High Quality Unigenes and Microsatellite Markers from Tissue Specific Transcriptome and Development of a Database in Clusterbean (Cyamopsis tetragonoloba, L. Taub)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Isolation
2.2. Library Preparation and RNA Sequencing
2.3. RNA-Seq Data Processing and De Novo Assembly
2.4. Functional Annotation of High Quality Unigenes
2.5. Differential Gene Expression Analysis
2.6. Simple Sequence Repeat Mining
2.7. Marker Validation and Diversity Analysis
2.8. Database Design
3. Results
3.1. Transcriptome Sequencing and De Novo Assembly
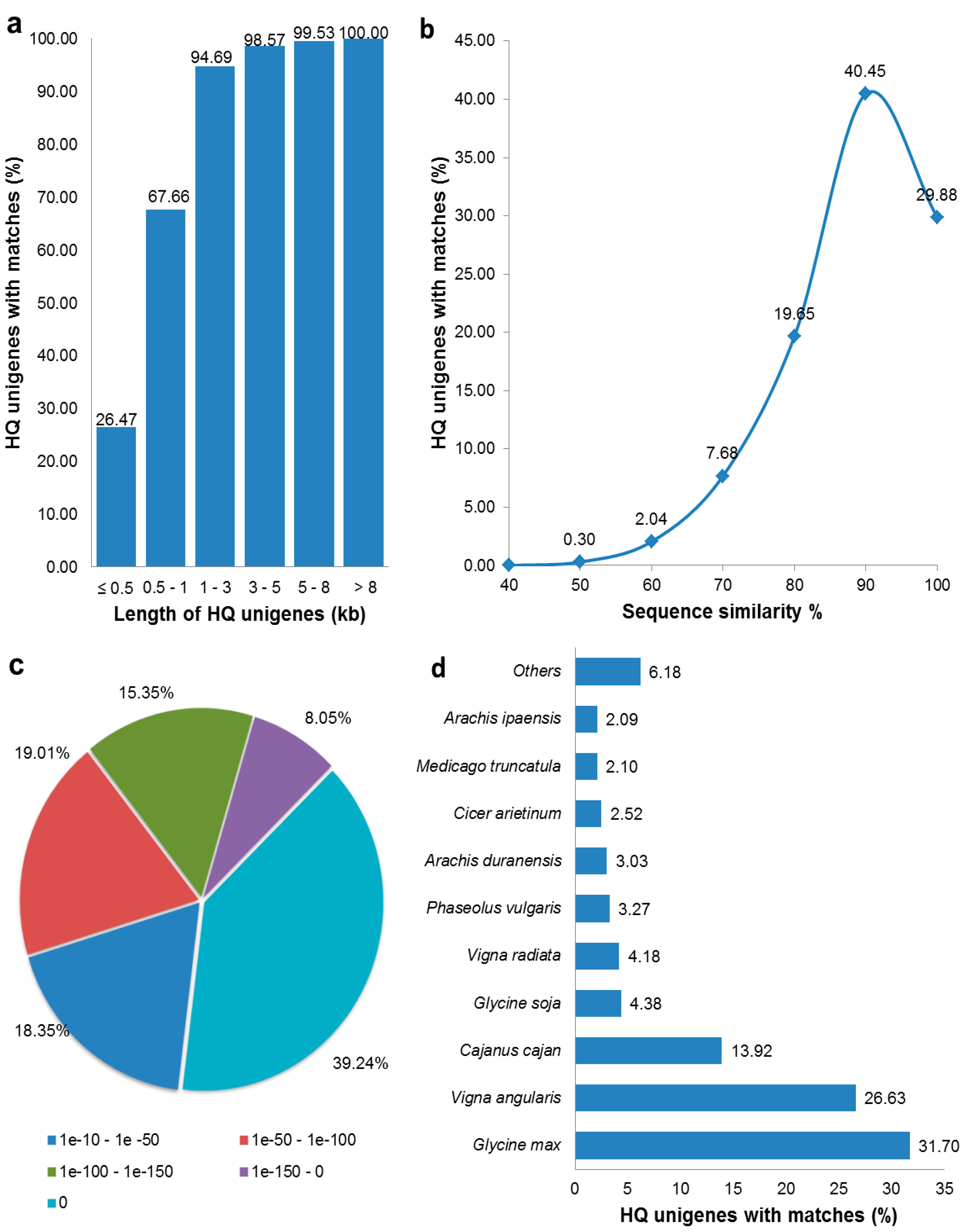
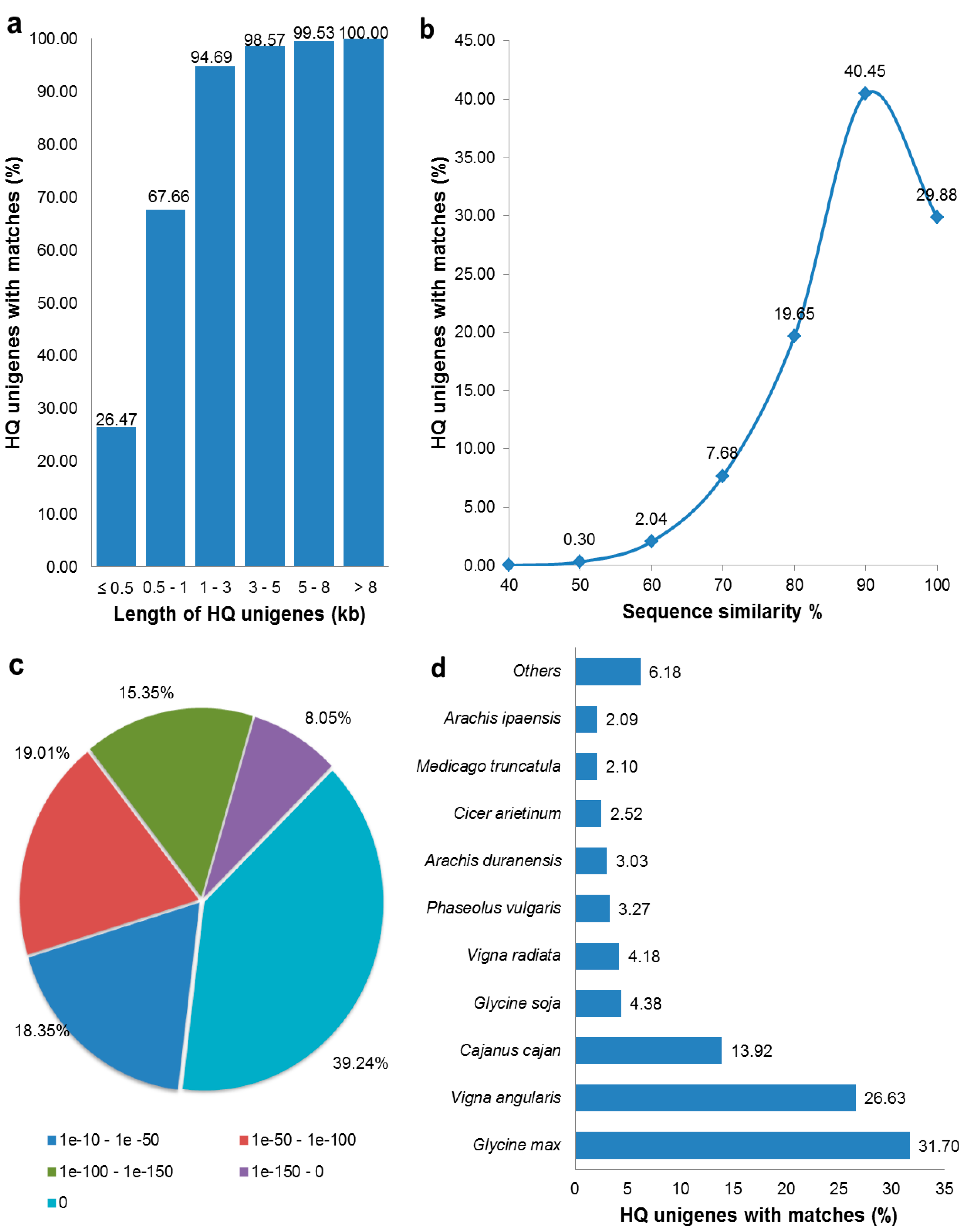
3.2. Functional Annotation of HQ Unigenes
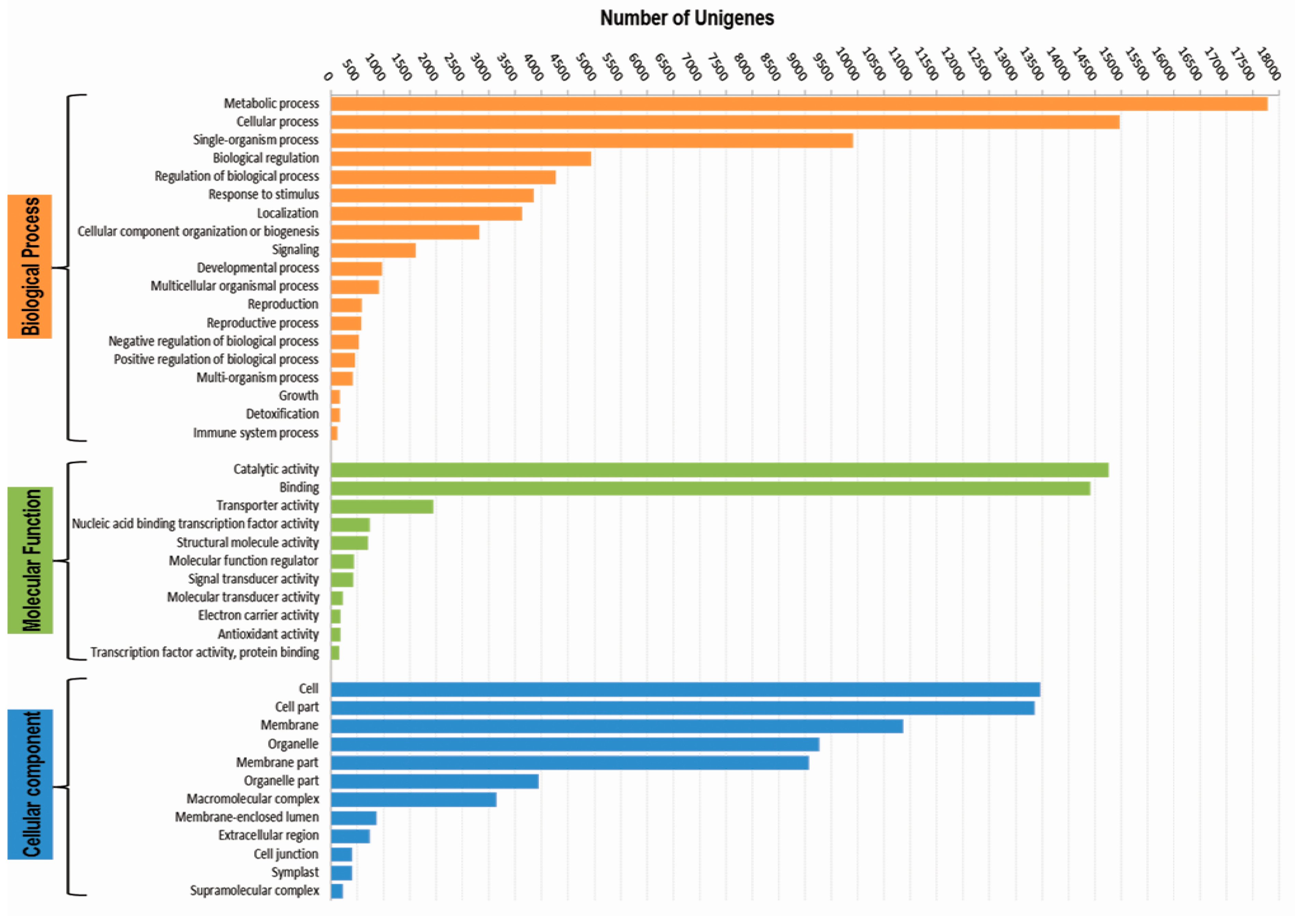
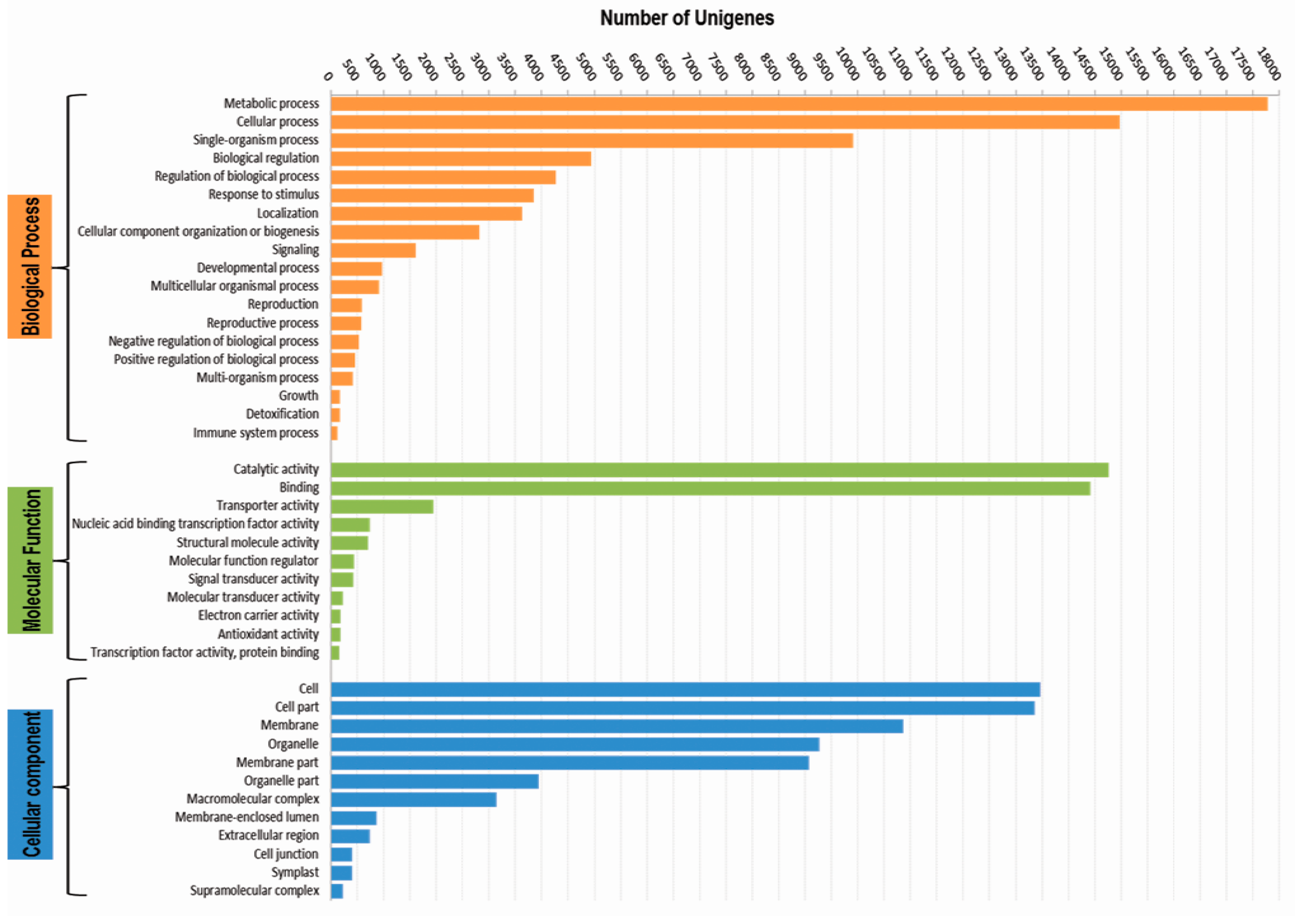
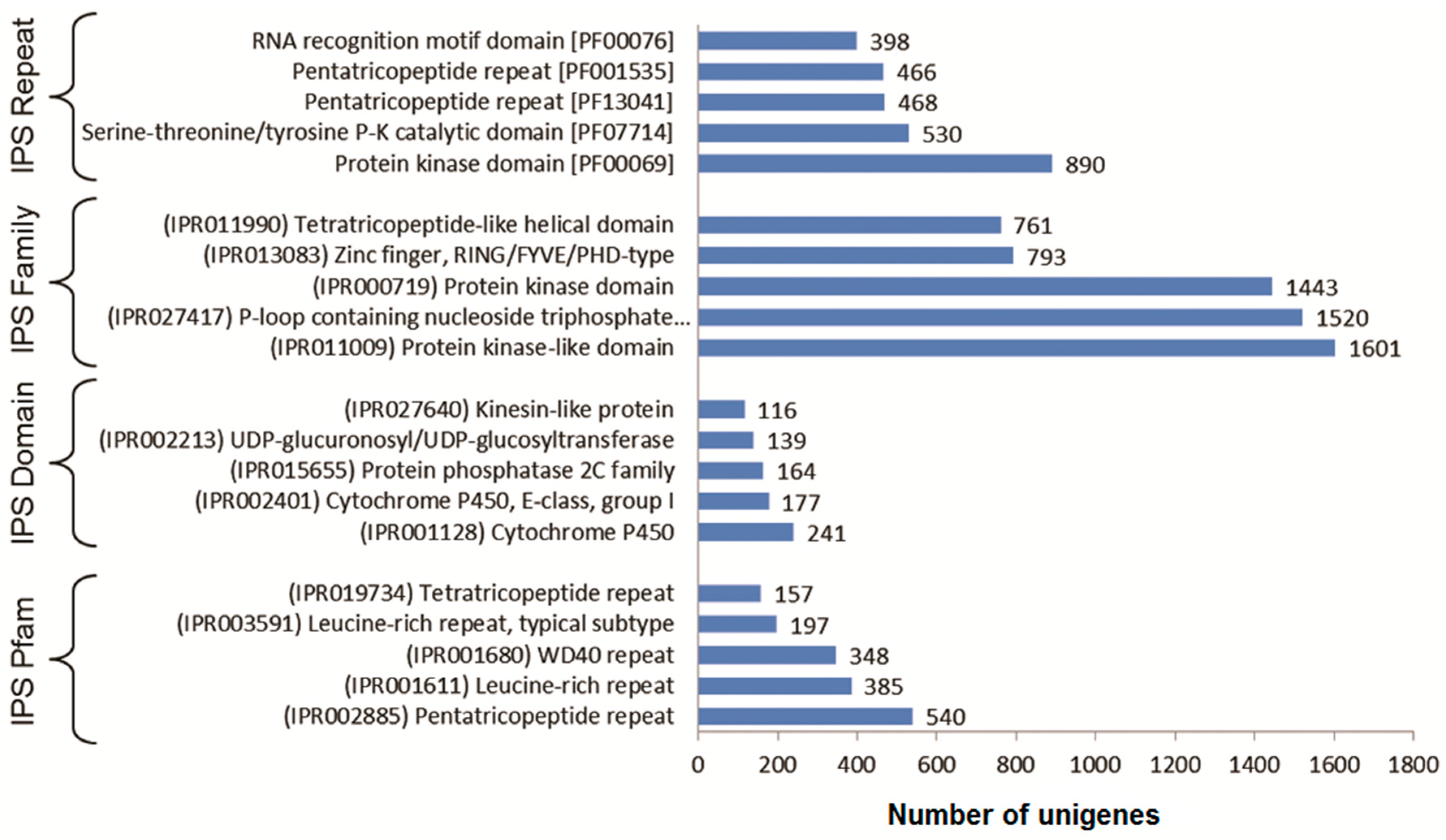
3.3. Functional Classification with Gene Ontology Terms, Enzyme Commission Numbers and InterproScan Search
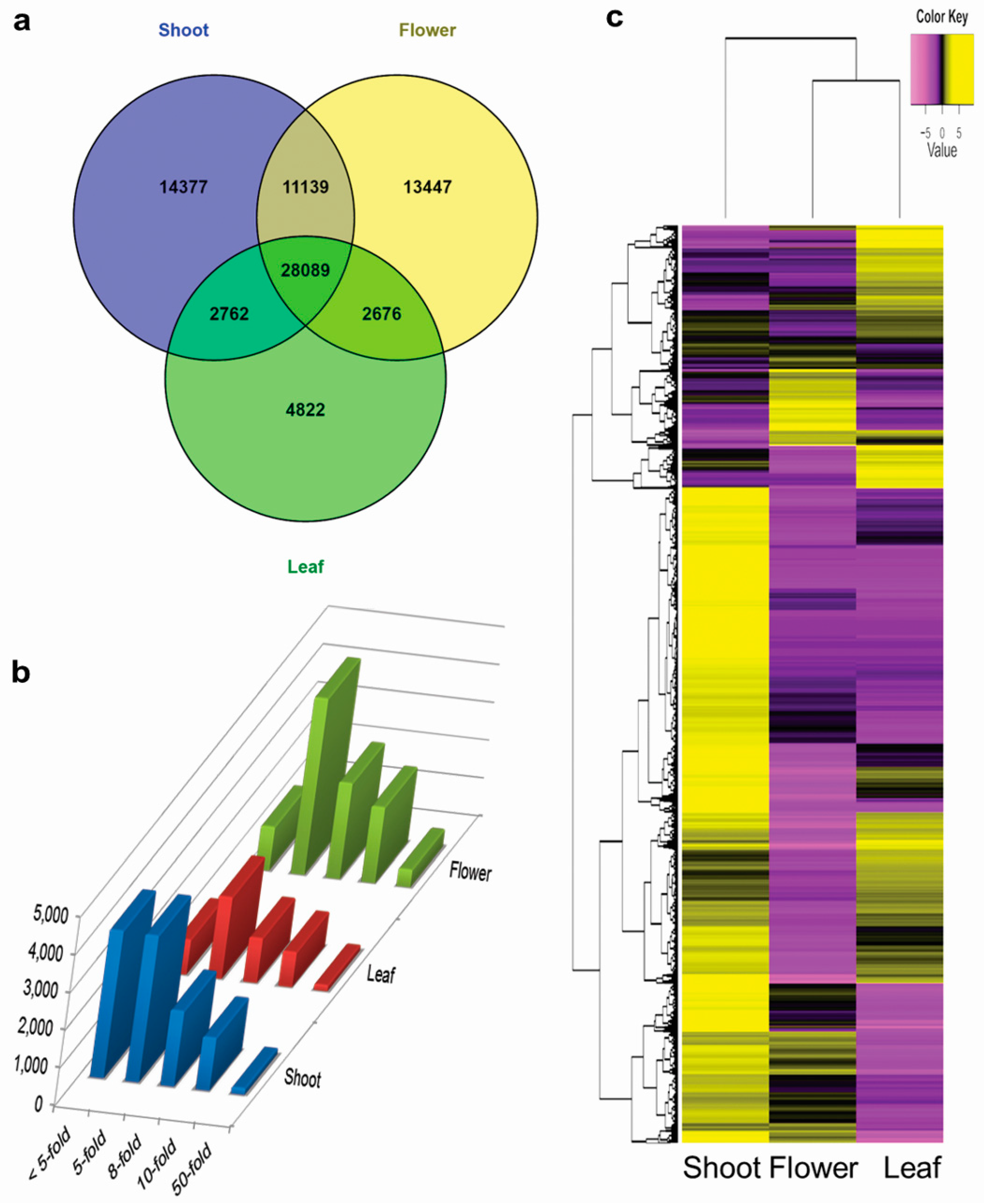
3.4. Tissue-Specific Transcriptome Analysis
3.5. Differential Expression of Genes
3.6. Simple Sequence Repeat Prediction
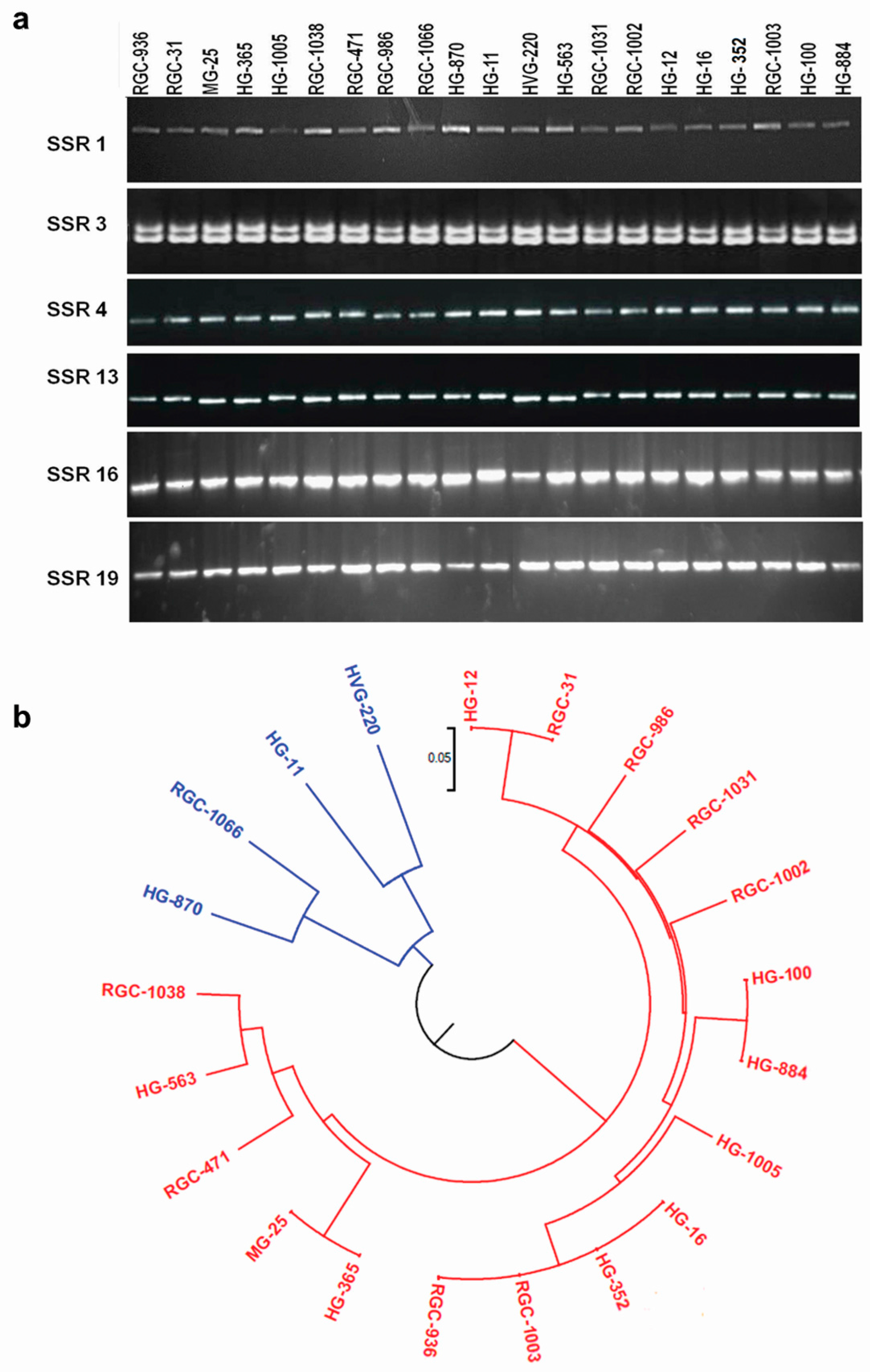
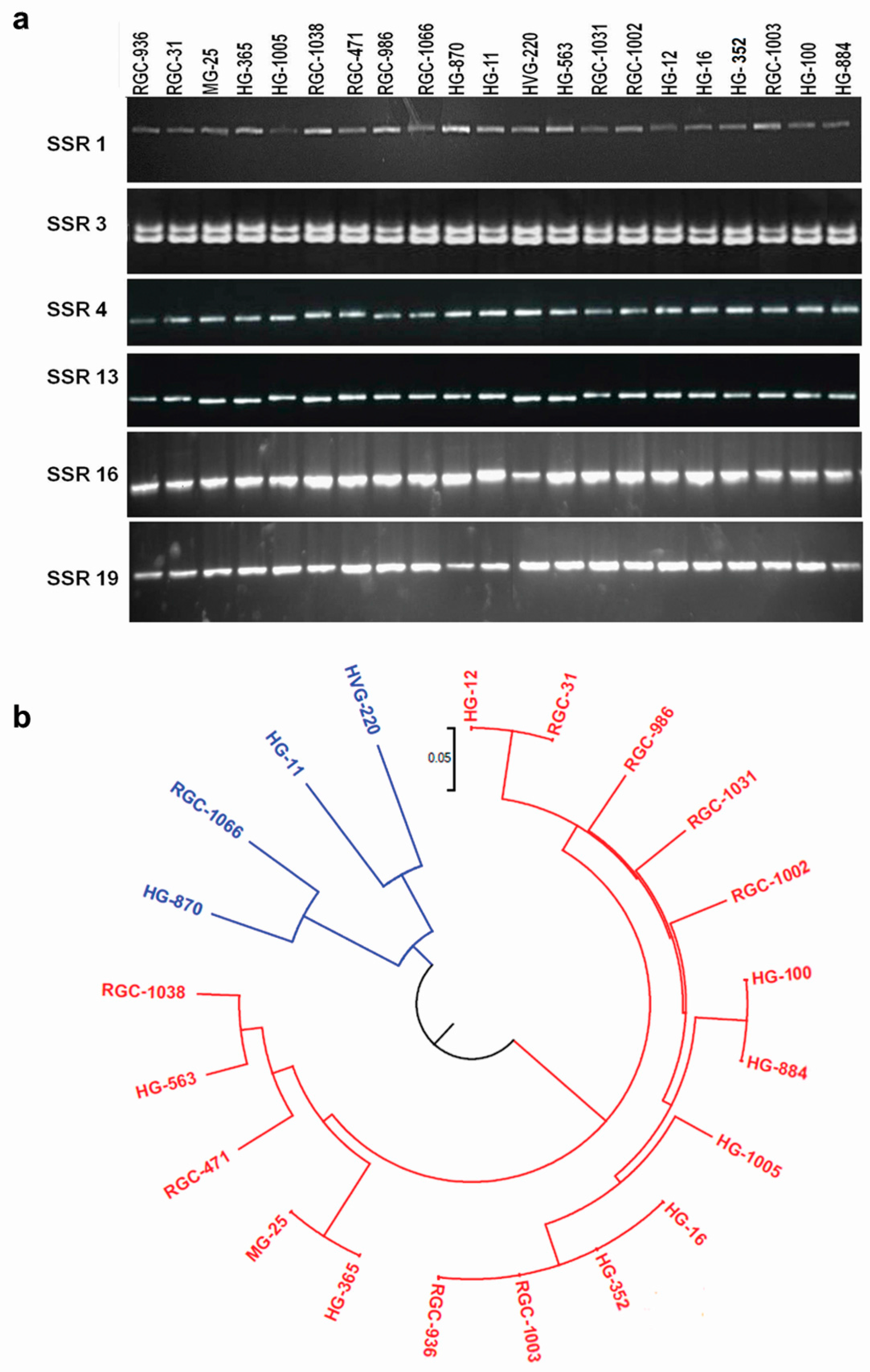
3.7. Marker Validation
3.8. Database for Clusterbean Unigenes and Microsatellite Markers
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krishnan, G.S.; Dwivedi, N.K.; Singh, J.P. Primitive weedy forms of guar, adak guar: Possible missing link in the domestication of guar Cyamopsis tetragonoloba (L.). Genet. Resour. Crop Evol. 2011, 58, 961–996. [Google Scholar] [CrossRef]
- Francois, L.E.; Donovan, T.J.; Maas, E.V. Salinity effects on emergence, vegetative growth, and seed yield of guar. Agron. J. 1990, 82, 587–592. [Google Scholar] [CrossRef]
- Ashraf, M.Y.; Akhtar, K.; Sarwar, G.; Ashraf, M. Evaluation of arid and semi-arid ecotypes of guar (Cyamopsis tetragonoloba L.) for salinity (NaCl) tolerance. J. Arid Environ. 2002, 52, 473–482. [Google Scholar] [CrossRef]
- Dhugga, K.S.; Barreiro, R.; Whitten, B.; Stecca, K.; Hazebroek, J.; Randhawa, G.S.; Dolan, M.; Kinney, A.J.; Tomes, D.; Nichols, S.; et al. Guar seed β-mannan synthase is a member of the cellulose synthase super gene family. Science 2004, 303, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.F.; Berhow, M.A.; Winkler-Moser, J.K.; Lee, E. Formulation of a biodegradable, odor-reducing cat litter from solvent-extracted corn dried distillers grains. Ind. Crops Prod. 2011, 34, 999–1002. [Google Scholar] [CrossRef]
- Lubbe, A.; Verpoorte, R. Cultivation of Medicinal and Aromatic Plants for Specialty Industrial Materials. Ind. Crop Prod. 2011, 34, 785–801. [Google Scholar] [CrossRef]
- Butt, M.S.; Shahzadi, N.; Sharif, M.K.; Shahzadi, N.; Sharif, M.K.; Nasir, M. Guar gum: A miracle therapy for hypercholesterolemia, hyperglycemia and obesity. Crit. Rev. Food Sci. Nutr. 2007, 47, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Mathur, N.K. Industrial Galactomannan Polysaccharides; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2012; p. 187. [Google Scholar]
- Sharma, P.; Gummagolmath, K.C. Reforming Guar Industry in India: Issues and Strategies. Agric. Econ. Res. Rev. 2012, 25, 37–48. [Google Scholar] [CrossRef]
- Rai, D.K. Trends and Economic Dynamics of Guar in India; Indian Council for Research on International Economic Relations: New Delhi, India, 2015. [Google Scholar]
- Kumawat, G.; Raje, R.S.; Bhutani, S.; Pal, J.K.; Mithra, S.V.; Gaikwad, K.; Sharma, T.R.; Singh, N.K. Molecular mapping of QTLs for plant type and earliness traits in pigeonpea (Cajanus cajan L. Millsp.). BMC Genet. 2012, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Gupta, D.K.; Jayaswal, P.K.; Mahato, A.K.; Dutta, S.; Singh, S.; Bhutani, S.; Dogra, V.; Singh, B.P.; Kumawat, G.; et al. The first draft of the pigeonpea genome sequence. J. Plant Biochem. Biotechnol. 2012, 21, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.A.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan L.), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2010, 30, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naoumkina, M.; Torres-Jerez, I.; Allen, S.; He, J.; Zhao, P.X.; Dixon, R.A.; May, G.D. Analysis of cDNA libraries from developing seeds of guar (Cyamopsis tetragonoloba (L.) Taub). BMC Plant Biol. 2007, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, U.K.; Pruthi, V.; Randhawa, G.S. RNA-Seq of Guar (Cyamopsis tetragonoloba, L. Taub.) Leaves: De novo Transcriptome Assembly, Functional Annotation and Development of Genomic Resources. Front. Plant Sci. 2017, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Kaila, T.; Chaduvla, P.K.; Rawal, H.C.; Saxena, S.; Tyagi, A.; Mithra, S.V.A.; Solanke, A.U.; Kalia, P.; Sharma, T.R.; Singh, N.K.; et al. Chloroplast Genome Sequence of Clusterbean (Cyamopsis tetragonoloba L.): Genome Structure and Comparative Analysis. Genes 2017, 8, 212. [Google Scholar] [CrossRef] [PubMed]
- Punia, A.; Yadav, R.; Arora, P.; Chaudhury, A. Molecular and morphophysiological characterization of superior cluster bean (Cymopsis tetragonoloba) varieties. J. Crop Sci. Biotechnol. 2009, 12, 143–148. [Google Scholar] [CrossRef]
- Pathak, R.; Singh, S.K.; Singh, M. Assessment of genetic diversity in clusterbean using nuclear rDNA and RAPD markers. J. Food Legum. 2011, 24, 180–183. [Google Scholar]
- Pathak, R.; Singh, S.K.; Singh, M.; Henry, A. Molecular assessment of genetic diversity in cluster bean (Cyamopsis tetragonoloba) genotypes. J. Genet. 2010, 89, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, V.; Raman, K.V.; Tiwari, K. A set of SCAR markers in cluster bean (Cyamopsis tetragonoloba L. Taub) genotypes. Adv. Biosci. Biotechnol. 2014, 5, 131–141. [Google Scholar] [CrossRef]
- Gresta, F.; Mercati, F.; Santonoceto, C.; Abenavoli, M.R.; Ceravolo, G.; Araniti, F.; Anastasi, U.; Sunseri, F. Morpho-agronomic and AFLP characterization to explore guar (Cyamopsis tetragonoloba L.) genotypes for the 1059 Mediterranean environment. Ind. Crop Prod. 2016, 86, 23–30. [Google Scholar] [CrossRef]
- Kumar, S.; Parekh, M.J.; Patel, C.B.; Zala, H.N.; Sharma, R.; Kulkarni, K.S.; Fougat, R.S.; Bhatt, R.K.; Sakure, A.A. Development and validation of EST-derived SSR markers and diversity analysis in cluster bean (Cyamopsis tetragonoloba). J. Plant Biochem. Biotechnol. 2016, 25, 263–269. [Google Scholar] [CrossRef]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F.; et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Volfovsky, N.; Town, C.D.; Troukhan, M.; Alexandrov, N.; Feldmann, K.A.; Flavell, R.B.; White, O.; Salzberg, S.L. Full-length messenger RNA sequences greatly improve genome annotation. Genome Biol. 2002, 3, research0029:1–research0029:12. [Google Scholar] [CrossRef]
- Cloonan, N.; Forrest, A.R.R.; Kolle, G.; Gardiner, B.B.A.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Marguerat, S.; Goodhead, I.; Bahler, J. Defining transcribed regions using RNA-Seq. Nat. Protoc. 2010, 5, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Paneru, B.; Al-Tobasei, R.; Abdouni, F.; Thorgaard, G.H.; Rexroad, C.E.; Yao, J. Transcriptome Assembly, Gene Annotation and Tissue Gene Expression Atlas of the Rainbow Trout. PLoS ONE 2015, 10, e0121778. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Shi, C.Y.; Yang, H.; Wei, C.L.; Yu, O.; Zhang, Z.Z.; Jiang, C.J.; Sun, J.; Li, Y.Y.; Chen, Q.; Xia, T.; et al. Deep sequencing of the C. sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.K.; Bhandari, D.C.; Dubas, B.S.; Agrawal, R.C.; Mandal, S.; Rana, R.S. Catalogue on Cluster Bean (Cyamopsis tetragonoloba (L.) Taub) Germplasm Part III; NBPGR: New Delhi, India, 1995. [Google Scholar]
- Patil, C.G. Nuclear DNA Amount Variation in Cyamopsis D.C. (Fabaceae). Cytologia 2004, 69, 59–62. [Google Scholar] [CrossRef]
- Nakasugi, K.; Crowhurst, R.N.; Bally, J.; Wood, C.C.; Hellens, R.P.; Waterhouse, P.M. De novo transcriptome sequence assembly and analysis of RNA silencing genes of Nicotiana benthamiana. PLoS ONE 2013, 8, e59534. [Google Scholar] [CrossRef] [PubMed]
- Mora-Ortiz, M.; Swain, M.T.; Vickers, M.J.; Hegarty, M.J.; Kelly, R.; Smith, L.M.J.; Skøt, L. De novo transcriptome assembly for gene identification, analysis, annotation, and molecular marker discovery in Onobrychis viciifolia. BMC Genom. 2016, 17, 756. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Patel, R.; Tyagi, A.; Jain, M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kudapa, H.; Azam, S.; Sharpe, A.G.; Taran, B.; Li, R.; Deonovic, B.; Cameron, C.; Farmer, A.D.; Cannon, S.B.; Varshney, R.K. Comprehensive transcriptome assembly of Chickpea (Cicer arietinum L.) using Sanger and Next Generation Sequencing platforms: Development and applications. PLoS ONE 2014, 9, e86039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liang, S.; Duan, J.; Wang, J.; Chen, S.; Cheng, Z.; Zhang, Q.; Liang, X.; Li, Y. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.). BMC Genom. 2012, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, L.; Wang, S.; Liu, C.; Blair, M.W.; Cheng, X. Transcriptome sequencing of mung bean (Vigna radiata L.) genes and the identification of EST-SSR markers. PLoS ONE 2015, 10, e0120273. [Google Scholar] [CrossRef]
- Liu, C.; Fan, B.; Cao, Z.; Su, Q.; Wang, Y.; Zhang, Z.; Wu, J.; Tian, J. A deep sequencing analysis of transcriptomes and the development of EST-SSR markers in mungbean (Vigna radiata). J. Genet. 2016, 95, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Dinkins, R.D.; Hunt, A.G. De novo transcriptome assembly and dynamic spatial gene expression analysis in red clover. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Barbara, T.; Palma-Silva, C.; Paggi, G.M.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Nepolean, T.; Singh, I.; Hossain, F.; Pandey, N.; Gupta, H.S. Molecular characterization and assessment of genetic diversity of inbred lines showing variability for drought tolerance in maize. J. Plant Biochem. Biotechnol. 2013, 22, 71. [Google Scholar] [CrossRef]
- Wu, J.; Wang, L.; Li, L.; Wang, S. De novo assembly of the common bean transcriptome using short reads for the discovery of drought-responsive genes. PLoS ONE 2014, 9, e109262. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library/Sample | Number of Raw Reads (Paired) | Number of HQ Reads (Paired) | Number of HQ Reads (Un-Paired) | HQ Reads (Bases) | Average Length (HQ Paired Reads) |
|---|---|---|---|---|---|
| Flower | 16,325,263 | 11,896,062 | 3,967,411 | 2,408,942,984 | 88.54 |
| Leaf | 41,575,280 | 30,676,700 | 9,701,552 | 6,192,249,152 | 88.91 |
| Shoot | 92,030,452 | 66,439,261 | 22,849,685 | 13,560,175,894 | 88.90 |
| Total | 149,930,995 | 109,012,023 | 36,518,648 | 22,161,368,030 |
| Assembly Statistics | Data |
|---|---|
| Transcripts | |
| Total Assembled | 127,706 (179.50 Mb) |
| Average Length | 1405.63 bp |
| GC% | 39.22 |
| ≥1000 bp | 64,606 (150.06 Mb) |
| ≥5000 bp | 2218 (138.75 Mb) |
| ≥10,000 bp | 53 (628.55 kb) |
| Largest Transcripts | 16,940 bp |
| N50 Length | 2263 bp |
| N75 Length | 2931 bp |
| L50 | 26,460 |
| L75 | 14,819 |
| Unigenes | |
| Total Number | 110,485 (152.13 Mb) |
| Average Length | 1376.95 bp |
| Number of HQ Unigenes | 48,007 (76.01 Mb) |
| Average Length (HQ Unigenes) | 1583.43 bp |
| N50 Length (HQ Unigenes) | 2179 bp |
| GC% (HQ Unigenes) | 39.87 |
| Annotation of HQ Unigenes | |
| Database Searched | Unigenes with significant hits |
| Against NCBI-Plant-Genes | 37,382 |
| Against SwissProt DB | 28,905 |
| Against Pfam | 34,752 |
| With Gene Ontology (GO) terms | 30,020 |
| With Enzyme Commission (EC) numbers | 9984 |
| All annotated transcript | 37,442 |
| With No Significant hit | 10,565 |
| Features | Transcripts | HQ Unigenes |
|---|---|---|
| Total number of sequences examined | 127,706 | 48,007 |
| Total size of examined sequences (bp) | 179,507,503 | 76,015,970 |
| Total number of identified SSRs | 17,593 | 8687 |
| Number of SSR containing sequences | 14,566 | 7047 |
| Number of sequences containing more than 1 SSR | 2430 | 1297 |
| Number of SSRs present in compound formation | 1137 | 590 |
| Frequency of SSRs | 1 SSR/10.20 kb | 1 SSR/8.75 kb |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rawal, H.C.; Kumar, S.; Mithra S.V., A.; Solanke, A.U.; Nigam, D.; Saxena, S.; Tyagi, A.; V., S.; Yadav, N.R.; Kalia, P.; et al. High Quality Unigenes and Microsatellite Markers from Tissue Specific Transcriptome and Development of a Database in Clusterbean (Cyamopsis tetragonoloba, L. Taub). Genes 2017, 8, 313. https://doi.org/10.3390/genes8110313
Rawal HC, Kumar S, Mithra S.V. A, Solanke AU, Nigam D, Saxena S, Tyagi A, V. S, Yadav NR, Kalia P, et al. High Quality Unigenes and Microsatellite Markers from Tissue Specific Transcriptome and Development of a Database in Clusterbean (Cyamopsis tetragonoloba, L. Taub). Genes. 2017; 8(11):313. https://doi.org/10.3390/genes8110313
Chicago/Turabian StyleRawal, Hukam C., Shrawan Kumar, Amitha Mithra S.V., Amolkumar U. Solanke, Deepti Nigam, Swati Saxena, Anshika Tyagi, Sureshkumar V., Neelam R. Yadav, Pritam Kalia, and et al. 2017. "High Quality Unigenes and Microsatellite Markers from Tissue Specific Transcriptome and Development of a Database in Clusterbean (Cyamopsis tetragonoloba, L. Taub)" Genes 8, no. 11: 313. https://doi.org/10.3390/genes8110313