Structural Characterization and Association of Ovine Dickkopf-1 Gene with Wool Production and Quality Traits in Chinese Merino

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Trait Measurements
2.2. Genomic DNA Isolation
2.3. RNA Isolation and Reverse Transcription
2.4. PCR Amplification
2.5. Sequencing and Sequence Analysis
2.6. Identification of Polymorphisms of the Ovine DKK1
2.7. Genotyping of the Ovine DKK1
2.8. Statistical Analysis
3. Results
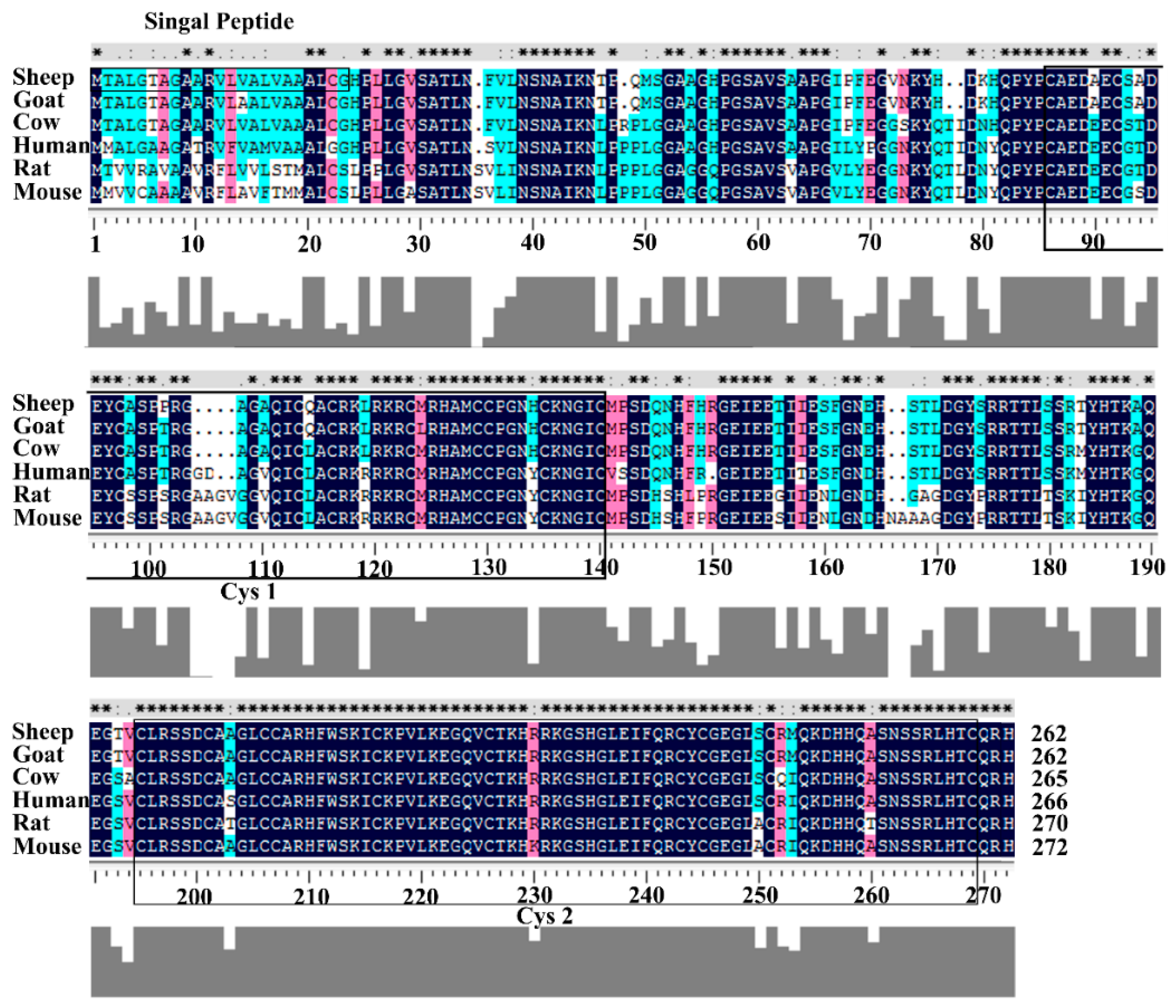
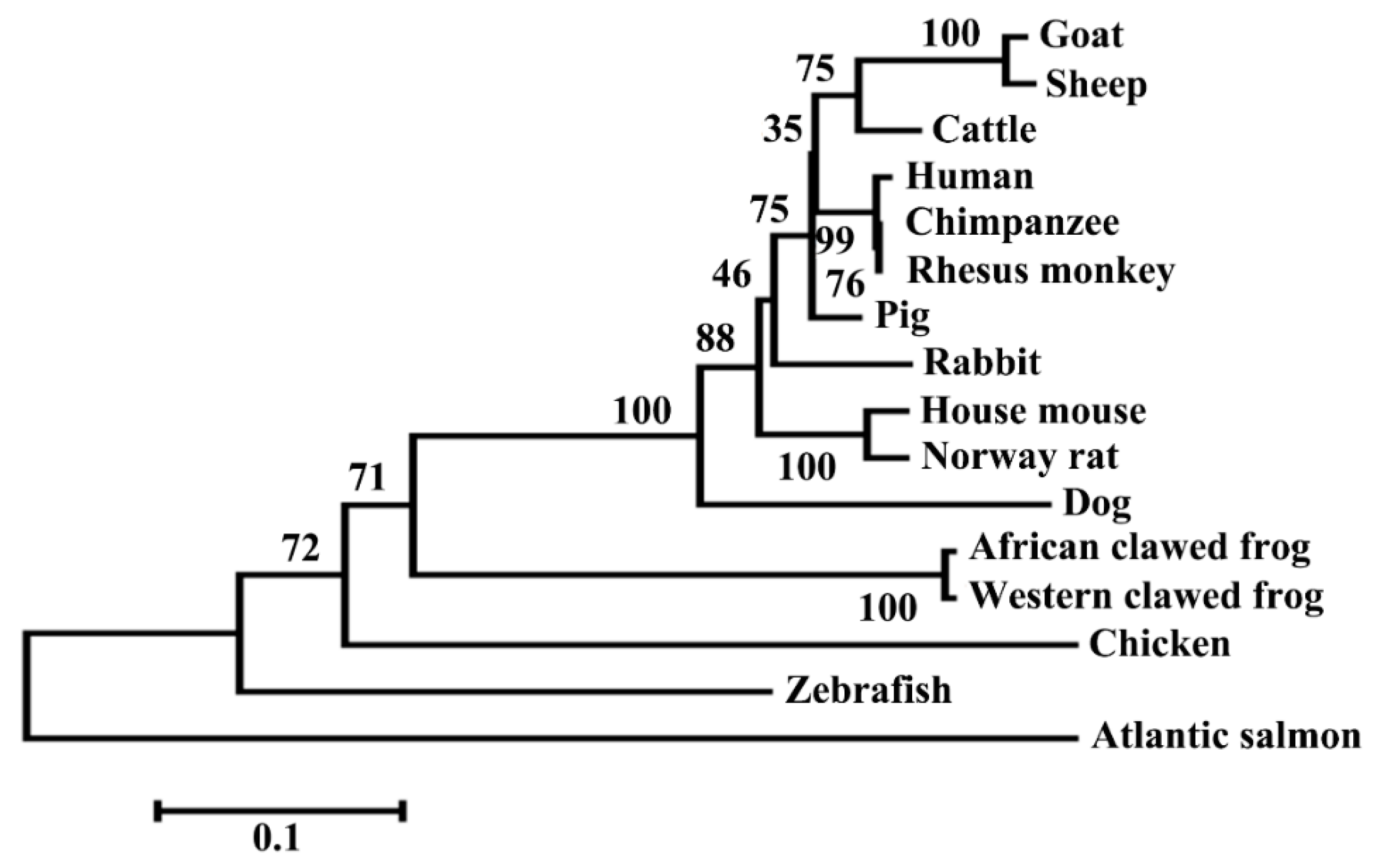
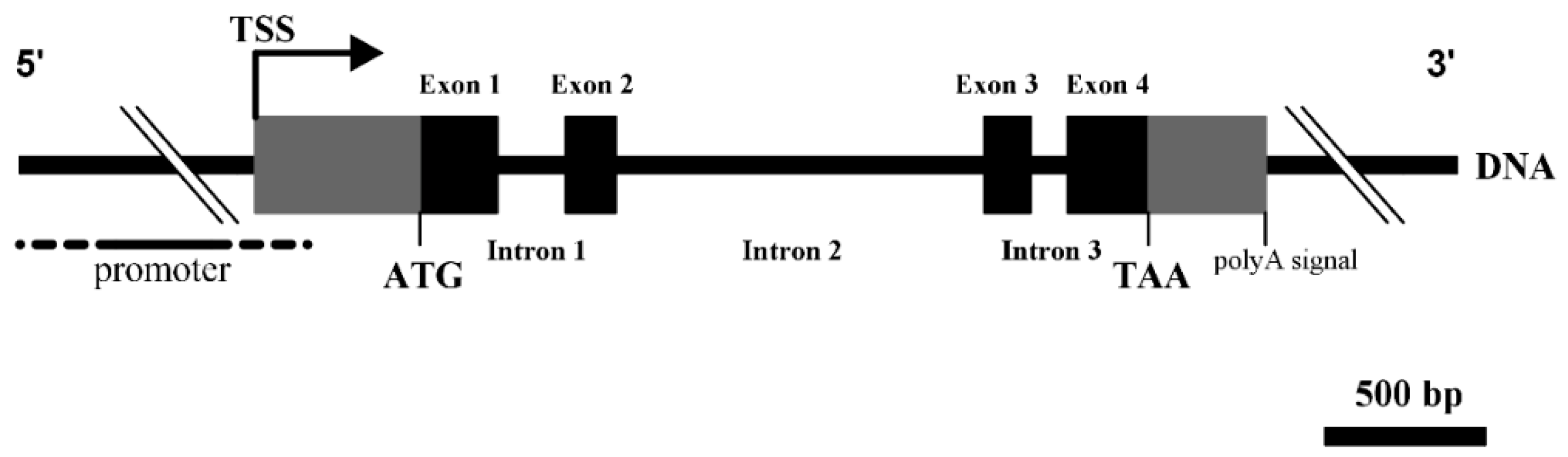
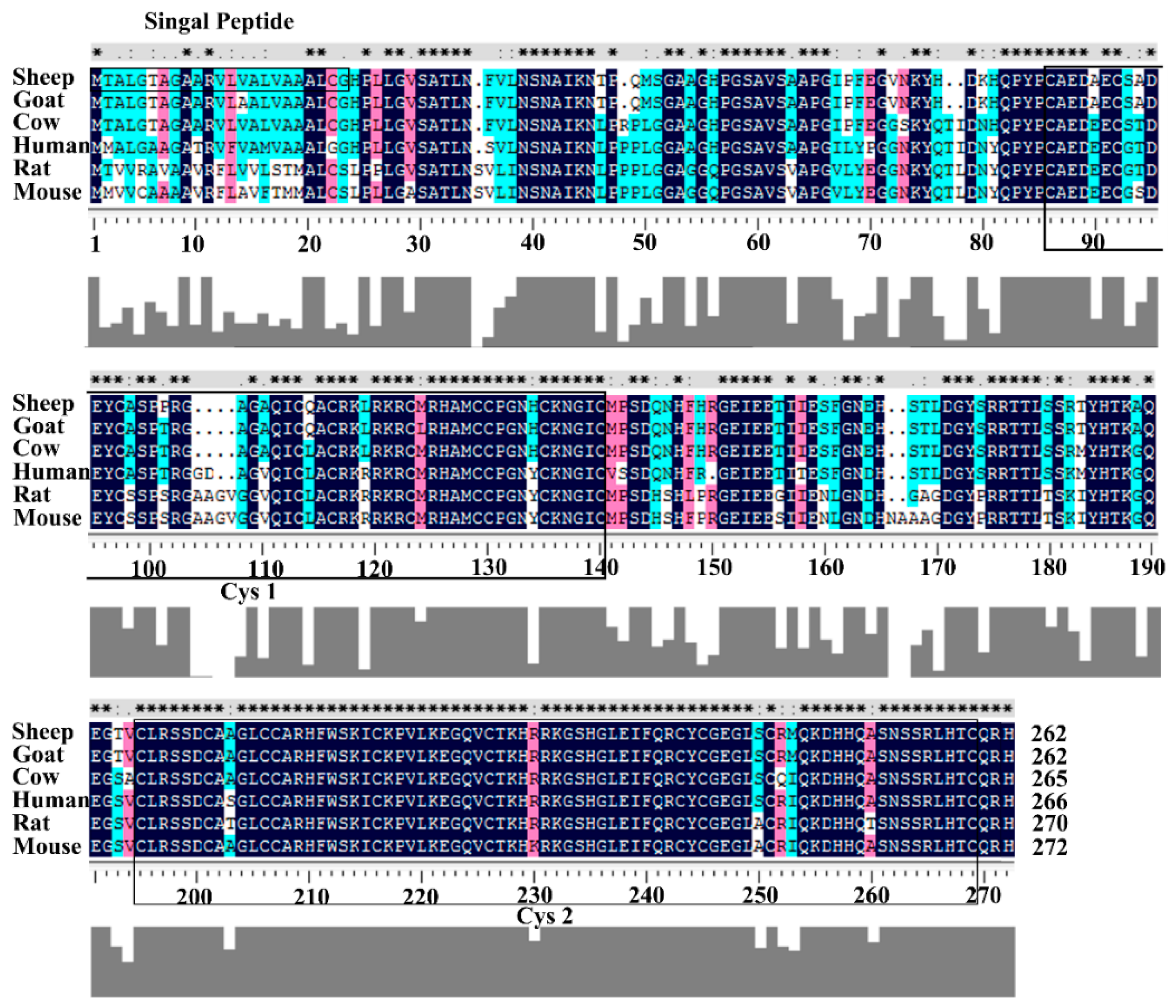
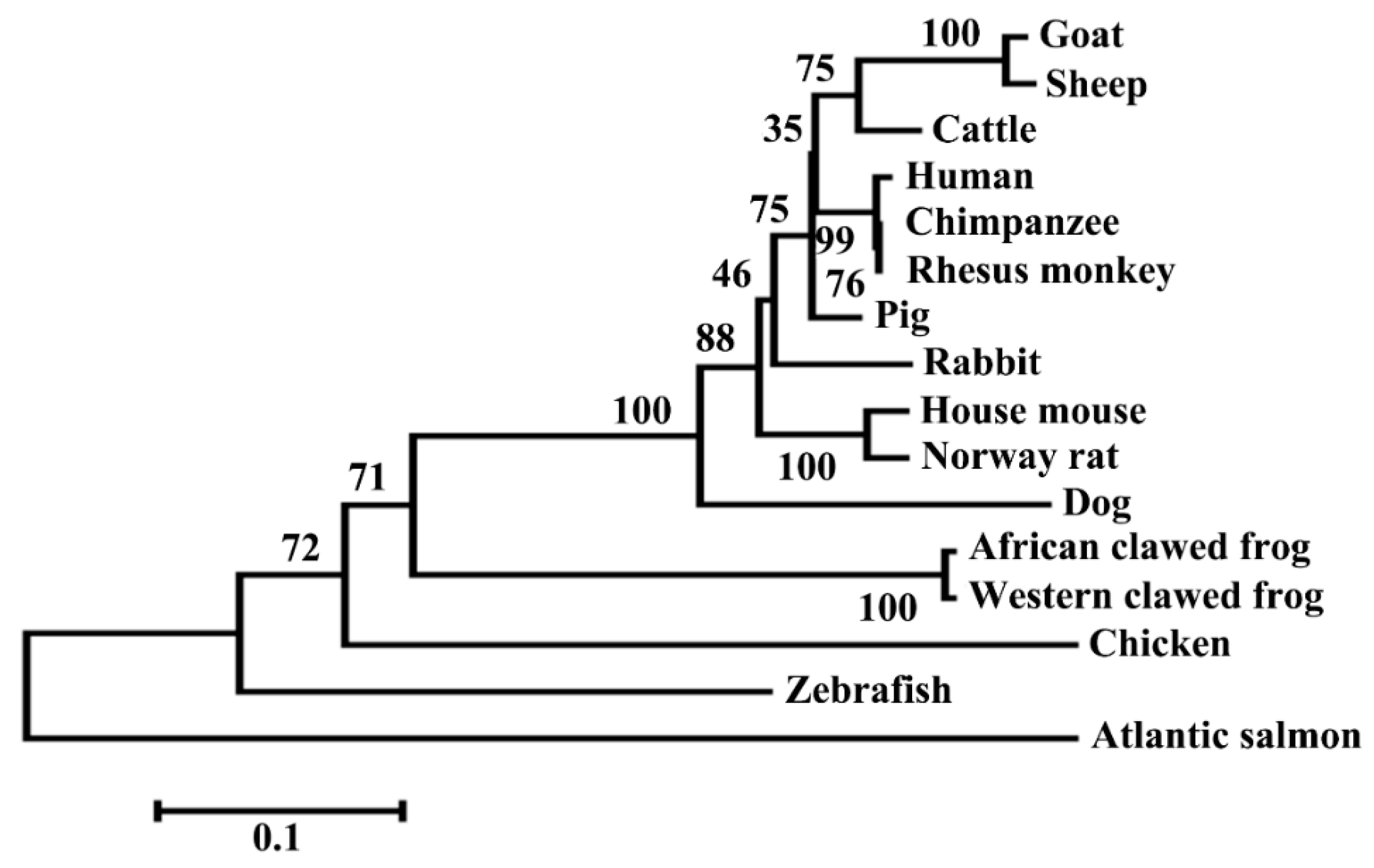
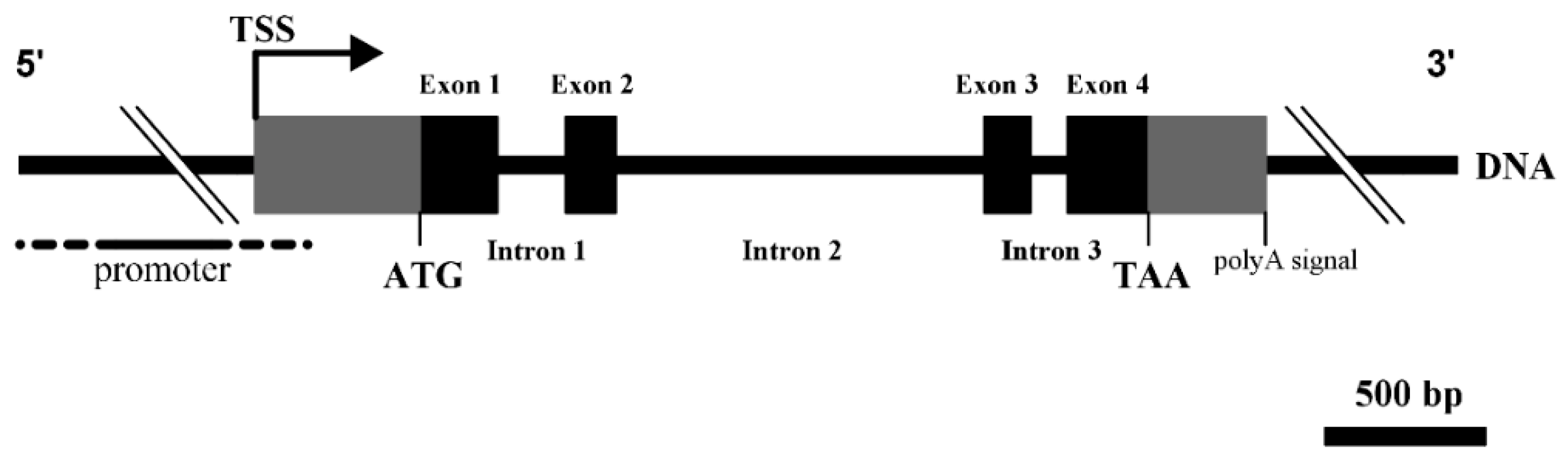
3.1. Structural Characterization of Ovine DKK1
3.2. Bioinformatics Analysis of Ovine DKK1 Promoter
3.3. Identification of SNP in DKK1
3.4. Allele, Genotype and Haplotype Frequencies of Ovine DKK1
3.5. Phenotypic and Genetic Correlations of the Wool Production and Quality Traits
3.6. Association of the Identified SNPs with Wool Production and Quality in Sheep
3.7. Association of DKK1 Haplotypes with Wool Quality Traits
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ling, Y.H.; Xiang, H.; Zhang, G.; Ding, J.P.; Zhang, Z.J.; Zhang, Y.H.; Han, J.L.; Ma, Y.H.; Zhang, X.R. Identification of complete linkage disequilibrium in the DSG4 gene and its association with wool length and crimp in Chinese indigenous sheep. Genet. Mol. Res. 2014, 13, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Forrest, R.H.; Itenge-Mweza, T.O.; Mckenzie, G.W.; Zhou, H.; Frampton, C.M.; Hickford, J.G.H. Polymorphism of the ovine β3-adrenergic receptor gene (ADRB3) and its association with wool mean staple strength and yield. Anim. Genet. 2009, 40, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Libro, R.; Bramanti, P.; Mazzon, E. The role of the Wnt canonical signaling in neurodegenerative diseases. Life Sci. 2016, 158, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Kandyba, E.; Kobielak, K. Wnt7b is an important intrinsic regulator of hair follicle stem cell homeostasis and hair follicle cycling. Stem Cells 2014, 32, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Zhang, Y.; Xu, M.; Yang, Y.; Ito, M.; Peng, T.; Cui, Z.; Nagy, A.; Hadjantonakis, A.K.; Lang, R.A.; et al. Distinct functions for Wnt/β-catenin in hair follicle stem cell proliferation and survival and interfollicular epidermal homeostasis. Cell Stem Cell 2013, 13, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Sick, S.; Reinker, S.; Timmer, J.; Schlake, T. WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science 2006, 314, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.R.; Choi, Y.S.; Millar, S.E.; Udey, M.C. Wnt signaling influences the development of murine epidermal langerhans cells. J. Investig. Dermatol. 2011, 131, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Guo, H.; Qiu, W.; Lai, X.; Yang, T.; Widelitz, R.B.; Chuong, C.M.; Lian, X.; Yang, L. Modulating hair follicle size with Wnt10b/DKK1 during hair regeneration. Exp. Dermatol. 2014, 23, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kwack, M.H.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Dickkopf 1 promotes regression of hair follicles. J. Investig. Dermatol. 2012, 132, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Lee, H.Y.; Kim, I.; Choi, K.; Park, J.; Yoon, S.K. Increased expression of Dkk1 by HR is associated with alteration of hair cycle in hairpoor mice. J. Dermatol. Sci. 2014, 74, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Müller-Röver, S.; Handjiski, B.; van der Veen, C.; Eichmüller, S.; Foitzik, K.; Mckay, I.A.; Stenn, K.S.; Paus, R. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Investig. Dermatol. 2001, 117, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Alonso, L.; Fuchs, E. The hair cycle. J. Cell Sci. 2006, 119, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.; Foitzik, K. Biology of the hair follicle: The basics. Semin. Cutan. Med. Surg. 2006, 25, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.Q.; Yang, Y.L.; Jian-Hong, N.I.; Wan, P.C.; Dai, R.; Zhou, P.; Zhang, Y.S.; Bai, D.P. Breeding of a new China merino strain with super-fine wool. Grass-Feed. Livest. 2010, 2, 1–7. [Google Scholar]
- National Research Council. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- McGregor, B. Comparative productivity and grazing behaviour of Huacaya alpacas and Peppin Merino sheep grazed on annual pastures. Small Rumin. Res. 2002, 44, 219–232. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. Cold Spring Harb. Protoc. 2006, 2006. [Google Scholar] [CrossRef]
- Chun, J. Computer Assisted Classification and Identification of Actinomycetes. Ph.D. Thesis, University of Newcastle upon Tyne, Newcastle upon Tyne, UK, 1995. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rong, E.G.; Yang, H.; Zhang, Z.W.; Wang, Z.P.; Yan, X.H.; Li, H.; Wang, N. Association of methionine synthase gene polymorphisms with wool production and quality traits in Chinese Merino population. J. Anim. Sci. 2015, 93, 4601–4609. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Acharya, M.; Mukherjee, S.; Ray, J.; Choudhury, S.; Khan, M.; Ray, K. Mutations in MYOC gene of Indian primary open angle glaucoma patients. Mol. Vis. 2002, 8, 442–448. [Google Scholar] [PubMed]
- Thomas, R.K.; Baker, A.C.; Debiasi, R.M.; Winckler, W.; Laframboise, T.; Lin, W.M.; Wang, M.; Feng, W.; Zander, T.; MacConaill, L.; et al. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 2007, 39, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.; Ziaugra, L.; Tabbaa, D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr. Protoc. Hum. Genet. 2009, 60, 2.12.1–2.12.18. [Google Scholar]
- Gilmour, A.; Gogel, B.; Cullis, B.; Thompson, R. ASReml User Guide Release 2.0; VSN International Ltd.: Hemel Hempstead, UK, 2006. [Google Scholar]
- Falconer, D.S.; Mackay, T.F.C. Introduction to quantitative genetics. Q. Rev. Biol. 1996, 49, 223–226. [Google Scholar]
- Buchanan, J.W.; Reecy, J.M.; Garrick, D.J.; Duan, Q.; Beitz, D.C.; Mateescu, R.G. Genetic parameters and genetic correlations among triacylglycerol and phospholipid fractions in Angus cattle. J. Anim. Sci. 2015, 93, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Brott, B.K.; Sokol, S.Y. Regulation of Wnt/LRP signaling by distinct domains of Dickkopf proteins. Mol. Cell. Biol. 2002, 22, 6100–6110. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, M.N.; Schwartz, D.R.; Vonica, A.; Brivanlou, A.H.; Cho, K.R.; Varmus, H.E. FGF-20 and DKK1 are transcriptional targets of β-catenin and FGF-20 is implicated in cancer and development. EMBO J. 2005, 24, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shou, J.; Chen, X. Dickkopf-1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene 2000, 19, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Li, Y.; Liu, Y.; Zhang, J.; Chen, K.; Huang, A.; Tang, H. HBx and SP1 upregulate DKK1 expression. Acta Biochim. Pol. 2017, 64, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Liu, Q.; Zhang, Y.Y.; Fan, X.C.; Xu, X.X.; Lv, Z.C.; Wei, M.L.; Jing, Y.; Mu, F.; Xu, R.F. Association of novel polymorphisms of forkhead box L2 and growth differentiation factor-9 genes with egg production traits in local Chinese Dagu hens. Poult. Sci. 2015, 94, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.R.; Li, Y.K.; Fu, C.Z.; Wang, J.L.; Wang, H.B.; Zan, L.S. Effects of bovine SMO gene polymorphisms on the body measurement and meat quality traits of Qinchuan cattle. Genet. Mol. Res. 2014, 13, 8105–8117. [Google Scholar] [CrossRef] [PubMed]
- Safari, E.; Fogarty, N.M.; Gilmour, A.R. A review of genetic parameter estimates for wool, growth, meat and reproduction traits in sheep. Livest. Prod. Sci. 2005, 92, 271–289. [Google Scholar] [CrossRef]
- Di, J.; Zhang, Y.; Tian, K.C.; Lazate; Liu, J.F.; Xu, X.M.; Zhang, Y.J.; Zhang, T.H. Estimation of (co)variance components and genetic parameters for growth and wool traits of Chinese superfine merino sheep with the use of a multi-trait animal model. Livest. Sci. 2011, 138, 278–288. [Google Scholar] [CrossRef]
- Notter, D.; Kuehn, L.; Kott, R. Genetic analysis of fibre characteristics in adult Targhee ewes and their relationship to breeding value estimates derived from yearling fleeces. Small Rumin. Res. 2007, 67, 164–172. [Google Scholar] [CrossRef]
- Safari, E.; Fogarty, N.; Gilmour, A.R.; Atkins, K.; Mortimer, S.; Swan, A.; Brien, F.; Greeff, J.; Van Der Werf, J. Genetic correlations among and between wool, growth and reproduction traits in Merino sheep. J. Anim. Breed. Genet. 2007, 124, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Liu, R.; Li, Q.; Tang, X.; Yu, M.; Li, X.; Cao, J.; Zhao, S. Identification of microRNAs in wool follicles during anagen, catagen and telogen phases in Tibetan sheep. PLoS ONE 2013, 8, e77801. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Yang, Z.; Darwish, H.; Zhang, Y.; Ge, Y.; Zhang, X.; Li, R.; Deng, X. Molecular cloning and characterization of the β-catenin gene from fine-wool sheep. Gene 2014, 546, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Nakajima, T.; Takahashi, K.; Kubo, R.; Yazawa, S.; Kominato, Y. The 3′ flanking region of the human ABO histo-blood group gene is involved in negative regulation of gene expression. Legal Med. 2011, 13, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, I.; Takafuji, K.; Ando, Y.; Satake, W.; Kanagawa, M.; Kobayashi, K.; Nagamori, S.; Shinohara, T.; Ito, C.; Yamamoto, M. YY1 binds to α-synuclein 3′-flanking region SNP and stimulates antisense noncoding RNA expression. J. Hum. Genet. 2013, 58, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Maiti, G.P.; Ghosh, A.; Mondal, P.; Baral, A.; Datta, S.; Samadder, S.; Nayak, S.P.; Chakrabarti, J.; Biswas, J.; Sikdar, N. The SNP rs1049430 in the 3′-1 UTR of SH3GL2 regulates its expression: Clinical and 2 prognostic implications in head and neck squamous cell carcinoma. Biochim. Biophys. Acta 2015, 1852, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Qian, X.; Zhang, C. U/G SNP rs111904020 in 3′ UTR of STAT3 regulated by miR-214 promotes hepatocellular carcinoma development in Chinese population. Tumor Biol. 2016, 37, 14629–14635. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Robles, A.I.; McClary, A.C.; Haznadar, M.; Bowman, E.D.; Pine, S.R.; Brown, D.; Khan, M.; Shiraishi, K.; Kohno, T. Identification of a functional SNP in the 3′ UTR of CXCR2 that is associated with reduced risk of lung cancer. Cancer Res. 2015, 75, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Popp, N.A.; Yu, D.; Green, B.; Chew, E.Y.; Ning, B.; Chan, C.C.; Tuo, J. Functional single nucleotide polymorphism in IL-17A 3′ untranslated region is targeted by miR-4480 in vitro and may be associated with age-related macular degeneration. Environ. Mol. Mutagen. 2016, 57, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Le, H.H.; Nott, A.; Moore, M.J. How introns influence and enhance eukaryotic gene expression. Trends Biochem. Sci. 2003, 28, 215. [Google Scholar] [CrossRef]
- Seo, S.; Takayama, K.; Uno, K.; Ohi, K.; Hashimoto, R.; Nishizawa, D.; Ikeda, K.; Ozaki, N.; Nabeshima, T.; Miyamoto, Y. Functional analysis of deep intronic SNP rs13438494 in intron 24 of PCLO gene. PLoS ONE 2013, 8, e76960. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Primer Name | Coverage | Sequence (5′-3′) | Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|---|
| DKK1-F1 | GCAAAGCGACACTCCTCC | 955 | 59.0 | |
| DKK1-R1 | GCTCTTACACCCCAGATTTTCGG | |||
| DKK1-F2 | chr22:6668693-6670458 | ATGAAACCGAACTCTTGACTTA | 1771 | 58.6 |
| DKK1-R2 | AACTTGATTAGGCAGACACG | |||
| DKK1-F3 | chr22:6667603-6668709 | GTCTGCYTAATCAAGTTCATCTAC | 1107 | 56.2 |
| DKK1-R3 | GGTTCTTGATV4GCGTTGGAAT | |||
| DKK1-F4 | chr22:6665566-6667623 | ATTCCAACGCB1ATCAAGAACC | 2100 | 56.2 |
| DKK1-R4 | CAGR2CCTTCY3CCRCAGTAACA | |||
| DKK1-F5 | chr22:6665250-6665656 | TCTCAAGGAAGGTCAAGTGT | 407 | 52.0 |
| DKK1-R5 | GGTTGCATTTACAGGTAAGT | |||
| DKK1-F6 | chr22:6663304-6665269 | ACTTACCTGTAAATGCAACC | 1966 | 46.9 |
| DKK1-R6 | TTATCCTACAACTATATCAGCAC |
| SNP ID | Location | db SNP rs # ID 1 | Alleles | Nomenclature 2 | MAF 3 | HW P 4 |
|---|---|---|---|---|---|---|
| SNP1 | Intron 2 | rs424404921 | G > A | g.909 G > A | 0.217 | 0.0546 |
| SNP2 | Intron 2 | rs411273297 | C > A | g.927 C > A | 0.217 | 0.0223 |
| SNP3 | Intron 2 | rs592810220 | Del CTG | g.1049 Del CTG | 0.235 | 2.9 × 10−37 |
| SNP4 | Intron 2 | rs409382156 | G > C | g.1147 G > C | 0.216 | 0.0070 |
| SNP5 | Intron 2 | rs419857384 | G > T | g.1321 G > T | 0.159 | 1.2 × 10−10 |
| SNP6 | Intron 3 | rs419902277 | G > A | g.2018 G > A | 0.385 | 0.4416 |
| SNP7 | exon 4 | rs401363941 | G > A | g.2062 G > A | 0.327 | 0.948 |
| SNP8 | 3′ UTR | rs428450289 | T > C | g.3093 T > C | 0.217 | 0.2552 |
| SNP9 | 3′ UTR | rs415015281 | C > T | g.3329 C > T | 0.490 | 0.4864 |
| SNP10 | 3′ Flanking Region | novel | T > C | g.4123 T > C | 0.355 | 1.2 × 10−19 |
| SNP11 | 3′ Flanking Region | novel | T > C | g.4443 T > C | 0.471 | 1.3 × 10−139 |
| Lines | SNP5 | SNP8 | SNP9 | SNP10 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | MFD (μm) | Genotype | MFD (μm) | FW (kg) | Genotype | Curvature (/2.5 cm) | Genotype | FDSD (μm) | CVFD | |
| A | GG | 21.08 ± 0.380 c | CC | 18.33 ± 1.362 d,e,f | 5.45 ± 0.515 a,c | CC | 11.56 ± 0.487 b,c,d,e | CC | 4.56 ± 0.150 a,b | 21.41 ± 0.608 a |
| GT | 20.72 ± 0.522 c,d,e | TC | 22.31 ± 0.531 a | 4.97 ± 0.191 a,b | TC | 11.11 ± 0.514 e,f | TC | 4.30 ± 0.144 a,b,c,d | 20.62 ± 0.585 a,b,c | |
| TT | 21.12 ± 0.823 b,c,d,e | TT | 20.36 ± 0.463 c,d,e | 4.44 ± 0.185 c,d,e | TT | 11.52 ± 0.681 b,c,d,e,f | TT | 4.09 ± 0.247 c,d,e | 19.56 ± 1.002 b,c,d,e,f | |
| B | GG | 21.54 ± 0.471 b,c | CC | NE | NE | CC | 13.50 ± 0.954 a,b | CC | 4.07 ± 0.193 c,d,e,g | 19.41 ± 0.784 b,c,d,e,f |
| GT | 19.08 ± 0.817 e,f | TC | 19.78 ± 0.946 c,d,e,f | 4.95 ± 0.371 a,b,c,d | TC | 11.06 ± 0.639 c,e,f | TC | 4.12 ± 0.192 b,c,d,e | 20.03 ± 0.778 a,b,c,d,e | |
| TT | 19.89 ± 1.234 c,d,e,f | TT | 20.90 ± 0.489 b,c,d | 4.95 ± 0.186 a,b | TT | 13.56 ± 1.022 a,b,c | TT | 4.00 ± 0.247 b,c,d,e,f,g | 20.11 ± 1.002 a,b,c,d,e,f | |
| PW | GG | 21.05 ± 0.454 c,d | CC | 22.59 ± 1.284 a,b,c | 4.58 ± 0.489 a,b,c,d,e,f | CC | 10.45 ± 0.765 e,f | CC | 4.64 ± 0.179 a | 20.76 ± 0.725 a,b,c,d |
| GT | 22.86 ± 0.693 a,b | TC | 22.09 ± 0.631 a,b | 4.64 ± 0.250 a,b,c,d,e | TC | 11.35 ± 0.409 d,e,f | TC | 4.27 ± 0.157 a,b,c,d | 19.33 ± 0.637 c,d,e,f | |
| TT | NE | TT | 21.29 ± 0.514 a,b,c | 4.56 ± 0.196 a,b,c,d,e | TT | 11.61 ± 0.856 b,c,d,e,f | TT | 4.10 ± 0.268 a,b,c,d,e,f,g | 18.04 ± 1.086 e,f | |
| PM | GG | 20.74 ± 0.504 c,d,e | CC | 20.73 ± 1.047 a,b,c,d,e,f | 4.19 ± 0.400 a,b,c,d,e,f | CC | 11.98 ± 0.687 b,c,d,e | CC | 4.41 ± 0.118 a,b,c | 21.09 ± 0.480 a,b |
| GT | 19.73 ± 1.373 c,d,e,f | TC | 21.56 ± 0.655 a,b,c | 3.67 ± 0.252 f,g | TC | 10.97 ± 0.564 e,f | TC | 3.73 ± 0.276 d,e,f,g | 17.20 ± 1.120 f | |
| TT | NE | TT | 21.03 ± 0.482 a,b,c,d | 3.29 ± 0.191 g | TT | 12.17 ± 0.505 b,c,d,e | TT | NE | NE | |
| U | GG | 23.74 ± 0.701 a | CC | NE | NE | CC | 8.88 ± 1.198 f | CC | NE | NE |
| GT | 19.86 ± 1.307 c,d,e,f | TC | 22.06 ± 0.744 a,b,c | 4.17 ± 0.283 d,e,f | TC | 14.62 ± 1.640 a,b,d | TC | NE | NE | |
| TT | NE | TT | 20.21 ± 1.070 a,b,c,d,e,f | 4.10 ± 0.407 b,d,e,f,g | TT | 17.15 ± 1.771 a | TT | NE | NE | |
| SF | GG | 18.79 ± 0.297 f | CC | NE | NE | CC | 15.44 ± 0.600 a | CC | 3.61 ± 0.129 f | 19.04 ± 0.522 d,e,f |
| GT | 19.86 ± 1.307 c,d,e,f | TC | 19.24 ± 0.494 e,f | 4.07 ± 0.198 e,f | TC | 15.32 ± 0.434 a | TC | 3.75 ± 0.159 e,f,g | 19.45 ± 0.644 c,d,e,f | |
| TT | 20.09 ± 1.275 b,c,d,e,f | TT | 18.81 ± 0.359 f | 4.66 ± 0.139 a,b,c,d | TT | 14.74 ± 0.523 a | TT | 3.59 ± 0.168 f,g | 19.37 ± 0.682 c,d,e,f | |
| p value | 0.0037 2 | 0.0139 | 0.0456 | 0.0186 | 0.0363 | 0.0053 | ||||
| Lines | Haplotype | MFD (μm) |
|---|---|---|
| A | H1: AAICGAGCCCC | NE |
| H2: AAICTAATCTT | 20.97 ± 0.628 a,b,c,d,e,g | |
| H3: GCDGGAATCTT | 22.43 ± 0.739 a,b | |
| H4: GCDGGGGCCCC | NE | |
| H5: GCDGGGGTTCC | 21.43 ± 0.670 a,b,c,d,e | |
| B | H1: AAICGAGCCCC | NE |
| H2: AAICTAATCTT | 18.43 ± 0.811 f | |
| H3: GCDGGAATCTT | 19.68 ± 0.654 d,e,f,g | |
| H4: GCDGGGGCCCC | 18.58 ± 1.369 d,e,f,g | |
| H5: GCDGGGGTTCC | 21.30 ± 0.881 a,b,c,d,e | |
| PW | H1: AAICGAGCCCC | 20.10 ± 1.024 b,d,e,f,g |
| H2: AAICTAATCTT | 23.21 ± 1.363 a,c | |
| H3: GCDGGAATCTT | 21.45 ± 0.627 a,b,c,d | |
| H4: GCDGGGGCCCC | 20.44 ± 1.624 a,b,c,d,e,f,g | |
| H5: GCDGGGGTTCC | 20.41 ± 0.811 a,b,c,d,e,f,g | |
| PM | H1: AAICGAGCCCC | 18.43 ± 1.106 f,g |
| H2: AAICTAATCTT | 21.05 ± 1.380 a,b,c,d,e,f,g | |
| H3: GCDGGAATCTT | 20.34 ± 0.692 c,d,e,f,g | |
| H4: GCDGGGGCCCC | 20.18 ± 0.678 d,e,f,g | |
| H5: GCDGGGGTTCC | 21.09 ± 0.679 a,b,c,d,e | |
| U | H1: AAICGAGCCCC | NE |
| H2: AAICTAATCTT | NE | |
| H3: GCDGGAATCTT | NE | |
| H4: GCDGGGGCCCC | NE | |
| H5: GCDGGGGTTCC | NE | |
| SF | H1: AAICGAGCCCC | 19.81 ± 0.722 d,e,f,g |
| H2: AAICTAATCTT | 20.16 ± 0.519 d,e,f,g | |
| H3: GCDGGAATCTT | 18.81 ± 0.538 f | |
| H4: GCDGGGGCCCC | NE | |
| H5: GCDGGGGTTCC | 19.89 ± 0.424 e,f,g | |
| p value | 0.014 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, F.; Rong, E.; Jing, Y.; Yang, H.; Ma, G.; Yan, X.; Wang, Z.; Li, Y.; Li, H.; Wang, N. Structural Characterization and Association of Ovine Dickkopf-1 Gene with Wool Production and Quality Traits in Chinese Merino. Genes 2017, 8, 400. https://doi.org/10.3390/genes8120400
Mu F, Rong E, Jing Y, Yang H, Ma G, Yan X, Wang Z, Li Y, Li H, Wang N. Structural Characterization and Association of Ovine Dickkopf-1 Gene with Wool Production and Quality Traits in Chinese Merino. Genes. 2017; 8(12):400. https://doi.org/10.3390/genes8120400
Chicago/Turabian StyleMu, Fang, Enguang Rong, Yang Jing, Hua Yang, Guangwei Ma, Xiaohong Yan, Zhipeng Wang, Yumao Li, Hui Li, and Ning Wang. 2017. "Structural Characterization and Association of Ovine Dickkopf-1 Gene with Wool Production and Quality Traits in Chinese Merino" Genes 8, no. 12: 400. https://doi.org/10.3390/genes8120400