
Fine Mapping and Transcriptome Analysis Reveal Candidate Genes Associated with Hybrid Lethality in Cabbage (Brassica Oleracea)

 and
and
Abstract
:1. Introduction
2. Materials and Methods
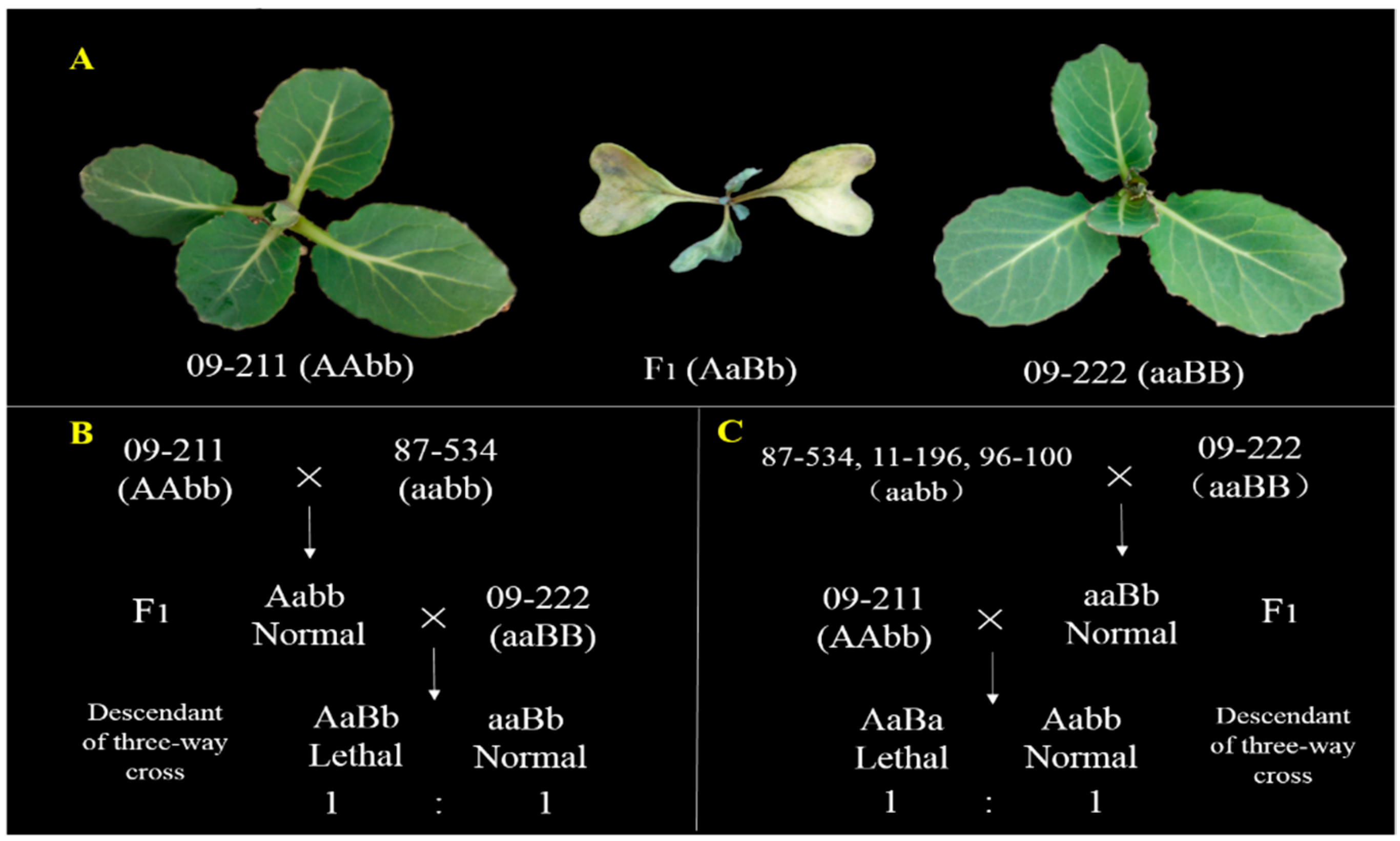
2.1. Plant Materials
2.2. Phenotyping
2.3. Genotyping
2.3.1. Genotyping InDel Primer Development
2.3.2. DNA Extraction
2.3.3. Bulked Segregation Analysis (BSA)
2.3.4. PCR Amplification and Electrophoresis
2.4. Sampling, RNA Isolation and RNA-Seq
2.5. RNA-Seq Data Analysis
2.6. Fine-Mapping Results and Candidate Gene Analysis
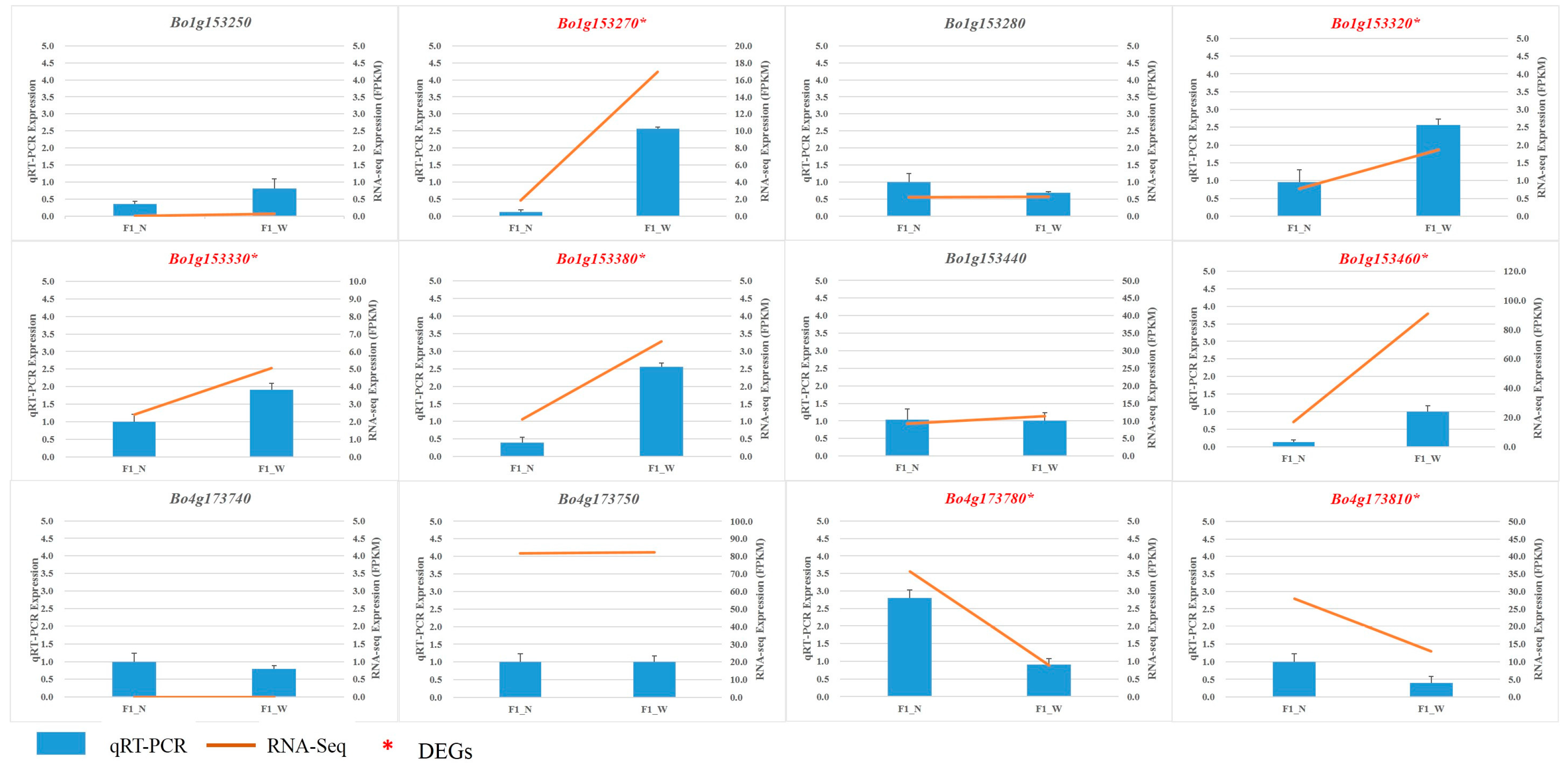
2.7. Quantitative Real-Time RT-PCR (qRT-PCR) Validation
3. Results
3.1. Genetic Analysis of BoHL1 and BoHL2 with New Populations
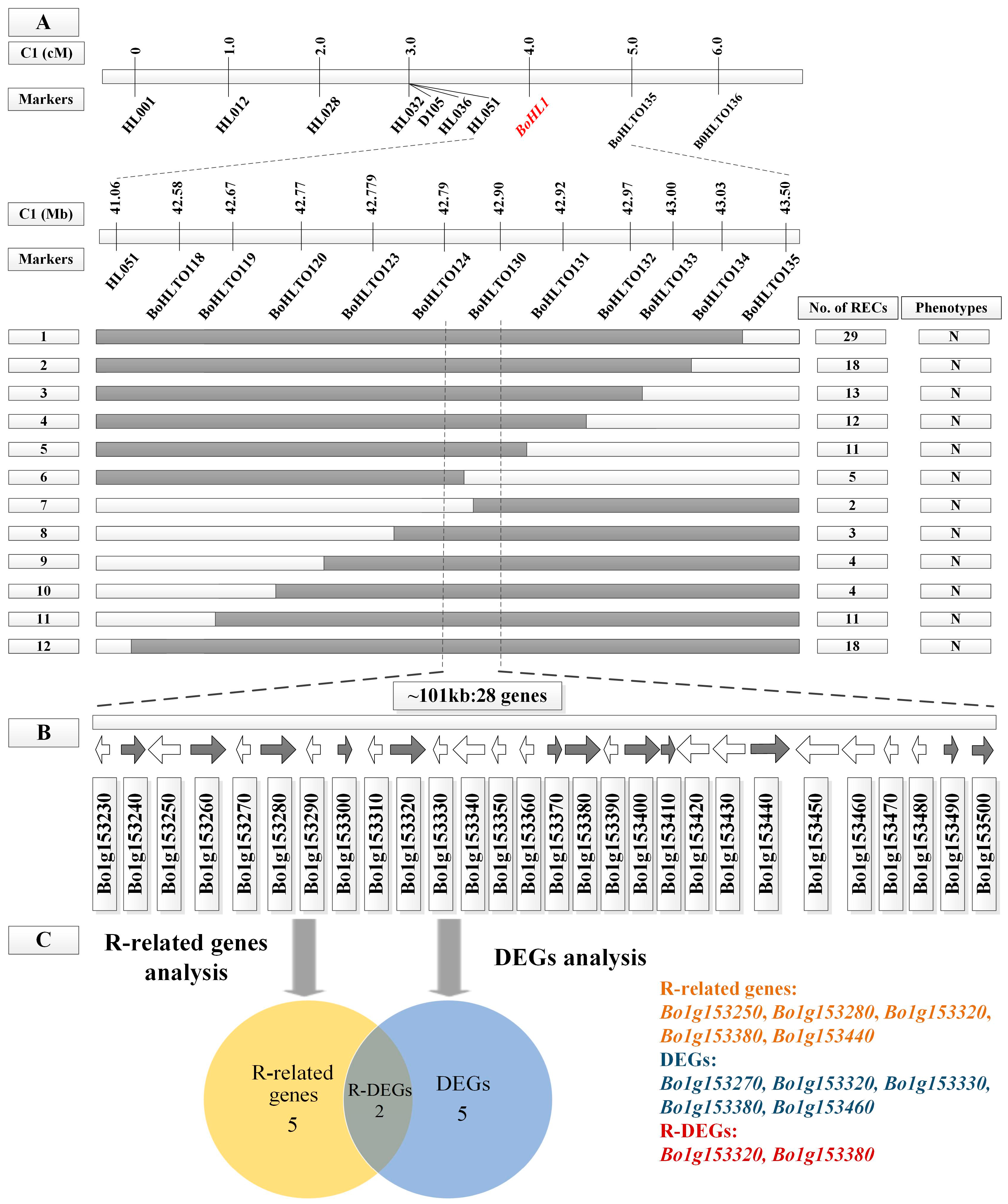
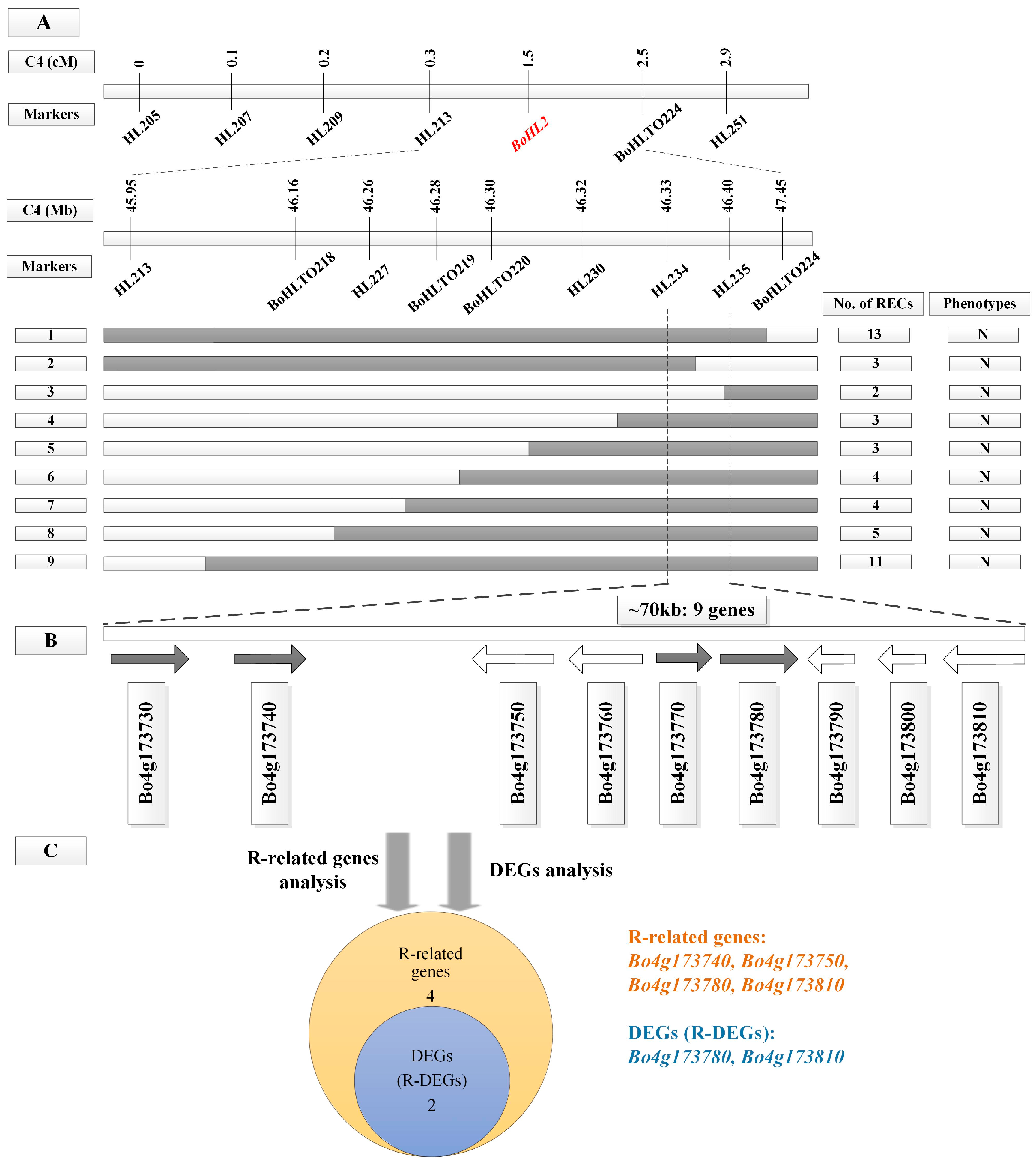
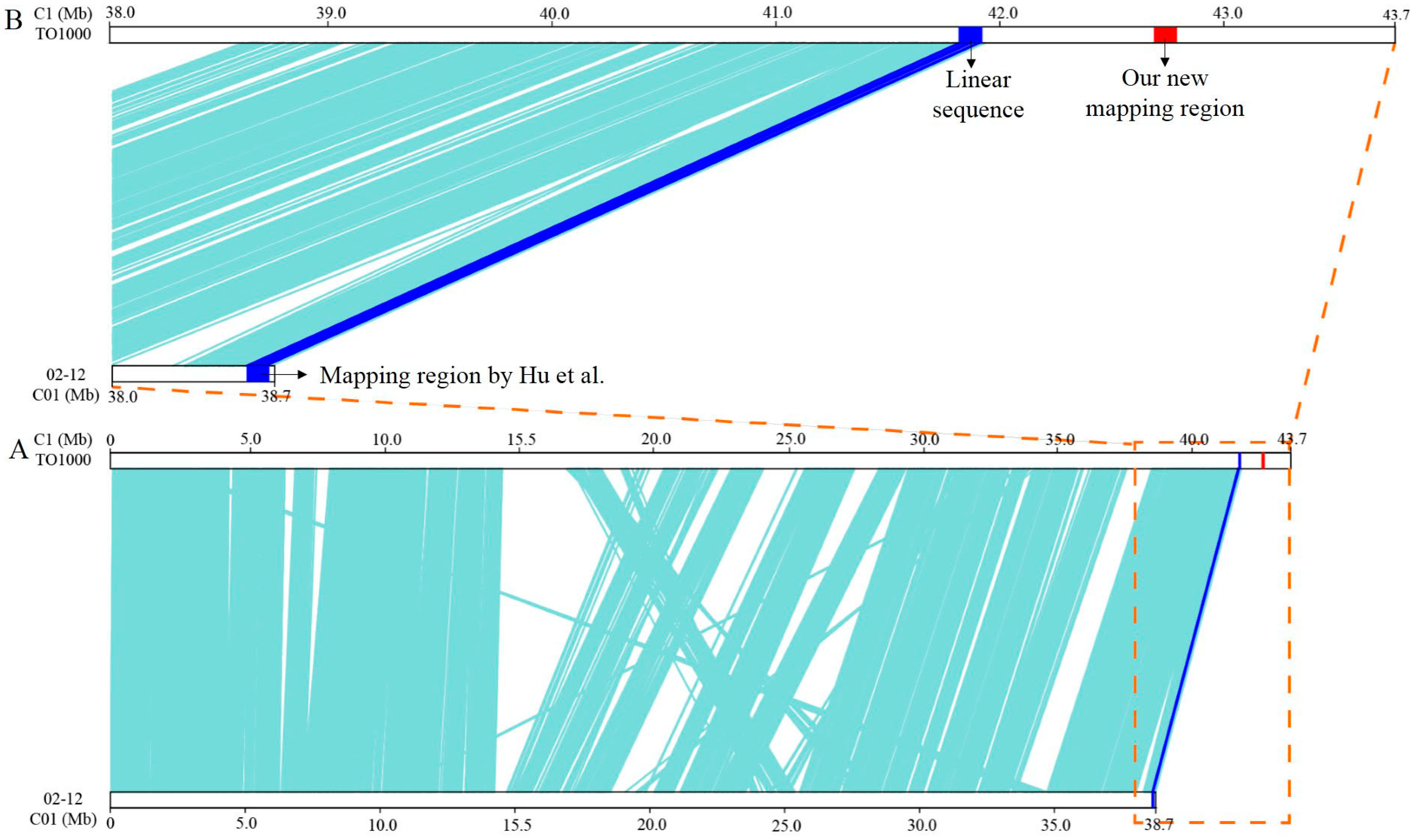
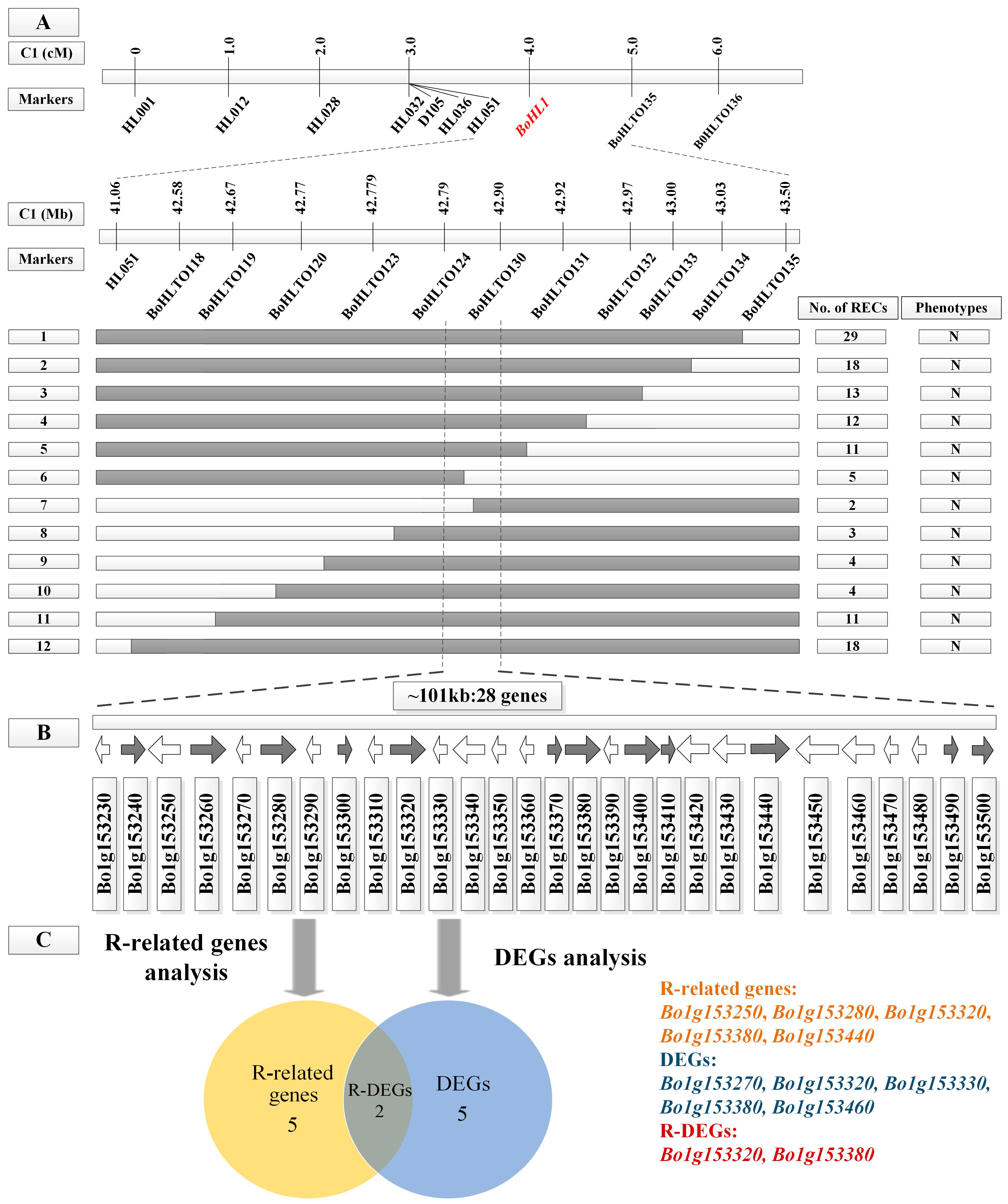
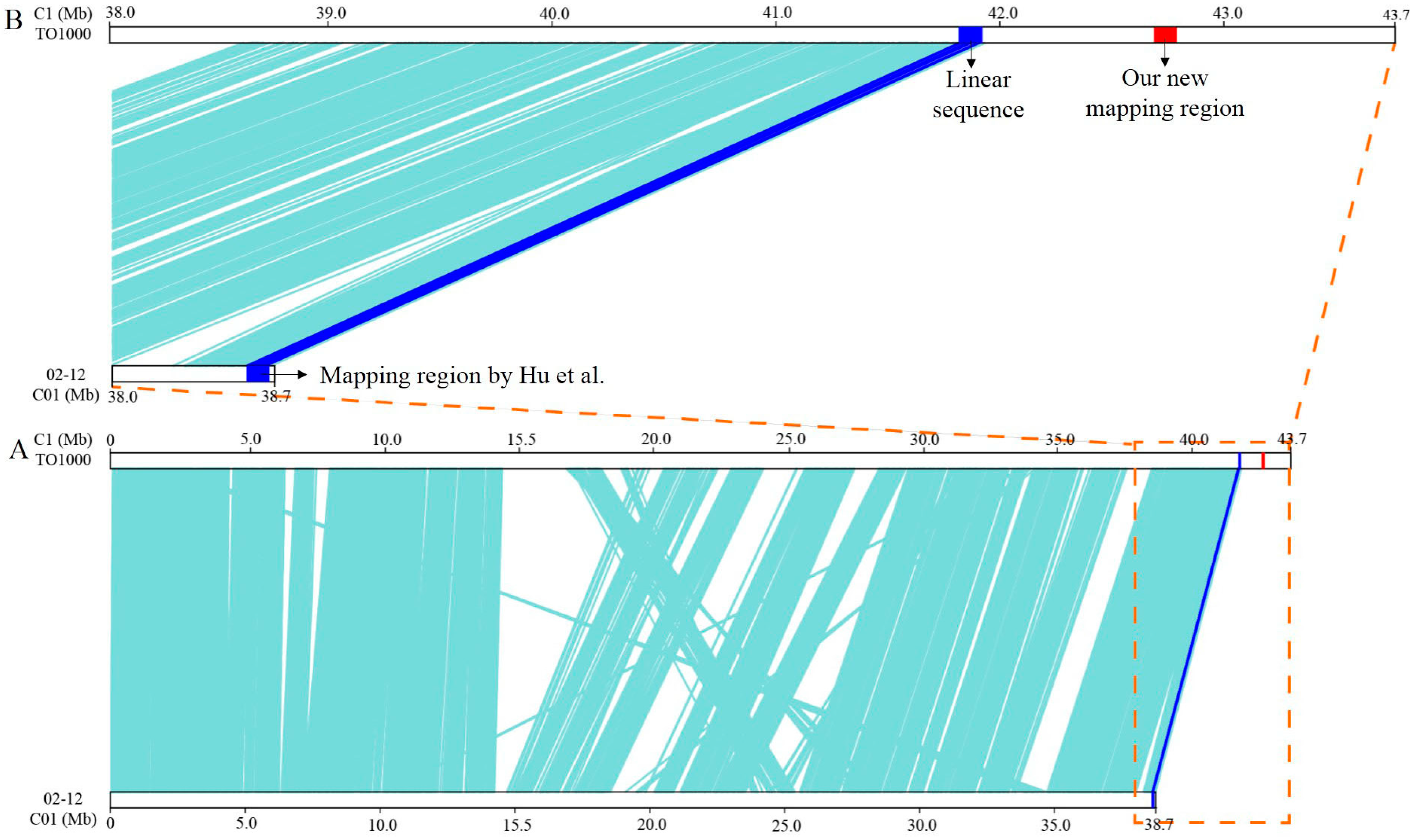
3.2. Fine Mapping of BoHL1 and BoHL2
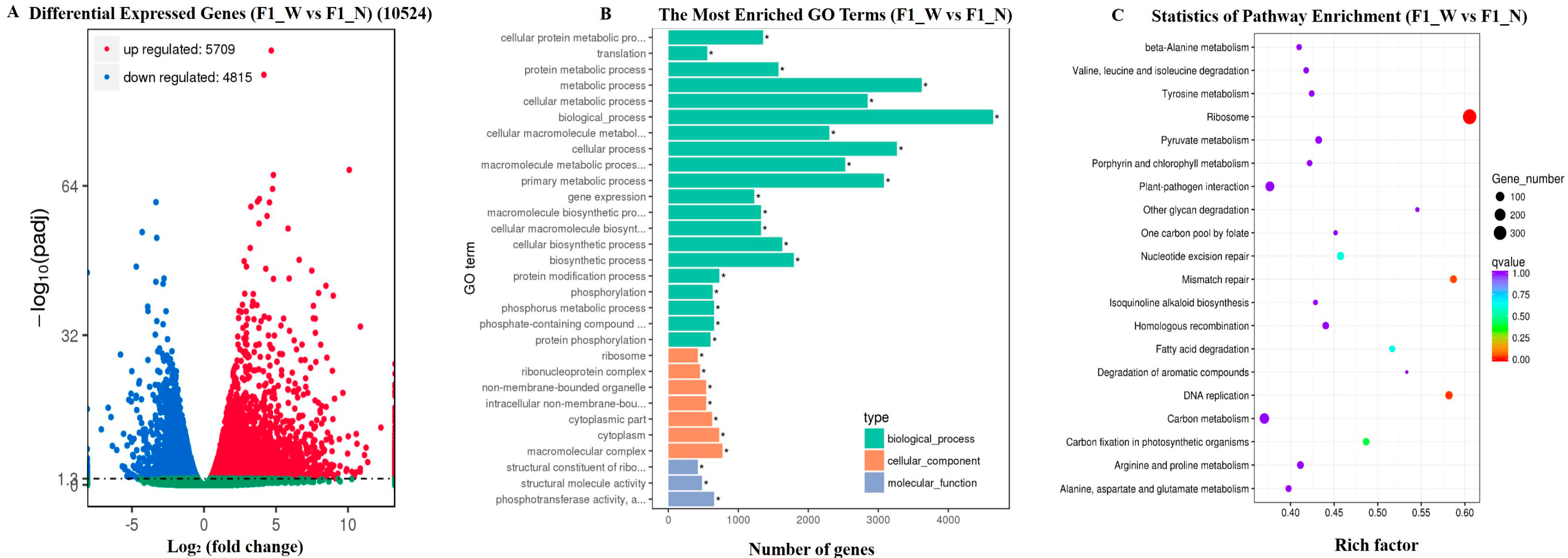
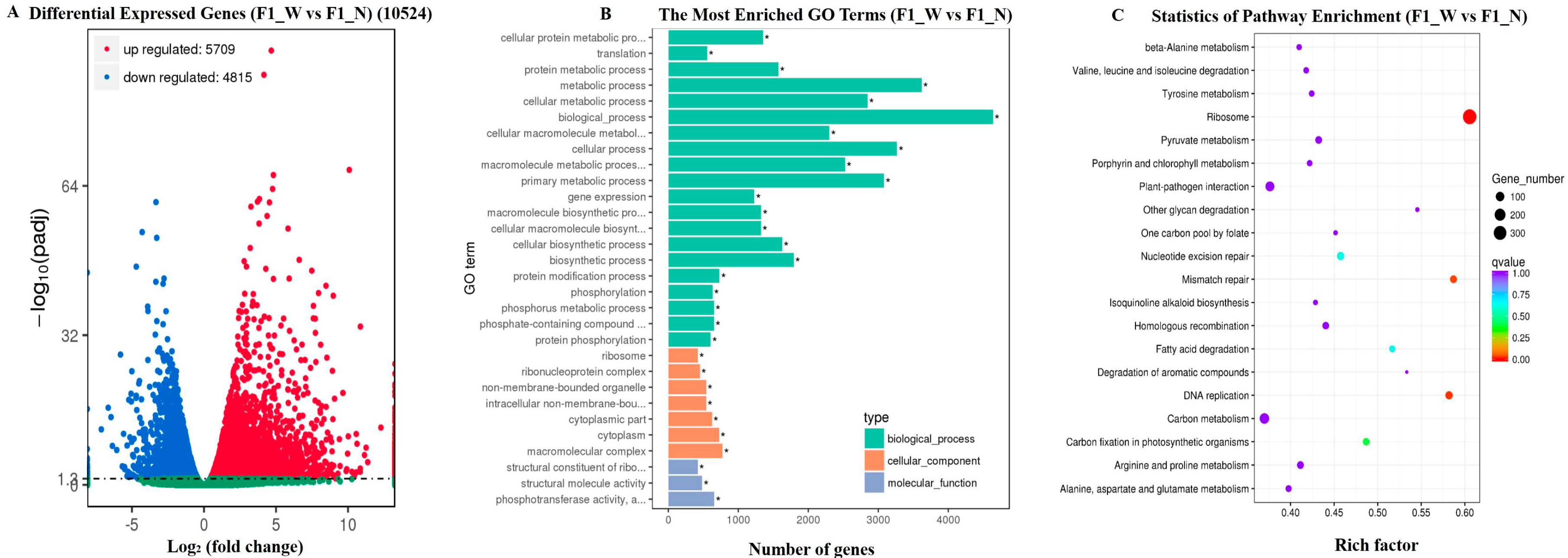
3.3. RNA-Seq Data Assembly and Analysis
3.4. Candidate Gene Analysis
3.4.1. Gene Analysis for the Candidate Regions
3.4.2. DEG Analysis for the Candidate Regions
3.4.3. Candidate Genes Analysis Using Re-Sequencing Data
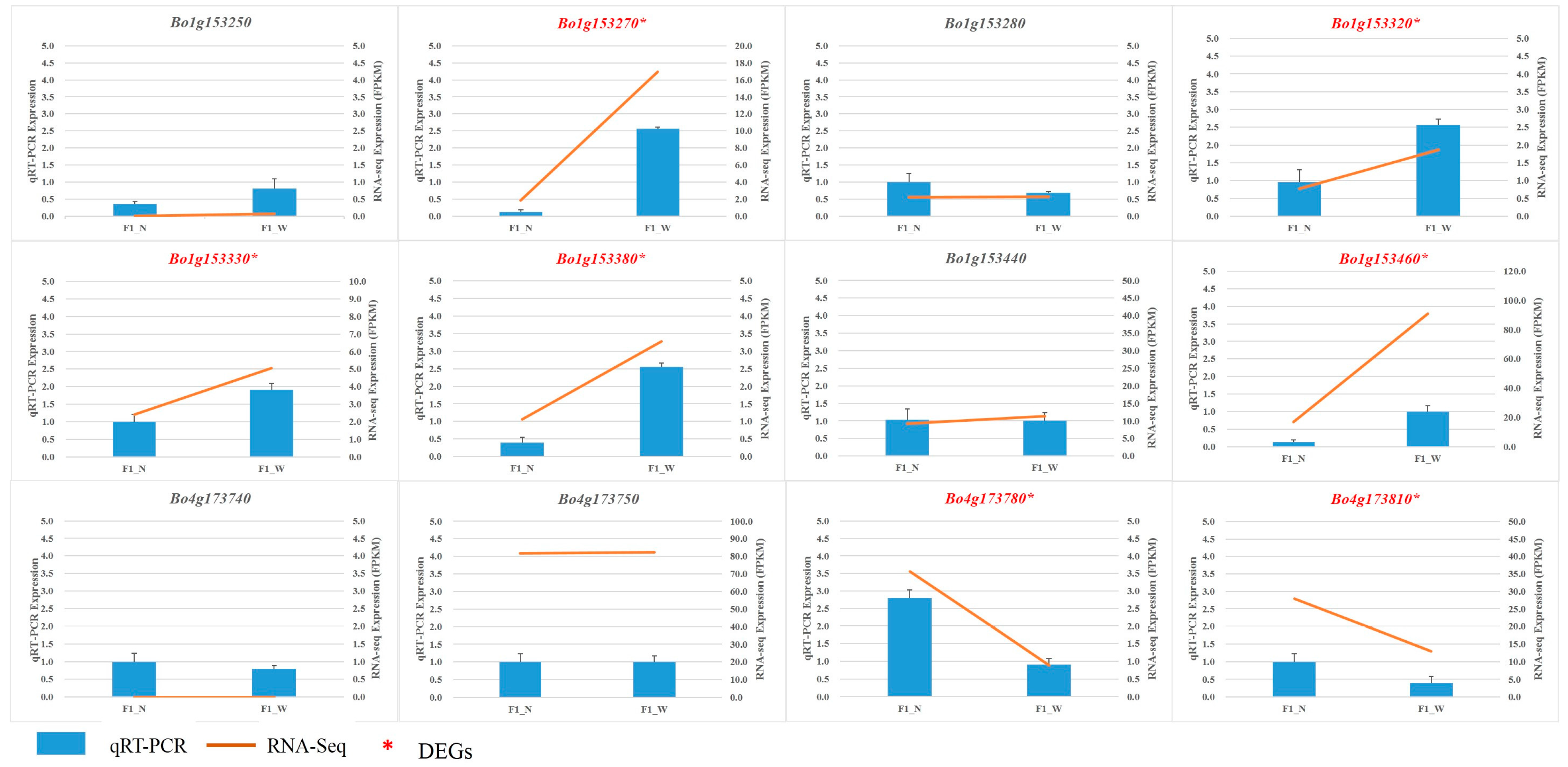
3.5. RNA Sequencing Validation by qRT-PCR
4. Discussion
4.1. Omics Technologies Promote Molecular Genetics Research in B. oleracea
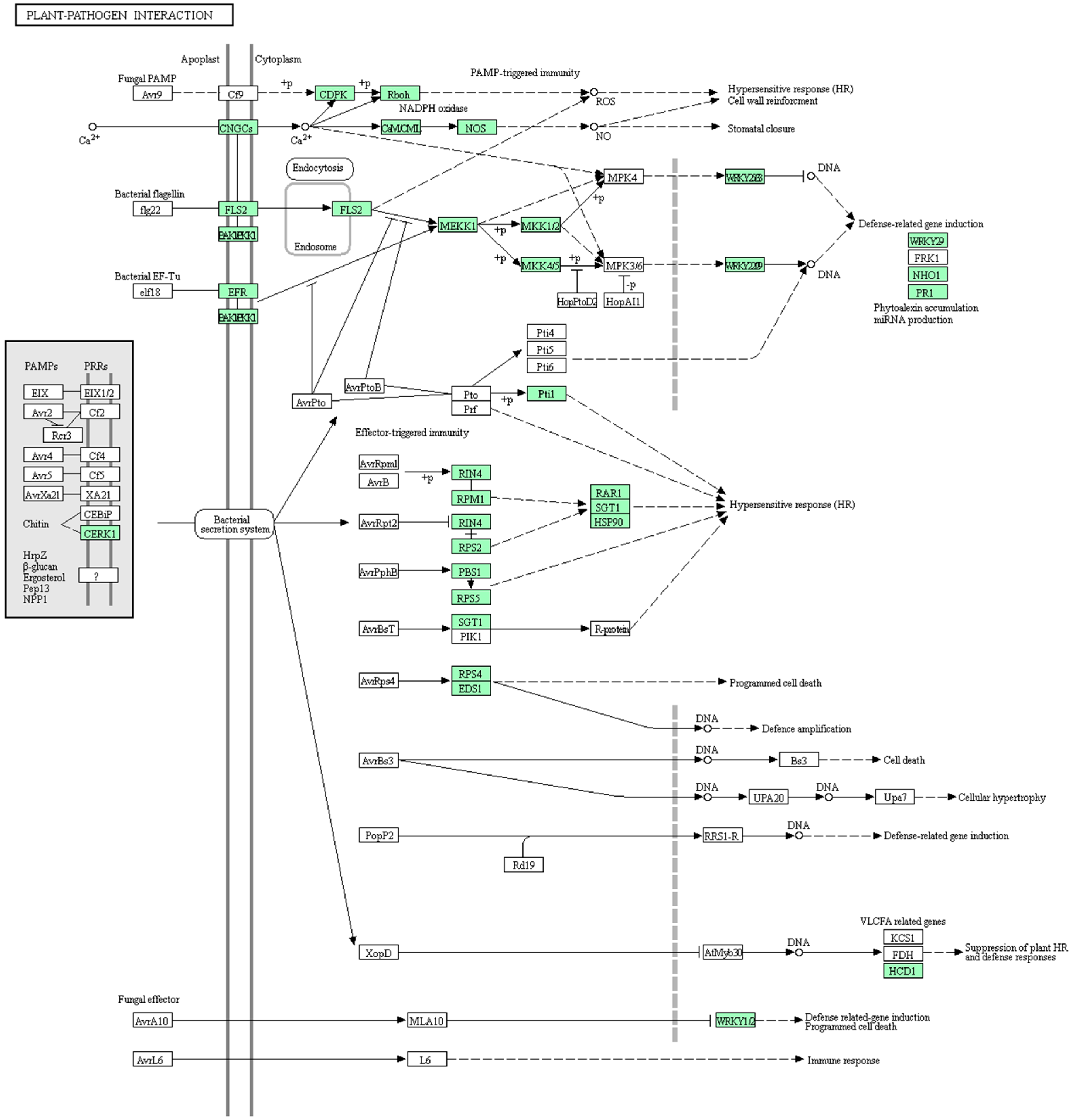
4.2. Mechanisms of HL In Plants
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HL | Hybrid lethality |
| DM | Dobzhansky-Muller |
| InDel | Insertion-deletion |
| SNP | Single nucleotide polymorphisms |
| COG | Cluster of orthologous groups |
| KEGG | Kyoto encyclopedia of genes and genomes |
| qRT-PCR | Quantitative real-time RT-PCR |
| DEGs | Differentially expressed genes |
| BSA | Bulked segregant analysis |
| TIR | Toll/interleukin-1 receptor homology |
| QTL | Quantitative trait loci |
| PAGE | Polyacrylamide gel electrophoresis |
| NBS-LRR | Nucleotide binding site-leucine rich repeat |
| ETI | Effector-triggered-immunity |
| HR | Hypersensitive response |
| PCD | Programmed cell death |
| SNP | Single nucleotide polymorphism |
| FPKM | Fragments per kilobase of exon model per million mapped reads |
| DM1 | Dangerous mix 1 |
| DM2 | Dangerous mix 2 |
References
- Orr, H.A.; Presgraves, D.C. Speciation by postzygotic isolation: Forces, genes and molecules. Bioessays 2000, 22, 1085–1094. [Google Scholar] [CrossRef]
- Bomblies, K.; Weigel, D. Hybrid necrosis: Autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 2007, 8, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Wiebe, G.A. Complementary factors in barley giving a lethal progeny. J. Hered. 1934, 25, 273–274. [Google Scholar] [CrossRef]
- Tezuka, T.; Kuboyama, T.; Matsuda, T.; Marubashi, W. Seven of eight species in Nicotiana section Suaveolentes have common factors leading to hybrid lethality in crosses with Nicotiana tabacum. Ann. Bot. 2010, 106, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Marubashi, W. Temperature-dependent programmed cell death detected in hybrids between Nicotiana langsdorffii and N. tabacum expressing lethality. Plant Biotechnol. J. 2004, 21, 151–154. [Google Scholar] [CrossRef]
- Stephens, S.G. The genetics of corky; the New World alleles and their possible role as an interspecific isolating mechanism. J. Genet. 1946, 47, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Marubashi, W. Overproduced ethylene causes programmed cell death leading to temperature-sensitive lethality in hybrid seedlings from the cross Nicotiana suaveolens x N. tabacum. Planta 2003, 217, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Dobzhansky, T. Genetics and the Origin of Species; Columbia University Press: New York, NY, USA, 1937. [Google Scholar]
- Muller, H.J. Isolating mechanisms, evolution and temperature. Biol. Symp. 1942, 6, 71–125. [Google Scholar]
- Bomblies, K.; Lempe, J.; Epple, P.; Warthmann, N.; Lanz, C.; Dangl, J.L.; Weigel, D. Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 2007, 5, e236. [Google Scholar] [CrossRef] [PubMed]
- Chae, E.; Bomblies, K.; Kim, S.T.; Karelina, D.; Zaidem, M.; Ossowski, S.; Martín-Pizarro, C.; Laitinen, R.A.; Rowan, B.A.; Tenenboim, H. Species-wide genetic incompatibility analysis identifies immune genes as Hot Spots of deleterious epistasis. Cell 2014, 159, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Jeuken, M.J.W.; Zhang, N.W.; Mchale, L.K.; Pelgrom, K.; Boer, E.D.; Lindhout, P.; Michelmore, R.W.; Visser, R.G.; Niks, R.E. Rin4 causes hybrid necrosis and race-specific resistance in an interspecific lettuce hybrid. Plant Cell 2009, 21, 3368. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Zhang, Y.Y.; Fang, Z.Y.; Xue, Y.Q.; Liu, Y.M.; Yang, L.M. A preliminary study on hybrid seeding lethality in cabbage (Brassica oleracea, L.). Acta Hortic Sin 2012, 39, 2660. (In Chinese) [Google Scholar]
- Hu, Y.; Xue, Y.Q.; Liu, J.S.; Fang, Z.Y.; Yang, L.M.; Zhang, Y.Y; Lv, H.H.; Liu, Y.M.; Li, Z.S.; Zhuang, M. Hybrid lethality caused by two complementary dominant genes in cabbage (Brassica oleracea, L.). Mol. Breed. 2016, 36, 73. [Google Scholar] [CrossRef]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. Brassica database. Available online: http://brassicadb.org/brad/ (accessed on 18 May 2017).
- Parkin, I.A.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L.; et al. TO1000DH genome. Available online: http://plants.ensembl.org/Brassica_oleracea/ (accessed on 18 May 2017).
- Lee, J.; Izzah, N.K.; Choi, B.S.; Joh, H.J.; Lee, S.C.; Perumal, S.; Seo, J.; Ahn, K.; Jo, E.J.; Choi, G.J.; et al. Genotyping-by-sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Res. 2016, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J.; Ahmad, S.; Lee, L.J. Current-generation high-throughput sequencing: Deepening insights into mammalian transcriptomes. Gene Dev. 2009, 23, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acid. Res. 1980, 8, 4321. [Google Scholar] [CrossRef] [PubMed]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.H.; Wang, Q.B.; Han, F.Q.; Liu, X.; Fang, Z.Y.; Yang, L.M.; Zhuang, M.; Liu, Y.M.; Li, Z.S.; Zhang, Y.Y. Genome-wide indel/SSR scanning reveals significant loci associated with excellent agronomic traits of a cabbage (Brassica oleracea) elite parental line ‘01–20’. Sci. Rep. 2017, 7, 41696. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pachter, L. Corrigendum: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated NR database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. A 1995, 57, 289–300. [Google Scholar]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acid. Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, M.J.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Liu, R.H.; Meng, J.L. MapDraw: A microsoft excel macro for drawing genetic linkage maps based on given genetic linkage data. Hereditas 2003, 25, 317–321. [Google Scholar] [PubMed]
- Li, H.B.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 −ΔΔ c t Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Flor, H.H. Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 2003, 9, 275–296. [Google Scholar] [CrossRef]
- Hulbert, S.H.; Webb, C.A.; Smith, S.M.; Sun, Q. Resistance gene complexes: Evolution and utilization. Annu. Rev. Phytopathol. 2003, 39, 285. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Dickerman, A.W.; Michelmore, R.W.; Sivaramakrishnan, S.; Sobral, B.W.; Young, N.D. Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J. 1999, 20, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Ea, V.D.; Jones, J.D. The NB-ARC domain: A novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. Curr. Biol. 1998, 8, 226–227. [Google Scholar]
- Luo, M.; Zhu, Y.; Yu, C.; Wu, Z.; Zou, Y. Cloning of the plant resistance genes and their structure and function. Hereditas 2000, 22, 429–433. [Google Scholar]
- Bai, C.; Sen, P.; Hofmann, K.; Ma, L.; Goebl, M.; Harper, J.W.; Elledge, S.J. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 1996, 86, 263–274. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198. [Google Scholar] [CrossRef]
- Francisco, M.; Soengas, P.; Velasco, P.; Bhadauria, V.; Cartea, M.E.; Rodríguez, V.M. Omics approach to identify factors involved in Brassica disease resistance. Curr. Issues Mol. Biol. 2015, 19, 31–42. [Google Scholar] [PubMed]
- Lv, H.H.; Fang, Z.Y.; Yang, L.M.; Zhang, Y.Y.; Wang, Q.B.; Liu, Y.M.; Zhuang, M.; Yang, Y.H.; Xie, B.Y.; Liu, B.; et al. Mapping and analysis of a novel candidate Fusarium wilt resistance gene FOC1 in Brassica oleracea. BMC Genom. 2014, 15, 1094. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, C.G.; Guimarães, G.A.; Nogueira, S.R.; MacLean, D.; Cook, D.R.; Steuernagel, B.; Baek, J.; Bouyioukos, C.; V A Melo, B.D.; Tristão, G.; et al. A pigeonpea gene confers resistance to Asian soybean rust in soybean. Nat. Biotechnol. 2016, 34, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z.; Sun, P.; Tang, J.; Liu, D.; et al. Fine-mapping and analysis of Cgl1, a gene conferring glossy trait in cabbage (Brassica oleracea L.var. capitata). Front. Plant Sci. 2017, 8, 239. [Google Scholar] [PubMed]
- Lee, J.; Izzah, N.K.; Jayakodi, M.; Perumal, S.; Joh, H.J.; Lee, H.J.; Lee, S.C.; Park, J.Y.; Yang, K.W.; Nou, I.S.; et al. Genome-wide SNP identification and QTL mapping for black rot resistance in cabbage. BMC Plant Biol. 2015, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.H.; Li, T.G.; Jia, S.R.; Huang, D.F. Bioinformatic analysis of the LRR-NBS gene family in rice. Sci. Agric. Sin. 2004, 37, 1–7. [Google Scholar]
- Abramovitch, R.B.; Anderson, J.C.; Martin, G.B. Bacterial elicitation and evasion of plant innate immunity. Nat. Rev. Mol. Cell. Biol. 2006, 7, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Heath, M.C. Hypersensitive response-related death. Plant Mol. Biol. 2000, 44, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Pontier, D.; Balagué, C.; Roby, D. The hypersensitive response. A programmed cell death associated with plant resistance. Comptes Rendus De Lacadémie Des Sciences Série III Sciences De La Vie 1998, 321, 721–734. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Jacobs, J.R.; Goodman, C.S.; Artavanis-Tsakonas, S. Slit: An extracellular protein necessary for development of midline glia and commissural axon pathways contains both EGF and LRR domains. Gene Dev. 1990, 4, 2169–2187. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID a | Gene Position on C1 b | Functional Annotation c | q-value d | Significant e |
|---|---|---|---|---|
| Bo1g153230 | 42,792,977–42,794,726 | Catalytic activity | 0.22844 | FALSE |
| Bo1g153240 | 42,799,211–42,800,882 | MYB domain | 0.57873 | FALSE |
| Bo1g153250 | 42,801,727–42,803,329 | Toll/interleukin-1 receptor homology (TIR) domain | 0.56028 | FALSE |
| P-loop containing nucleoside triphosphate hydrolase | ||||
| Winged helix-turn-helix DNA-binding domain | ||||
| Bo1g153260 | 42,805,899–42,808,534 | Ubiquitin-related domain | 0.74850 | FALSE |
| Bo1g153270 | 42,809,711–42,810,514 | Secondary shoot formation | 0.00123 | TRUE |
| Strigolactone biosynthetic process | ||||
| Bo1g153280 | 42,812,792–42,816,443 | Toll/interleukin-1 receptor homology (TIR) domain | 0.87254 | FALSE |
| P-loop-containing nucleoside triphosphate hydrolase | ||||
| Leucine-rich repeat domain, L domain-like | ||||
| Bo1g153290 | 42,816,826–42,818,076 | NAD(P)-binding domain | 1 | FALSE |
| Glucose/ribitol dehydrogenase | ||||
| Bo1g153300 | 42,819,805–42,820,996 | NAD(P)-binding domain | 0.84117 | FALSE |
| Bo1g153310 | 42,823,170–42,823,680 | NAD(P)-binding domain | N/A | NA |
| Bo1g153320 | 42,828,669–42,832,299 | Toll/interleukin-1 receptor homology (TIR) domain | 0.00778 | TRUE |
| P-loop containing nucleoside triphosphate hydrolase | ||||
| Leucine-rich repeat domain, L domain-like | ||||
| Bo1g153330 | 42,832,541–42,833,708 | Oxidoreductase activity | 0.00825 | TRUE |
| Oxidation-reduction process | ||||
| NAD(P)-binding domain | ||||
| Bo1g153340 | 42,835,185–42,836,860 | Harbinger transposase-derived protein | N/A | N/A |
| Bo1g153350 | 428,376,66–42,838,484 | Myb-like domain | N/A | N/A |
| Bo1g153360 | 42,838,840–42,839,506 | No comment | N/A | N/A |
| Bo1g153370 | 42,843,786–42,845,295 | Agenet domain, plant type | N/A | N/A |
| Bo1g153380 | 42,846,297–42,849,389 | Toll/interleukin-1 receptor homology (TIR) domain | 1.43E–07 | TRUE |
| Ploop-containing nucleoside triphosphate hydrolase | ||||
| Winged helix-turn-helix DNA-binding domain | ||||
| Leucine-rich repeat domain, L domain-like | ||||
| Bo1g153390 | 42,849,475–42,850,408 | NAD(P)-binding domain | 0.04255 | FALSE |
| Glucose/ribitol dehydrogenase | ||||
| Bo1g153400 | 42,852,450–428,54,117 | Armadillo-type fold | N/A | N/A |
| Bo1g153410 | 42,854,711–42,855,688 | No comment | 0.96769 | FALSE |
| Bo1g153420 | 42,858,540–42,861,395 | T-complex protein 1, theta subunit | 0.40463 | FALSE |
| Bo1g153430 | 42,862,213–42,864,216 | YTH domain | 0.93226 | FALSE |
| Bo1g153440 | 42,869,749–42,873,374 | Protein kinase-like domain | 0.09861 | FALSE |
| Bo1g153450 | 42,881,692–42,883,434 | NAD(P)-binding domain | 0.74443 | FALSE |
| Bo1g153460 | 42,891,683–42,892,718 | No comment | 2.55E–32 | TRUE |
| Bo1g153470 | 42,894,992–42,896,857 | Thioredoxin-like fold | 0.53912 | FALSE |
| Bo1g153480 | 42,898,263–42,899,388 | No comment | 0.87865 | FALSE |
| Bo1g153490 | 42900,367–42,900,597 | Response to auxin | 0.92167 | FALSE |
| Bo1g153500 | 42,901,824–42,905,154 | GDP-fucose protein O-fucosyltransferase | 0.25833 | FALSE |
| Gene ID a | Gene Position on C4 b | Functional Annotation c | q-value d | Significant e |
|---|---|---|---|---|
| Bo4g173730 | 46,336,886–46,339,234 | Biosynthetic process | 0.44732 | FALSE |
| Catalytic activity | ||||
| Bo4g173740 | 46,341,684–46,342,808 | F-box domain | N/A | N/A |
| Bo4g173750 | 46,364,288–46,367,249 | Protein kinase-like domain | 0.69186 | FALSE |
| Bo4g173760 | 46,369,761–46,371,466 | Protein of unknown function DUF688 | 0.37224 | FALSE |
| Bo4g173770 | 46,376,730–46,377,865 | Domain of unknown function DUF1985 | N/A | N/A |
| Bo4g173780 | 46,383,190–46,387,379 | Protein kinase-like domain | 3.13E–08 | TRUE |
| Serine-threonine/tyrosine-protein | ||||
| Kinase catalytic domain | ||||
| Bo4g173790 | 46,388,006–46,389,868 | No comment | 0.13635 | FALSE |
| Bo4g173800 | 46,395,063–46,395,976 | No comment | 0.07896 | FALSE |
| Bo4g173810 | 46,405,666–46,407,281 | Nucleic acid-binding, OB-fold | 0.00004 | TRUE |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Z.; Hu, Y.; Zhang, X.; Xue, Y.; Fang, Z.; Yang, L.; Zhang, Y.; Liu, Y.; Li, Z.; Liu, X.; et al. Fine Mapping and Transcriptome Analysis Reveal Candidate Genes Associated with Hybrid Lethality in Cabbage (Brassica Oleracea). Genes 2017, 8, 147. https://doi.org/10.3390/genes8060147
Xiao Z, Hu Y, Zhang X, Xue Y, Fang Z, Yang L, Zhang Y, Liu Y, Li Z, Liu X, et al. Fine Mapping and Transcriptome Analysis Reveal Candidate Genes Associated with Hybrid Lethality in Cabbage (Brassica Oleracea). Genes. 2017; 8(6):147. https://doi.org/10.3390/genes8060147
Chicago/Turabian StyleXiao, Zhiliang, Yang Hu, Xiaoli Zhang, Yuqian Xue, Zhiyuan Fang, Limei Yang, Yangyong Zhang, Yumei Liu, Zhansheng Li, Xing Liu, and et al. 2017. "Fine Mapping and Transcriptome Analysis Reveal Candidate Genes Associated with Hybrid Lethality in Cabbage (Brassica Oleracea)" Genes 8, no. 6: 147. https://doi.org/10.3390/genes8060147