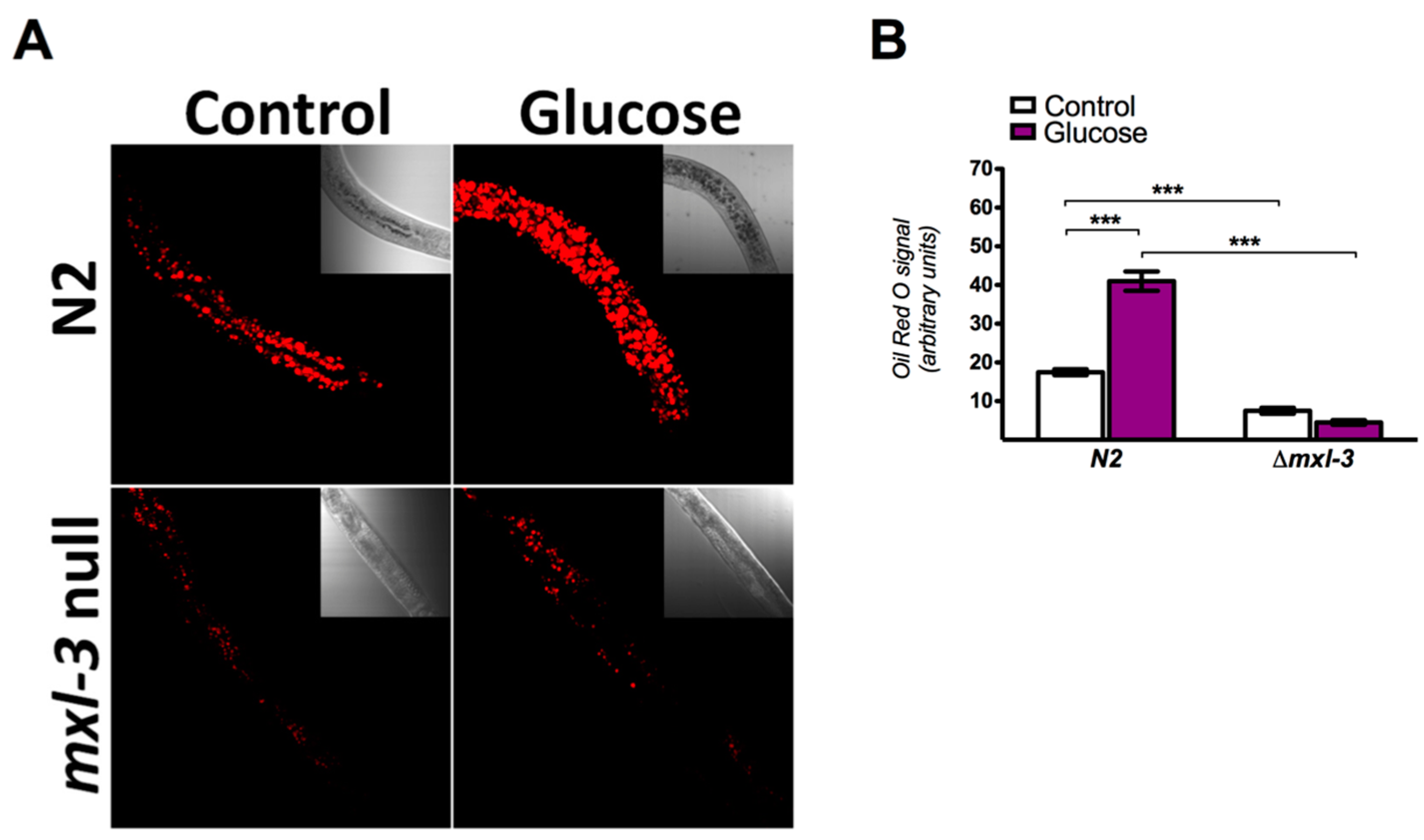
3.1. MXL-3 Is Involved in Lipid Accumulation Due to High Glucose Exposure in C. elegans
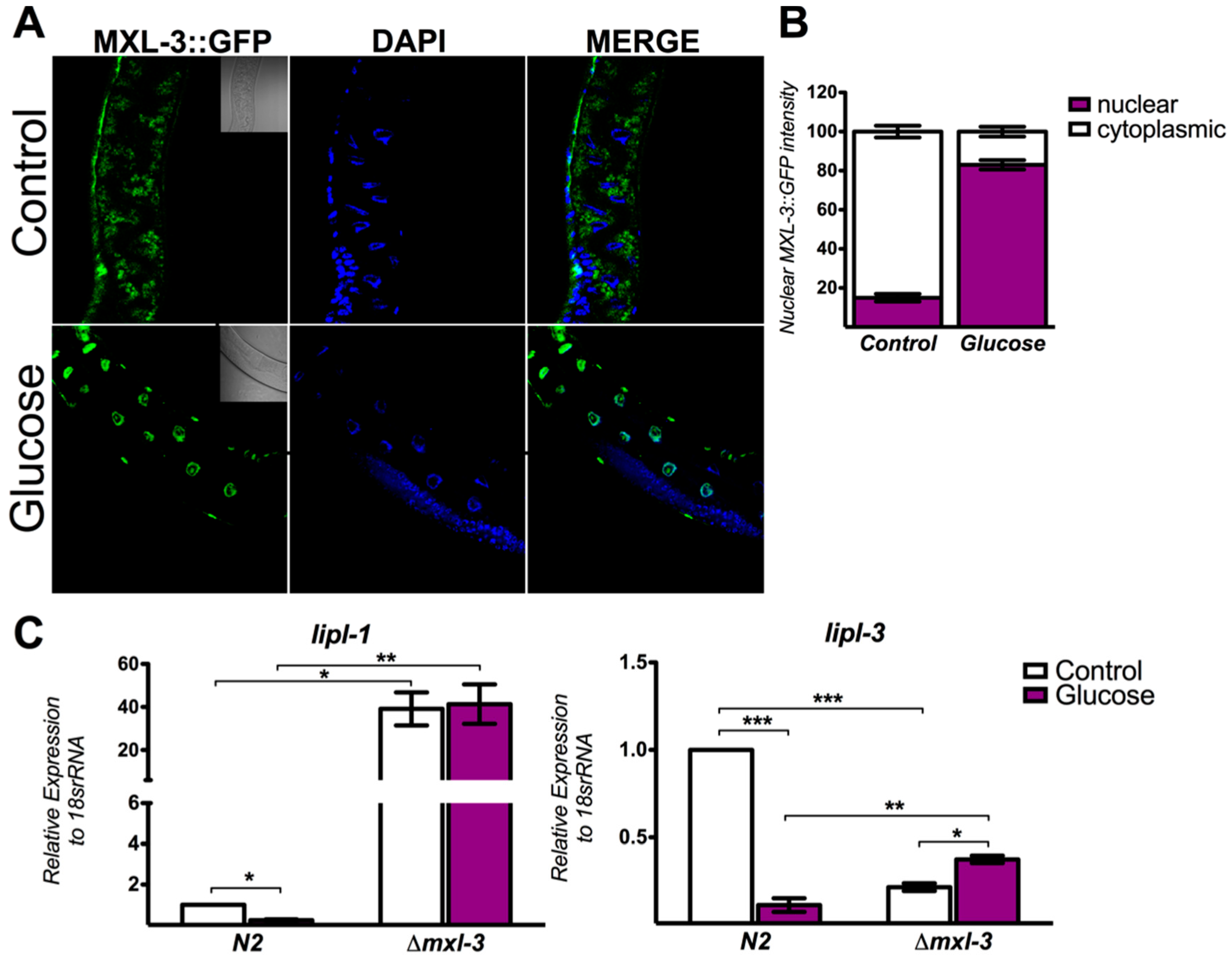
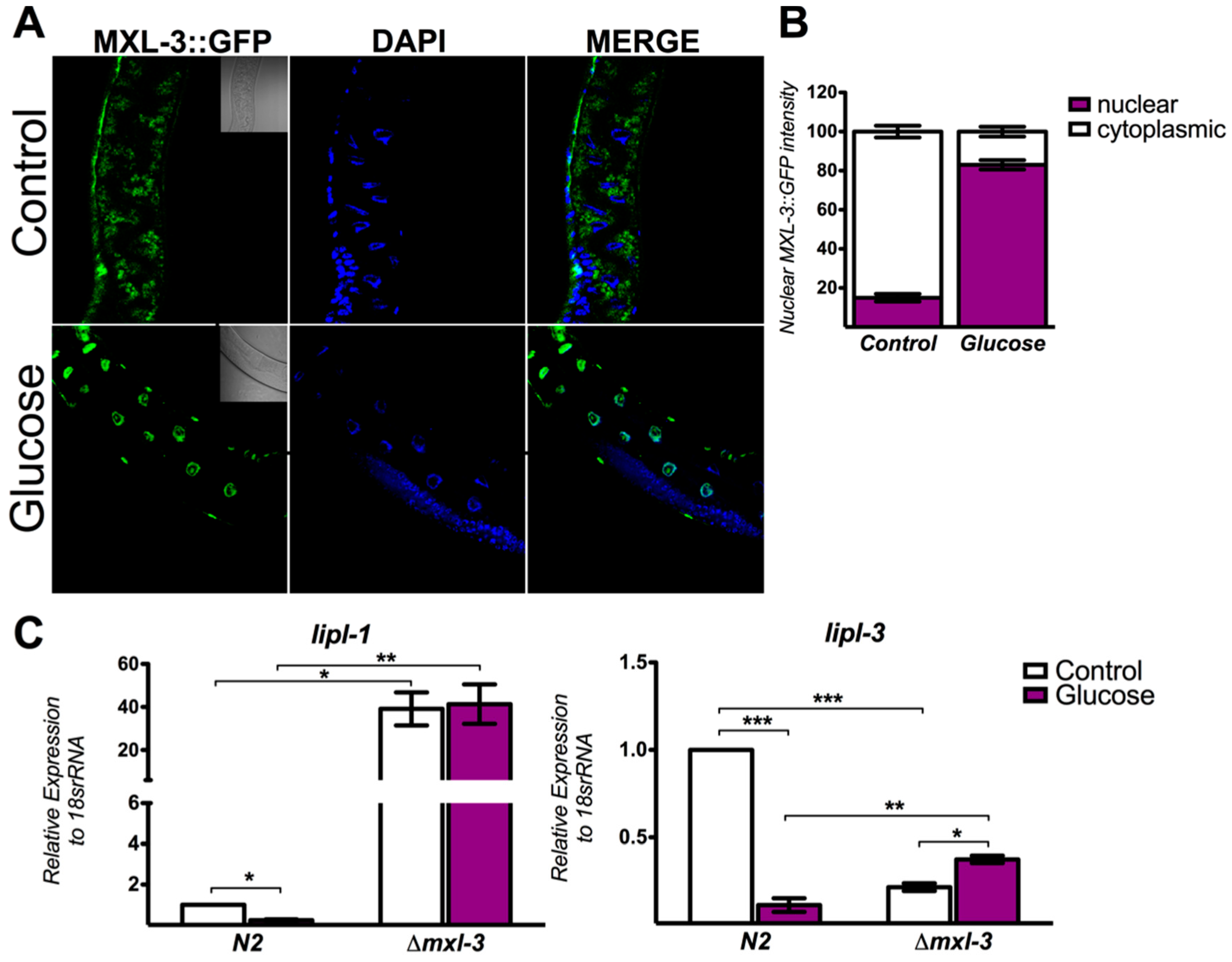
3.2. MXL-3 Actively Shuttles between the Cytoplasm and Nucleus in Response to High Glucose
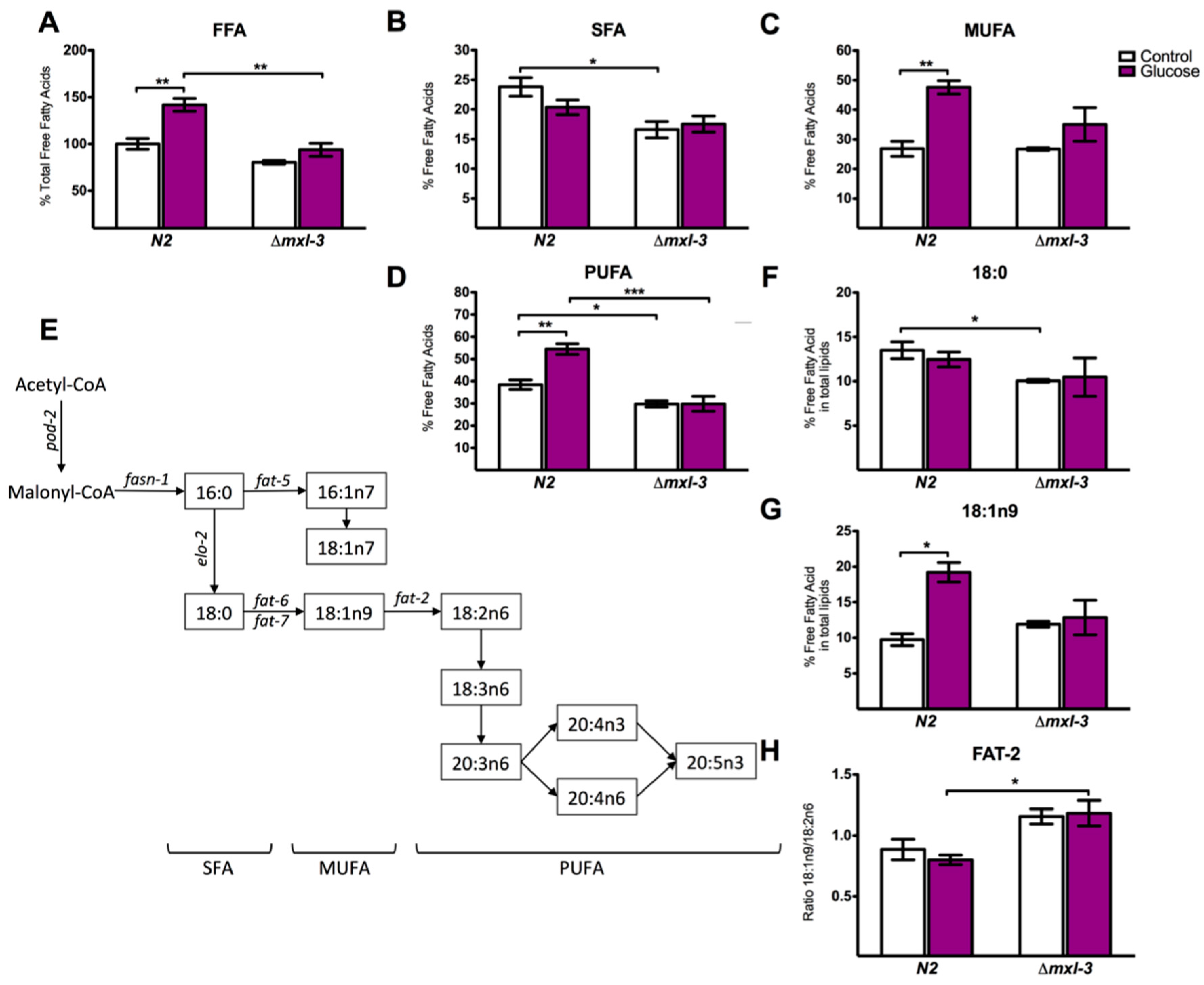
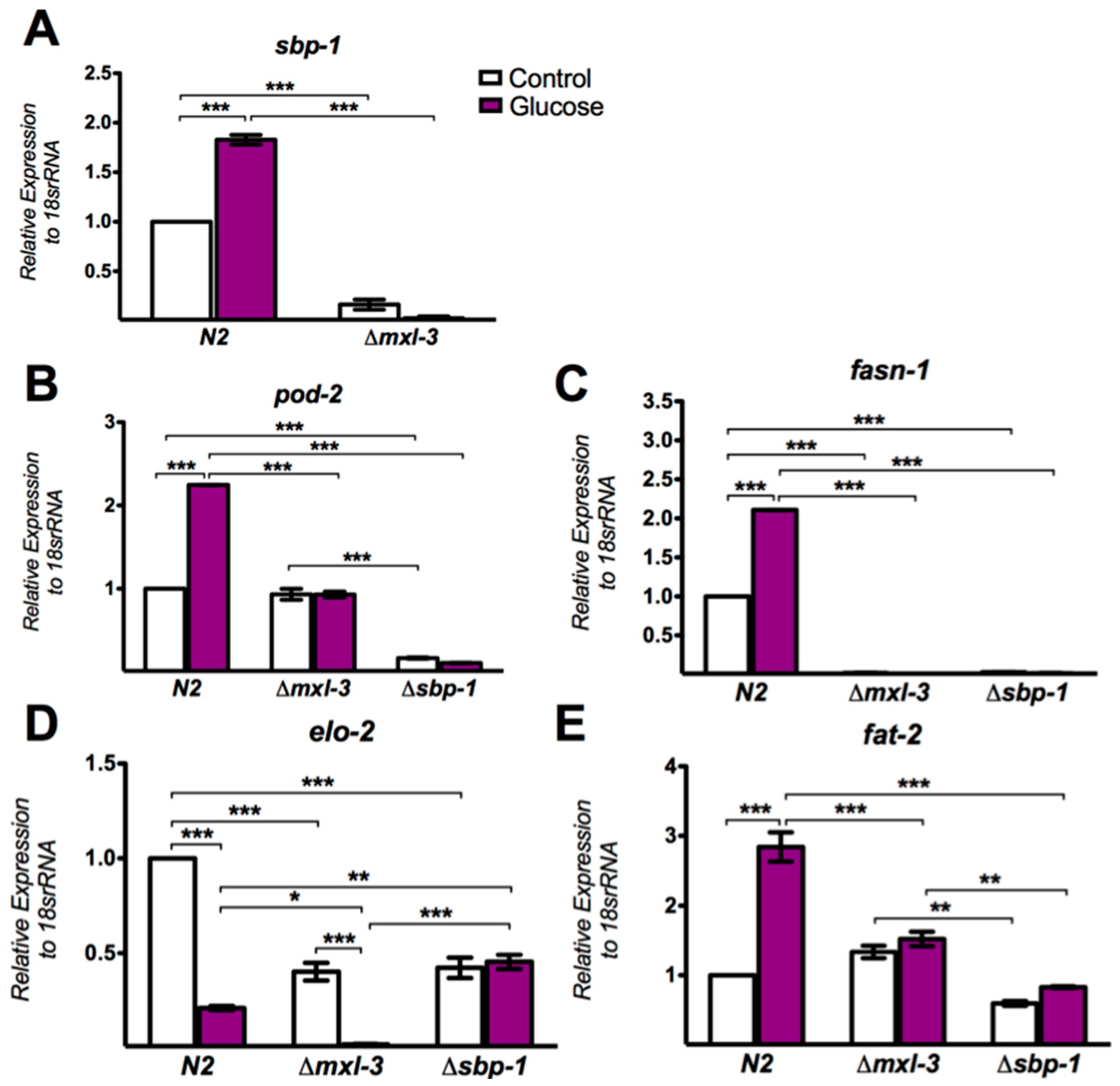
3.3. MXL-3 Regulates Lipid Synthesis Through SBP-1
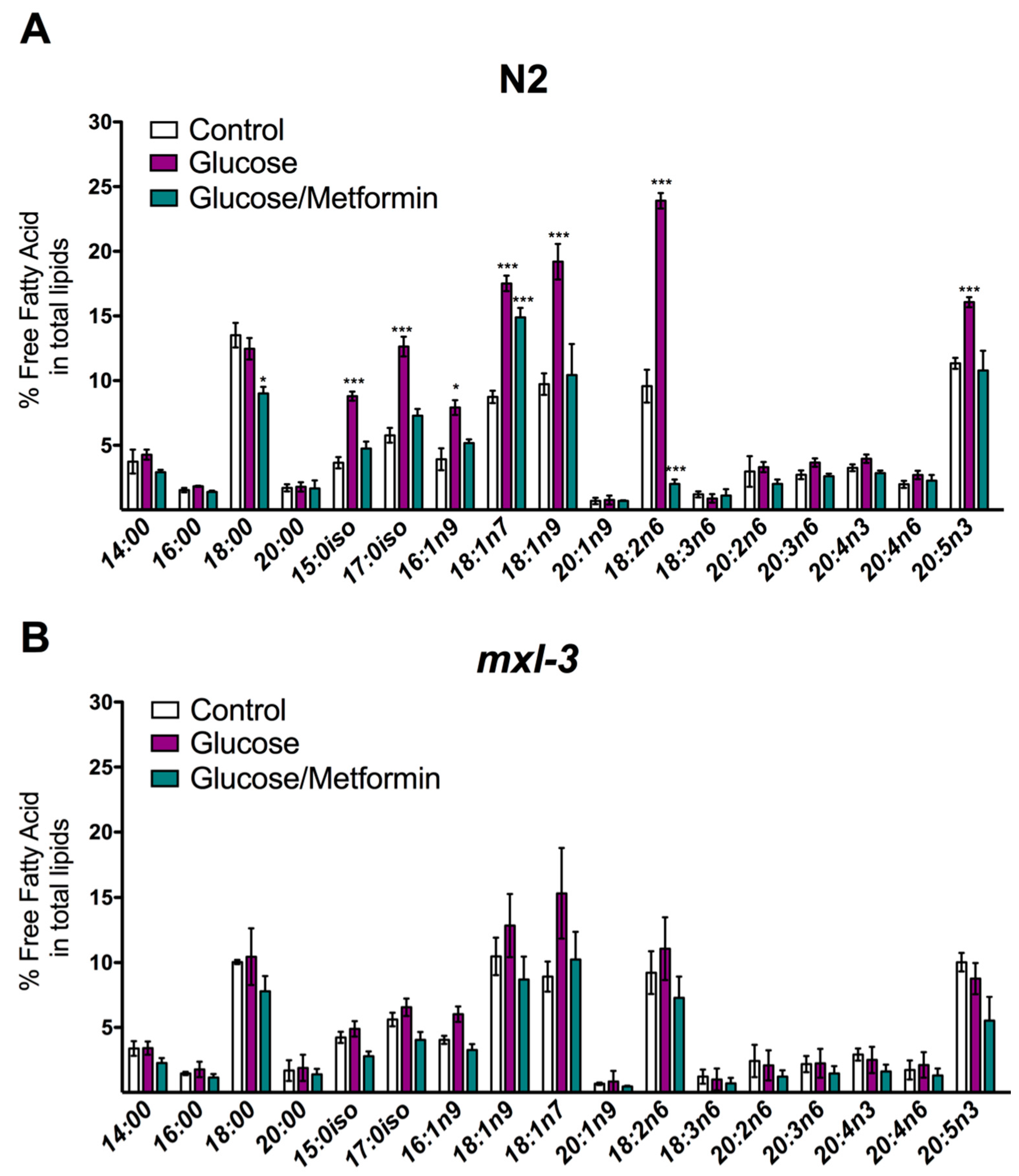
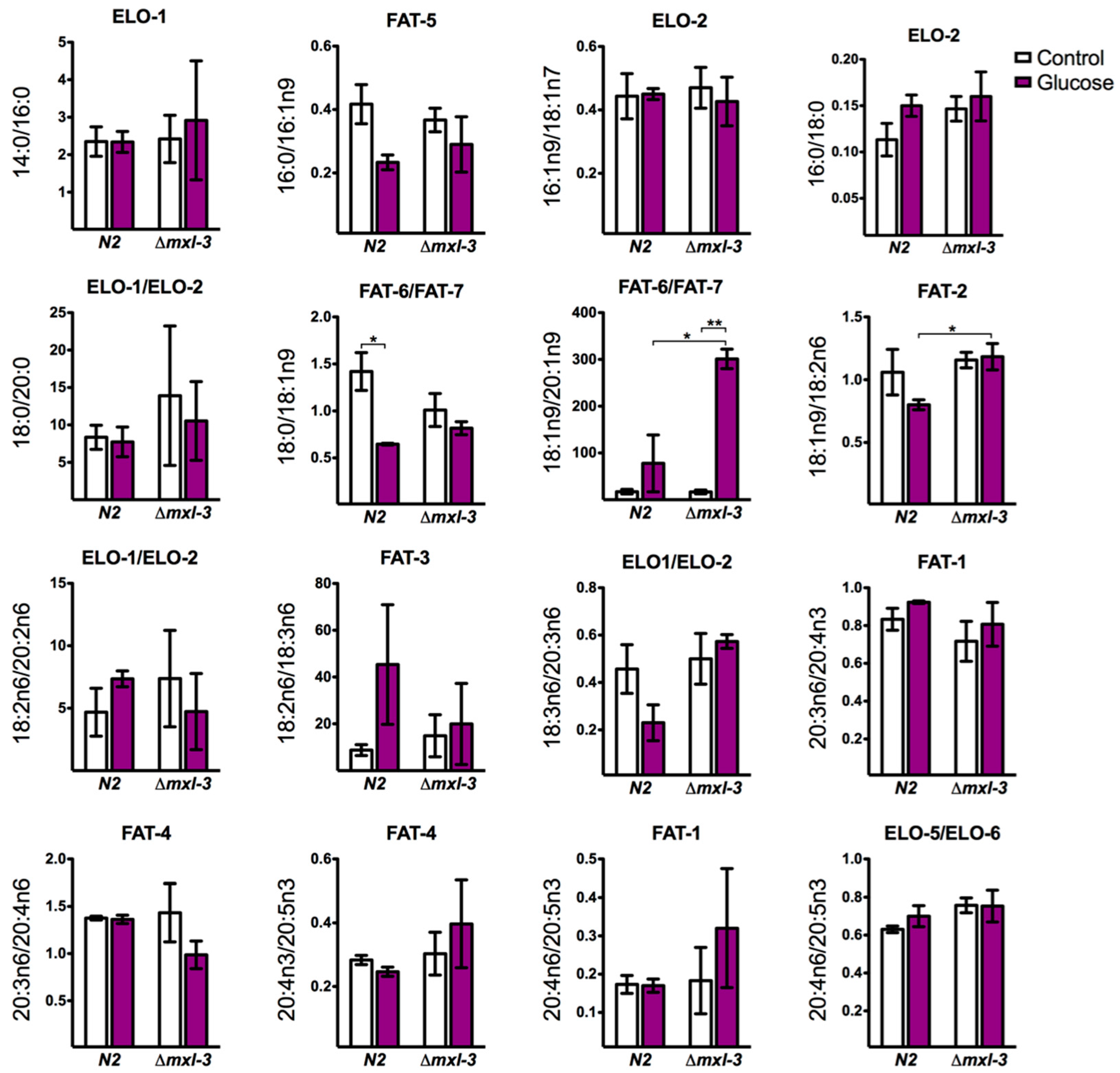
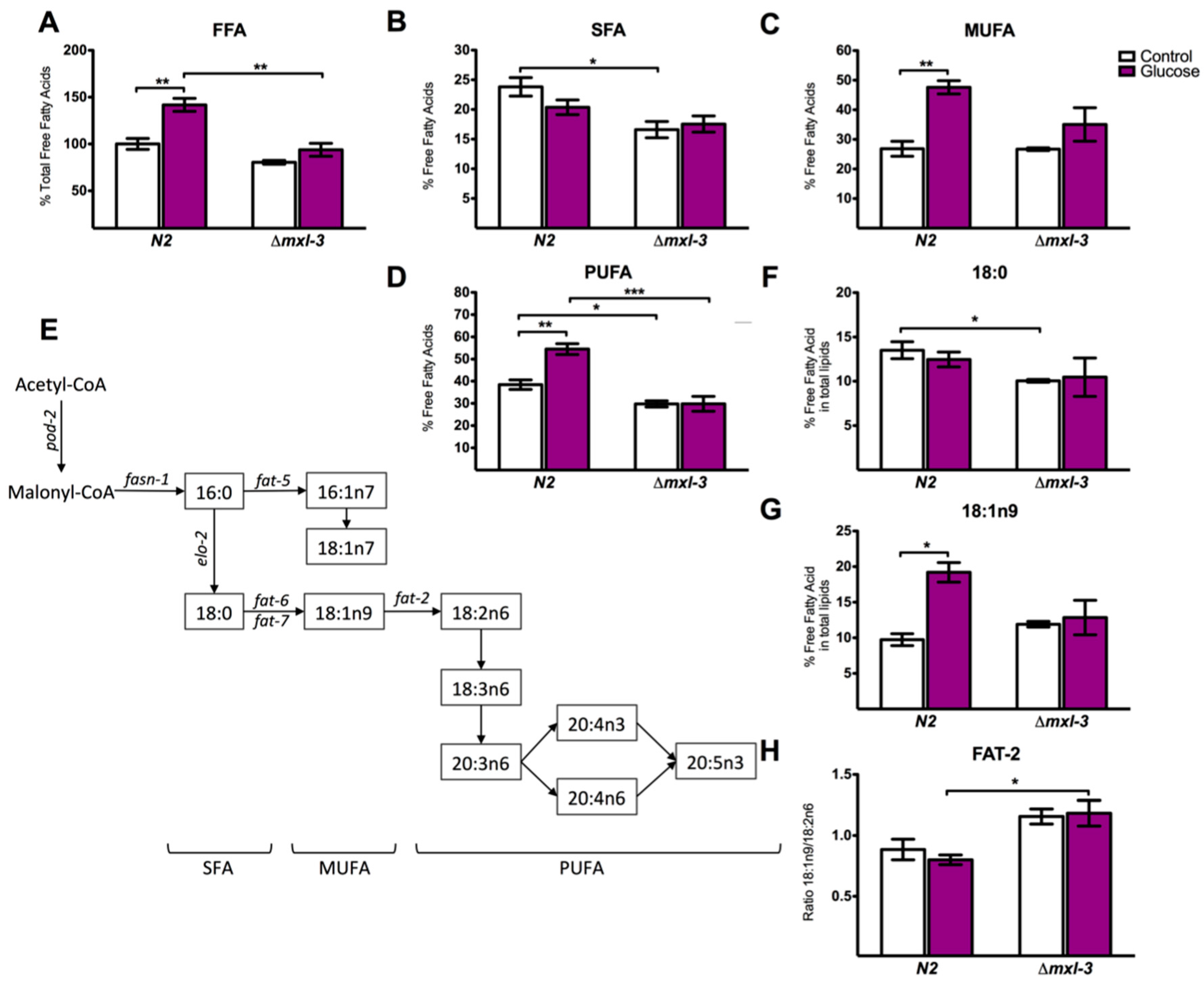
3.4. MXL-3 Is Required to Maintain Fat Stores and De Novo Fat Synthesis
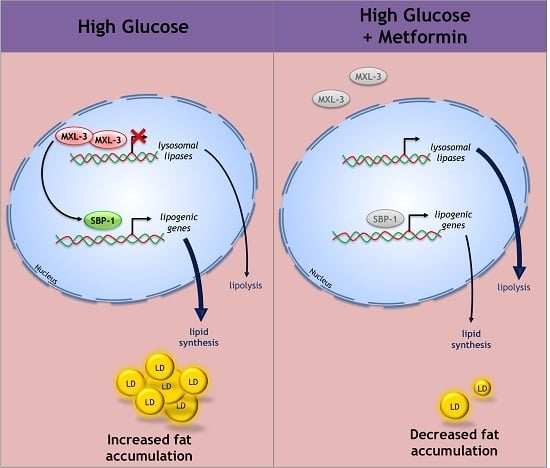
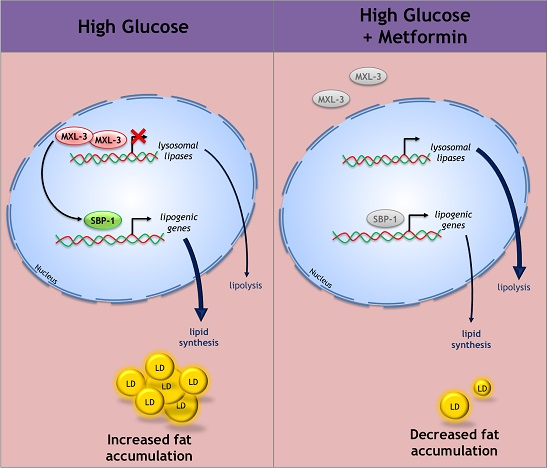
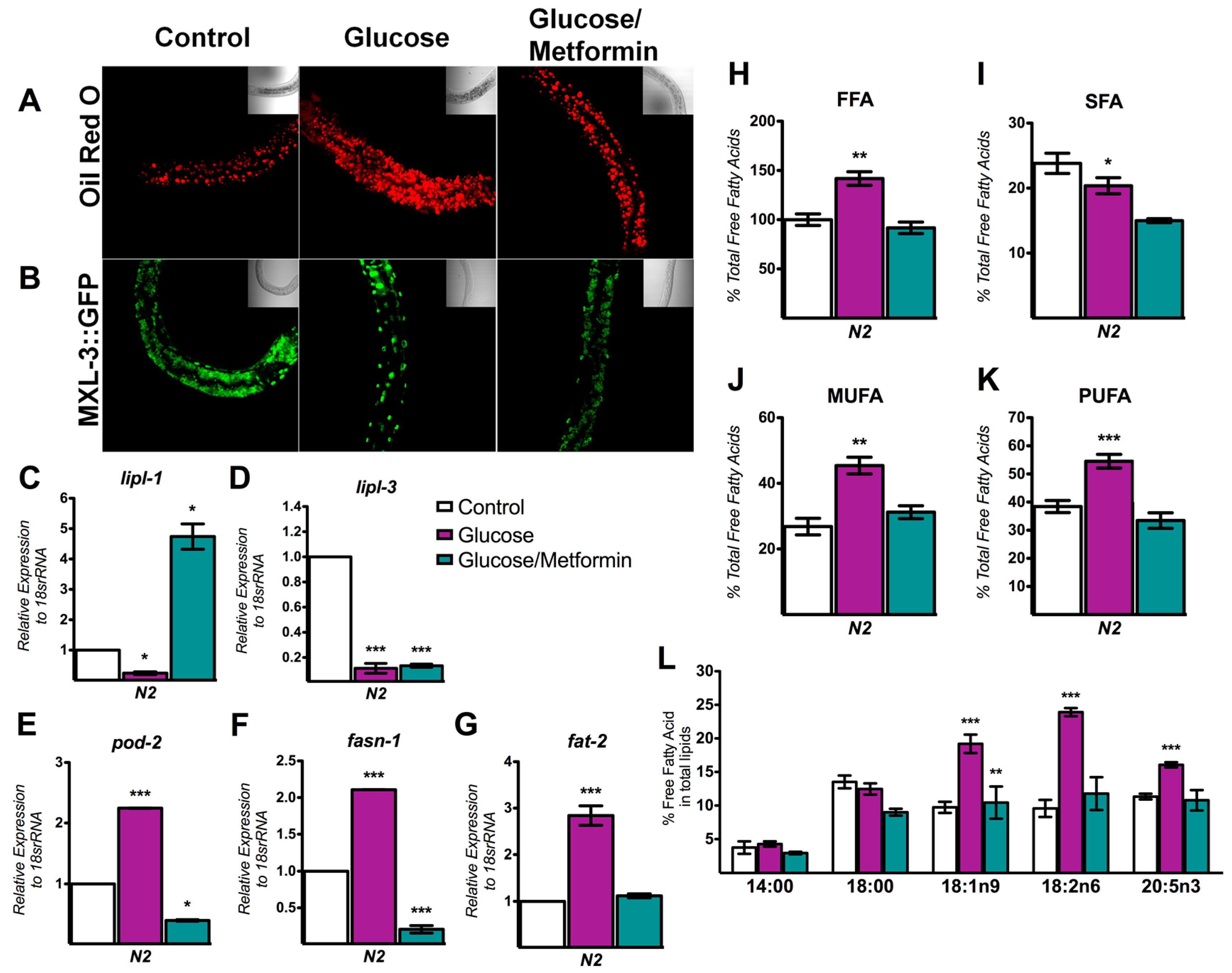
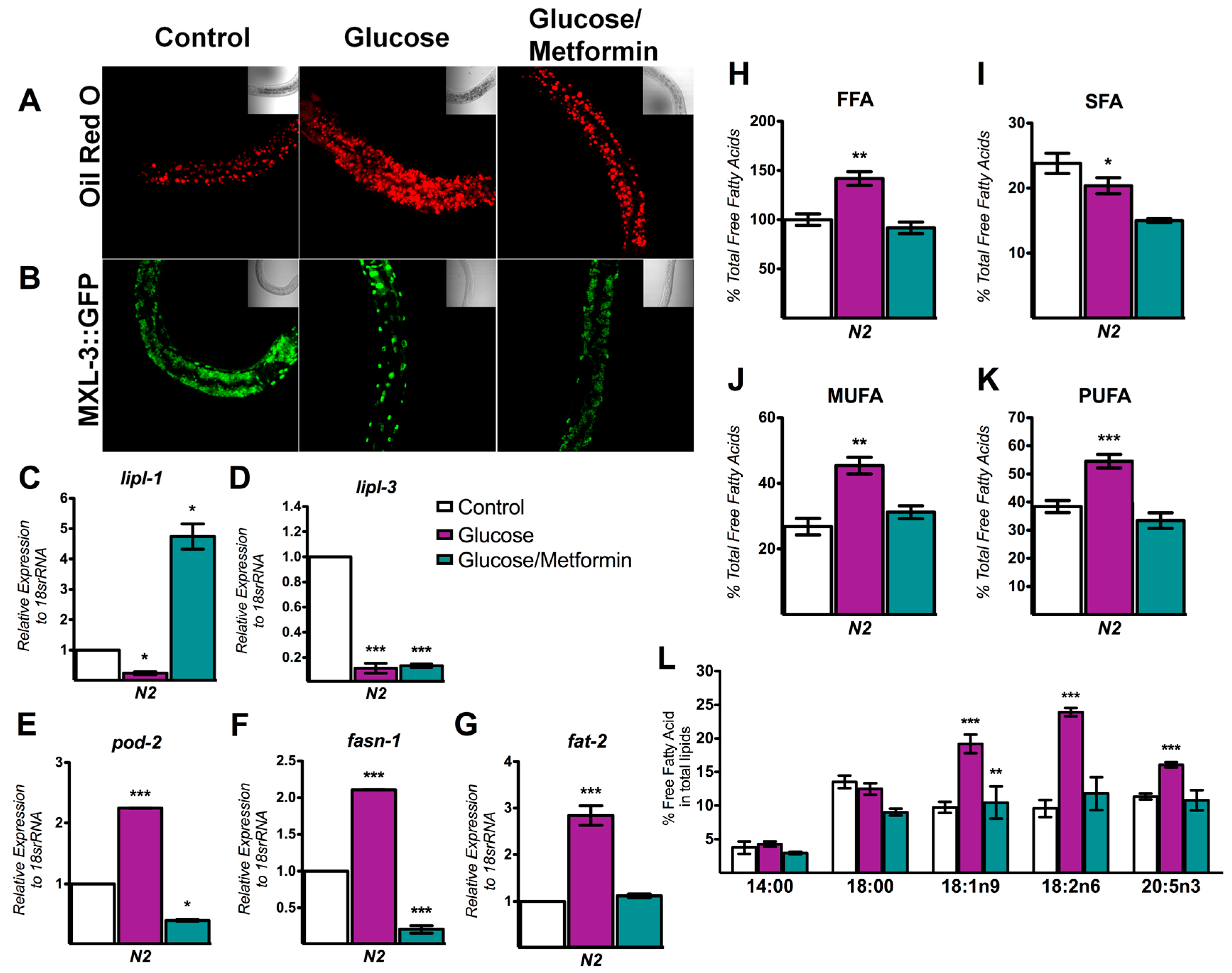
3.5. Metformin Decreases Lipid Overload through MXL-3
4. Discussion
4.1. MXL-3 Senses Nutrient Availability and Regulates the Obese Phenotype during High Glucose Exposure
4.2. MXL-3 Converges in the Complex Lipid Biosynthesis Signaling Pathway
4.3. MXL-3 Is a New Player That Mediates the Protective Effect of Metformin against Lipid Accumulation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Muniesa, P.; Martinez-Gonzalez, M.A.; Hu, F.B.; Despres, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Prim. 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef]
- Serlie, M.J.; Ter Horst, K.W.; Brown, A.W. Addendum: Hypercaloric diets with high meal frequency, but not increased meal size, increase intrahepatic triglycerides: A randomized controlled trial. Hepatology 2016, 64, 1814–1816. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Murphy, C.T.; Kenyon, C. Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 2009, 10, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Schlotterer, A.; Kukudov, G.; Bozorgmehr, F.; Hutter, H.; Du, X.; Oikonomou, D.; Ibrahim, Y.; Pfisterer, F.; Rabbani, N.; Thornalley, P.; et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes 2009, 58, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.T.; Ashrafi, K. Caenorhabditis elegans as an emerging model for studying the basic biology of obesity. Dis. Model. Mech. 2009, 2, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jeong, D.E.; Son, H.G.; Yamaoka, Y.; Kim, H.; Seo, K.; Khan, A.A.; Roh, T.Y.; Moon, D.W.; Lee, Y.; et al. SREBP and MDT-15 protect C. elegans from glucose-induced accelerated aging by preventing accumulation of saturated fat. Genes Dev. 2015, 29, 2490–2503. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Vervoorts, J. Regulation of gene transcription by the oncoprotein MYC. Gene 2012, 494, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.Y.; Watson, J.D.; Yan, P.S.; Barsyte-Lovejoy, D.; Khosravi, F.; Wong, W.W.; Farnham, P.J.; Huang, T.H.; Penn, L.Z. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 2003, 13, 882–886. [Google Scholar] [CrossRef]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef] [PubMed]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerstrom, L.; Westermark, U.K.; Larsson, K.; Munksgaard Persson, M.; Hultenby, K.; Lehtio, J.; Einvik, C.; et al. Myc inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, K.; Liu, X.; Huang, L.; Zhao, D.; Li, L.; Gao, M.; Pei, D.; Wang, C.; Liu, X. Srebp-1 interacts with c-Myc to enhance somatic cell reprogramming. Stem Cells 2016, 34, 83–92. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J.; Ruvkun, G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 2013, 15, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Llop, J.R.; Farrell, S.F.; Yuan, J.; Stolzenburg, L.R.; Samuelson, A.V. The Caenorhabditis elegans Myc-Mondo/Mad complexes integrate diverse longevity signals. PLoS Genet. 2014, 10, e1004278. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, L.T.; Pons, C.; Arda, H.E.; Giese, G.E.; Myers, C.L.; Walhout, A.J. Transcription factor activity mapping of a tissue-specific in vivo gene regulatory network. Cell Syst. 2015, 1, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Moreno-Arriola, E.; El Hafidi, M.; Ortega-Cuellar, D.; Carvajal, K. AMP-activated protein kinase regulates oxidative metabolism in Caenorhabditis elegans through the NHR-49 and MDT-15 transcriptional regulators. PLoS ONE 2016, 11, e0148089. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J.; Soukas, A.A.; Carr, C.E.; Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 2009, 10, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Soukas, A.A.; Kane, E.A.; Carr, C.E.; Melo, J.A.; Ruvkun, G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 2009, 23, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Steinbaugh, M.J.; Narasimhan, S.D.; Robida-Stubbs, S.; Moronetti Mazzeo, L.E.; Dreyfuss, J.M.; Hourihan, J.M.; Raghavan, P.; Operana, T.N.; Esmaillie, R.; Blackwell, T.K. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Harlow, E.; Lane, D. Fixing Caenorhabditis elegans in paraformaldehyde. CSH Protoc. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Ly, K.; Reid, S.J.; Snell, R.G. Rapid RNA analysis of individual Caenorhabditis elegans. MethodsX 2015, 2, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Watts, J.L.; Browse, J. Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 5854–5859. [Google Scholar] [CrossRef] [PubMed]
- Pino, E.C.; Webster, C.M.; Carr, C.E.; Soukas, A.A. Biochemical and high throughput microscopic assessment of fat mass in Caenorhabditis elegans. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, T.; Horikawa, M.; Shimamura, S.; Hashimoto, T.; Sakamoto, K. Fat accumulation in Caenorhabditis elegans is mediated by SREBP homolog SBP-1. Genes Nutr. 2010, 5, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Aa, M.P.; Elst, M.A.; van de Garde, E.M.; van Mil, E.G.; Knibbe, C.A.; van der Vorst, M.M. Long-term treatment with metformin in obese, insulin-resistant adolescents: Results of a randomized double-blinded placebo-controlled trial. Nutr. Diabetes 2016, 6, e228. [Google Scholar] [CrossRef] [PubMed]
- Steiger, D.; Furrer, M.; Schwinkendorf, D.; Gallant, P. Max-independent functions of Myc in Drosophila melanogaster. Nat. Genet. 2008, 40, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.L. Fat synthesis and adiposity regulation in Caenorhabditis elegans. Trends Endocrinol. Metab. 2009, 20, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Grove, C.A.; De Masi, F.; Barrasa, M.I.; Newburger, D.E.; Alkema, M.J.; Bulyk, M.L.; Walhout, A.J. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 2009, 138, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heide, L.P.; Hoekman, M.F.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S.; Andrali, S.S.; Cantrell, J.E. Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 2010, 1799, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, M.; Kim, R.; Fukushima, H.; Sasaki, K.; Suzuki, K.; Okamura, M.; Ishii, Y.; Kawashima, T.; Kamijo, S.; Takemoto-Kimura, S.; et al. Region-specific activation of CRTC1-CREB signaling mediates long-term fear memory. Neuron 2014, 84, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Gruner, M.; Grubbs, J.; McDonagh, A.; Valdes, D.; Winbush, A.; van der Linden, A.M. Cell-autonomous and non-cell-autonomous regulation of a feeding state-dependent chemoreceptor gene via MEF-2 and bHLH transcription factors. PLoS Genet. 2016, 12, e1006237. [Google Scholar] [CrossRef] [PubMed]
- Tullet, J.M.; Araiz, C.; Sanders, M.J.; Au, C.; Benedetto, A.; Papatheodorou, I.; Clark, E.; Schmeisser, K.; Jones, D.; Schuster, E.F.; et al. DAF-16/FoxO directly regulates an atypical AMP-activated protein kinase gamma isoform to mediate the effects of insulin/IGF-1 signaling on aging in Caenorhabditis elegans. PLoS Genet. 2014, 10, e1004109. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejia-Martinez, F.; Franco-Juarez, B.; Moreno-Arriola, E.; Hernández-Vázquez, A.; Martinez-Avila, M.; Gómez-Manzo, S.; Marcial-Quino, J.; Carvajal, K.; Velazquez-Arellano, A.; Ortega-Cuellar, D. The MXL-3/SBP-1 Axis Is Responsible for Glucose-Dependent Fat Accumulation in C. elegans. Genes 2017, 8, 307. https://doi.org/10.3390/genes8110307
Mejia-Martinez F, Franco-Juarez B, Moreno-Arriola E, Hernández-Vázquez A, Martinez-Avila M, Gómez-Manzo S, Marcial-Quino J, Carvajal K, Velazquez-Arellano A, Ortega-Cuellar D. The MXL-3/SBP-1 Axis Is Responsible for Glucose-Dependent Fat Accumulation in C. elegans. Genes. 2017; 8(11):307. https://doi.org/10.3390/genes8110307
Chicago/Turabian StyleMejia-Martinez, Fanny, Berenice Franco-Juarez, Elizabeth Moreno-Arriola, Alain Hernández-Vázquez, Marco Martinez-Avila, Saul Gómez-Manzo, Jaime Marcial-Quino, Karla Carvajal, Antonio Velazquez-Arellano, and Daniel Ortega-Cuellar. 2017. "The MXL-3/SBP-1 Axis Is Responsible for Glucose-Dependent Fat Accumulation in C. elegans" Genes 8, no. 11: 307. https://doi.org/10.3390/genes8110307