
Targeting Splicing in the Treatment of Human Disease




Abstract
:1. Introduction
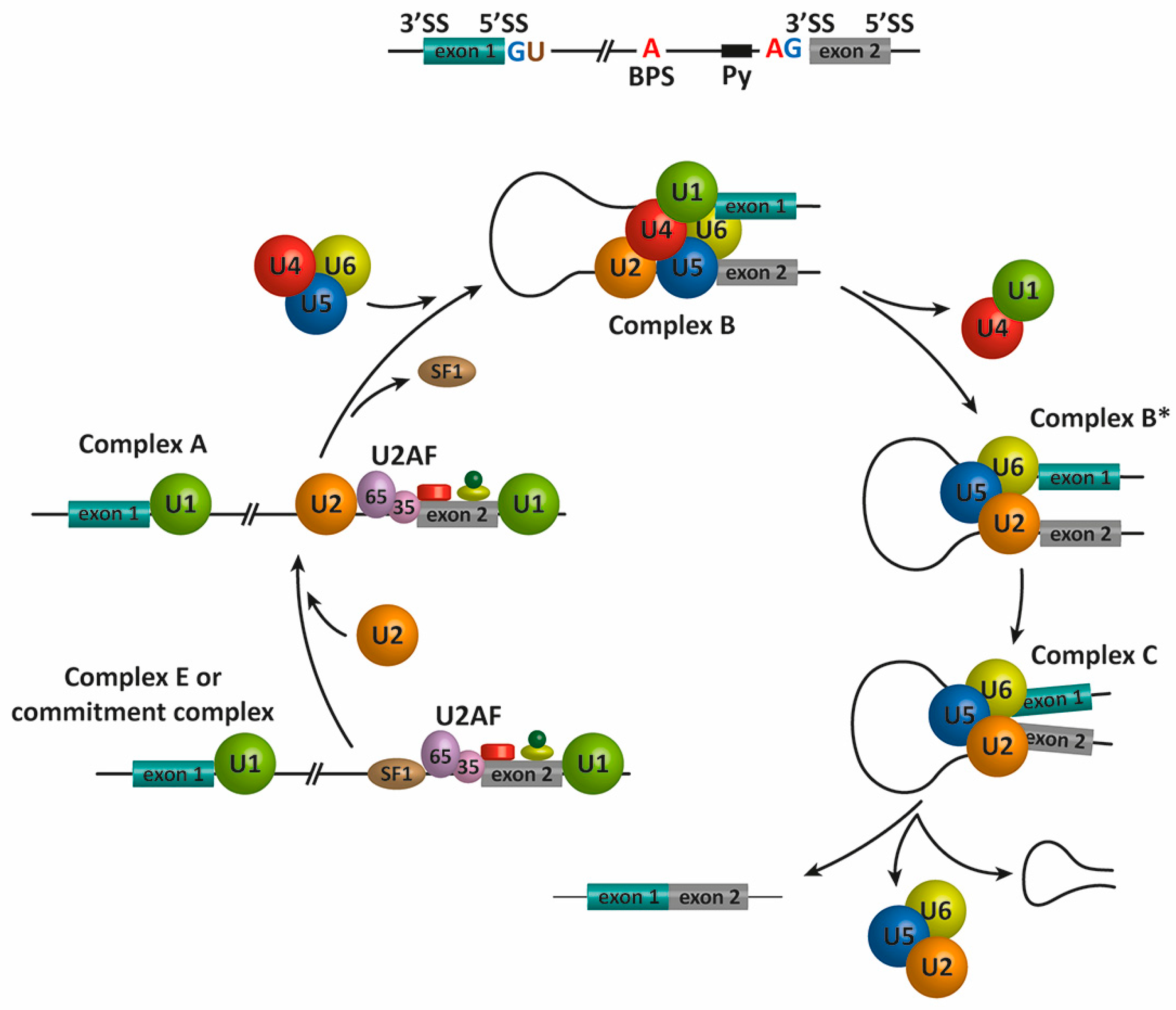
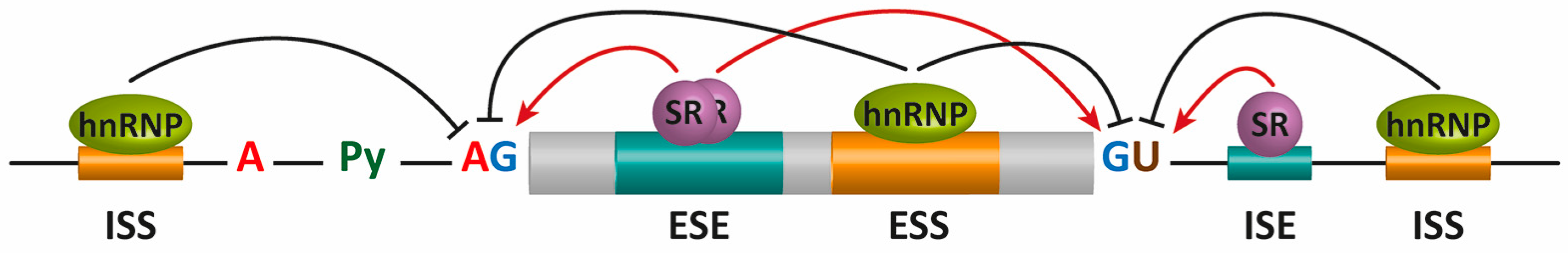
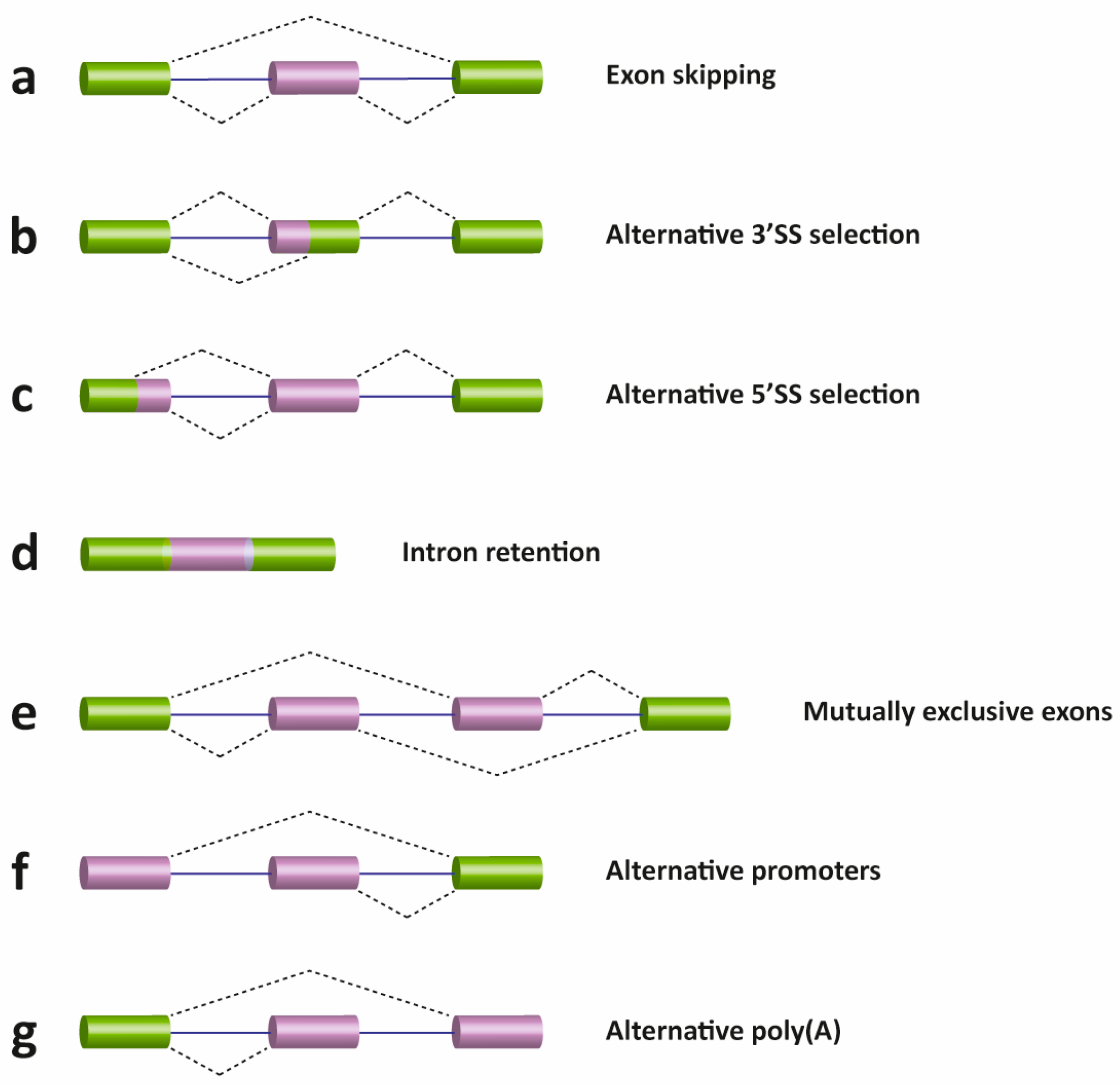
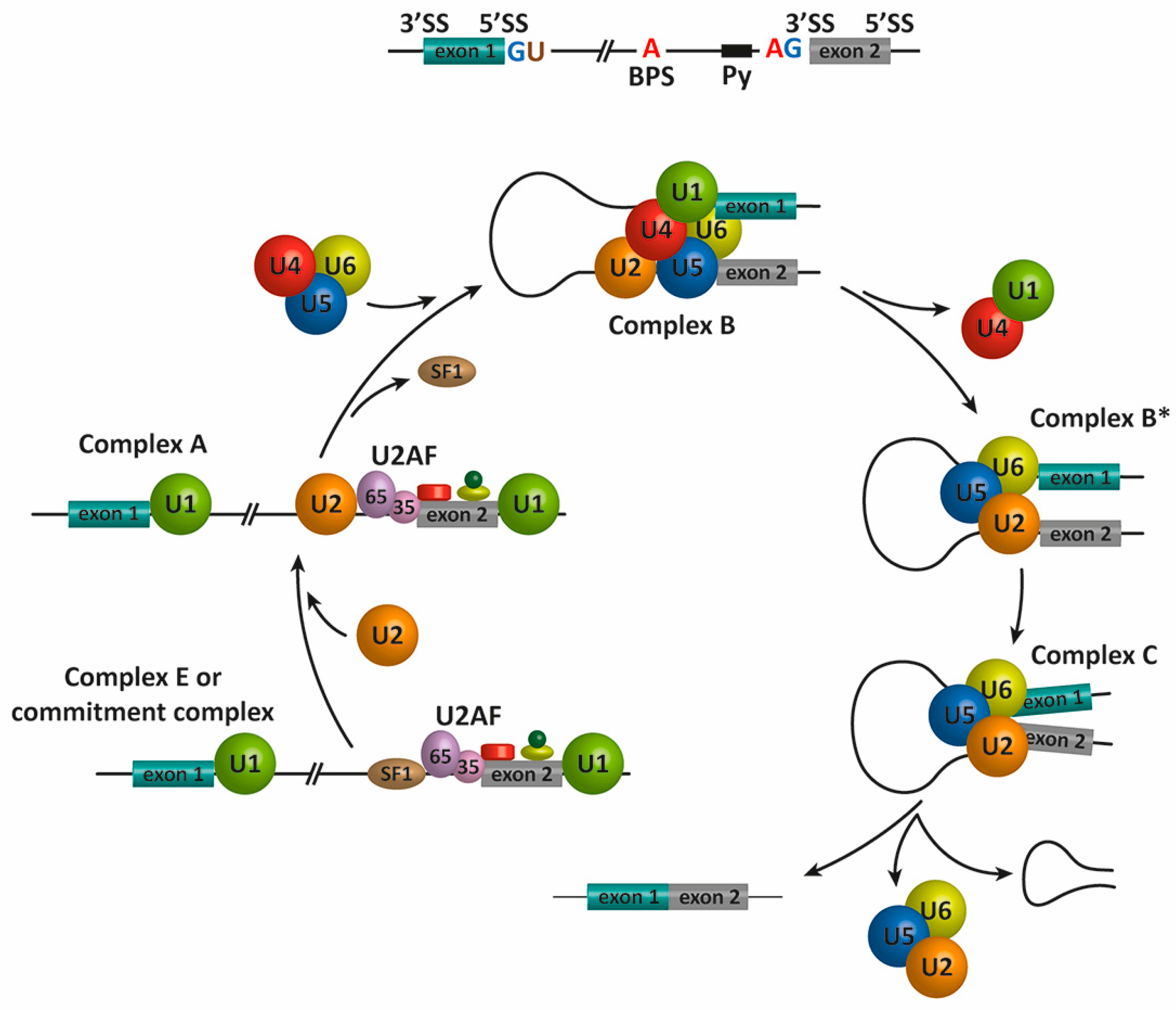
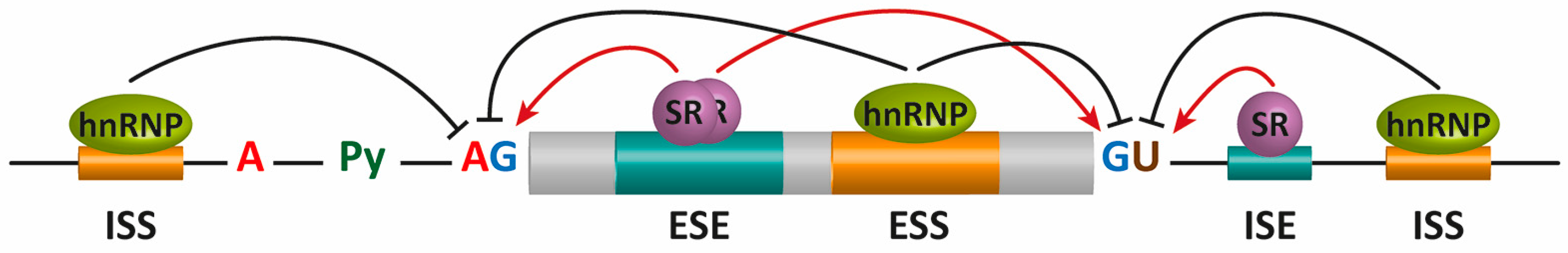
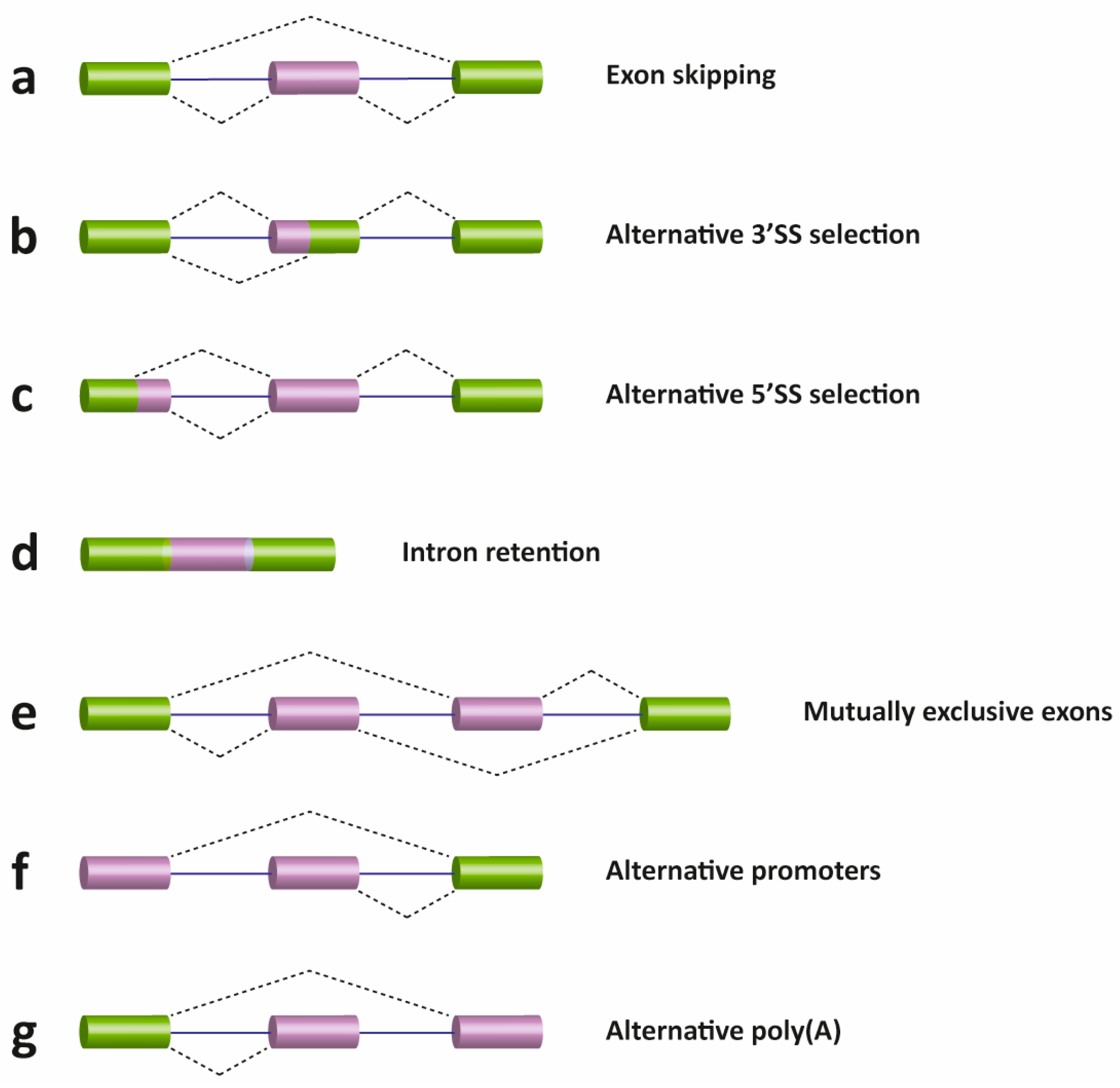
2. Pre-mRNA Splicing
3. Connections between Splicing and Human Disease
4. Therapeutic Approaches
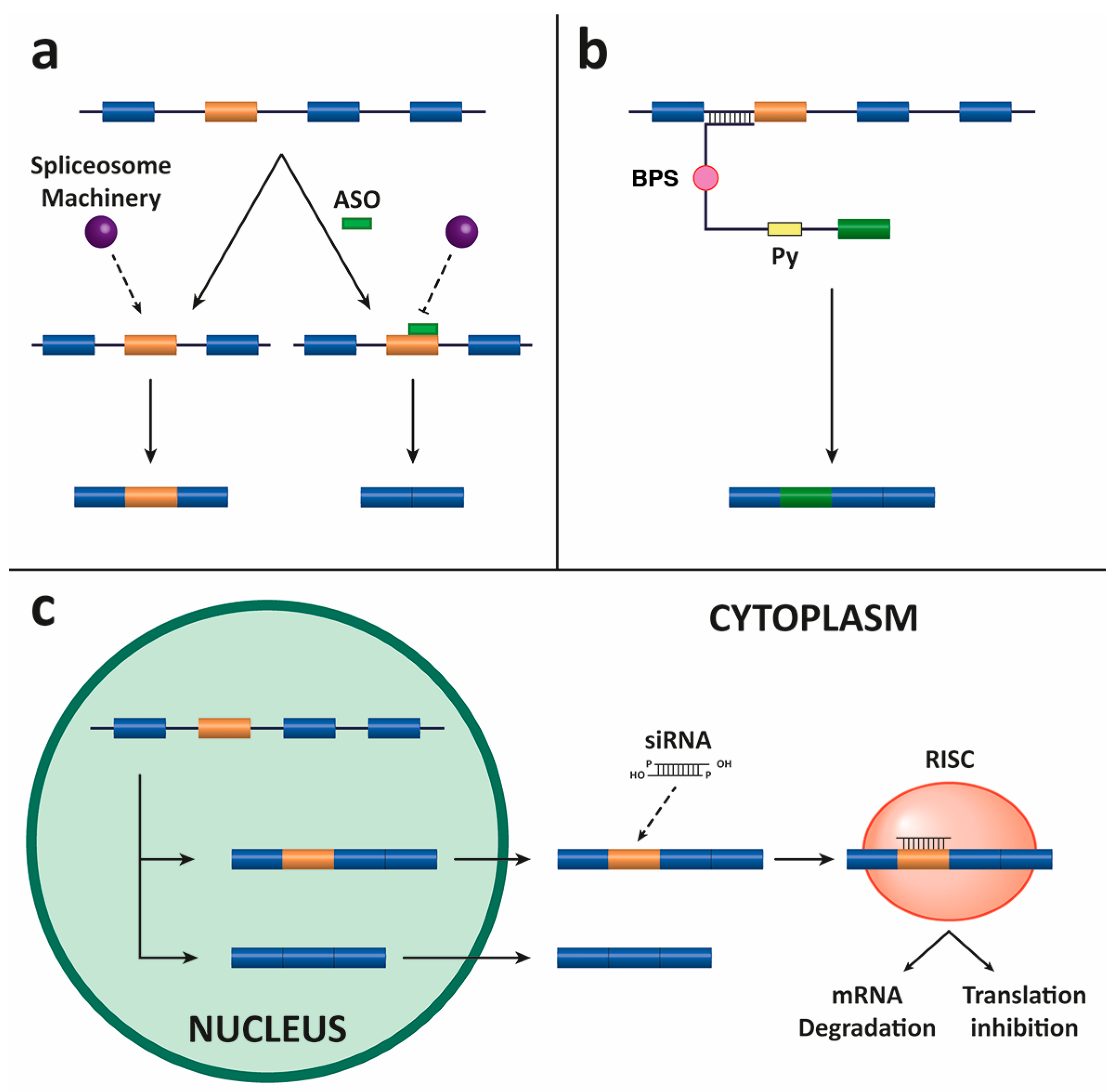
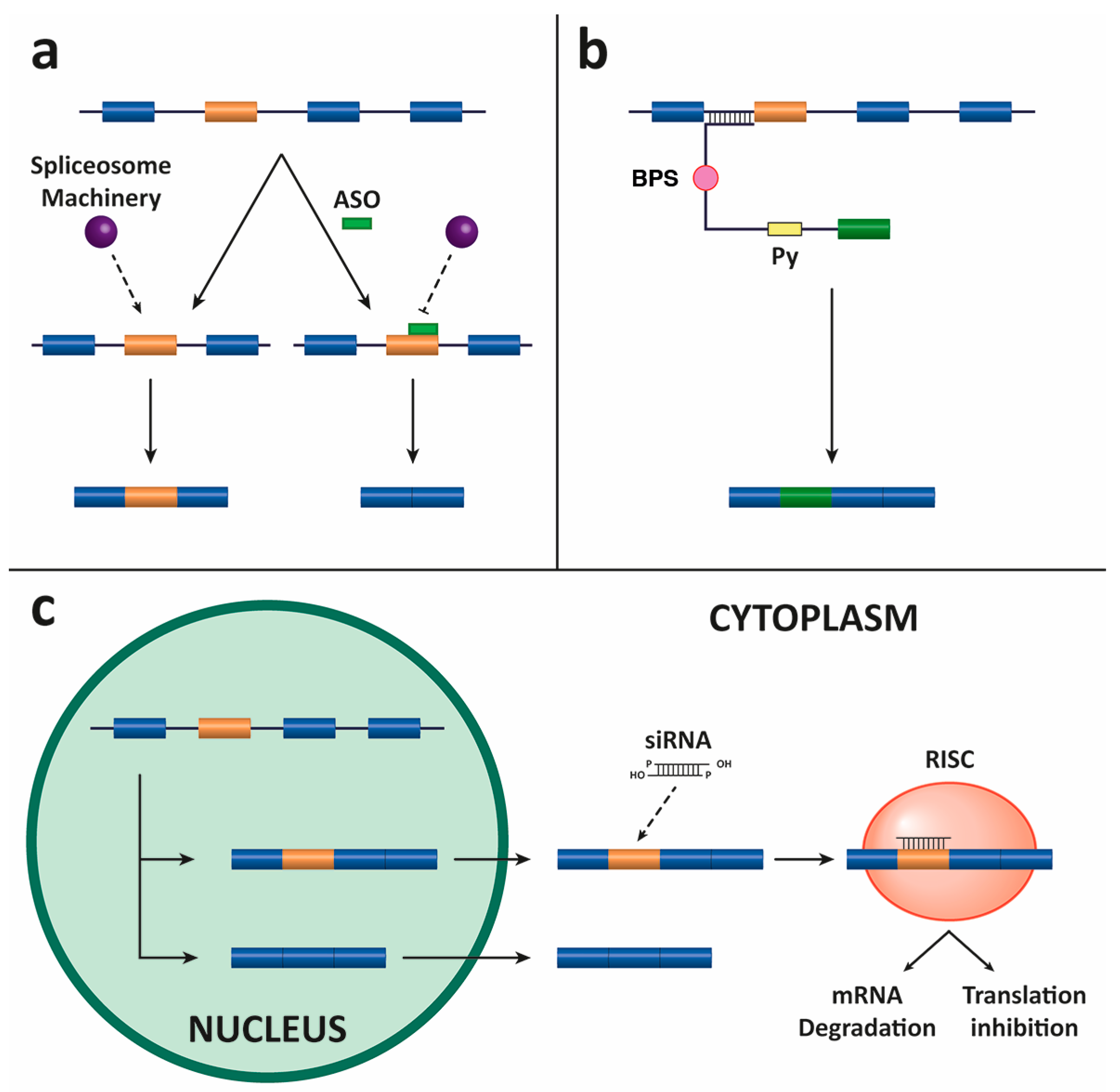
4.1. Antisense Oligonucleotides (ASOs)
4.2. Spliceosome-Mediated RNA Trans-Splicing (SMaRT)
4.3. Small Interfering RNAs (siRNAs)
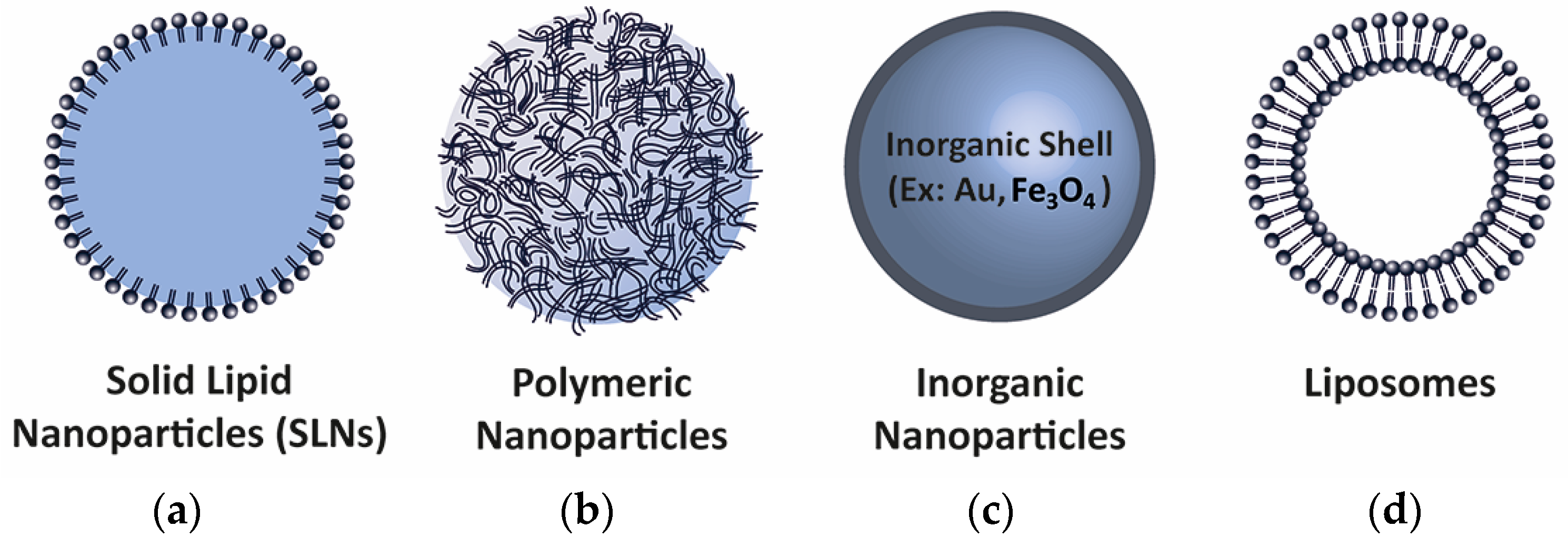
5. Delivery Methods
5.1. Viral Methods
5.2. Non-Viral Methods
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AS | alternative splicing |
| pre-mRNA | precursor messenger RNA |
| mRNA | messenger RNA |
| snRNPs | small nuclear ribonucleoproteins |
| SS | splice sites |
| Py | Polypyrimidine |
| BPS | branch point sequence |
| ISS, ISE, ESS, and ESE | exonic and intronic splicing silencers or enhancers |
| ASO | antisense RNA |
| SMaRT | spliceosome-mediated RNA trans-splicing |
| PTM | pre-trans-splicing molecule |
| siRNAs | small interfering RNAs |
References
- Consortium IHGS. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- The Human Proteome Map. Available online: www.humanproteomemap.org (accessed on 16 December 2016).
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Carstens, R.P. Functional roles of alternative splicing factors in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, A.A.; Friedman, L.J.; Gallagher, S.S.; Crawford, D.J.; Anderson, E.G.; Wombacher, R.; Ramirez, N.; Cornish, V.W.; Gelles, J.; Moore, M.J. Ordered and dynamic assembly of single spliceosomes. Science 2011, 331, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Berglund, J.A.; Abovich, N.; Rosbash, M. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev. 1998, 12, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Berglund, J.A.; Chua, K.; Abovich, N.; Reed, R.; Rosbash, M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 1997, 89, 781–787. [Google Scholar] [CrossRef]
- MacMillan, A.M.; Query, C.C.; Allerson, C.R.; Chen, S.; Verdine, G.L.; Sharp, P.A. Dynamic association of proteins with the pre-mRNA branch region. Genes Dev. 1994, 8, 3008–3020. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.S.; Manley, J.L. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev. 1995, 9, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, P.L.; Guthrie, C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 1998, 8, 847–855. [Google Scholar] [CrossRef]
- Schwer, B.; Gross, C.H. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J. 1998, 17, 2086–2094. [Google Scholar] [CrossRef] [PubMed]
- Fourmann, J.B.; Schmitzova, J.; Christian, H.; Urlaub, H.; Ficner, R.; Boon, K.L.; Fabrizio, P.; Lührmann, R. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev. 2013, 27, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.W.; Valcarcel, J. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem. Sci. 2000, 25, 381–388. [Google Scholar] [CrossRef]
- Singh, R.; Valcarcel, J. Building specificity with nonspecific RNA-binding proteins. Nat. Struct. Mol. Biol. 2005, 12, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M.; Valcarcel, J. A simple principle to explain the evolution of pre-mRNA splicing. Genes Dev. 2006, 20, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Norcliffe-Kaufmann, L.; Kaufmann, H. Familial dysautonomia (Riley-Day syndrome): When baroreceptor feedback fails. Auton. Neurosci. 2012, 172, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.B.; Sorensen, S.; Cartegni, L.; Corydon, T.J.; Doktor, T.K.; Schroeder, L.D.; Reinert, L.S.; Elpeleg, O.; Krainer, A.R.; Gregersen, N.; et al. Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: A synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am. J. Hum. Genet. 2007, 80, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar] [PubMed]
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Genes and mutations causing autosomal dominant retinitis pigmentosa. Cold Spring Harb. Perspect Med. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Disset, A.; Bourgeois, C.F.; Benmalek, N.; Claustres, M.; Stevenin, J.; Tuffery-Giraud, S. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum. Mol. Genet. 2006, 15, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Edery, P.; Marcaillou, C.; Sahbatou, M.; Labalme, A.; Chastang, J.; Touraine, R.; Tubacher, E.; Senni, F.; Bober, M.B.; Nampoothiri, S.; et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 2011, 332, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi-Ikeda, M.; Kobayashi, K.; Kanagawa, M.; Yu, C.C.; Mori, K.; Oda, T.; Kuga, A.; Kurahashi, H.; Akman, H.O.; DiMauro, S.; et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature 2011, 478, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tucker, H.M.; Grear, K.E.; Simpson, J.F.; Manning, A.K.; Cupples, L.A.; Estus, S. A common polymorphism decreases low-density lipoprotein receptor exon 12 splicing efficiency and associates with increased cholesterol. Hum. Mol. Genet. 2007, 16, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.S.; Trapnell, B.C.; Curristin, S.; Cutting, G.R.; Crystal, R.G. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat. Genet. 1993, 3, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.J. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA 2007, 13, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Blanco, M.A.; Baraniak, A.P.; Lasda, E.L. Alternative splicing in disease and therapy. Nat. Biotechnol. 2004, 22, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Skordis, L.A.; Dunckley, M.G.; Yue, B.; Eperon, I.C.; Muntoni, F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. USA 2003, 100, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Villemaire, J.; Dion, I.; Elela, S.A.; Chabot, B. Reprogramming alternative pre-messenger RNA splicing through the use of protein-binding antisense oligonucleotides. J. Biol. Chem. 2003, 278, 50031–50039. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, J.P.; Lucier, J.F.; Lamarche, A.A.; Shkreta, L.; Gendron, D.; Lapointe, E.; Thibault, P.; Paquet, E.; Perreault, J.P.; Abou Elela, S.; et al. Redirecting splicing with bifunctional oligonucleotides. Nucleic Acids Res. 2014, 42, e40. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Takeshima, Y.; Nishio, H.; Sakamoto, H.; Nakamura, H.; Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J. Clin. Investig. 1995, 95, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First global approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Pyle, A.D. Exon skipping therapy. Cell 2016, 167, 1144. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003, 10, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; de Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Marquis, J.; Meyer, K.; Angehrn, L.; Kampfer, S.S.; Rothen-Rutishauser, B.; Schümperli, D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol. Ther. 2007, 15, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Marquis, J.; Trub, J.; Nlend Nlend, R.; Verp, S.; Ruepp, M.D.; Imboden, H.; Barde, I.; Trono, D.; Schümperli, D. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum. Mol. Genet. 2009, 18, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Nlend, R.N.; Schumperli, D. Antisense genes to induce exon inclusion. Methods Mol. Biol. 2012, 867, 325–347. [Google Scholar] [PubMed]
- Goto, M.; Sawamura, D.; Nishie, W.; Sakai, K.; McMillan, J.R.; Akiyama, M.; Shimizu, H. Targeted skipping of a single exon harboring a premature termination codon mutation: Implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J. Investig. Dermatol. 2006, 126, 2614–2620. [Google Scholar] [CrossRef] [PubMed]
- Kalbfuss, B.; Mabon, S.A.; Misteli, T. Correction of alternative splicing of tau in frontotemporal dementia and parkinsonism linked to chromosome 17. J. Biol. Chem. 2001, 276, 42986–42993. [Google Scholar] [CrossRef] [PubMed]
- Khoo, B.; Roca, X.; Chew, S.L.; Krainer, A.R. Antisense oligonucleotide-induced alternative splicing of the APOB mRNA generates a novel isoform of APOB. BMC Mol. Biol. 2007, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Igreja, S.; Clarke, L.A.; Botelho, H.M.; Marques, L.; Amaral, M.D. Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat. 2007, 37, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, S.G.; Clark, R.H.; Puttaraju, M.; Kole, J.; Cohn, J.A.; Mitchell, L.G.; Garcia-Blanco, M.A. 5’ exon replacement and repair by spliceosome-mediated RNA trans-splicing. RNA 2003, 9, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-mediated trans-splicing: The therapeutic cut and paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Maire, S.; Gaillard, M.C.; Sahel, J.A.; Hantraye, P.; Bemelmans, A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Puttaraju, M.; Jamison, S.F.; Mansfield, S.G.; Garcia-Blanco, M.A.; Mitchell, L.G. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat. Biotechnol. 1999, 17, 246–252. [Google Scholar] [PubMed]
- Mansfield, S.G.; Kole, J.; Puttaraju, M.; Yang, C.C.; Garcia-Blanco, M.A.; Cohn, J.A.; Mitchell, L.G. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Ther. 2000, 7, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Puttaraju, M.; DiPasquale, J.; Baker, C.C.; Mitchell, L.G.; Garcia-Blanco, M.A. Messenger RNA repair and restoration of protein function by spliceosome-mediated RNA trans-splicing. Mol. Ther. 2001, 4, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, T.; Garcia-Blanco, M.A.; Mansfield, S.G.; Grover, A.C.; Hutton, M.; Yu, Q.; Zhou, J.; Anderton, B.H.; Gallo, J.M. Reprogramming of tau alternative splicing by spliceosome-mediated RNA trans-splicing: Implications for tauopathies. Proc. Natl. Acad. Sci. USA 2005, 102, 15659–15664. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, T.; Anthony, K.; Garcia-Blanco, M.A.; Mansfield, S.G.; Anderton, B.H.; Gallo, J.M. Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum. Mol. Genet. 2009, 18, 3266–3273. [Google Scholar] [CrossRef] [PubMed]
- Avale, M.E.; Rodriguez-Martin, T.; Gallo, J.M. Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum. Mol. Genet. 2013, 22, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Hainzl, S.; Kocher, T.; Huttner, C.; Klausegger, A.; Gruber, C.; Mayr, E.; Wally, V.; Bauer, J.W.; Murauer, E.M. Trans-splicing improvement by the combined application of antisense strategies. Int. J. Mol. Sci. 2015, 16, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Coady, T.H.; Lorson, C.L. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J. Neurosci. 2010, 30, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Allo, M.; Buggiano, V.; Fededa, J.P.; Petrillo, E.; Schor, I.; de la Mata, M.; Agirre, E.; Plass, M.; Eyras, E.; Elela, S.A.; et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, V.; Zou, Y.; Ko, D.; Bonnemann, C.G. siRNA-mediated allele-specific silencing of a COL6A3 mutation in a cellular model of dominant ullrich muscular dystrophy. Mol. Ther. Nucleic Acids 2014, 3, e147. [Google Scholar] [CrossRef] [PubMed]
- Ryther, R.C.; Flynt, A.S.; Harris, B.D.; Phillips, J.A., 3rd; Patton, J.G. GH1 splicing is regulated by multiple enhancers whose mutation produces a dominant-negative GH isoform that can be degraded by allele-specific small interfering RNA (siRNA). Endocrinology 2004, 145, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Thrasher, A. Gene therapy: Progress and predictions. Proc. Biol. Sci. 2015, 282, 20143003. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, D.; Elaissari, A.; Fessi, H. Gene therapy and DNA delivery systems. Int. J. Pharm. 2014, 459, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Investig. 2012, 122, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Tomono, T.; Kajita, M.; Yano, K.; Ogihara, T. Adenovirus vector infection of non-small-cell lung cancer cells is a trigger for multi-drug resistance mediated by P-glycoprotein. Biochem. Biophys. Res. Commun. 2016, 476, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, J.C.; Wright, J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2016, 25, R42–R52. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Jackson, C.S.; Oprea, I.; Ozturk, F.; Pepper, M.S.; Diaconu, I.; Braicu, C.; Raduly, L.Z.; Calin, G.A.; Berindan-Neagoe, I. Progresses towards safe and efficient gene therapy vectors. Oncotarget 2015, 6, 30675–30703. [Google Scholar] [PubMed]
- Geib, T.; Hertel, K.J. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS ONE 2009, 4, e8204. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Cheng, S.H. Prospects for the gene therapy of spinal muscular atrophy. Trends Mol. Med. 2011, 17, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Lorain, S.; Peccate, C.; Le Hir, M.; Griffith, G.; Philippi, S.; Précigout, G.; Mamchaoui, K.; Jollet, A.; Voit, T.; Garcia, L. Dystrophin rescue by trans-splicing: A strategy for DMD genotypes not eligible for exon skipping approaches. Nucleic Acids Res. 2013, 41, 8391–8402. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, M.; Goyenvalle, A.; Peccate, C.; Precigout, G.; Davies, K.E.; Voit, T.; Garcia, L.; Lorain, S. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol. Ther. 2013, 21, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Wright, J.; Babbs, A.; Wilkins, V.; Garcia, L.; Davies, K.E. Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Babbs, A.; van Ommen, G.J.; Garcia, L.; Davies, K.E. Enhanced exon-skipping induced by U7 snRNA carrying a splicing silencer sequence: Promising tool for DMD therapy. Mol. Ther. 2009, 17, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Yue, Y.; Liu, M.; Ghosh, A.; Engelhardt, J.F.; Chamberlain, J.S.; Duan, D. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat. Biotechnol. 2005, 23, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lou, H.H.; Boyer, J.L.; Limberis, M.P.; Vandenberghe, L.H.; Hackett, N.R.; Leopold, P.L.; Wilson, J.M.; Crystal, R.G. Functional cystic fibrosis transmembrane conductance regulator expression in cystic fibrosis airway epithelial cells by AAV6.2-mediated segmental trans-splicing. Hum. Gene Ther. 2009, 20, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, M.; Zhang, L.N.; Yan, Z.; Zak, R.; Ding, W.; Mansfield, S.G.; Mitchell, L.G.; Engelhardt, J.F. Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum. Gene Ther. 2005, 16, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.J.; Dai, X.; Boye, S.E.; Barone, I.; Boye, S.L.; Mao, S.; Everhart, D.; Dinculescu, A.; Liu, L.; Umino, Y.; et al. Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol. Ther. 2011, 19, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Schon, C.; Biel, M.; Michalakis, S. Retinal gene delivery by adeno-associated virus (AAV) vectors: Strategies and applications. Eur. J. Pharm. Biopharm. 2015, 95, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Lee, Y.M.; Chen, P.W.; Byrne, B.J.; Hwu, W.L. Mutation-adapted U1 snRNA corrects a splicing error of the dopa decarboxylase gene. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tal-Goldberg, T.; Lorain, S.; Mitrani-Rosenbaum, S. Correction of the Middle Eastern M712T mutation causing GNE myopathy by trans-splicing. Neuromol. Med. 2014, 16, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.; Neveling, K.; Borkens, S.; Schneider, H.; Freund, M.; Grassman, E.; Theiss, S.; Wawer, A.; Burdach, S.; Auerbach, A.D.; et al. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am. J. Hum. Genet. 2010, 87, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.S.; Suh, M.S.; Kim, S.; Shim, G.; Lee, S.; Han, S.S.; Lee, K.E.; Jeon, H.; Choi, H.G.; Choi, Y.; et al. Cationic drug-derived nanoparticles for multifunctional delivery of anticancer siRNA. Biomaterials 2011, 32, 9785–9795. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold nanoparticles for nucleic acid delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Kinoh, H.; Ishii, T.; Matsui, A.; Tockary, T.A.; Takeda, K.M.; Uchida, H.; Osada, K.; Itaka, K.; Kataoka, K. Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 2016, 82, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, Y.; Peng, H.; Chen, Y.; Zhu, P.; Huang, Y. Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv. Drug Deliv. Rev. 2015, 81, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Al-Dosari, M.S.; Gao, X. Nonviral gene delivery: Principle, limitations, and recent progress. AAPS J. 2009, 11, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Vankayala, R.; Chiang, C.S.; Chao, J.I.; Yuan, C.J.; Lin, S.Y.; Hwang, K.C. A general strategy to achieve ultra-high gene transfection efficiency using lipid-nanoparticle composites. Biomaterials 2014, 35, 8261–8272. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.K.; Kwok, P.C. Production methods for nanodrug particles using the bottom-up approach. Adv. Drug Deliv. Rev. 2011, 63, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Hanes, J.; Ensign, L.M. Nanoparticles for oral delivery: Design, evaluation and state-of-the-art. J. Control. Release 2016, 240, 504–526. [Google Scholar] [CrossRef] [PubMed]
- Prow, T.W.; Grice, J.E.; Lin, L.L.; Faye, R.; Butler, M.; Becker, W.; Wurm, E.M.; Yoong, C.; Robertson, T.A.; Soyer, H.P.; et al. Nanoparticles and microparticles for skin drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 470–491. [Google Scholar] [CrossRef] [PubMed]
- Kundu, A.K.; Chandra, P.K.; Hazari, S.; Pramar, Y.V.; Dash, S.; Mandal, T.K. Development and optimization of nanosomal formulations for siRNA delivery to the liver. Eur. J. Pharm. Biopharm. 2012, 80, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Zimmer, A.; Pardeike, J. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) for pulmonary application: A review of the state of the art. Eur. J. Pharm. Biopharm. 2014, 86, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Zhang, M.; Liao, R.; Wood, T.; Nance, E. Systems-level thinking for nanoparticle-mediated therapeutic delivery to neurological diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.; Babu, S.M.; Beg, S.; Jena, J. Nanoparticles for cancer targeting: current and future directions. Curr. Drug Deliv. 2016, 13, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, S.; Li, J.; Kuang, Y.; He, X.; Guo, Y.; Ma, H.; Zhang, Y.; Ji, B.; Jiang, C. Brain-targeted co-delivery of therapeutic gene and peptide by multifunctional nanoparticles in Alzheimer’s disease mice. Biomaterials 2016, 80, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef] [PubMed]
- Kralovicova, J.; Moreno, P.M.; Cross, N.C.; Pego, A.P.; Vorechovsky, I. Antisense oligonucleotides modulating activation of a nonsense-mediated RNA decay switch exon in the ATM gene. Nucleic Acid Ther. 2016, 26, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, P.; Sabatelli, P.; Fabris, M.; Braghetta, P.; Bassi, E.; Spitali, P.; Vattemi, G.; Tomelleri, G.; Mari, L.; Perrone, D.; et al. Cationic PMMA nanoparticles bind and deliver antisense oligoribonucleotides allowing restoration of dystrophin expression in the mdx mouse. Mol. Ther. 2009, 17, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Sabatelli, P.; Fabris, M.; Bassi, E.; Falzarano, S.; Vattemi, G.; Perrone, D.; Gualandi, F.; Maraldi, N.M.; Merlini, L.; et al. Dystrophin restoration in skeletal, heart and skin arrector pili smooth muscle of mdx mice by ZM2 NP-AON complexes. Gene Ther. 2010, 17, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Feng, L. Targeted delivery of a splice-switching oligonucleotide by cationic polyplexes of RGD-oligonucleotide conjugate. Mol. Pharm. 2012, 9, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Juliano, R.L. Enhanced delivery of antisense oligonucleotides with fluorophore-conjugated PAMAM dendrimers. Nucleic Acids Res. 2000, 28, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Sazani, P.; Juliano, R.L. PAMAM dendrimers as delivery agents for antisense oligonucleotides. Pharm. Res. 1999, 16, 1799–1804. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Regulatory Element Mutated | Mechanism | Splicing Effect | References | |
|---|---|---|---|---|---|
| Familial dysautonomia (FD) | Cis | T > C mutation at position 6 of intron 20 of the IKBKAP gene | Exon skipping; introduction of a premature termination codon (PTC) | [23] | |
| Spinal muscular atrophy (SMA) | Cis | C > T mutation at position 6 of exon 7 of the SMN2 gene | Alteration of a putative ESE | [24] | |
| Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency | Cis | c362C > T mutation in exon 5 of the MCAD gene | Exon skipping | [25] | |
| Hutchinson-Gilford progeria syndrome (HGPS) | Cis | c1824C > T mutation in exon 11 of LMNA gene | Activation of a cryptic splice site | [26] | |
| Myotonic dystrophy | Type 1 (DM1) | Cis | Expanded CTG tract in the 3′ UTR region of the DMPK gene | Misregulation of trans-acting factors | [27] |
| Type 2 (DM2) | Cis | Expanded CCCTG tract in intron 1 of the ZNF9 gene | Misregulation of trans-acting factors | [27] | |
| Autosomal dominant retinitis pigmentosa (RP) | Trans | Mutations in genes of the core spliceosome (PRPF31, PRPF8, PRPF3, RP9) | Disruption of basal spliceosome function | [28] | |
| Duchenne muscular dystrophy (DMD) | Cis | T > A mutation in exon 31 of the Distrophin gene | Creation of a PTC and introduction of ESS | [29] | |
| Microcephalic steodysplastic primordial dwarfism type 1 (MOPD1) or Taybi-Linder syndrome (TALS) | Trans | Mutations in the gene encoding the U4atac snRNA | Reduced splicing efficiency and increased intron retention | [30] | |
| Frontotemporal dementia with parkinsonism-17 (FTDP-17) | Cis | Mutations within and downstream exon 10 of the MAPT gene | Disruption of Tau protein balance | [31] | |
| Fukuyama congenital muscular dystrophy (FCMD) | Cis | SVA insertion in the 3′ UTR of the FKTN gene | Inclusion of a new exon | [32] | |
| Amyotrophic lateral sclerosis (ALS) | Trans | Mutations in TDP-43 | Altered gene splicing | [33] | |
| Hypercholesterolemia | Cis | rs688T > C mutation in exon 12 of the LDLR gene | Alteration of ESE and exon skipping | [34] | |
| Cystic fibrosis (CF) | Cis | Longer (UG)n tract at the exon 9 3′ SS of the CFTR gene | Exon skipping | [35] | |
| Disease | Therapeutic Approach | Target Gene | Regulated Exon |
|---|---|---|---|
| DMD | ASO | DMD | 51 |
| SMA | ASO | SMN2 | 7 |
| Dystrophic epidermolysis bullosa (DEB) | ASO | COL7A1 | 70 |
| FTDP-17 | ASO | MAPT | 10 |
| SMaRT | MAPT | 1 | |
| Atherosclerosis | ASO | APOB | 27 |
| CF | ASO | CFTR | 16 |
| SMaRT | CFTR | 10 | |
| Ullrich congenital muscular dystrophy (UCMD) | siRNA | COL6A3 | 16 |
| Growth hormone deficiency (GHD) type II | siRNA | GH1 | 3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suñé-Pou, M.; Prieto-Sánchez, S.; Boyero-Corral, S.; Moreno-Castro, C.; El Yousfi, Y.; Suñé-Negre, J.M.; Hernández-Munain, C.; Suñé, C. Targeting Splicing in the Treatment of Human Disease. Genes 2017, 8, 87. https://doi.org/10.3390/genes8030087
Suñé-Pou M, Prieto-Sánchez S, Boyero-Corral S, Moreno-Castro C, El Yousfi Y, Suñé-Negre JM, Hernández-Munain C, Suñé C. Targeting Splicing in the Treatment of Human Disease. Genes. 2017; 8(3):87. https://doi.org/10.3390/genes8030087
Chicago/Turabian StyleSuñé-Pou, Marc, Silvia Prieto-Sánchez, Sofía Boyero-Corral, Cristina Moreno-Castro, Younes El Yousfi, Josep Mª Suñé-Negre, Cristina Hernández-Munain, and Carlos Suñé. 2017. "Targeting Splicing in the Treatment of Human Disease" Genes 8, no. 3: 87. https://doi.org/10.3390/genes8030087