Next Generation Sequencing of Chromosome-Specific Libraries Sheds Light on Genome Evolution in Paleotetraploid Sterlet (Acipenser ruthenus)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Origin
2.2. Chromosome Preparation, Staining, Probe Generation and Painting
2.3. Next Generation Sequencing
2.4. Alignment of the Sterlet Chromosome-Specific Library to the Spotted Gar Genome
2.5. Alignment of the Dog Chromosome-Specific Library to the Genomes of Vertebrate Species
3. Results
3.1. Characterization of Microdissection-Derived Libraries
3.2. Sequencing and Alignment
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Evolution by Gene Duplication; Springer-Verlag: Berlin, Germany, 1970; ISBN 978-3-642-86659-3. [Google Scholar]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Vandepoele, K.; De Vos, W.; Taylor, J.S.; Meyer, A.; Van de Peer, Y. Major events in the genome evolution of vertebrates: Paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc. Natl. Acad. Sci. USA 2004, 101, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Murat, F.; Van de Peer, Y.; Salse, J. Decoding plant and animal genome plasticity from differential paleo-evolutionary patterns and processes. Genome Biol. Evol. 2012, 4, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Keinath, M.C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 2015, 251, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Glover, N.M.; Redestig, H.; Dessimoz, C. Homoeologs: What are they and how do we infer them? Trends Plant Sci. 2016, 21, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.K.; et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Romanenko, S.A.; Biltueva, L.S.; Serdyukova, N.A.; Kulemzina, A.I.; Beklemisheva, V.R.; Gladkikh, O.L.; Lemskaya, N.A.; Interesova, E.A.; Korentovich, M.A.; Vorobieva, N.V.; et al. Segmental paleotetraploidy revealed in sterlet (Acipenser ruthenus) genome by chromosome painting. Mol. Cytogenet. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, V.A.; Romanenko, S.A.; Beklemisheva, V.R.; Biltueva, L.S.; Makunin, A.I.; Lemskaya, N.A.; Kulemzina, A.I.; Stanyon, R.; Graphodatsky, A.S. Evolutionary plasticity of acipenseriform genomes. Chromosoma 2016, 125, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Biltueva, L.S.; Prokopov, D.Y.; Makunin, A.I.; Komissarov, A.S.; Kudryavtseva, A.V.; Lemskaya, N.A.; Vorobieva, N.V.; Serdyukova, N.A.; Romanenko, S.A.; Gladkikh, O.L.; et al. Genomic organization and physical mapping of tandemly arranged repetitive DNAs in sterlet (Acipenser ruthenus). Cytogenet. Genome Res. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Makunin, A.I.; Kichigin, I.G.; Larkin, D.M.; O’Brien, P.C.; Ferguson-Smith, M.A.; Yang, F.; Proskuryakova, A.A.; Vorobieva, N.V.; Chernyaeva, E.N.; O’Brien, S.J.; et al. Contrasting origin of B chromosomes in two cervids (Siberian roe deer and grey brocket deer) unravelled by chromosomespecific DNA sequencing. BMC Genomics 2016, 17, 618. [Google Scholar] [CrossRef] [PubMed]
- Bemis, W.E.; Findeis, E.K.; Grande, L. An overview of Acipenseriformes. Environ. Biol. Fishes 1997, 48, 25–71. [Google Scholar] [CrossRef]
- Betancur-R., R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; López, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanciangco, M.; Cureton II, J.C.; et al. The tree of life and a new classification of bony fishes. PLoS Curr. 2013, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Belfiore, N.M.; Pitra, C.; Svirsky, V.; Jenneckens, I. Genome duplication events and functional reduction of ploidy levels in sturgeon (Acipenser, Huso and Scaphirhynchus). Genetics 2001, 158, 1203–1215. [Google Scholar] [PubMed]
- Nakatani, Y.; Takeda, H.; Kohara, Y.; Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 2007, 17, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Ludwig, A.; Wang, D.; Diogo, R.; Wei, Q.; He, S. Age and biogeography of major clades in sturgeons and paddlefishes (Pisces: Acipenseriformes). Mol. Phylogenet Evol. 2007, 42, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Nam, Y.K.; Noh, J.K.; Park, C.H.; Chapman, F.A. Karyotype of North American shortnose sturgeon Acipenser brevirostrum with the highest chromosome number in the Acipenseriformes. Ichthyol. Res. 2005, 52, 94–97. [Google Scholar] [CrossRef]
- Yang, F.; Trifonov, V.; Ng, B.L.; Kosyakova, N.; Carter, N.P. Generation of Paint Probes from Flow-Sorted and Microdissected Chromosomes. In Fluorescence in Situ Hybridization (FISH); Liehr, T., Ed.; Springer Protocols Handbooks; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; 1–3arXiv:1303.3997. [Google Scholar]
- Yang, F.; O’Brien, P.C.M.; Milne, B.S.; Graphodatsky, A.S.; Solanky, N.; Trifonov, V.; Rens, W.; Sargan, D.; Ferguson-Smith, M.A. A Complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps. Genomics 1999, 62, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC genome browser database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef] [PubMed]
- Barbazuk, W.B.; Korf, I.; Kadavi, C.; Heyen, J.; Tate, S.; Wun, E.; Bedell, J.A.; McPherson, J.D.; Johnson, S.L. The syntenic relationship of the zebrafish and human genomes. Genome Res. 2000, 10, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Larkin, D.M.; Everts-van der Wind, A.; Bourque, G.; Tesler, G.; Auvil, L.; Beever, J.E.; Chowdhary, B.P.; Galibert, F.; Gatzke, L.; et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 2005, 309, 613–617. [Google Scholar] [CrossRef] [PubMed]
- De La Herrán, R.; Fontana, F.; Lanfredi, M.; Congiu, L.; Leis, M.; Rossi, R.; Ruiz Rejón, C.; Ruiz Rejón, M.; Garrido-Ramos, M.A. Slow rates of evolution and sequence homogenization in an ancient Satellite DNA family of sturgeons. Mol. Biol. Evol. 2001, 18, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Sire, J.Y.; Donoghue, P.C.; Vickaryous, M.K. Origin and evolution of the integumentary skeleton in non-tetrapod vertebrates. J. Anat. 2009, 214, 409–440. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Lanfredi, M.; Rossi, R.; Bronzi, P.; Arlati, G. Karyotypic characterization of Acipenser gueldenstaedti with C-, AgNO3, and fluorescence banding techniques. Ital. J. Zool. 1996, 63, 113–118. [Google Scholar] [CrossRef]
- Amores, A.; Catchen, J.; Nanda, I.; Warren, W.; Walter, R.; Schartl, M.; Postlethwait, J.H. A RAD-tag genetic map for the platyfish (Xiphophorus maculatus) reveals mechanisms of karyotype evolution among teleost fish. Genetics 2014, 197, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.; Paiz, L.M.; Baumgärtner, L.; Margarido, V.P.; Vieira, M.M.R. Comparative Cytogenetics of the Black Ghost Knifefish (Gymnotiformes: Apteronotidae): Evidence of Chromosomal Fusion and Pericentric Inversions in Karyotypes of Two Apteronotus Species. Zebrafish 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Evans, B.J.; Bogart, J.P. Polyploidy in Amphibia. Cytogenet. Genome Res. 2015, 145, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Proskuryakova, A.A.; Kulemzina, A.I.; Perelman, P.L.; Makunin, A.I.; Larkin, D.M.; Farré, M.; Kukekova, A.V.; Lynn Johnson, J.; Lemskaya, N.A.; Beklemisheva, V.R.; et al. X Chromosome evolution in Cetartiodactyla. Genes 2017, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Romanenko, S.A.; Serdyukova, N.A.; Perelman, P.L.; Pavlova, S.V.; Bulatova, N.S.; Golenishchev, F.N.; Stanyon, R.; Graphodatsky, A.S. Intrachromosomal rearrangements in rodents from the perspective of comparative region-specific painting. Genes 2017, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, T.; Nanda, I.; Volobouev, V.; Yang, F.; Schartl, M.; Haaf, T.; Schmid, M. Cross-species chromosome painting corroborates microchromosome fusion during karyotype evolution of birds. Cytogenet. Genome Res. 2009, 126, 281–304. [Google Scholar] [CrossRef] [PubMed]
- Kasai, F.; O’Brien, P.C.; Martin, S.; Ferguson-Smith, M.A. Extensive homology of chicken macrochromosomes in the karyotypes of Trachemys scripta elegans and Crocodylus niloticus revealed by chromosome painting despite long divergence times. Cytogenet. Genome Res. 2012, 136, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Kichigin, I.G.; Giovannotti, M.; Makunin, A.I.; Ng, B.L.; Kabilov, M.R.; Tupikin, A.E.; Barucchi, V.C.; Splendiani, A.; Ruggeri, P.; Rens, W.; et al. Evolutionary dynamics of Anolis sex chromosomes revealed by sequencing of flow sorting-derived microchromosome-specific DNA. Mol. Genet. Genom. 2016, 291, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
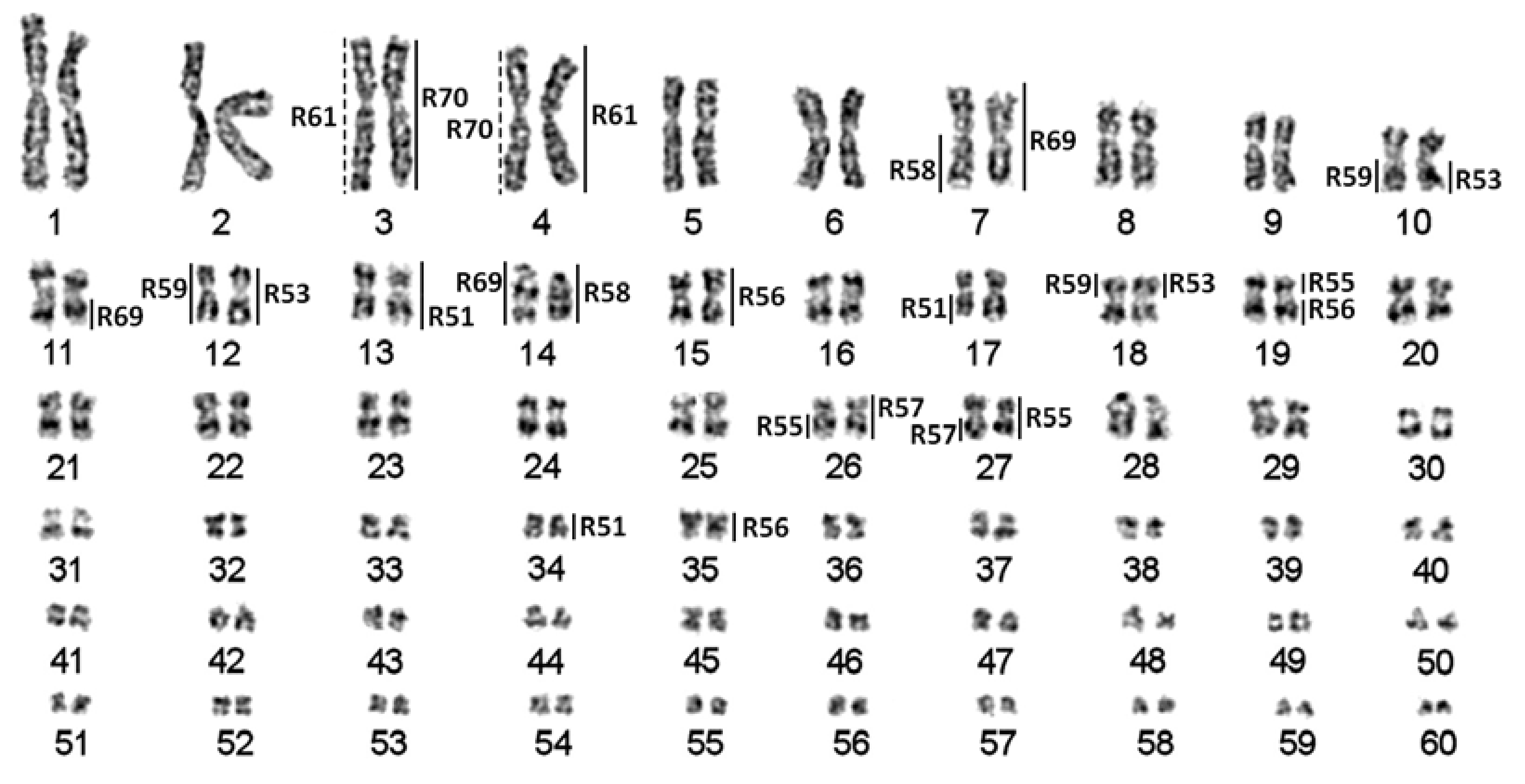

{kind=link}
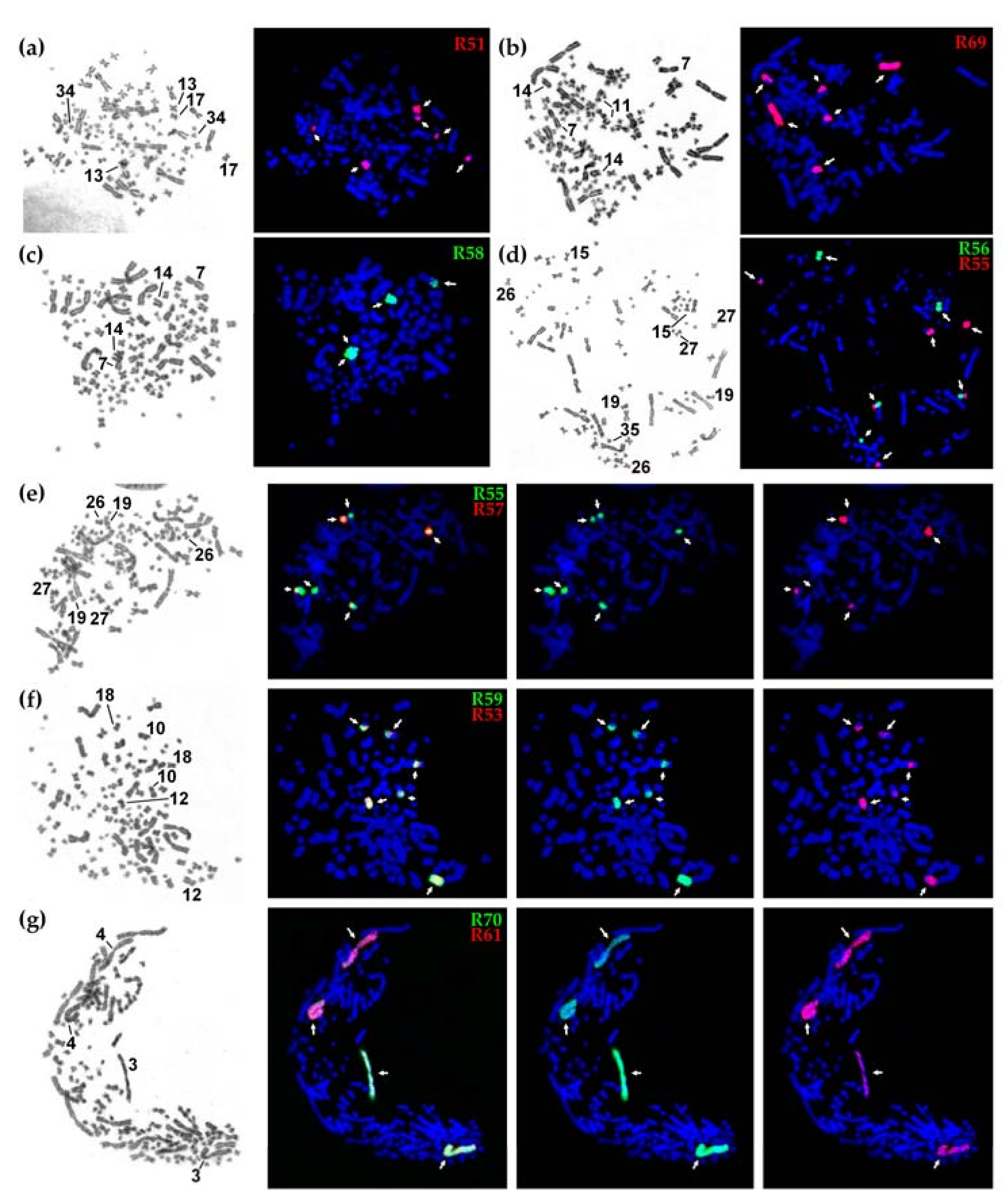
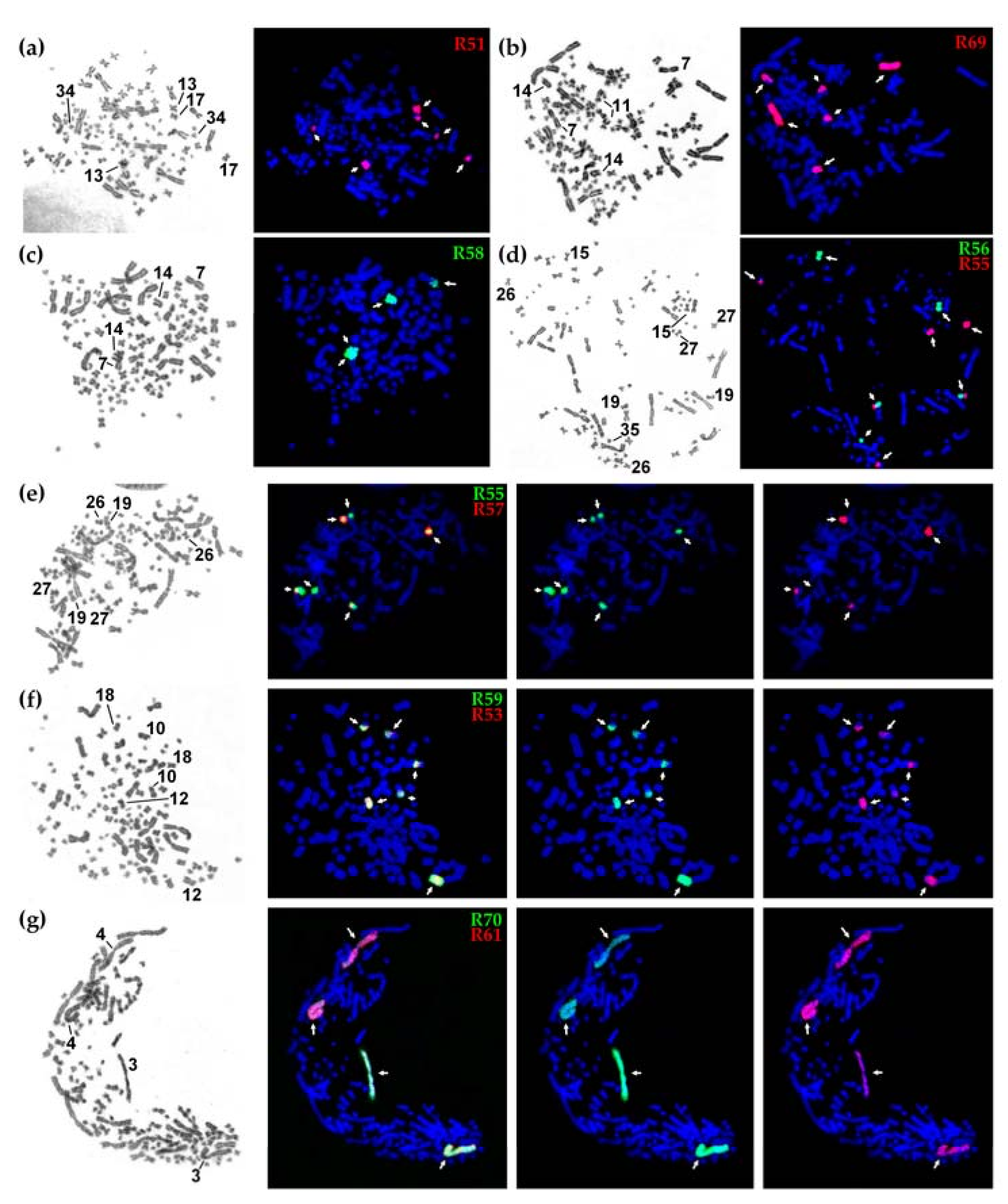{kind=link}
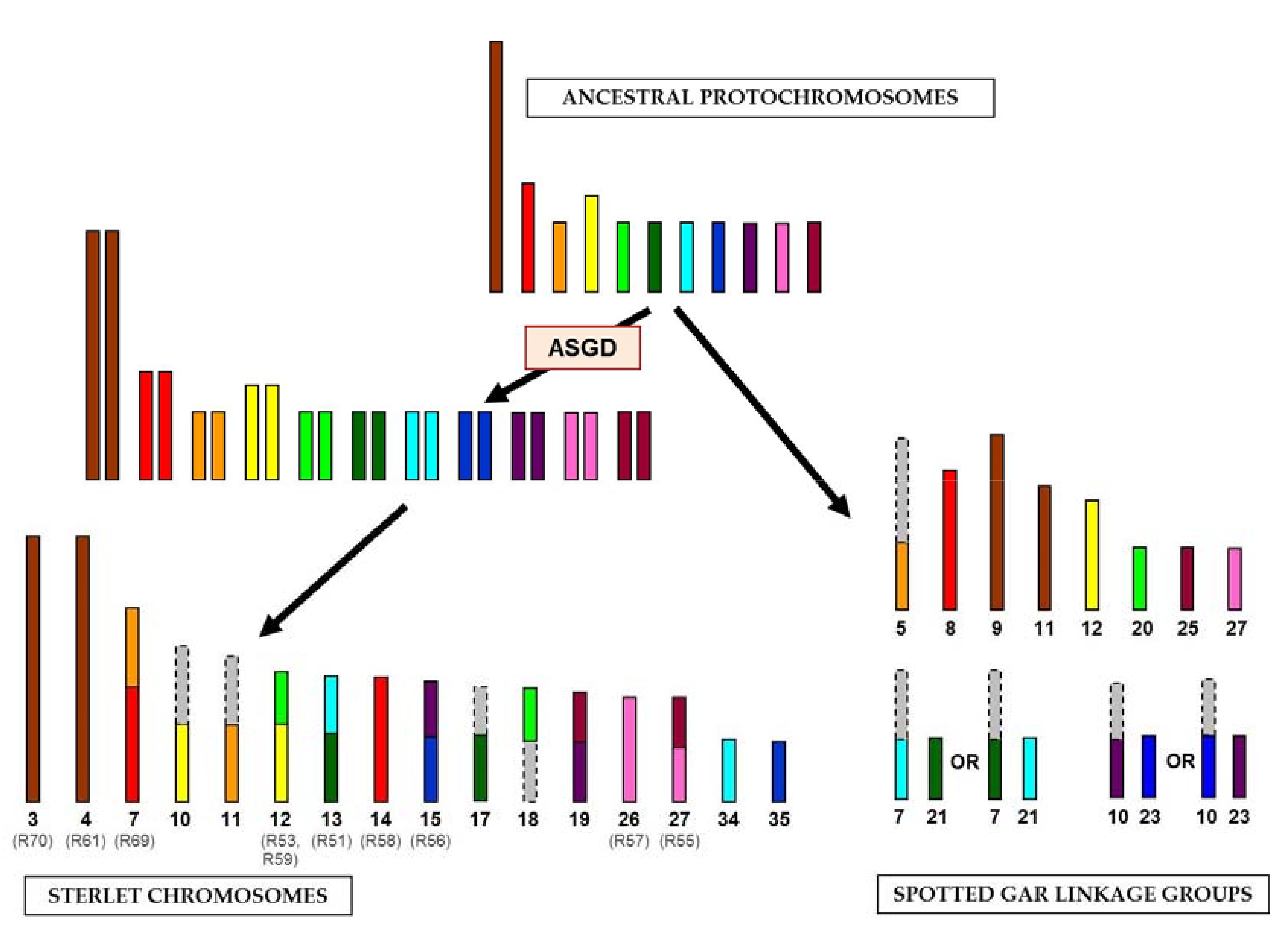
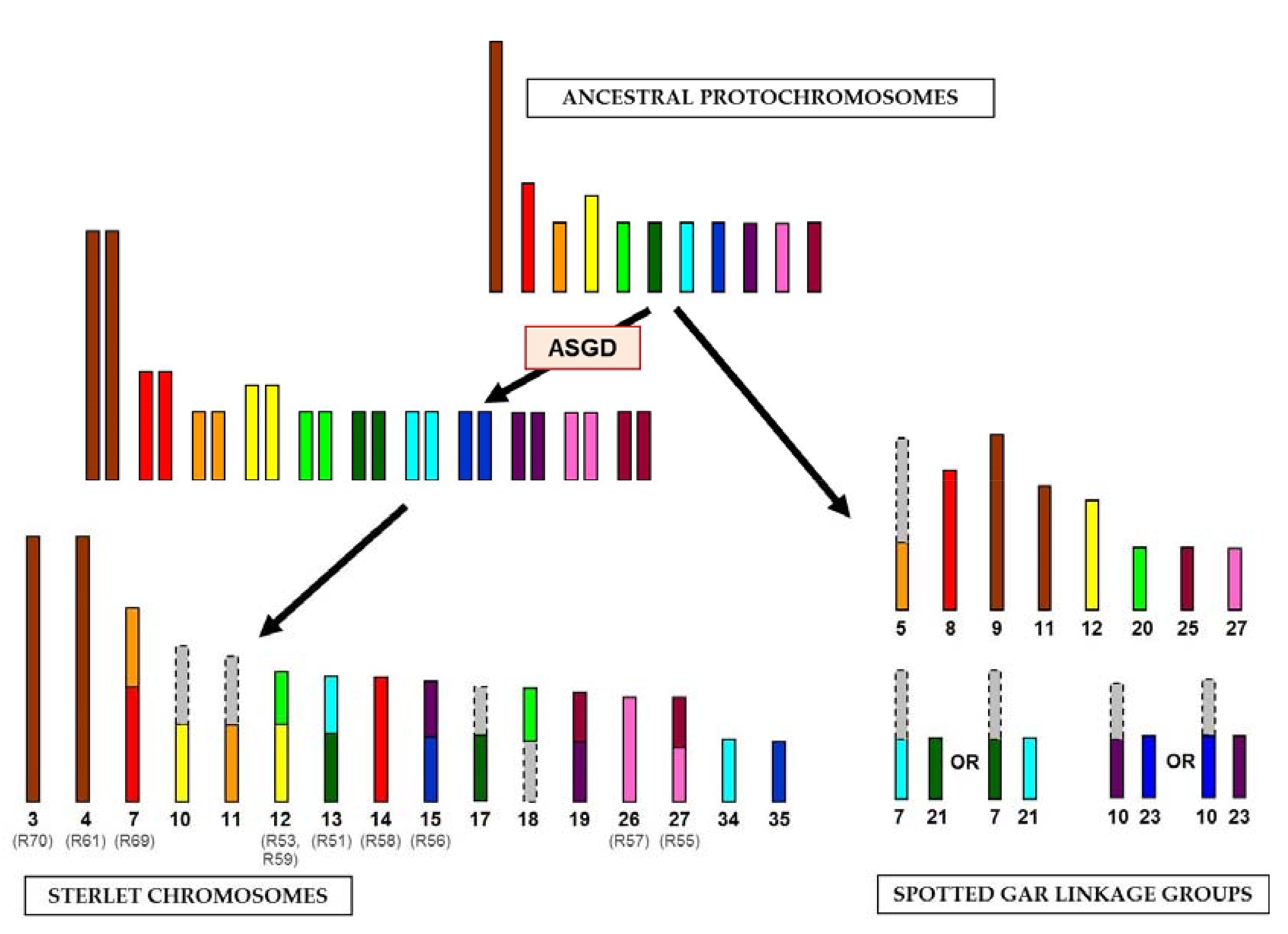{kind=link}
| Library | Total Reads | First Round of Sequencing | Second Round of Sequencing | Combined Alignment | ||||
|---|---|---|---|---|---|---|---|---|
| # Reads | # Aligned Reads, Bowtie 2 | # Aligned Reads, BWA-MEM | # Reads | # Aligned Reads, Bowtie 2 | # Aligned Reads, BWA-MEM | # Aligned Reads, BWA-MEM | ||
| R51 | 136,912 | 136,912 | 1806 | 66,894 | - | - | - | - |
| R53 | 291,544 | 85,926 | 1015 | 43,365 | 205,618 | 1582 | 92,913 | 136,288 |
| R55 | 100,975 | 100,975 | 1077 | 47,303 | - | - | - | - |
| R56 | 109,161 | 109,161 | 1064 | 57,278 | - | - | - | - |
| R57 | 66,669 | 66,669 | 968 | 30,541 | - | - | - | - |
| R58 | 369,795 | 83,882 | 1493 | 37,870 | 285,913 | 4102 | 118,337 | 156,226 |
| R59 | 98,724 | 98,724 | 1493 | 44,889 | - | - | - | - |
| R61 | 208,810 | 78,743 | 1702 | 35,949 | 130,067 | 2364 | 57,796 | 93,743 |
| R69 | 225,362 | 64,839 | 1048 | 29,142 | 160,523 | 1980 | 66,237 | 95,380 |
| R70 | 213,732 | 42,616 | 826 | 19,336 | 171,116 | 2456 | 69,026 | 88,356 |
| Libraries | ARUT Microdissected Chromosome | FISH Signals | Top Target Regions of LOC | Chicken Chromosomes [10] | Ancestral Amniote Chromosomes [19] | Ancestral Bony Vertebrate Chromosomes [19] |
|---|---|---|---|---|---|---|
| R51 | ARUT13 | ARUT13, ARUT17q, ARUT34 | LOC7 (distal region) | 5 | 7 or 13 or 14 | 8 or 15 or 16 |
| LOC21 | 17 | 3 (part) or 26 | 3 | |||
| R53, R59 | ARUT12 | ARUT10q, ARUT12, ARUT18p | LOC12 | 7 | 4 (part) | 2 (part) |
| LOC20 | 15 | 11 | 12 | |||
| R55 | ARUT27 | ARUT19p, ARUT26q, ARUT27 | LOC25 | 21 | 22 | 25 |
| LOC27 | 5 | 7 or 13 or 14 | 8 or 15 or 16 | |||
| R56 | ARUT15 | ARUT15, ARUT19q, ARUT35 | LOC10 (distal region) | 18 | 1 (part) | 6 (part) |
| LOC23 | 11 | 12 | 14 | |||
| R57 | ARUT26 | ARUT26, ARUT27q | LOC27 | 5 | 7 or 13 or 14 | 8 or 15 or 16 |
| R58 | ARUT14 | ARUT7q, ARUT14 | LOC8 | 1 | 1 or 4 | 2 (part) or 5 or 23 |
| R61 | ARUT4 | ARUT3 (weak), ARUT4 | LOC9 | 2 | 2 | 1 |
| LOC11 | ||||||
| LOC16 | 3 | 5 or 1 or 13 or 14 | 4 or 15 or 16 | |||
| R69 | ARUT7 | ARUT7, ARUT14, ARUT10q | LOC5 (distal region) | 6 | 6 | 17 |
| LOC8 | 1 | 1 or 4 | 2 (part) or 5 or 23 | |||
| R70 | ARUT3 | ARUT3, ARUT4 (weak) | LOC9 | 2 | 2 | 1 |
| LOC11 | ||||||
| LOC19 | 28 | 20 | 22 | |||
| LOC25 | 21 | 22 | 25 |
| Alignment Results | Reference Genomes | |||||||
|---|---|---|---|---|---|---|---|---|
| CFA | FCA | BTA | HSA | MDO | GGA | LOC | DRE | |
| Aligned reads | 330,701 | 213,039 | 172,411 | 180,466 | 137,621 | 193,115 | 208,142 | 216,574 |
| Collapsed positions | 36,073 | 34,781 | 25,688 | 32,951 | 17,452 | 24,822 | 26,229 | 29,875 |
| Recovery, bp | 2,743,657 | 2,178,208 | 919,150 | 1,909,100 | 424,206 | 519,302 | 537,072 | 620,913 |
| Reads per position | 9.2 | 6.1 | 6.7 | 5.5 | 7.9 | 7.8 | 7.9 | 7.2 |
| Position mean size, bp | 76 | 63 | 36 | 58 | 24 | 21 | 20 | 21 |
| Identified regions | 12 | B2 | 9, 23 | 6 | 2 | 27, 5, 3, 2 | 16, 7, 8, 1, 11 | 4 |
| Syntenic regions for CFA12 | 12 | B2 [25,26] | 9, 23 [26] | 6 [25] | 2 [26] | 3, 26 [26] | 1, 16, 9, 11 [10] | 13, 16, 19, 20, 21, 23 [27,28] |
| Approximate divergence time, Mya | 0 | 55 [29] | 85 [29] | 90 [29] | 180 [19] | 350 [4] | 450 [4] | 450 [4] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreyushkova, D.A.; Makunin, A.I.; Beklemisheva, V.R.; Romanenko, S.A.; Druzhkova, A.S.; Biltueva, L.B.; Serdyukova, N.A.; Graphodatsky, A.S.; Trifonov, V.A. Next Generation Sequencing of Chromosome-Specific Libraries Sheds Light on Genome Evolution in Paleotetraploid Sterlet (Acipenser ruthenus). Genes 2017, 8, 318. https://doi.org/10.3390/genes8110318
Andreyushkova DA, Makunin AI, Beklemisheva VR, Romanenko SA, Druzhkova AS, Biltueva LB, Serdyukova NA, Graphodatsky AS, Trifonov VA. Next Generation Sequencing of Chromosome-Specific Libraries Sheds Light on Genome Evolution in Paleotetraploid Sterlet (Acipenser ruthenus). Genes. 2017; 8(11):318. https://doi.org/10.3390/genes8110318
Chicago/Turabian StyleAndreyushkova, Daria A., Alexey I. Makunin, Violetta R. Beklemisheva, Svetlana A. Romanenko, Anna S. Druzhkova, Larisa B. Biltueva, Natalya A. Serdyukova, Alexander S. Graphodatsky, and Vladimir A. Trifonov. 2017. "Next Generation Sequencing of Chromosome-Specific Libraries Sheds Light on Genome Evolution in Paleotetraploid Sterlet (Acipenser ruthenus)" Genes 8, no. 11: 318. https://doi.org/10.3390/genes8110318