
The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance

1
Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta University, Augusta, GA 30912, USA
2
Tumor Signaling and Angiogenesis Program, Georgia Cancer Center, Augusta University, Augusta, GA 30912, USA
3
Biochemistry and Cancer Biology Program, The Graduate School, Augusta University, Augusta, GA 30912, USA
*
Author to whom correspondence should be addressed.
Genes 2017, 8(6), 158; https://doi.org/10.3390/genes8060158
Submission received: 28 February 2017
/
Revised: 31 May 2017
/
Accepted: 1 June 2017
/
Published: 7 June 2017
(This article belongs to the Special Issue MYC Networks)
{kind=link}
Abstract
:Cancer is associated with genomic instability and aging. Genomic instability stimulates tumorigenesis, whereas deregulation of oncogenes accelerates DNA replication and increases genomic instability. It is therefore reasonable to assume a positive feedback loop between genomic instability and oncogenic stress. Consistent with this premise, overexpression of the MYC transcription factor increases the phosphorylation of serine 139 in histone H2AX (member X of the core histone H2A family), which forms so-called γH2AX, the most widely recognized surrogate biomarker of double-stranded DNA breaks (DSBs). Paradoxically, oncogenic MYC can also promote the resistance of cancer cells to chemotherapeutic DNA-damaging agents such as cisplatin, clearly implying an antagonistic role of MYC in genomic instability. In this review, we summarize the underlying mechanisms of the conflicting functions of MYC in genomic instability and discuss when and how the oncoprotein exerts the contradictory roles in induction of DSBs and protection of cancer-cell genomes.
1. Introduction: MYC as a Transcription Factor
The MYC family of transcription factors directly and indirectly regulates various genes involved in cell proliferation [1], cellular metabolism [2], induction of apoptosis [3], blocking of differentiation [4], and initiation and promotion of cancer [5,6,7]. There are three MYC proto-oncoproteins, c-MYC, N-MYC, and L-MYC, which are encoded by separate genes and have different tissue distribution and oncogenic potency [8,9,10]. Deregulated expression of c-MYC is associated with the development of most human malignancies [11,12,13], whereas overproduction of N-MYC is frequently associated with neuroblastomas and gliomas [14,15]. In contrast, L-MYC expression is evident in both neonatal and adult lung tissues and most frequently overexpressed in lung cancer, including small-cell lung carcinomas [9], suggesting a non-redundant biology sustained by each member of the MYC family of transcription factors. However, in the absence of c-MYC, the majority of c-MYC-dependent gene transcription [16] and c-MYC-dependent tissue development [17] were compensated by the re-establishment of N-MYC, implying a functional redundancy as sequence-specific transcription factors due to the structural similarity of each MYC protein.
The MYC transcription factor contains the amino-terminal (N-terminal) transcriptional activation domain (TAD), followed by the central region, the nuclear localization motif, and the carboxyl-terminal (C-terminal) basic-helix-loop-helix-leucine zipper (bHLH-LZ) domain. The N-terminal TAD region harbors at least two conserved segments, termed MYC boxes 1 and 2 (MB1 and MB2). Depending on the availability of MB1- and/or MB2-interacting coactivators and corepressors, such as the transformation/transcription domain–associated protein (TRRAP) [18], the pocket protein p107 [19], and a pro-apoptotic corepressor bridging integrator 1 (BIN1) [20,21], the MYC TAD region is essential for fine-tuning MYC-dependent transcriptional activation and repression [10,22].
The C-terminal bHLH-LZ region is required for hetero-dimerization with MYC-associated factor X (MAX), which allows MYC to recognize the MYC-binding consensus sequence “CACGTG”, termed the Enhancer box (E-box). MYC–MAX heterodimer formation is crucial for MYC-dependent gene transcription, cell proliferation, apoptosis, and oncogenic transformation [23,24], but MYC may also function as a transcription factor in a MAX-independent manner [25,26,27,28], implying the presence of a non-MAX heterodimeric binding partner of MYC to recognize the E-box. Furthermore, MYC directly interacts with MYC-interacting Zinc finger protein 1 (MIZ1) [29], which recognizes an initiator (INR) element, PyPyAN[T/A]PyPy (Py: Pyrimidine; N: any nucleotide) [30], in a core promoter region. Through direct MYC–MIZ1 interaction via an INR sequence of several growth-arresting genes, MYC cancels MIZ1-dependent transactivation [29,31,32,33]. In addition to a new emerging research area regarding the transcription-independent function of MYC, particularly in protein translation and DNA replication [34], it is important to identify downstream effectors of MYC-dependent gene transcription to better understand the multiple roles of MYC in various cell behaviors, such as cell proliferation survival, apoptosis, genomic instability, and cancer.
2. MYC in Cancer Development and Cancer Stemness
The critical involvement of MYC in cell growth was evident from the study showing rapid and sustained induction of MYC, following mitogenic stimulation of quiescent cells [35,36]. Furthermore, homozygous inactivation of Myc in immortalized rat fibroblasts resulted in prolonged doubling time and accumulation of cells in the G1 and G2/M phases in the cell cycle [37]. MYC abundance is consistently elevated during embryogenesis and in highly proliferative tissues, such as skin epidermis and gut of adults [38]. The first evidence for MYC as an immortalizing factor came from a classical RAS co-transformation assay using primary rodent fibroblasts as an in vitro cancer model system [39]. A number of studies have since been conducted providing evidence that MYC overproduction was sufficient to induce tumorigenesis both in vitro and in vivo [40,41,42]. Accordingly, an elevated or deregulated level of MYC is a hallmark of the vast majority of human malignancies and is usually recognized as a poor prognostic marker, indicating a strong link between deregulation or overexpression of the MYC gene and advanced cancer development.
It is important that MYC has been identified as one of the “Yamanaka” factors, along with three other distinct transcription factors, SOX2, OCT4, and KLF4, which are all essential for the establishment of inducible pluripotent stem (iPS) cells by reprograming transcriptional networks of fully differentiated somatic cells [43,44,45]. The growth behaviors of iPS cells are normally sustained properly. However, presumably because of ectopically carrying the MYC allele, iPS cells share several cellular properties with cancer cells naturally overexpressing MYC, such as malfunctioned senescence and cellular immortality [46,47]. MYC-expressing iPS cells could be therefore oncogenically transformed once proper or strict biological control of MYC activity has disappeared. Intriguingly, recent findings demonstrated that oncogenic MYC preserves cancer stemness, which enables cancer cells to survive longer with increasing metastatic potential even under genotoxic conditions [6,48]. Given that MYC participates in transcriptional reprogramming that facilitates the creation of iPS cells from differentiated cells [43,44,45], it may be possible for MYC-overexpressing cancer cells to acquire traits similar to those of cancer-stem cells [49,50,51]. It is undoubtedly envisioned that the failure of stringent regulation of MYC activity is associated with aberrant growth behaviors leading to the development of cancer stemness.
3. Oncogenic Addiction to MYC as an “Achilles’ Heel” of MYC-Driven Cancer Cells
Oncogene addiction represents a dependency of cancer cells on a single oncogenic protein (or pathway) for maintaining their survival and malignant properties [52,53]. Because of the strong correlation between MYC overexpression and tumorigenesis, it is anticipated that survival of cancer cells would be greatly compromised if MYC is inactivated. The depletion of MYC by the co-transfection of antisense RNA of c-MYC in cell lines derived from human tumors shows inhibition of proliferation and induction of differentiation [54,55], serving as the first experimental evidence of MYC addiction in cancer cells in vitro. Using in vivo model systems with conditional transgenic mice, inactivation of MYC consistently resulted in tumor regression in vivo in hematopoietic and solid tumors [56,57]. Of note, brief inactivation of MYC is sufficient to induce long-term loss of neoplastic phenotypes [58], indicating that oncogenic addiction elicited by MYC serves as an “Achilles’ heel” of MYC-expressing cancers [53,59]. However, because of the limited availability of a MYC-specific small-molecule inhibitor with little or no adverse effect in clinical settings [60,61], a strategy for therapeutically targeting MYC addiction seems fascinating, but remains frustratingly difficult.
4. MYC-Induced Replication Stress, Genomic Instability, and Oncogenic Transformation
During cell division, chromosomal DNA is entirely replicated, but errors in this process may result in various forms of genetic alterations, such as point mutations and gene amplifications [62]. A process prone to genomic alterations is termed as genomic instability, which could be one of the major causes of cancer initiation [63]. Chromosomal alteration, a genomic instability at the chromosomal level, has long been recognized to be the hallmark of human cancer [11,64]. It is well acknowledged that a tumor cell is the progeny of a single genetically unstable cell, which continuously acquires chromosomal abnormalities over time [65]. The genomic instability driving oncogenesis can be further fueled by errors in DNA replication. Some cells may accumulate more genetic changes in their chromosomes, which subsequently provide selective advantages for growth [66,67]. The majority of cancers exhibit deregulated expression of MYC as a result of either activation of upstream regulatory signals, such as WNT-dependent MYC expression [68] and Src-dependent MYC activation [69], or chromosomal instability, which causes gene amplification, insertional mutagenesis, and gene translocation [70,71,72]. For example, gene amplification of the c-MYC locus is one of the most representative gene abnormalities in prostate cancer [73].
In contrast, deregulated MYC could be a trigger of chromosomal instability presumably as a consequence of MYC-induced aberrant DNA replication and cell division [74]. Deregulated MYC expression may result in uncontrolled activation of a number of downstream genes, which promote the cell cycle and DNA synthesis, and ultimately initiates and promotes genomic instability [75]. In agreement with this premise, MYC induction is often associated with a variety of chromosomal changes, for example, generation of extrachromosomal elements [76], centromere and telomere fusions [77], chromosome and chromatid breaks, translocations, deletions, inversions, and aneuploidy [74,75,77,78,79].
The close relationship between MYC and chromosomal instability was further proven by the studies showing MYC-mediated induction of nonrandom amplification and rearrangement of the genes encoding several growth-promoting genes, such as dihydrofolate reductase (DHFR), carbamyl-P synthetase, aspartate transcarbamylase, dihydro-orotase, cyclin D2, and the R2 subunit of ribinucleotide reductase [80,81,82,83,84]. Interestingly, human pre-invasive cervical cancer shows DHFR amplification in association with MYC overexpression [85]. Unless apoptosis or cellular senescence is activated, MYC-induced chromosomal rearrangements would provide a proliferative advantage to MYC-overexpressing cells, which may subsequently induce even more genomic instability, including double stranded DNA breaks (DSBs). Consistent with this idea, deregulated MYC upregulates the formation of γH2AX foci, a surrogate biomarker of DSBs [86,87]. In normal human foreskin fibroblasts, MYC overexpression is sufficient to increase the formation of γH2AX foci [88].
A possible mechanism by which MYC induces DSBs is through accumulation of reactive oxygen species (ROS) [89,90]. However, MYC-induced DSBs also occur in a manner independent of ROS production, implying that a different mechanism may also contribute to MYC-mediated production of DSBs [91]. For example, several studies support the possibility that overexpression of MYC represses cellular DSB repair potentials [88,92,93]. It is important that replication stress causes aberrant replication intermediates and MYC is actively involved in DNA replication, particularly initiation of replication [94,95]. Overexpressed MYC can function as an illegitimate replication-licensing factor [75]. MYC is also capable of overriding cell-cycle checkpoints, thereby triggering genomic instability during DNA replication [74]. Deregulated MYC may therefore induce DSBs by multiple mechanisms, such as accumulation of ROS, an increase in replication stress [96], and decrease in DSB repair capacity [88,92,93]. The tumorigenic nature of MYC is thus attributable to not only its pivotal roles in the upregulation of genes promoting cell proliferation, but also its ability to promote genomic instability through chromosomal aberrations and DSBs.
In normal fibroblasts, oncogenic MYC-induced replication stress provokes genomic instability, which then stimulates cellular checkpoint mechanisms, including cell-cycle arrest, apoptosis, and premature senescence [97,98]. Hence, oncogene-induced checkpoints are thought to be cellular defense mechanisms that halt or delay the onset of tumorigenesis as long as the levels of DNA damage are not devastating [96]. Because oncogenic RAS [98,99] and oncogenic RAF , which functions downstream of the RAS protein family [100], can also induce premature senescence in normal fibroblasts, oncogene-induced checkpoint mechanisms are not MYC-specific functions, but broadly triggered by oncogenic stress and subsequent acceleration of cell-cycle progression and DNA hyper-replication [97,98]. Because tumorigenesis is promoted by genomic instability [96], any malfunction of the checkpoint machinery could be a potential cause of cellular transformation.
Using rodent primary embryo fibroblasts as an in vitro model system, oncogenic foci formation can be experimentally achieved by the co-transfection of two independent oncogenes; one is a nuclear oncogene (such as c-MYC and adenovirus E1A) and the other is a cytoplasmic oncogene (such as activated RAS) [39,101]. Since then, the requirement of concurrent activation of multiple oncogenes for cellular transformation has been experimentally established [102], which implies a functional collaboration between two independent oncogenic pathways to circumvent or inactivate cellular checkpoint mechanisms. BIN1 was identified as one of the 17 cellular anticancer proteins that promote apoptosis and senescence in response to oncogenic RAF [100]. Intriguingly, overexpression of c-MYC broadly represses the BIN1 transcription in a manner dependent on MIZ1 [33]. It will be interesting to investigate whether oncogenic MYC represses or inactivates any other checkpoint-related cellular factors activated by oncogenic RAS and RAF.
5. MYC in DNA Repair and Cancer Chemoresistance
Current cancer treatments rely greatly on conventional chemotherapeutic drugs. Most of the clinically approved chemotherapeutic agents cause cytotoxicity in rapidly proliferating cells, such as cancer cells and hair-follicle cells, by inducing DNA damage. However, tumor cells often become refractory to a chemotherapeutic agent, a phenomenon known as chemoresistance. Cancer cells may have intrinsic drug-resistance or acquire it during genotoxic treatment [103]. The mechanisms by which advanced cancer cells increase chemoresistance could be attributable to (a) an increase in the amount (and/or activity) of cellular DNA repair machinery [104,105]; (b) suppression or inactivation of pro-apoptotic machinery [106,107]; and/or (c) detoxification and enhanced efflux of genotoxic chemicals [108,109]. Clinical evidence shows that once cancer cells acquire resistance to first-line chemotherapeutic drugs such as cisplatin, cancer cells naturally develop cross-resistance to a range of other chemotherapeutic agents, ultimately leading to treatment failure in over 90% of patients with metastatic diseases [108,110]. The major clinical challenge for effective chemotherapy is therefore the rapid elimination of chemosensitive cancer cells before chemoresistance appears.
In general, accumulation of genomic instability renders the cancer-cell genome vulnerable to DNA-damaging agents [111,112]. MYC-induced genomic instability, which increases cancer incidence (see above), might thus serve as a double-edged sword to eradicate cancer cells overexpressing MYC in combination with therapeutic DNA-damaging agents. Paradoxically, it is well known that the abundance of MYC oncoprotein closely correlates with chemoresistance in many tumor types, including prostate cancer, ovarian cancer, melanoma, lung cancer, and hepatocellular carcinoma both in vitro and in vivo [33,113,114,115,116]. In patients with ovarian cancer, high levels of MYC are coupled to tumor recurrence, poor overall survival, and cisplatin resistance [117].
Moreover, c-MYC expression is elevated in tumor-cell fractions that have survived platinum-based chemotherapy in vivo [113]. In embryonal rhabdomyosarcoma cell lines, MYC protects cancer cells from radiation-induced apoptosis and DNA damage, while promoting radiation-induced DNA repair [118]. In light of these reports, oncogenic MYC seems to be an attractive target to overcome chemoresistance in a wide range of human cancers. Consistent with this premise, antisense oligodeoxynucleotides or the silencing of MYC by small-interfering RNA increases cisplatin sensitivity in inherently cisplatin-resistant melanoma cell lines in vitro and reduces tumor formation in xenograft models of cisplatin-resistant ovarian cancer cells [117,119]. Moreover, the small-molecule MYC inhibitor, 10058-F4, was reported to be effective in anti-tumor treatment, such as for leukemia [120], prostate cancer [33], and hepatocellular carcinoma [116]. These in vitro and in vivo observations unfailingly suggest that oncogenic MYC protects cancer-cell genomes from therapeutic DNA-damaging agents.
As we discussed above, there is solid evidence that oncogenic MYC increases replication stress and genomic instability, which then initiates an early step leading to oncogenic transformation if cellular checkpoint mechanisms are inactivated. However, the maintenance of genomic integrity during DNA replication is one of the most fundamental biological traits for the preservation of genetic information, even though MYC, a powerful inducer of DSBs, always promotes the G1/S phase transition in the cell cycle. Notably, in addition to MYC, the E2F1 transcription factor is another preeminent transcription factor, single transfection of which is sufficient to promote G1/S transition in quiescent (i.e., serum-starved) fibroblasts [121]. Because E2F1 is well known to actively promote DSB repair via physically interacting with Nijmegen Breakage Syndrome Protein 1 (NBS1), a key component of the MRN (MRE11/RAD50/NBS1) DNA-end-binding complex [122,123,124], and transcriptionally stimulates the human c-myc gene promoter [125,126], there may be a possible collaboration between these two master cell-cycle-promoting transcription factors not only in the G1/S phase transition [121], but also in the synchronized activation of DSB-repair machinery. Hence, it is logical to hypothesize that similar to E2F1, MYC facilitates a DSB repair-promoting function to counterbalance (or, at least, mitigate) an unavoidable effect of MYC on replication stress-induced genomic instability during DNA replication.
Consistent with this premise, MYC physically associates with the 5′-flanking promoter region of various DSB repair-related genes, such as NBS1, Ku70, Rad51, BRCA2, Rad50, and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), and activates their transcription [127,128,129,130]. Possible involvement of MYC in DNA repair is evident, in addition, from the finding that the silencing of MYC was correlated with a decrease in kinase activities of ataxia-telangiectasia mutated serine/threonine protein kinase (ATM) and DNA-PKCs, which consequently resulted in a reduction in irradiation-induced DSB repair efficiency [131]. Consistently, there was a significant reduction in the capability of DSB repair in HeLa cells after the silencing of MYC [131]. It is pertinent to experimentally establish the effect of the impairment of endogenous MYC on DSB repair pathway(s) in various cultured cancer model systems. Interestingly, cancer stem cells are well known to survive longer, even under genotoxic circumstances, such as chemo and radiation therapy [132,133,134], and robustly upregulate cellular DNA-repair activity [135,136]. Because oncogenic MYC transcriptionally stimulates a number of DSB repair genes [127,128,129,130] and is responsible for the maintenance of cancer stemness [137,138], we propose that, under devastating genotoxic stress and/or during the maintenance of cancer stemness, oncogenic MYC protects the cancer-cell genome from DNA damage, thereby allowing advanced cancer (or cancer stem) cells to survive longer.
If so, inhibiting DNA repair following treatment with DNA-damaging agents could be a promising approach for eradicating MYC-induced chemoresistance. Small-molecule inhibitors that target DNA repair proteins, such as poly (ADP-ribose) polymerase (PARP) [139,140], DNA-PKCs [141,142], and ATM [143,144], are emerging as attractive targets for chemotherapy [145]. PARP inhibitors have been most widely tested as an anticancer therapeutic option [146]. Our group has previously shown that BIN1, originally identified as a MYC-interacting tumor suppressor [21], functions as a cellular inhibitor of PARP1 in vitro and that MYC induces cisplatin resistance by liberating intrinsic PARP1 activity through directly suppressing BIN1 levels [33]. We have also recently shown that PARP inhibition increases BIN1 levels, which play an important role in mediating E2F1-induced apoptosis, particularly under serum-starved conditions [147]. Because reduced blood supply is frequently observed in solid tumor tissues, especially following chemotherapy [148,149], induction of endogenous BIN1 expression by PARP inhibitors, such as olaparib, may be combined with conventional chemotherapy to reverse MYC-dependent chemoresistance.
6. Conclusions
Given that rapidly proliferating cancer cells, which naturally produce oncogenic MYC, accumulate more genomic instability, it is logical to assume that late-stage or recurrent cancer cells, which are expected to carry high levels of MYC, must have accumulated more DNA damage. Advanced (i.e., late-stage) cancer cells should subsequently be more susceptible to DNA-damaging chemotherapeutic drugs. However, the opposite occurs; late-stage cancer cells survive longer even during/after DNA-damaging chemotherapy [113,119]. This evidence clearly suggests that, in advanced cancer cells, oncogenic MYC develops a highly efficient DNA repair system and offsets the genomic instability induced by chemo and radiation therapy [118].
To better understand the functional discrepancy of the formation of γH2AX foci by oncogenic MYC in chemoresistant cancer cells, we must remember that most of the evidence that supports the MYC-induced DSBs in cancer cells mainly derives from the efficient formation of γH2AX foci by experimentally overexpressed MYC. Although γH2AX is generally considered to be the most reliable biomarker of DSBs, there are several reports showing DSB-independent functions of γH2AX [150,151,152]. ATM kinase is indeed responsible for the formation of γH2AX in response to DSBs, but the same enzyme can also be activated in a DSB-independent manner [153], such as hypoxia [154], where γH2AX foci can be formed in a manner independent of DSBs.
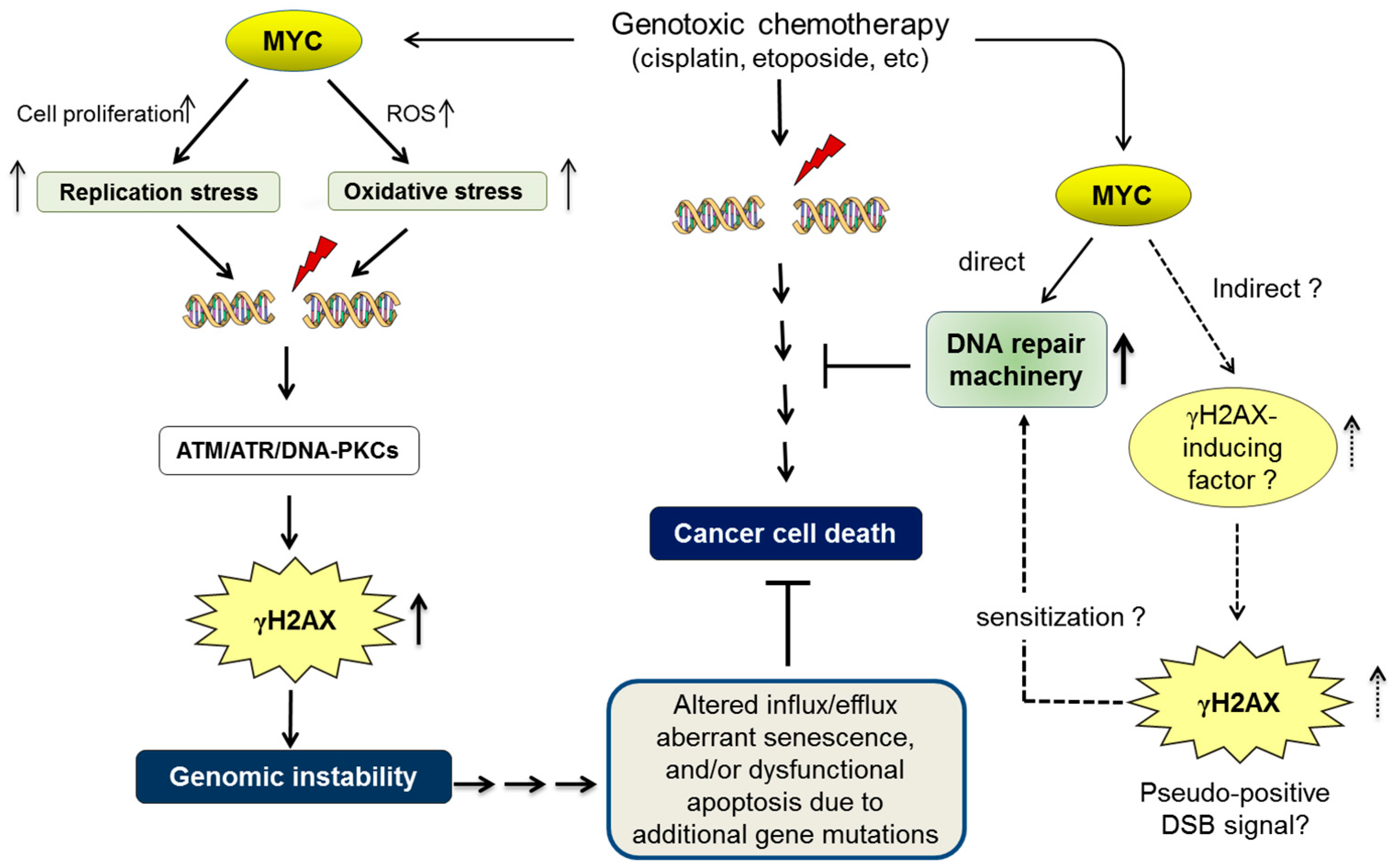
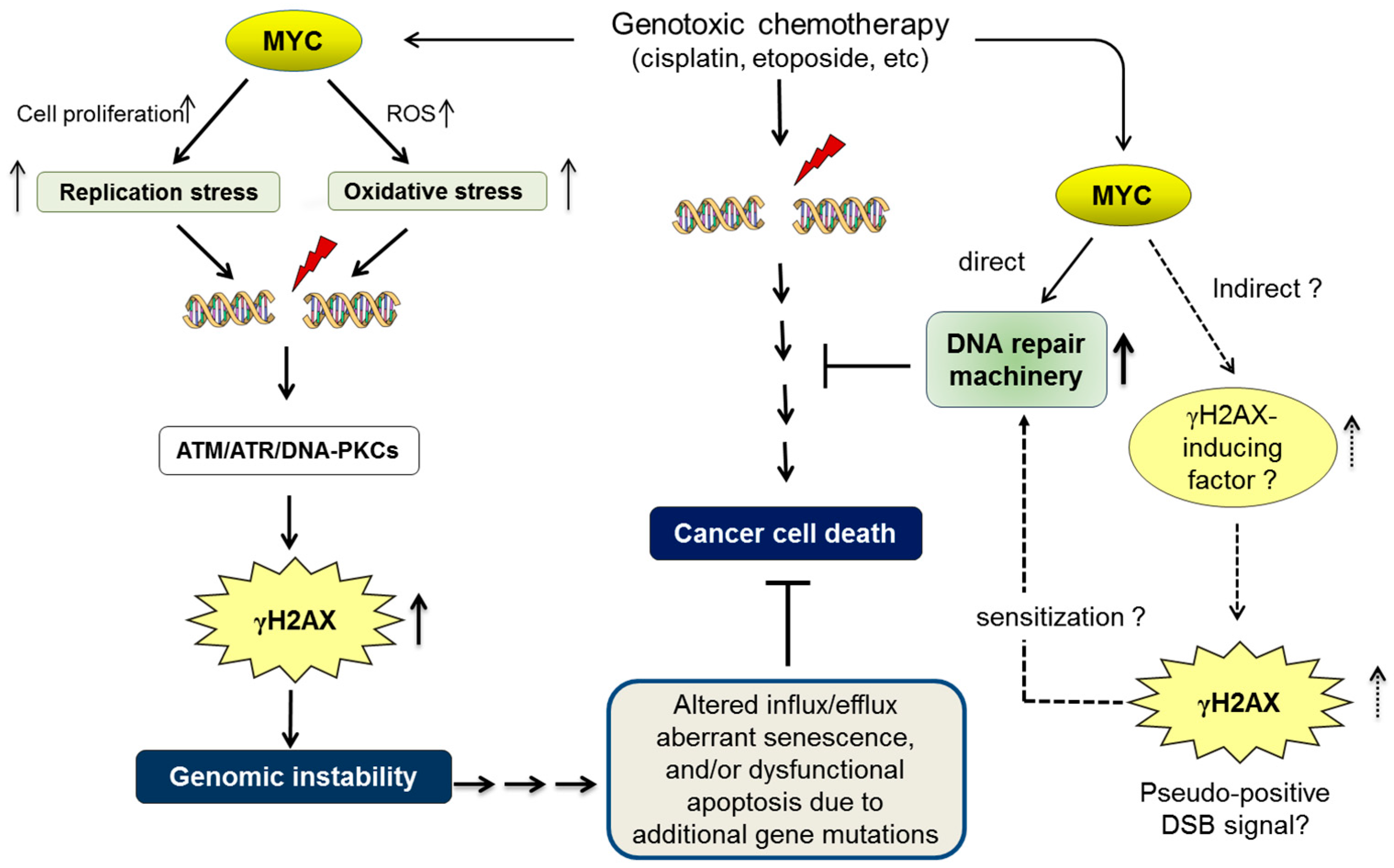
If MYC increases γH2AX foci in a DSB-independent manner, one possible model is to stimulate a γH2AX-inducing factor independent of DSBs. Consistent with this idea, it has been reported that MYC is required for the activation of ATM and formation of γH2AX foci [155], whereas suppression of ATM dramatically sensitizes tumors to DNA-damaging chemotherapy [156]. We propose that upregulation of the γH2AX-inducing factor (such as ATM) by MYC in cancer cells might result in persistent formation of γH2AX foci, which serves as a pseudo-positive DSB signal that sustains the activation of DNA-repair machinery (Figure 1).
Deregulated MYC induces replication stress and oxidative stress due to reactive oxygen species (ROS), which subsequently contribute to promoting double-stranded DNA breaks (DSBs). DSBs upregulate the phosphorylation of histone H2AX (forming γH2AX) by activating the sensors of DNA damage, such as ataxia-telangiectasia mutated serine/threonine protein kinase (ATM), ATM- and Rad3-related protein kinase (ATR), and/or DNA-dependent protein kinase (DNA-PK). γH2AX is widely recognized as a surrogate biomarker of DSBs. A MYC-dependent increase in γH2AX foci may therefore be indirect evidence of increased genomic instability by MYC. Genomic instability is a consequence of oncogenic MYC, but it may also act as an upstream factor that further promotes malignant phenotypes. For example, additional gene mutations due to genomic instability may have negative influence on cancer-cell death machinery by altering the influx/efflux, cellular senescence, and apoptotic/survival signals [106,107,108,109]. Although genotoxic chemotherapeutic drugs effectively kill cancer cells by inducing deleterious DSBs, it is widely recognized that high levels of MYC eventually render cancer cells resistant to genotoxic drugs [117]. In addition to the direct upregulation of DNA-repair machinery [130], we propose that deregulated MYC induces chemoresistance indirectly by activating a γH2AX-inducing factor, such as ATM [131,155], which subsequently increases γH2AX foci as a pseudo-positive DSB signal and sensitizes the cellular DNA-repair machinery.
However, it is our consensus that the underlying mechanisms of cancer chemosensitivity depend, at least in part, on intrinsic tumour suppressor mechanisms, such as TP53 [107] and BIN1 [33], by which DSB-induced apoptosis or senescence can be elicited. Moreover, cancer chemoresistance relies, to some extent, on enhanced efflux and/or detoxification of anticancer drugs [109,157]. Therefore, in addition to the development of DSB-repair function by MYC in late-stage cancer cells, deregulated MYC, which leads to increased genomic instability, at least partly, via replication stress (see above), offers a chance for cancer cells to accumulate more genetic mutation and develop ways of evading the cytotoxic effects of anticancer drugs by, for example, downregulating tumour suppressor functions and/or upregulating drug efflux and detoxification (Figure 1).
In this review, we have discussed the dual roles of MYC in the development of genomic instability and chemoresistance in cancer cells. Further work, such as identification of downstream MYC effectors that liberate ATM-dependent formation of γH2AX foci, is important to reconcile the contradictory nature of this enigmatic oncoprotein in response to DNA damage.
Acknowledgments
This work was supported by grants from the USA National Institutes of Health (NIH) (R01CA140379) and the 2017 Biochemistry and Molecular Biology Translational Pilot Research Award (to D.S.). A.K. is the recipient of the Georgia Cancer Center Postdoctoral Research Excellence Award. W.P.F. was supported by the Augusta University Biochemistry and Cancer Biology Graduate Program.
Conflicts of Interest
D. Sakamuro holds a 1999 USA Patent with G.C. Prendergast No. 6410238, “Box-dependent Myc-interacting protein (Bin1) compositions and uses thereof” (The Wistar Institute of Anatomy and Biology, Philadelphia, PA, USA).
References
- Eilers, M.; Schirm, S.; Bishop, J.M. The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 1991, 10, 133–141. [Google Scholar] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-MYC protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Freytag, S.O.; Geddes, T.J. Reciprocal regulation of adipogenesis by Myc and C/EBPalpha. Science 1992, 256, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [PubMed]
- Tansey, W.P. Mammalian MYC Proteins and Cancer. New J. Sci. 2014, 2014, 1–27. [Google Scholar] [CrossRef]
- Nisen, P.D.; Zimmerman, K.A.; Cotter, S.V.; Gilbert, F.; Alt, F.W. Enhanced expression of the N-myc gene in Wilm’s tumors. Cancer Res. 1986, 46, 6217–6222. [Google Scholar] [PubMed]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Morgenbesser, D.; DePinho, R. Myc family oncoproteins function through a common pathway to transform normal cells in culture: Cross-interference by Max and trans-acting dominant mutants. Genes Dev. 1992, 6, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Evan, G.I.; Littlewood, T.D. The role of c-Myc in cell growth. Curr. Opin. Genet. Dev. 1993, 3, 44–49. [Google Scholar] [CrossRef]
- Henriksson, M.; Luscher, B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. 2014, 12, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.; Mateyak, M.; Dugan, K.; Obaya, A.; Adachi1, S.; Sedivy, J.; Cole, M. c-myc null cells misregulate cad and gadd45 but not other proposed c-Myc targets. Genes Dev. 1998, 12, 3797–3802. [Google Scholar] [CrossRef] [PubMed]
- Malynn, B.A.; Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [PubMed]
- McMahon, S.B.; Van Buskirk, H.A.; Dugan, K.A.; Copeland, T.D.; Cole, M.D. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 1998, 94, 363–374. [Google Scholar] [CrossRef]
- Gu, W.; Bhatia, K.; Magrath, I.T.; Dang, C.V.; Dalla-Favera, R. Binding and suppression of the Myc transcriptional activation domain by p107. Science 1994, 264, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Sakamuro, D.; Elliott, K.J.; Wechsler-Reya, R.; Prendergast, G.C. BIN1 is a novel MYC-interacting protein with features of a tumor suppressor. Nat. Genet. 1996, 14, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Sakamuro, D.; Prendergast, G.C. New Myc-interacting proteins: A second Myc network emerges. Oncogene 1999, 18, 2942–2954. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.; Birrer, M.J.; Kato, G.J.; Dosaka-Akita, H.; Dang, C.V. Activation domains of L-Myc and c-Myc determine their transforming potencies in rat embryo cells. Mol. Cell. Biol. 1992, 12, 3130–3137. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Ziff, E.B. A new bind for Myc. Trends Genet. 1992, 8, 91–96. [Google Scholar] [CrossRef]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Hopewell, R.; Ziff, E.B. The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner. Max. Mol. Cell. Biol. 1995, 15, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Steiger, D.; Furrer, M.; Schwinkendorf, D.; Gallant, P. Max independent functions of Myc in Drosophila melanogaster. Nat. Genet. 2008, 40, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Wert, M.; Kennedy, S.; Palfrey, H.C.; Hay, N. Myc drives apoptosis in PC12 cells in the absence of Max. Oncogene 2001, 20, 3746–3750. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Cole, M.D. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol. Cell. Biol. 2007, 27, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Peukert, K.; Staller, P.; Schneider, A.; Carmichael, G.; Hänel, F.; Eilers, M. An alternative pathway for gene regulation by Myc. EMBO J. 1997, 16, 5672–5686. [Google Scholar] [CrossRef] [PubMed]
- Javahery, R.; Khachi, A.; Lo, K.; Zenzie-Gregory, B.; Smale, S.T. DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol. Cell. Biol. 1994, 14, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massagué, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. c-MYC suppresses BIN1 to release poly (ADP-ribose) polymerase 1: A mechanism by which cancer cells acquire cisplatin resistance. Sci. Signal. 2011, 4, Ra19. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.D.; Cowling, V.H. Transcription-Independent Functions of MYC: Regulation of Translation and DNA Replication. Nat. Rev. Mol. Cell Biol. 2008, 9, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Campisi, J.; Gray, H.E.; Pardee, A.B.; Dean, M.; Sonenshein, G.E. Cell-cycle control of c-myc but not c-ras expression is lost following chemical transformation. Cell 1984, 36, 241–247. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Obaya, A.J.; Adachi, S.; Sedivy, J.M. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997, 8, 1039–1048. [Google Scholar] [PubMed]
- Pfeifer-Ohlsson, S.; Rydnert, J.; Goustin, A.S.; Larsson, E.; Bethsholtz, C.; Ohlsson, R. Cell-type-specific pattern of myc protooncogene expression in developing human embryos. Proc. Natl. Acad. Sci. USA 1985, 82, 5050–5054. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Leder, A.; Pattengale, P.K.; Kuo, A.; Stewart, T.A.; Leder, P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: Multiple neoplasms and normal development. Cell 1986, 45, 485–495. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Wilson, A.; Trumpp, A. Myc’s other life: Stem cells and beyond. Curr. Opin. Cell Biol. 2009, 21, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.M.; Dalton, S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell 2009, 5, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.W.; Barrilleaux, B.L.; Varlakhanova, N.; Bush, K.M.; Chan, V.; Knoepfler, P.S. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013, 22, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Wolfera, A.; Wittnera, B.S.; Irimiab, D.; Flavinb, R.J.; Lupienb, M.; Gunawardaneb, R.N.; Meyerg, C.A.; Lightcaph, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.N.; Singh, A.M.; Dalton, S. Myc sustains pluripotency by repressing primitive endoderm specification. Cell Stem Cell 2010, 7, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.C.; Knabenhans, C.; Macdonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004, 18, 2747–2763. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Disorders in cell circuitry during multistage carcinogenesis: The role of homeostasis. Carcinogenesis 2000, 21, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Addiction to oncogenes—The Achilles’ heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Imamoto, F. Transcriptional control of the endogenous MYC protooncogene by antisense RNA. Proc. Natl. Acad. Sci. USA 1987, 84, 7363–7367. [Google Scholar] [CrossRef] [PubMed]
- Loke, S.L.; Stein, C.; Zhang, X.; Avigan, M.; Cohen, J.; Neckers, L.M. Delivery of c-myc antisense phosphorothioate oligodeoxynucleotides to hematopoietic cells in culture by liposome fusion: Specific reduction in c-myc protein expression correlates with inhibition of cell growth and DNA synthesis. Curr. Top. Microbiol. Immunol. 1988, 141, 282–289. [Google Scholar] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell. 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Anderton, B.; Goga, A. Taking on Challenging Targets: Making MYC Druggable. Am. Soc. Clin. Oncol. Educ. Book 2014, e497–e502. [Google Scholar] [CrossRef] [PubMed]
- Darmon, E.; Leach, D.R.F. Bacterial Genome Instability. Mol. Biol. Rev. 2014, 78, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Popescu, N.C.; Zimonjic, D.B. Chromosome-mediated alterations of the MYC gene in human cancer. J. Cell. Mol. Med. 2002, 6, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar] [PubMed]
- Hartwell, L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992, 71, 543–546. [Google Scholar] [CrossRef]
- Juan, J.; Muraguchi, T.; Iezza, G.; Sears, R.C.; McMahon, M. Diminished WNT → β-catenin → c-MYC signaling is a barrier for malignant progression of BRAFV600E-induced lung tumors. Genes Dev. 2014, 28, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.V.; Courtneidge, S.A. Myc but not Fos rescue of PDGF signalling block caused by kinase-inactive Src. Nature 1995, 378, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Taub, A.; Kirsch, I.; Morton, C.; Lenoir, G.M.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed]
- Shen-Ong, G.L.C.; Keath, E.J.; Piccoli, S.P.; Cole, M.D. Novel myc oncogene RNA from abortive immunoglobulin-gene recombination in mouse plasmacytomas. Cell 1982, 31, 443–480. [Google Scholar] [CrossRef]
- Pear, W.S.; Wahlstrom, G.; Nelson, S.F.; Axelson, H.; Szeles, A.; Wiener, F.; Bazin, H.; Klein, G.; Sumegi, J. 6;7 chromosomal translocation in spontaneously arising rat immunocytomas Evidence for c-myc breakpoint clustering and correlation between isotype expression and the c-myc target. Mol. Cell. Biol. 1988, 8, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, N.; Yegnasubramanian, S.; De Marzo, A. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Mai, S. c-MYC-Induced Genomic Instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef] [PubMed]
- Kuttler, F.; Mai, S. Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. Semin. Cancer Biol. 2007, 17, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Fluri, M.; Siwarski, D.; Huppi, K. Genomic instability in MycER-activated Rat1A MycER cells. Chromosome Res. 1996, 4, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, L.D.; Torrey, T.A.; Kim, J.S.; Coleman, A.E.; Kovalchuk, A.L.; Xiang, S.; Ried, T.; Morse, H.C., III; Janz, S. Genomic instability in mouse Burkitt lymphoma is dominated by illegitimate genetic recombinations, not point mutations. Oncogene 2002, 21, 7235–7240. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.G.; Graves, H.A.; Guffei, A.; Ricca, T.I.; Mortara, R.A.; Jasiulionis, M.G.; Mai, S. Telomere-centromere-driven genomic instability contributes to karyotype evolution in a mouse model of melanoma. Neoplasia 2010, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Wahl, G.M. c-Myc, Genome Instability, and Tumorigenesis: The Devil Is in the Details. CTMI 2006, 302, 169–203. [Google Scholar]
- Mai, S. Overexpression of c-myc precedes amplification of the gene encoding dihydrofolate reductase. Gene 1994, 148, 253–260. [Google Scholar] [CrossRef]
- Mai, S.; Hanley-Hyde, J.; Fluri, M. c-Myc overexpression associated DHFR gene amplification in hamster, rat, mouse and human cell lines. Oncogene 1996, 12, 277–288. [Google Scholar] [PubMed]
- Mai, S.; Hanley-Hyde, J.; Rainey, G.J.; Kuschak, T.I.; Paul, J.T.; Littlewood, T.D.; Mischak, H.; Stevens, L.M.; Henderson, D.W.; Mushinski, J.F. Chromosomal and extrachromosomal instability of the cyclin D2 gene is induced by Myc overexpression. Neoplasia 1999, 1, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Kuschak, T.I.; Taylor, C.; McMillan-Ward, E.; Israels, S.; Henderson, D.W.; Mushinski, J.F.; Wright, J.A.; Mai, S. The ribonucleotide reductase R2 gene is a non-transcribed target of c-Myc-induced genomic instability. Gene 1999, 238, 351–365. [Google Scholar] [CrossRef]
- Guijon, F.B.; Greulich-Bode, K.; Paraskevas, M.; Baker, P.; Mai, S. Premalignant cervical lesions are characterized by dihydrofolate reductase gene amplification and c-Myc overexpression: Possible biomarkers. J. Low. Genit. Tract. Dis. 2007, 11, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Pilch, D.R.; Redon, C.; Bonner, W.M. Histone H2AX in DNA damage and repair. Cancer Biol. Ther. 2003, 2, 233–235. [Google Scholar] [PubMed]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell. 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Ray, S.; Atkuri, K.R.; Deb-Basu, D.; Adler, A.S.; Chang, H.Y.; Herzenberg, L.A.; Felsher, D.W. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006, 66, 6598–6605. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Owonikoko, T.; Ramalingam, S.S.; Doetsch, P.W.; Xiao, Z.; Khuri, F.; Curran, W.; Deng, X. c-Myc suppression of DNA double-strand break repair. Neoplasia 2012, 14, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, S.; Amente, S.; Napolitano, G.; Di Palo, G.; Lania, L.; Majello, B. MYC impairs resolution of site-specific DNA double-strand breaks repair. Mutat. Res. 2015, 774, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Henriksson, M.; Sümegi, J.; Klein, G.; Hammarskjöld, M.L. Elevated c-myc expression facilitates the replication of SV40 DNA in human lymphoma cells. Nature 1987, 330, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Amati, B. Two sides of the Myc-induced DNA damage response: From tumor suppression to tumor maintenance. Cell Div. 2012, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ruley, H.E. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature 1983, 304, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S.; Kobayashi, H.; Graham, C.H. Intrinsic or acquired drug resistance and metastasis: Are they linked phenotypes? J. Cell. Biochem. 1994, 56, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Dijt, F.J.; Fichtinger-Schepman, A.M.; Berends, F.; Reedijk, J. Formation and repair of cisplatin-induced adducts to DNA in cultured normal and repair-deficient human fibroblasts. Cancer Res. 1988, 48, 6058–6062. [Google Scholar] [PubMed]
- Dabholkar, M.; Bostick-Bruton, F.; Weber, C.; Bohr, V.A.; Egwuagu, C.; Reed, E. ERCC1 and ERCC2 expression in malignant tissues from ovarian cancer patients. J. Natl. Cancer Inst. 1992, 84, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, E.; Silcocks, P.; Smith, J.; Hancock, B.W.; Goyns, M.H. P53 mutation in a series of epithelial ovarian cancer from the U.K. and its prognostic significance. Eur. J. Cancer 1994, 30A, 1701–1704. [Google Scholar] [CrossRef]
- Perego, P.; Giarola, M.; Righetti, S.C.; Supino, R.; Caserini, C.; Delia, D.; Pierotti, M.A.; Miyashita, T.; Reed, J.C.; Zunino, F. Association between cisplatin resistance and mutation of p53 gene and reduced Bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562. [Google Scholar] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gómez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.L.; White, J.D.; Esdale, W.J.; Burton, M.A.; DeCruz, E.E. Tumour cells surviving in vivo cisplatin chemotherapy display elevated c-myc expression. Br. J. Cancer 1996, 73, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, C.; Biroccio, A.; Benassi, B.; Stringaro, A.; Stoppacciaro, A.; Semple, S.C.; Zupi, G. Encapsulation of c-myc antisense oligodeoxynucleotides in lipid particles improves antitumoral efficacy in vivo in a human melanoma line. Cancer Gene Ther. 2001, 8, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.C.; Mata, J.E.; Reddy, M.T.; Devi, G.R.; Iversen, P.L. Resistance to chemotherapeutic drugs overcome by c-Myc inhibition in a Lewis lung carcinoma murine model. Anticancer Drugs 2003, 14, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Liu, J.D.; Chow, J.M.; Liu, C.R.; Liu, H.E. Small-molecule c-Myc inhibitor, 10058-F4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anticancer Drugs 2007, 18, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Reyes-González, J.M.; Armaiz-Peña, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarría-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejía, P.E. Targeting c-MYC in Platinum-Resistant Ovarian Cancer. Mol. Cancer. Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanità, P.; Delle Monache, S.; Musio, D.; De Felice, F.; Di Cesare, E.; et al. c-Myc Sustains Transformed Phenotype and Promotes Radioresistance of Embryonal Rhabdomyosarcoma Cell Lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, C.; Biroccio, A.; Candiloro, A.; Citro, G.; Fornari, C.; Mottolese, M.; Del Bufalo, D.; Zupi, G. Increase of cisplatin sensitivity by c-myc antisense oligodeoxynucleotides in a human metastatic melanoma inherently resistant to cisplatin. Clin. Cancer Res. 1999, 5, 2588–2595. [Google Scholar] [PubMed]
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. E2F and cell proliferation: A world turned upside down. Cell 1996, 85, 457–459. [Google Scholar] [CrossRef]
- Maser, R.S.; Mirzoeva, O.K.; Wells, J.; Olivares, H.; Williams, B.R.; Zinkel, R.A.; Farnham, P.J.; Petrini, J.H. Mre11 complex and DNA replication: Linkage to E2F and sites of DNA synthesis. Mol. Cell. Biol. 2001, 21, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Frame, F.M.; Rogoff, H.A.; Pickering, M.T.; Cress, W.D.; Kowalik, T.F. E2F1 induces MRN foci formation and a cell cycle checkpoint response in human fibroblasts. Oncogene 2006, 25, 3258–3266. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, F.; Weaks, R.L.; Biswas, A.K.; Guo, R.; Li, Y.; Johnson, D.G. E2F1 promotes recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011, 10, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Thalmeier, K.; Synovzik, H.; Mertz, R.; Winnacker, E.L.; Lipp, M. Nuclear factor E2F mediates basic transcription and trans-activation of E1a of the human MYC promoter. Genes Dev. 1989, 3, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Mudryj, M.; Hiebert, S.W.; Nevins, J.R. A role for the adenovirus inducible E2F transcription factor in a proliferation dependent signal transduction pathway. EMBO J. 1990, 9, 2179–2184. [Google Scholar] [PubMed]
- Chiang, Y.C.; Teng, S.C.; Su, Y.N.; Hsieh, F.J.; Wu, K.J. c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. J. Biol. Chem. 2003, 278, 19286–19291. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.R.; Zhao, H.; Coackley, C.L.; Penn, L.Z.; Bristow, R.G. Tumor cell kill by c-MYC depletion: Role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Fan, R.; Chen, Q.; He, Y.; Song, M.; Shang, Z.; Zhang, S.; Zhu, W.; Cao, J.; Guan, H.; et al. The involvement of c-Myc in the DNA double-strand break repair via regulating radiation-induced phosphorylation of ATM and DNA-PKcs activity. Mol. Cell. Biochem. 2015, 406, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Rich, J.N. Survival of the Fittest: Cancer Stem Cells in Therapeutic Resistance and Angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Joo, K.M.; Jin, J.; Nam, D. Cancer Stem Cells and Their Mechanism of Chemo-Radiation Resistance. Int. J. Stem Cells. 2009, 2, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Wei, X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine 2012, 7, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Wei, X. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World J. Biol. Chem. 2015, 6, 57–64. [Google Scholar]
- Skvortsov, S.; Debbage, P.; Lukas, P.; Skvortsova, I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin. Cancer Biol. 2015, 31, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc Is Required for Maintenance of Glioma Cancer Stem Cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Kristeleit, R.S. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: A current practice and future directions. Br. J. Cancer 2016, 115, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Kaye, S.B.; Yap, T.A. PARP inhibitors: The race is on. Br. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R. Small molecules, inhibitors of DNA-PK, Targeting DNA repair, and beyond. Front Pharmacol. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Boehler, C.; Guirouilh Barbat, J.; Bonnet, M.E.; Illuzi, G.; Ronde, P.; Gauthier, L.R.; Magroun, N.; Rajendran, A.; Lopez, B.S.; et al. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014, 42, 5616–5632. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Macurek, L. Targeting the checkpoint to kill cancer cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Helleday, T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006, 34, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Wiggans, A.J.; Cass, G.K.S.; Bryant, A.; Lawrie, T.A.; Morrison, J. Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Kumari, A.; Iwasaki, T.; Pyndiah, S.; Cassimere, E.K.; Palani, C.D.; Sakamuro, D. Regulation of E2F1-induced apoptosis by poly(ADP-ribosyl)ation. Cell Death Differ. 2015, 22, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Denekamp, J.; Dasu, A.; Waites, A. Vasculature and microenvironmental gradients: The missing links in novel approaches to cancer therapy? Adv. Enzyme Regul. 1998, 38, 281–299. [Google Scholar] [CrossRef]
- Harrison, L.; Blackwell, K. Hypoxia and anemia: Factors in decreased sensitivity to radiation therapy and chemotherapy? Oncologist 2004, 9, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wrann, S.; Kaufmann, M.R.; Wirthner, R.; Stiehl, D.P.; Wenger, R.H. HIF mediated and DNA damage independent histone H2AX phosphorylation in chronic hypoxia. Biol. Chem. 2013, 394, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.D.; Guan, H.; Zhang, S.M.; Tang, Y.; Rang, W.Q.; Zhou, P.K. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Giachino, C. Multiple facets of histone variant H2AX: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed]
- Olcina, M.M.; Grand, R.J.; Hammond, E.M. ATM activation in hypoxia—Causes and consequences. Mol. Cell. Oncol. 2014, 1, e29903. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Albihn, A.; Tronnersjö, S.; Yan, Q.; Guidi, R.; Stenerlöw, B.; Sterzenbach, T.; Josenhans, C.; Fox, J.G.; Schauer, D.B.; et al. Myc is required for activation of the ATM-Dependent checkpoints in response to DNA damage. PLoS ONE 2010, 5, e8924. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
MYC induces genomic instability and chemoresistance.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kumari, A.; Folk, W.P.; Sakamuro, D. The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance. Genes 2017, 8, 158. https://doi.org/10.3390/genes8060158
AMA Style
Kumari A, Folk WP, Sakamuro D. The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance. Genes. 2017; 8(6):158. https://doi.org/10.3390/genes8060158
Chicago/Turabian StyleKumari, Alpana, Watson P. Folk, and Daitoku Sakamuro. 2017. "The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance" Genes 8, no. 6: 158. https://doi.org/10.3390/genes8060158
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.