
A Genetic Population Isolate in The Netherlands Showing Extensive Haplotype Sharing and Long Regions of Homozygosity

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Populations and Genotype Data
2.2. Data Analysis
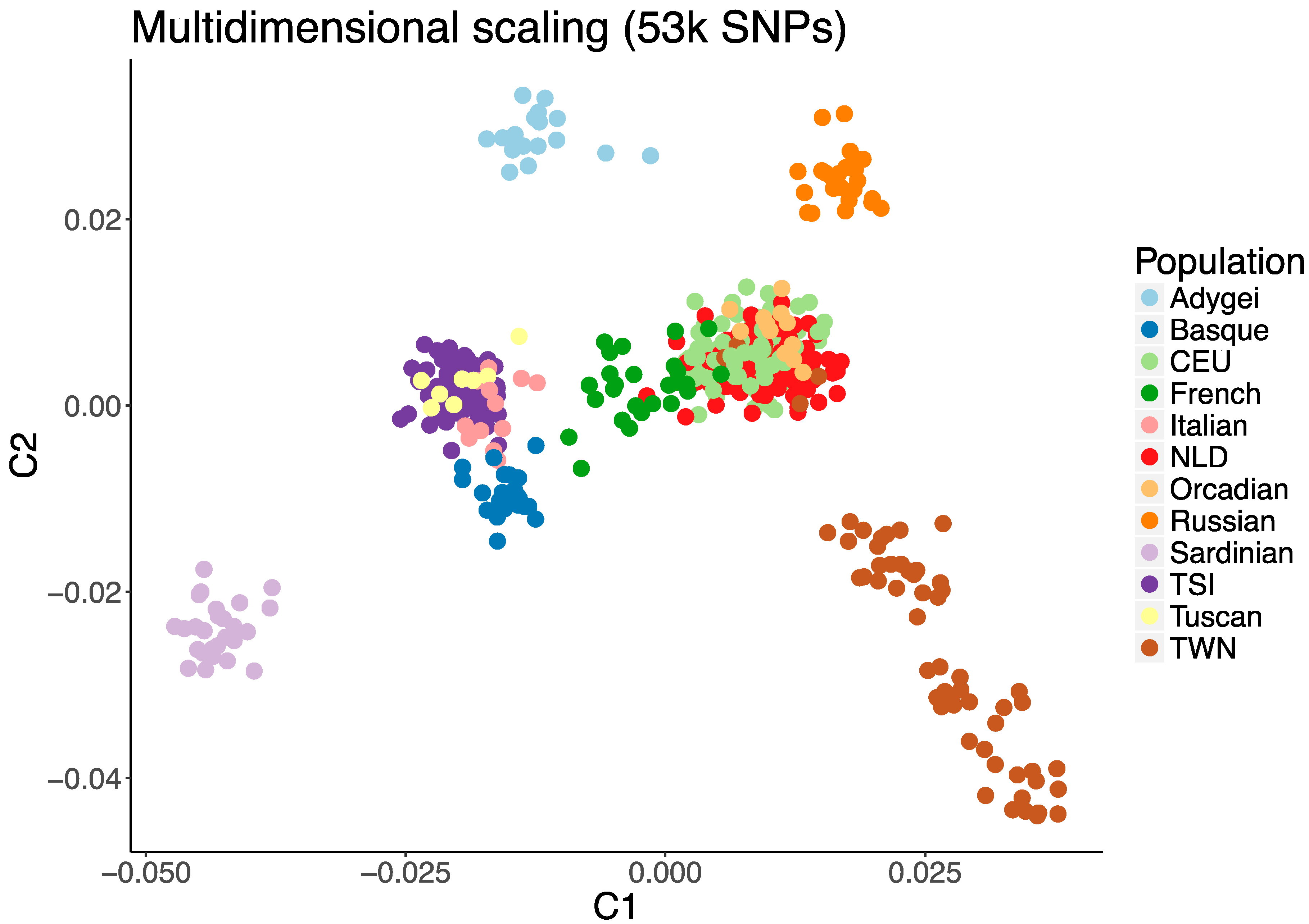
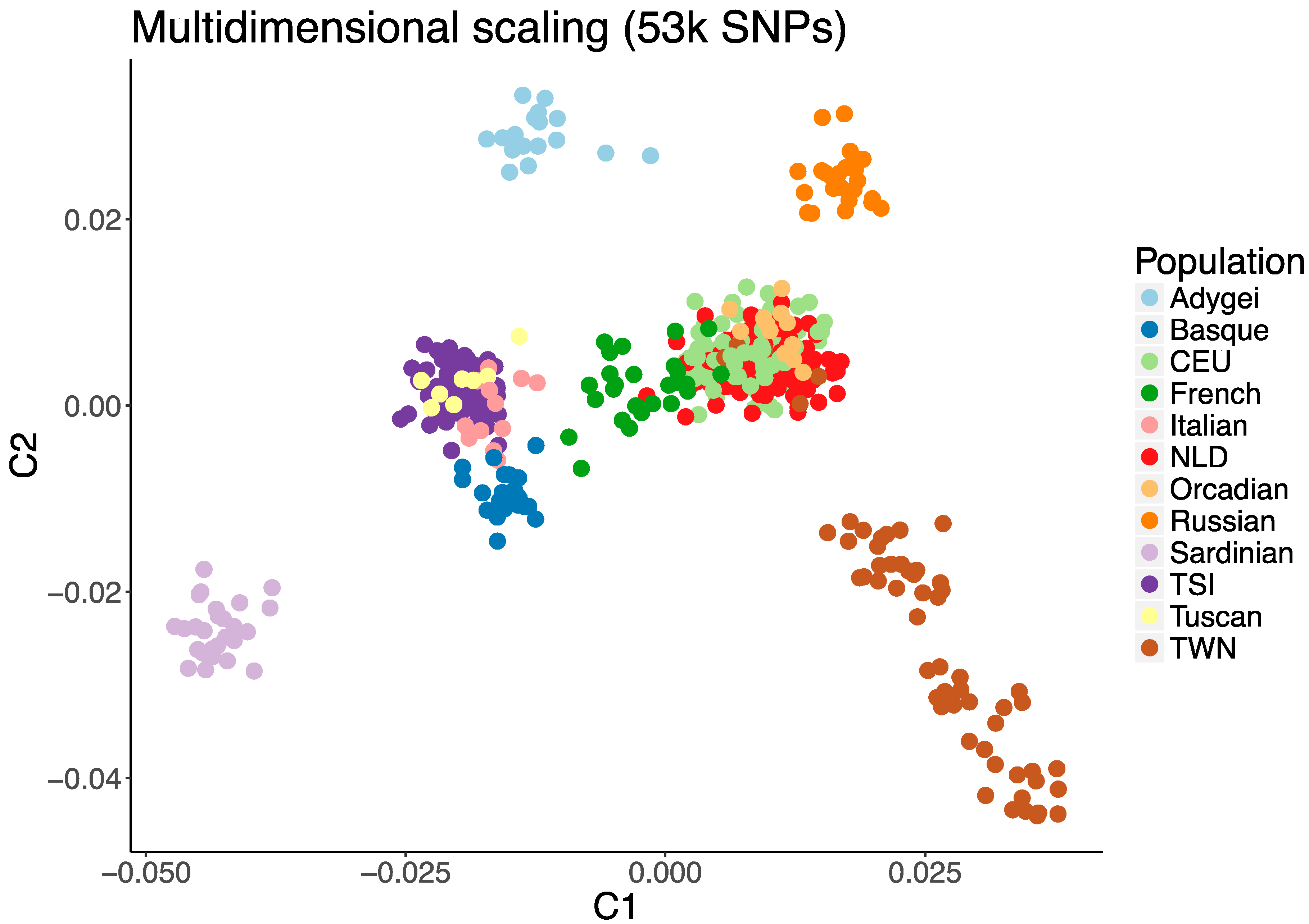
2.3. Measure of Population Stratification
2.4. Pairwise Genetic Distance
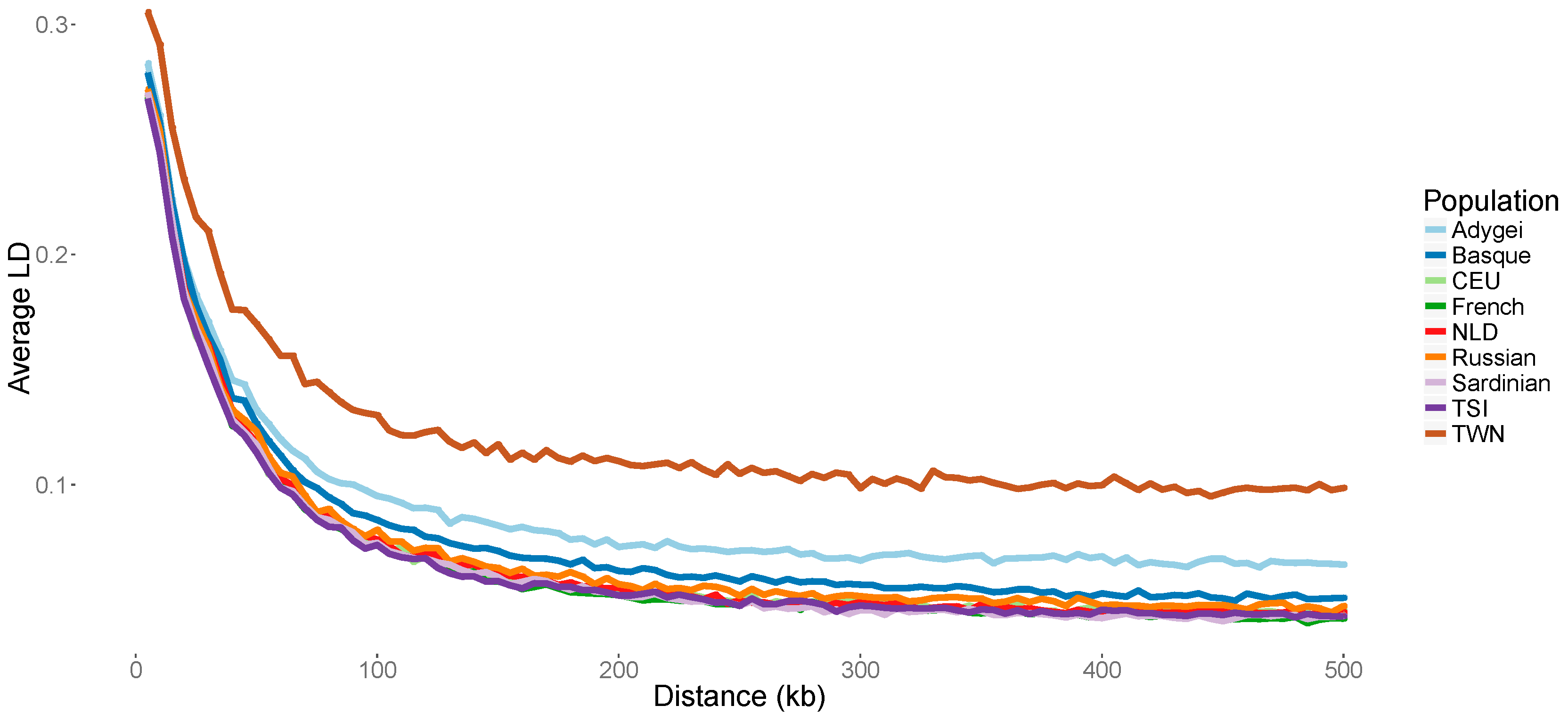
2.5. Decay of Linkage Disequilibrium (LD)
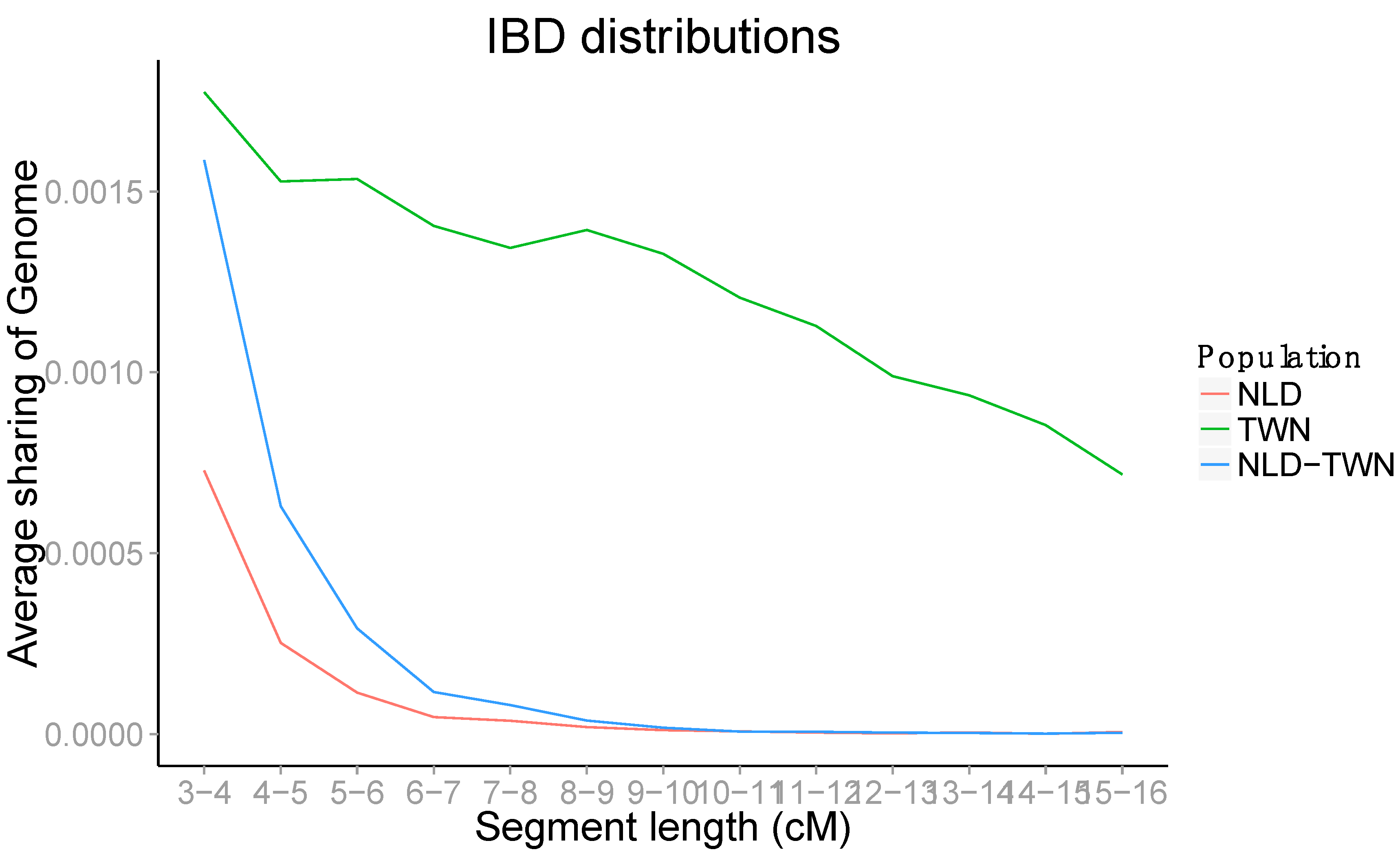
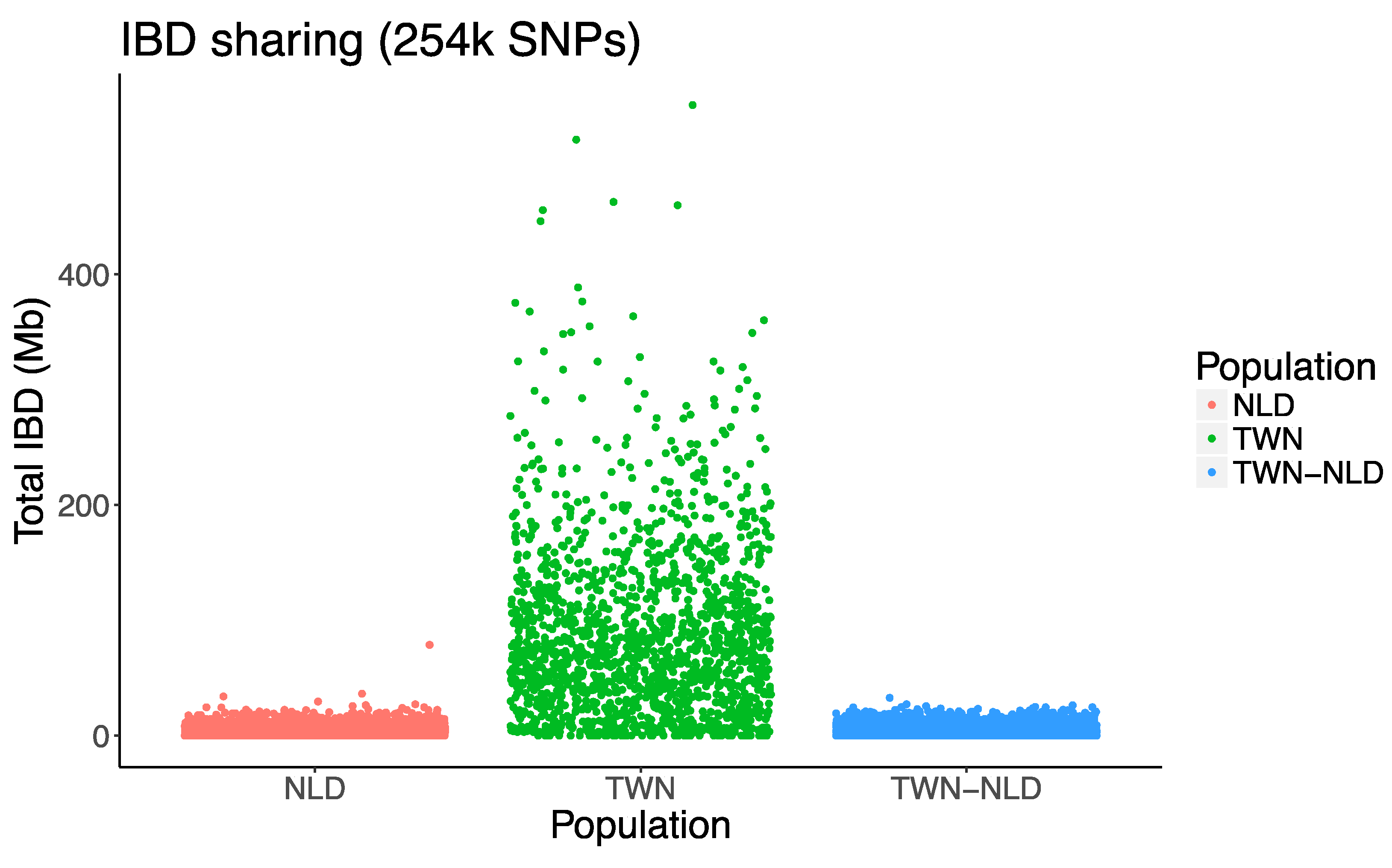
2.6. IBD Sharing
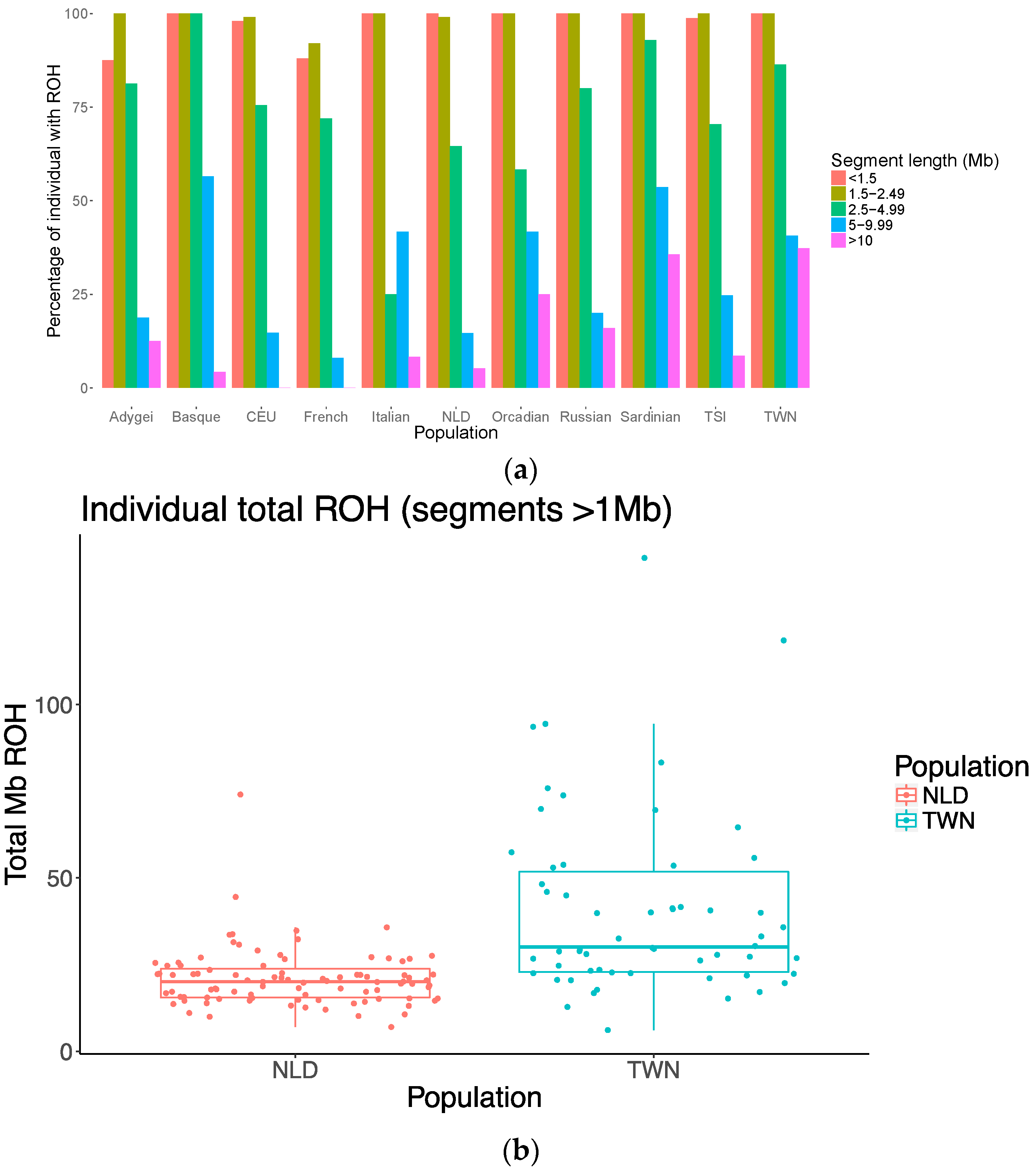
2.7. Runs of Homozygosity (ROH)
2.8. Effective Population Size
3. Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Quality Control Pipeline
Appendix A.1. Part 1
Appendix A.2. Part 2a
Appendix A.3. Part 2b
Appendix B
References
- Peltonen, L.; Palotie, A.; Lange, K. Use of population isolates for mapping complex traits. Nat. Rev. Genet. 2000, 1, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Service, S.; DeYoung, J.; Karayiorgou, M.; Roos, J.L.; Pretorious, H.; Bedoya, G.; Ospina, J.; Ruiz-Linares, A.; Macedo, A.; Palha, J.A.; et al. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat. Genet. 2006, 38, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Axenovich, T.I.; Zorkoltseva, I.V.; Belonogova, N.M.; Struchalin, M.V.; Kirichenko, A.V.; Kayser, M.; Oostra, B.A.; van Duijn, C.M.; Aulchenko, Y.S. Linkage analysis of adult height in a large pedigree from a Dutch genetically isolated population. Hum. Genet. 2009, 126, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Panoutsopoulou, K.; Tachmazidou, I.; Zeggini, E. In search of low-frequency and rare variants affecting complex traits. Hum. Mol. Genet. 2013, 22, R16–R21. [Google Scholar] [CrossRef] [PubMed]
- Aulchenko, Y.S.; Heutink, P.; Mackay, I.; Bertoli-Avella, A.M.; Pullen, J.; Vaessen, N.; Rademaker, T.A.M.; Sandkuijl, L.A.; Cardon, L.; Oostra, B.; et al. Linkage disequilibrium in young genetically isolated Dutch population. Eur. J. Hum. Genet. 2004, 12, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; Mckeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [PubMed]
- Kristiansson, K.; Naukkarinen, J.; Peltonen, L. Isolated populations and complex disease gene identification. Genome Biol. 2008, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Ophoff, R.A.; Escamilla, M.A.; Service, S.K.; Spesny, M.; Meshi, D.B.; Poon, W.; Molina, J.; Fournier, E.; Gallegos, A.; Mathews, C.; et al. Genomewide linkage disequilibrium mapping of severe bipolar disorder in a population isolate. Am. J. Hum. Genet. 2002, 71, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Sulem, P.; Gudbjartsson, D.F.; Walters, G.B.; Helgadottir, H.T.; Helgason, A.; Gudjonsson, S.A.; Zanon, C.; Besenbacher, S.; Bjornsdottir, G.; Magnusson, O.T.; et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat. Genet. 2011, 43, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Pollin, T.I.; Damcott, C.M.; Shen, H.; Ott, S.H.; Shelton, J.; Horenstein, R.B.; Post, W.; McLenithan, J.C.; Bielak, L.F.; Peyser, P.A.; et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008, 322, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Tachmazidou, I.; Dedoussis, G.; Southam, L.; Farmaki, A.-E.; Ritchie, G.R.S.; Xifara, D.K.; Matchan, A.; Hatzikotoulas, K.; Rayner, N.W.; Chen, Y.; et al. A rare functional cardioprotective APOC3 variant has risen in frequency in distinct population isolates. Nat. Commun. 2013, 4, 2872. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Masson, G.; Agnarsson, B.A.; Benediktsdottir, K.R.; Sigurdsson, A.; Magnusson, O.T.; Gudjonsson, S.A.; Magnusdottir, D.N.; et al. A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat. Genet. 2012, 44, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E. Next-generation association studies for complex traits. Nat. Genet. 2011, 43, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Gaál, E.I.; Kettunen, J.; Lappalainen, T.; Menelaou, A.; Anttila, V.; van’t Hof, F.N.G.; von und zu Fraunberg, M.; Helisalmi, S.; Hiltunen, M.; et al. High risk population isolate reveals low frequency variants predisposing to intracranial aneurysms. PLoS Genet. 2014, 10, e1004134. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Carmona, L.G.; Ophoff, R.; Service, S.; Hartiala, J.; Molina, J.; Leon, P.; Ospina, J.; Bedoya, G.; Freimer, N.; Ruiz-Linares, A. Genetic demography of Antioquia (Colombia) and the Central Valley of Costa Rica. Hum. Genet. 2003, 112, 534–541. [Google Scholar] [PubMed]
- Colonna, V.; Pistis, G.; Bomba, L.; Mona, S.; Matullo, G.; Boano, R.; Sala, C.; Viganò, F.; Torroni, A.; Achilli, A.; et al. Small effective population size and genetic homogeneity in the Val Borbera isolate. Eur. J. Hum. Genet. 2013, 21, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Tenesa, A.; Wright, A.F.; Knott, S.A.; Carothers, A.D.; Hayward, C.; Angius, A.; Persico, I.; Maestrale, G.; Hastie, N.D.; Pirastu, M.; et al. Extent of linkage disequilibrium in a Sardinian sub-isolate: Sampling and methodological considerations. Hum. Mol. Genet. 2004, 13, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Houwen, R.H.; Baharloo, S.; Blankenship, K.; Raeymaekers, P.; Juyn, J.; Sandkuijl, L.A.; Freimer, N.B. Genome screening by searching for shared segments: Mapping a gene for benign recurrent intrahepatic cholestasis. Nat. Genet. 1994, 8, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Somers, M.; Ophoff, R.A.; Aukes, M.F.; Cantor, R.M.; Boks, M.P.; Dauwan, M.; de Visser, K.L.; Kahn, R.S.; Sommer, I.E. Linkage Analysis in a Dutch Population Isolate Shows No Major Gene for Left-Handedness or Atypical Language Lateralization. J. Neurosci. 2015, 35, 8730–8736. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, T.D.; Yu, F.; Yang, H.; Chang, L.-Y.; Huang, W.; Liu, B.; Shen, Y.; et al. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar]
- Cann, H.M.; de Toma, C.; Cazes, L.; Legrand, M.F.; Morel, V.; Piouffre, L.; Bodmer, J.; Bodmer, W.F.; Bonne-Tamir, B.; Cambon-Thomsen, A.; et al. A human genome diversity cell line panel. Science 2002, 296, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Absher, D.M.; Tang, H.; Southwick, A.M.; Casto, A.M.; Ramachandran, S.; Cann, H.M.; Barsh, G.S.; Feldman, M.; Cavalli-Sforza, L.L.; et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008, 319, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, K.R.; de Jong, S.; Boks, M.P.; Langeveld, T.; Colas, F.; Veldink, J.H.; de Kovel, C.G.; Janson, E.; Strengman, E.; Langfelder, P.; et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics 2012, 13, 636. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Sabatti, C.; Risch, N. Homozygosity and linkage disequilibrium. Genetics 2002, 160, 1707–1719. [Google Scholar] [PubMed]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Lowe, J.K.; Stoffel, M.; Daly, M.J.; Altshuler, D.; Breslow, J.L.; Friedman, J.M.; Pe’er, I. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009, 19, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Palamara, P.F.; Pe’er, I. Inference of historical migration rates via haplotype sharing. Bioinformatics 2013, 29, i180–i188. [Google Scholar] [CrossRef] [PubMed]
- Francioli, L.C.; Menelaou, A.; Pulit, S.L.; van Dijk, F.; Palamara, P.F.; Elbers, C.C.; Neerincx, P.B.T.; Ye, K.; Guryev, V.; Kloosterman, W.P.; et al. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Novembre, J.; Johnson, T.; Bryc, K.; Kutalik, Z.; Boyko, A.R.; Auton, A.; Indap, A.; King, K.S.; Bergmann, S.; Nelson, M.R.; et al. Genes mirror geography within Europe. Nature 2008, 456, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Leslie, S.; Winney, B.; Hellenthal, G.; Davison, D.; Boumertit, A.; Day, T.; Hutnik, K.; Royrvik, E.C.; Cunliffe, B. Wellcome Trust Case Control Consortium 2. The fine-scale genetic structure of the British population. Nature 2015, 519, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Pistis, G.; Piras, I.; Pirastu, N.; Persico, I.; Sassu, A.; Picciau, A.; Prodi, D.; Fraumene, C.; Mocci, E.; Manias, M.T.; et al. High differentiation among eight villages in a secluded area of Sardinia revealed by genome-wide high density SNPs analysis. PLoS ONE 2009, 4, e4654. [Google Scholar] [CrossRef] [PubMed]
- Angius, A.; Hyland, F.C.L.; Persico, I.; Pirastu, N.; Woodage, T.; Pirastu, M.; De La Vega, F.M. Patterns of linkage disequilibrium between SNPs in a Sardinian population isolate and the selection of markers for association studies. Hum. Hered. 2007, 65, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Calò, C.M.; Melis, A.; Vona, G.; Piras, I.S. Review Synthetic Article: Sardinian Population (Italy): A Genetic Review. Int. J. Mod. Anthropol. 2008, 1, 39–64. [Google Scholar] [CrossRef]
- Esko, T.; Mezzavilla, M.; Nelis, M.; Borel, C.; Debniak, T.; Jakkula, E.; Julia, A.; Karachanak, S.; Khrunin, A.; Kisfali, P.; et al. Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. Eur. J. Hum. Genet. 2013, 21, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Garagnani, P.; Laayouni, H.; González-Neira, A.; Sikora, M.; Luiselli, D.; Bertranpetit, J.; Calafell, F. Isolated populations as treasure troves in genetic epidemiology: The case of the Basques. Eur. J. Hum. Genet. 2009, 17, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Palamara, P.F.; Lencz, T.; Darvasi, A.; Pe’er, I. Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 2012, 91, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Abdellaoui, A.; Hottenga, J.-J.; de Knijff, P.; Nivard, M.G.; Xiao, X.; Scheet, P.; Brooks, A.; A Ehli, E.; Hu, Y.; Davies, G.E.; et al. Population structure, migration, and diversifying selection in the Netherlands. Eur. J. Hum. Genet. 2013, 21, 1277–1285. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adygei | Basque | CEU | NLD | French | Russian | Sard | TSI | TWN | |
|---|---|---|---|---|---|---|---|---|---|
| Adygei | - | 0.0182 (0.0179–0.0185) | 0.0101 (0.0099–0.0103) | 0.0108 (0.0105–0.0110) | 0.0095 (0.0092–0.0098) | 0.0116 (0.0112–0.0119) | 0.0191 (0.0188–0.0194) | 0.0078 (0.0075–0.0080) | 0.01557 (0.0153–0.0159) |
| Basque | - | - | 0.0081 (0.0079–0.0083) | 0.0087 (0.0086–0.0089) | 0.0068 (0.0066–0.0070) | 0.0142 (0.0140–0.0146) | 0.0129 (0.0127–0.0132) | 0.0081 (0.0080–0.0083) | 0.0135 (0.0133–0.0137) |
| CEU | - | - | - | 0.0002 (0.0002–0.0002) | 0.0004 (0.0003–0.0005) | 0.0043 (0.0042–0.0044) | 0.0119 (0.0117–0.0121) | 0.0035 (0.0034–0.0036) | 0.0044 (0.0043–0.0045) |
| NLD | - | - | - | - | 0.0008 (0.0007–0.0010) | 0.0047 (0.0046–0.0049) | 0.0126 (0.0124–0.0128) | 0.0040 (0.0039–0.0041) | 0.0040 (0.0039–0.0041) |
| French | - | - | - | - | - | 0.0053 (0.0051–0.0055) | 0.0091 (0.0089–0.0093) | 0.0018 (0.0017–0.0020) | 0.0053 (0.0052–0.0055) |
| Russian | - | - | - | - | - | - | 0.0197 (0.0194–0.0200) | 0.0090 (0.0089–0.0092) | 0.0089 (0.0088–0.0091) |
| Sard | - | - | - | - | - | - | - | 0.0067 (0.0065–0.0069) | 0.0175 (0.0173–0.0178) |
| TSI | - | - | - | - | - | - | - | - | 0.0086 (0.0084–0.0087) |
| # Samples | # Pairs | # IBD Segments | Mean/Median/SD # IBD Segments/Pair | Mean/Median/SD Size (Mb)/Segment | Mean/Median/SD Size (Mb)/Pair | |
|---|---|---|---|---|---|---|
| NLD | 92 | 4186 | 4548 | 1.09/1/1.05 | 3.99/3.57/1.36 | 4.34/3.5/4.57 |
| TWN | 58 | 1653 | 15444 | 9.34/8/6.45 | 9.34/7.22/8.20 | 87.25/71.99/73.88 |
| p-value | <0.0001 | <0.0001 | <0.0001 |
| # Samples | # Pairs | # IBD Segments | Mean/Median/SD # IBD Segments/Pair | Mean/Median/SD Size (Mb)/Segment | Mean/Median/SD Size (Mb)/Pair | |
|---|---|---|---|---|---|---|
| NLD-TWN | 92 | 2622 | 3822 | 1.45/1/1.12 | 3.92/3.57/1.09 | 5.68/3.64/4.67 |
| # Samples | # Pairs | # IBD Segments | Mean/Median/SD # Segments/Pair | Mean/Median/SD Size (Mb)/Segment | Mean/Median/SD Size (MB)/Pair | |
|---|---|---|---|---|---|---|
| NLD | 92 | 4186 | 2648 | 0.63/0/0.80 | 3.93/3.57/1.36 | 2.48/0/3.41 |
| TWN | 58 | 1653 | 9802 | 6.05/5/4.43 | 10.01/7.22/8.20 | 59.37/46.85/55.63 |
| p-value | <0.0001 | <0.0001 | <0.0001 |
| # Samples | Mean/Median/SD # ROH Segments/Pair | Mean/Median/SD Size (Mb)/Segment | Mean/Median/SD ROH Total Length (Mb) | |
|---|---|---|---|---|
| NLD | 92 | 11.92/11/2.97 | 1.73/1.64/0.38 | 20.99/20.04/8.55 |
| TWN | 58 | 15.17/14/4.80 | 2.64/2.13/1.34 | 40.94/30.09/26.51 |
| p-value | <0.0001 | <0.0001 | <0.0001 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somers, M.; Olde Loohuis, L.M.; Aukes, M.F.; Pasaniuc, B.; De Visser, K.C.L.; Kahn, R.S.; Sommer, I.E.; Ophoff, R.A. A Genetic Population Isolate in The Netherlands Showing Extensive Haplotype Sharing and Long Regions of Homozygosity. Genes 2017, 8, 133. https://doi.org/10.3390/genes8050133
Somers M, Olde Loohuis LM, Aukes MF, Pasaniuc B, De Visser KCL, Kahn RS, Sommer IE, Ophoff RA. A Genetic Population Isolate in The Netherlands Showing Extensive Haplotype Sharing and Long Regions of Homozygosity. Genes. 2017; 8(5):133. https://doi.org/10.3390/genes8050133
Chicago/Turabian StyleSomers, Metten, Loes M. Olde Loohuis, Maartje F. Aukes, Bogdan Pasaniuc, Kees C. L. De Visser, René S. Kahn, Iris E. Sommer, and Roel A. Ophoff. 2017. "A Genetic Population Isolate in The Netherlands Showing Extensive Haplotype Sharing and Long Regions of Homozygosity" Genes 8, no. 5: 133. https://doi.org/10.3390/genes8050133