Genes and Gut Bacteria Involved in Luminal Butyrate Reduction Caused by Diet and Loperamide

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Design for Animal Studies
2.2. Mucosal Studies
2.3. Butyrate Quantification
2.4. Fecal Microbiome Analysis
2.5. Statistical Analysis
3. Results
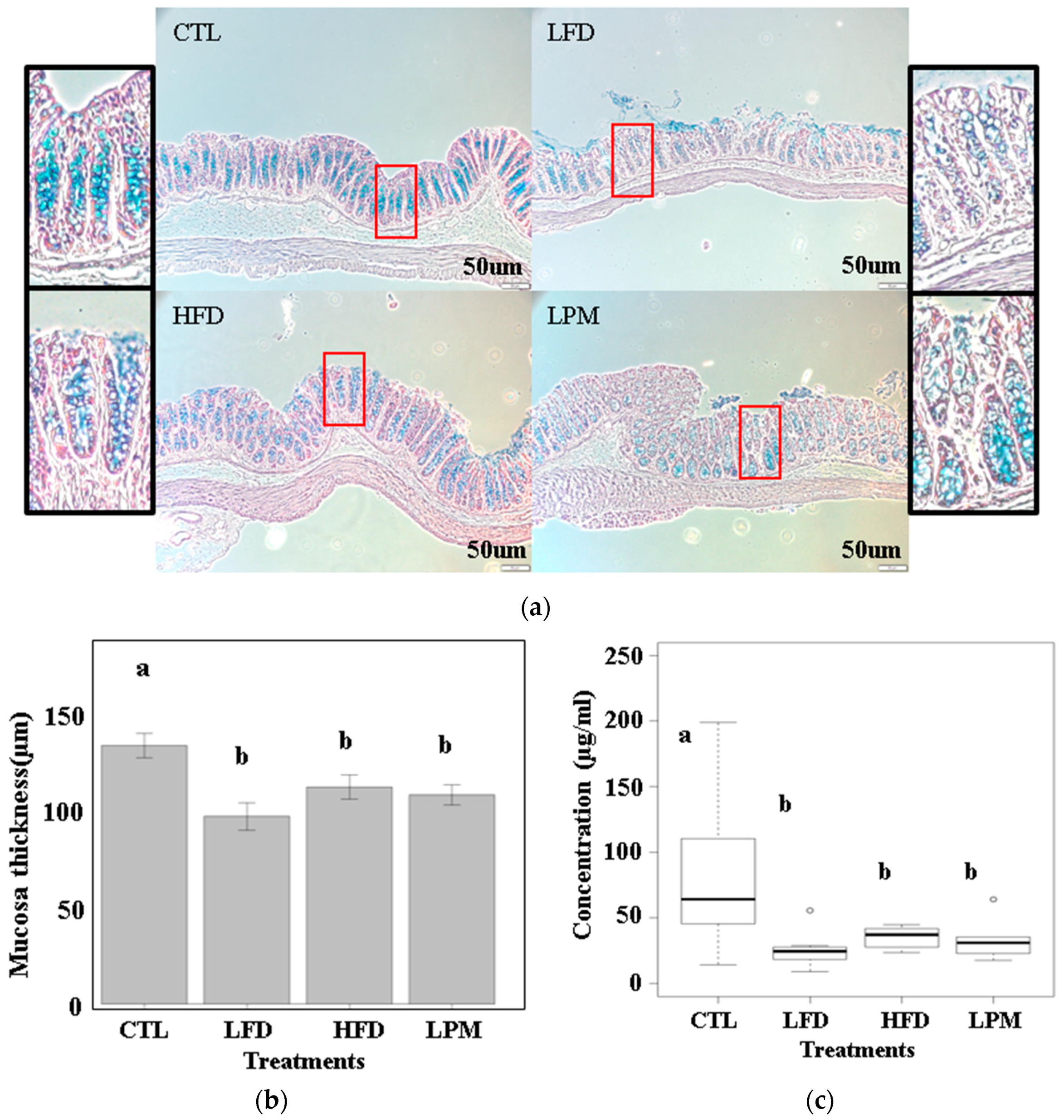
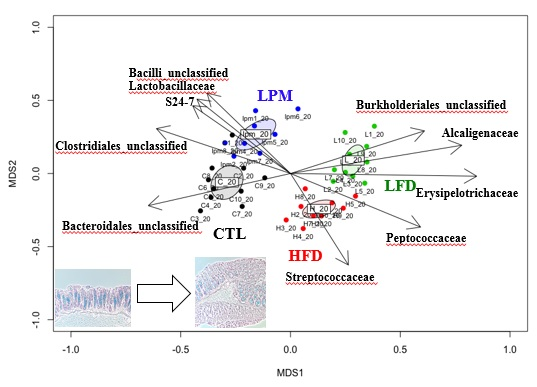
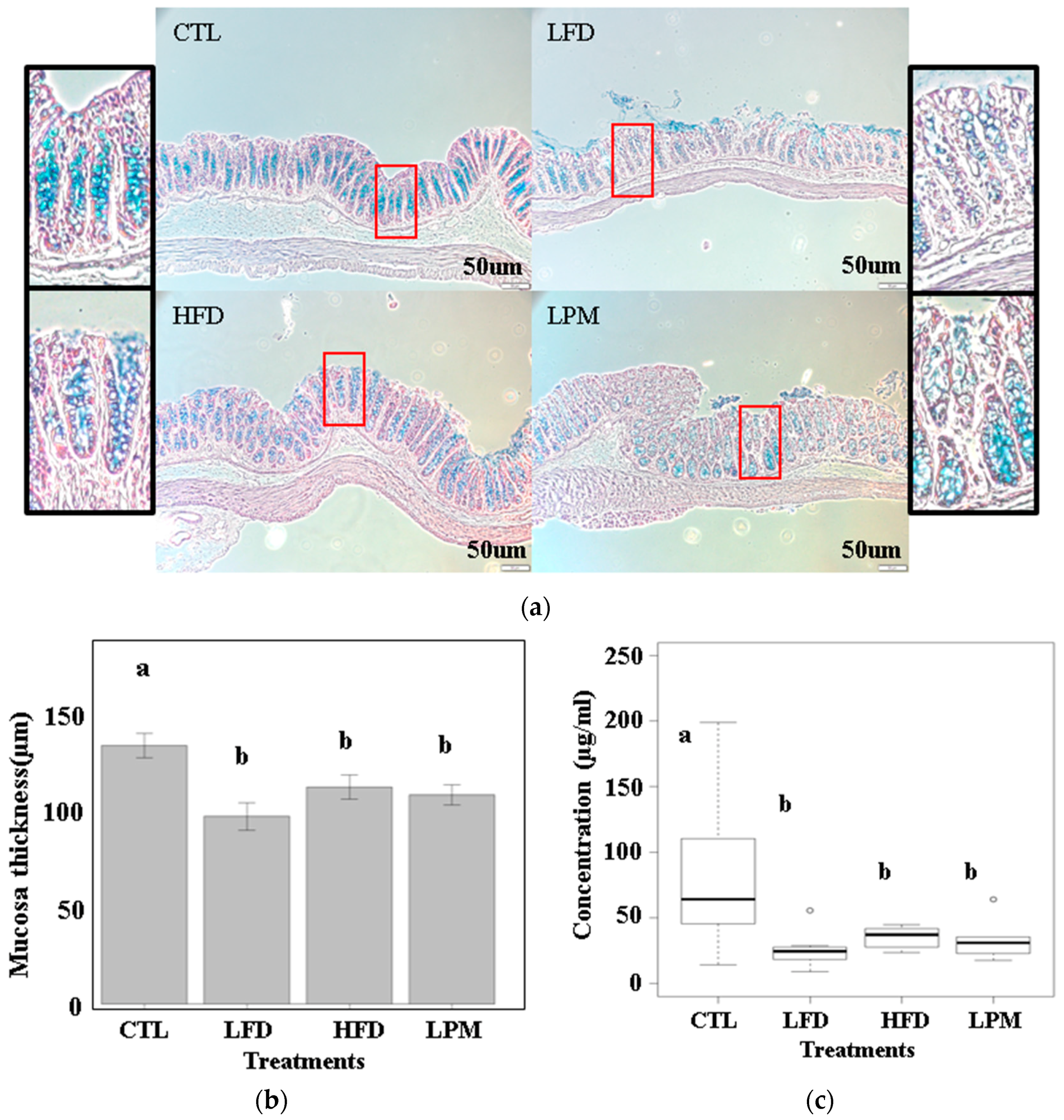
3.1. Bioparametric and Histological Changes
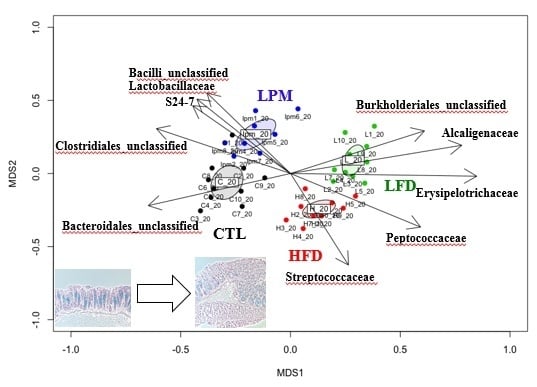
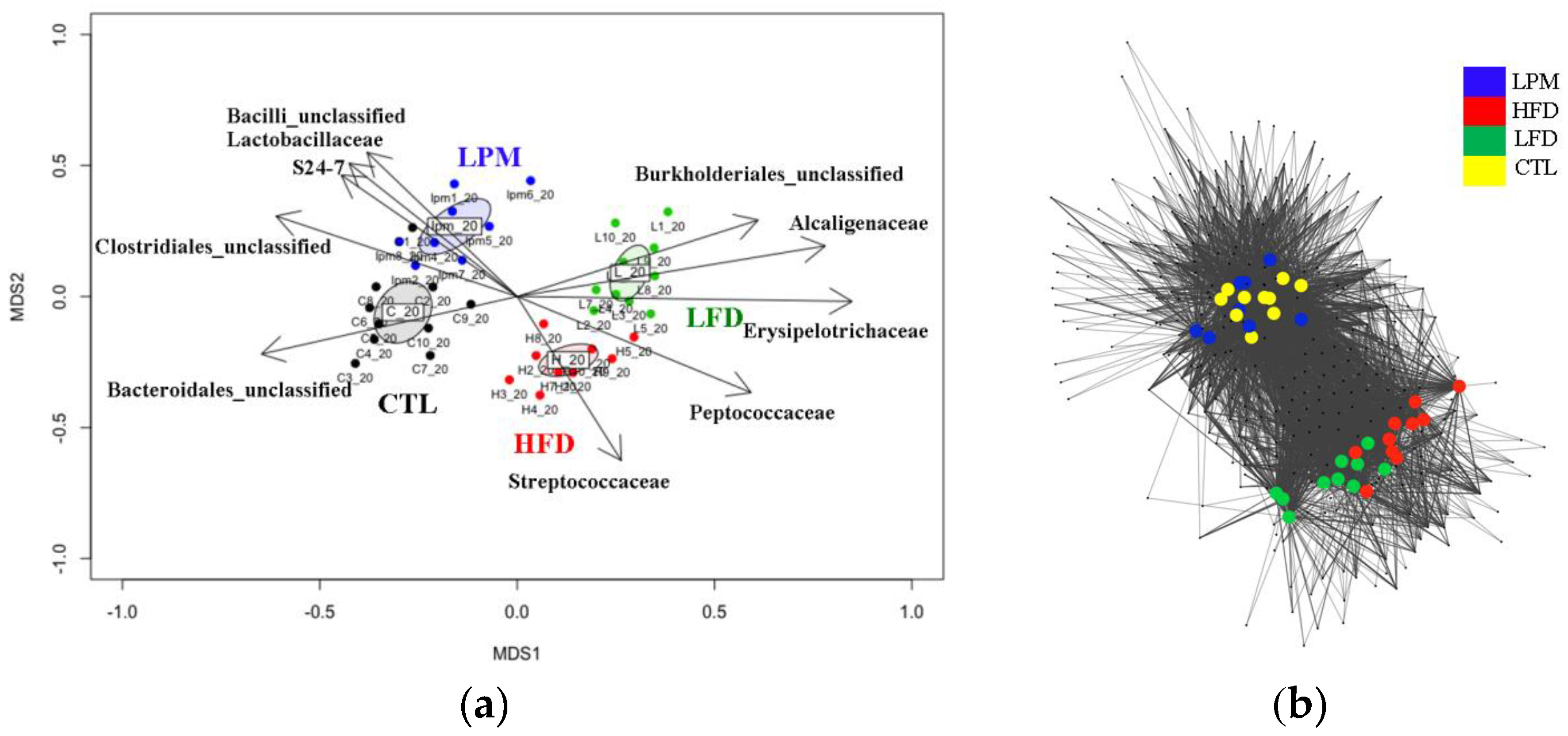
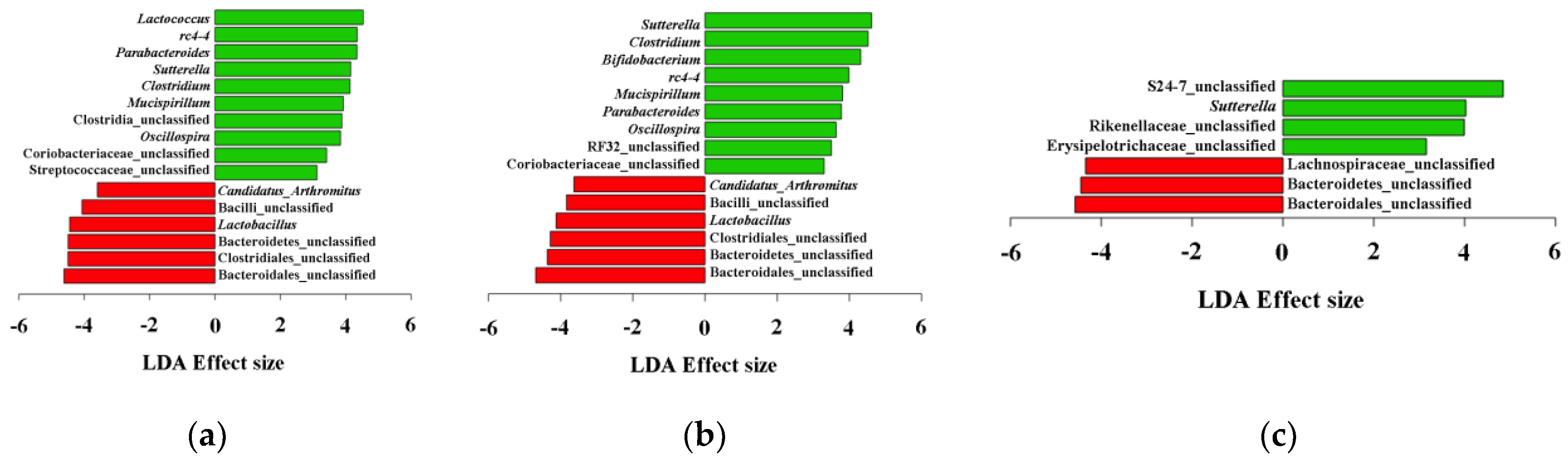
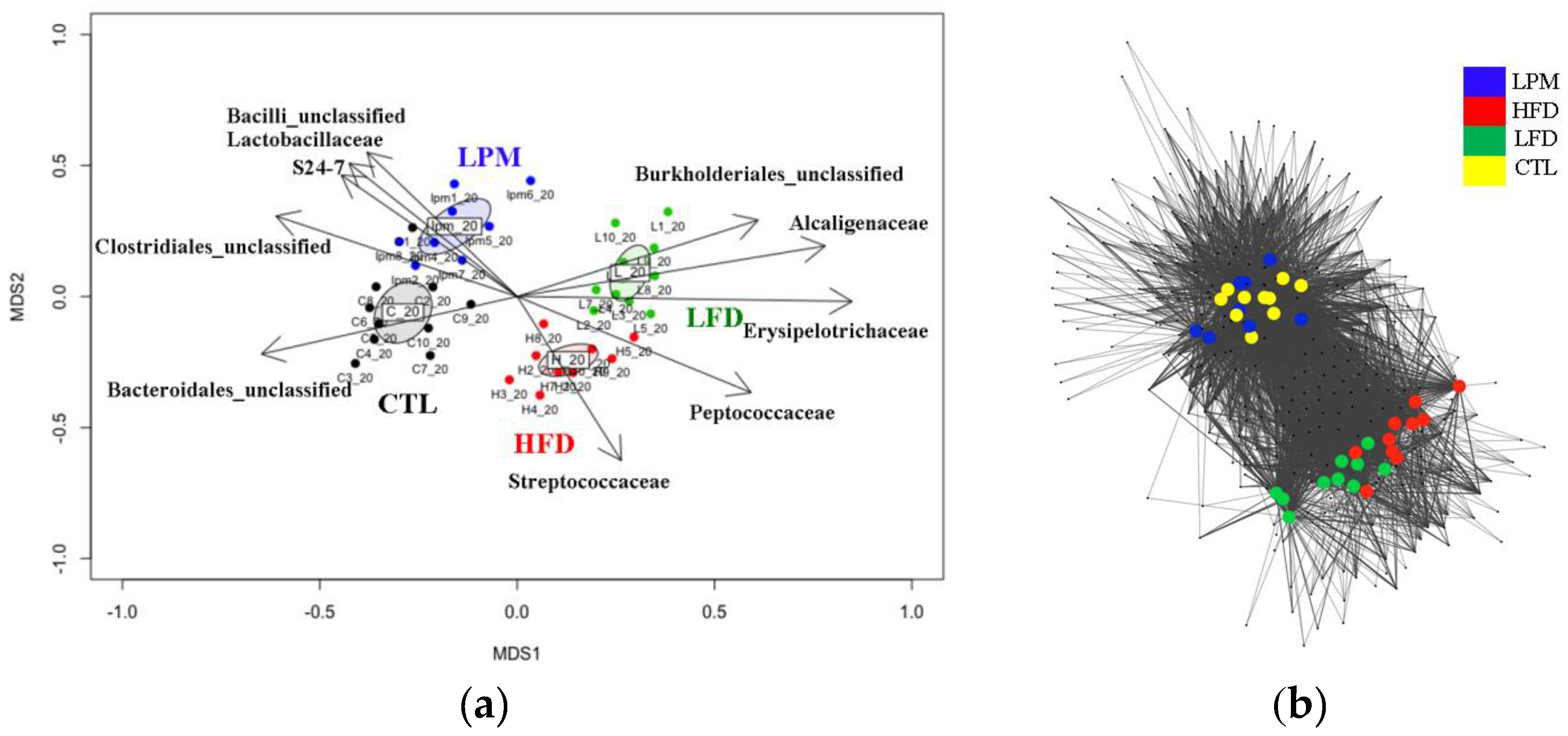
3.2. Gut Microbial Community Comparison
3.3. Analysis of Genes Involved in the Butanoate Metabolism Pathway
4. Discussion
4.1. Duration of Feeding Trial for Reduction in Butyrate Synthesis
4.2. Mechanisms in Butyrate Reduction Induced by HFD, LFD, and LPM Treatments
4.2.1. Butyrate Reduction Mechanism(s) Induced by HFD
4.2.2. Butyrate Reduction Mechanisms Induced by LFD
4.2.3. Butyrate Reduction Mechanisms Induced by LPM
4.3. Other Bacteria Involved in Gut Dysbiosis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [PubMed]
- Tang, Y.; Chen, Y.; Jiang, H.; Robbins, G.T.; Nie, D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int. J. Cancer 2011, 128, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G.B. Protective role of sodium butyrate, a HDAC inhibitor on beta-cell proliferation, function and glucose homeostasis through modulation of p38/ERK MARK mapk and apoptotic pathways: Study in juvenile diabetic rat. Chem. Biol. Interact. 2014, 213, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Everard, A.; Gomez-Valades, A.G.; Matamoros, S.; Ramirez, S.; Delzenne, N.M.; Gomis, R.; Claret, M.; Cani, P.D. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci. Rep. 2015, 5, 16643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruas-Madiedo, P.; Gueimonde, M.; Fernandez-Garcia, M.; de los Reyes-Gavilan, C.G.; Margolles, A. Mucin degradation by bifidobacterium strains isolated from the human intestinal microbiota. Appl. Environ. Microbiol. 2008, 74, 1936–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin glycan foraging in the human gut microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Jakobsdottir, G.; Xu, J.; Molin, G.; Ahrne, S.; Nyman, M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, L.; Yan, S.; Jiang, T.; Fang, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Effects of different oligosaccharides at various dosages on the composition of gut microbiota and short-chain fatty acids in mice with constipation. Food Funct. 2017, 8, 1966–1978. [Google Scholar] [CrossRef] [PubMed]
- Tottey, W.; Feria-Gervasio, D.; Gaci, N.; Laillet, B.; Pujos, E.; Martin, J.F.; Sebedio, J.L.; Sion, B.; Jarrige, J.F.; Alric, M.; et al. Colonic transit time is a driven force of the gut microbiota composition and metabolism: In vitro evidence. J. Neurogastroenterol. Motil. 2017, 23, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lay, C.; Dore, J.; Rigottier-Gois, L. Separation of bacteria of the Clostridium leptum subgroup from the human colonic microbiota by fluorescence-activated cell sorting or group-specific PCR using 16s rRNA gene oligonucleotides. FEMS Microbiol. Ecol. 2007, 60, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Kaur, H.; Mande, S.S. Comparative in silico analysis of butyrate production pathways in gut commensals and pathogens. Front. Microbiol. 2016, 7, 1945. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Go, J.; Sung, J.E.; Lee, H.A.; Yun, W.B.; Hong, J.T.; Hwang, D.Y. Uridine stimulate laxative effect in the loperamide-induced constipation of SD rats through regulation of the mAChRs signaling pathway and mucin secretion. BMC Gastroenterol. 2017, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Li, P.E.; Lo, C.C.; Anderson, J.J.; Davenport, K.W.; Bishop-Lilly, K.A.; Xu, Y.; Ahmed, S.; Feng, S.; Mokashi, V.P.; Chain, P.S. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucl. Acids Res. 2017, 45, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. Idba-ud: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nuleic Acids Res. 2007, 35. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pop, M. Metapath: Identifying differentially abundant metabolic pathways in metagenomic datasets. BMC Proc. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Roewer, L.; Kayser, M.; Dieltjes, P.; Nagy, M.; Bakker, E.; Krawczak, M.; de Knijff, P. Analysis of molecular variance (amova) of Y-chromosome-specific microsatellites in two closely related human populations. Hum. Mol. Genet. 1996, 5, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anitha, M.; Reichardt, F.; Tabatabavakili, S.; Nezami, B.G.; Chassaing, B.; Mwangi, S.; Vijay-Kumar, M.; Gewirtz, A.; Srinivasan, S. Intestinal dysbiosis contributes to the delayed gastrointestinal transit in high-fat diet fed mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Kakino, M.; Tazawa, S.; Maruyama, H.; Tsuruma, K.; Araki, Y.; Shimazawa, M.; Hara, H. Laxative effects of agarwood on low-fiber diet-induced constipation in rats. BMC Complement. Altern. Med. 2010, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J. Appl. Microbiol. 2007, 102, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Flint, H.J. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 2004, 70, 5810–5817. [Google Scholar] [CrossRef] [PubMed]
- Shankar, V.; Homer, D.; Rigsbee, L.; Khamis, H.J.; Michail, S.; Raymer, M.; Reo, N.V.; Paliy, O. The networks of human gut microbe-metabolite associations are different between health and irritable bowel syndrome. ISME J. 2015, 9, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, M.; Gustafson, Y.; Haglin, L.; Eriksson, S. The feasibility of serving liquid yoghurt supplemented with probiotic bacteria, lactobacillus rhamnosus LB 21, and Lactococcus lactis L1A—A pilot study among old people with dementia in a residential care facility. J. Nutr. Health Aging 2009, 13, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 2012, 36, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Solem, C.; Dehli, T.; Jensen, P.R. Rewiring Lactococcus lactis for ethanol production. Appl. Environ. Microbiol. 2013, 79, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Zietak, M.; Kovatcheva-Datchary, P.; Markiewicz, L.H.; Stahlman, M.; Kozak, L.P.; Backhed, F. Altered microbiota contributes to reduced diet-induced obesity upon cold exposure. Cell Metab. 2016, 23, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, R.; Adriany, T.; Verbrugghe, K.; De Vuyst, L. Kinetic analysis of Bifidobacterial metabolism reveals a minor role for succinic acid in the regeneration of NAD+ through its growth-associated production. Appl. Environ. Microbiol. 2006, 72, 5204–5210. [Google Scholar] [CrossRef] [PubMed]
- Touw, K.; Ringus, D.L.; Hubert, N.; Wang, Y.; Leone, V.A.; Nadimpalli, A.; Theriault, B.R.; Huang, Y.E.; Tune, J.D.; Herring, P.B.; et al. Mutual reinforcement of pathophysiological host-microbe interactions in intestinal stasis models. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, K.L.; Wood, D.L.; Lachner, N.; Gellatly, S.L.; Daly, J.N.; Parsons, J.D.; Dal’Molin, C.G.; Palfreyman, R.W.; Nielsen, L.K.; Cooper, M.A.; et al. Genomic characterization of the uncultured Bacteroidales family S24–7 inhabiting the guts of homeothermic animals. Microbiome 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.L.; Hornig, M.; Parekh, T.; Lipkin, W.I. Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Increased abundance of Sutterella spp. And Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Han, S.; Wang, P.; Wen, B.; Jian, W.; Guo, W.; Yu, Z.; Du, D.; Fu, X.; Kong, F.; et al. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci. Rep. 2015, 5, 9342. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Jeraldo, P.R.; Kurti, A.; Miller, M.E.; Cook, M.D.; Whitlock, K.; Goldenfeld, N.; Woods, J.A.; White, B.A.; Chia, N.; et al. Diet and exercise orthogonally alter the gut microbiome and reveal independent associations with anxiety and cognition. Mol. Neurodegener. 2014, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.R.; O’Rourke, J.L.; Neilan, B.A.; Vandamme, P.; On, S.L.; Fox, J.G.; Lee, A. Mucispirillum schaedleri gen. Nov., sp. Nov., a spiral-shaped bacterium colonizing the mucus layer of the gastrointestinal tract of laboratory rodents. Int. J. Syst. Evolut. Microbiol. 2005, 55, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Loy, A.; Pfann, C.; Steinberger, M.; Hanson, B.; Herp, S.; Brugiroux, S.; Gomes Neto, J.C.; Boekschoten, M.V.; Schwab, C.; Urich, T.; et al. Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota. mSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, V.; Kaakoush, N.O.; Maloney, C.A.; Raipuria, M.; Huinao, K.D.; Mitchell, H.M.; Morris, M.J. Changes in gut microbiota in rats fed a high fat diet correlate with obesity-associated metabolic parameters. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Konikoff, T.; Gophna, U. Oscillospira: A central, enigmatic component of the human gut microbiota. Trends Microbiol. 2016, 24, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospira and related bacteria—From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Li, S.; Gan, R.Y.; Zhou, T.; Xu, D.P.; Li, H.B. Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Mean | Cumulative Fraction | Genus | ||
|---|---|---|---|---|---|
| Total | HFD–LFD | CTL–LPM | |||
| CTL–LPM | 6.88 | 4.66 | 9.58 | 0.11 | Clostridiales unclassified |
| CTL–LPM | 4.16 | 2.42 | 6.69 | 0.2 | Lactobacillus |
| HFD–LFD | 40.29 | 42.01 | 37.8 | 0.29 | Bacteroides |
| HFD–LFD | 2.17 | 5.11 | 0.99 | 0.38 | Clostridium (Erysipelotrichaceae) |
| CTL–LPM | 1.6 | 0.7 | 4.09 | 0.46 | Bacteroidales unclassified |
| HFD–LFD | 2.33 | 3.86 | 1.27 | 0.51 | Sutterella |
| CTL–LPM | 1.38 | 0.63 | 3.09 | 0.57 | Bacilli unclassified |
| HFD–LFD | 3.32 | 4.15 | 2.23 | 0.61 | Parabacteroides |
| HFD–LFD | 2.51 | 3.41 | 1.56 | 0.65 | rc4-4 |
| CTL–LPM | 0.69 | 0.25 | 1.82 | 0.68 | S24-7 unclassified |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, N.; Eom, T.; Gupta, S.K.; Jeong, S.-Y.; Jeong, D.-Y.; Kim, Y.S.; Lee, J.-H.; Sadowsky, M.J.; Unno, T. Genes and Gut Bacteria Involved in Luminal Butyrate Reduction Caused by Diet and Loperamide. Genes 2017, 8, 350. https://doi.org/10.3390/genes8120350
Hwang N, Eom T, Gupta SK, Jeong S-Y, Jeong D-Y, Kim YS, Lee J-H, Sadowsky MJ, Unno T. Genes and Gut Bacteria Involved in Luminal Butyrate Reduction Caused by Diet and Loperamide. Genes. 2017; 8(12):350. https://doi.org/10.3390/genes8120350
Chicago/Turabian StyleHwang, Nakwon, Taekil Eom, Sachin K. Gupta, Seong-Yeop Jeong, Do-Youn Jeong, Yong Sung Kim, Ji-Hoon Lee, Michael J. Sadowsky, and Tatsuya Unno. 2017. "Genes and Gut Bacteria Involved in Luminal Butyrate Reduction Caused by Diet and Loperamide" Genes 8, no. 12: 350. https://doi.org/10.3390/genes8120350