
Comparative Analysis of Genome Wide DNA Methylation Profiles for the Genic Male Sterile Cabbage Line 01-20S and Its Maintainer Line


Abstract
:1. Introduction
2. Results
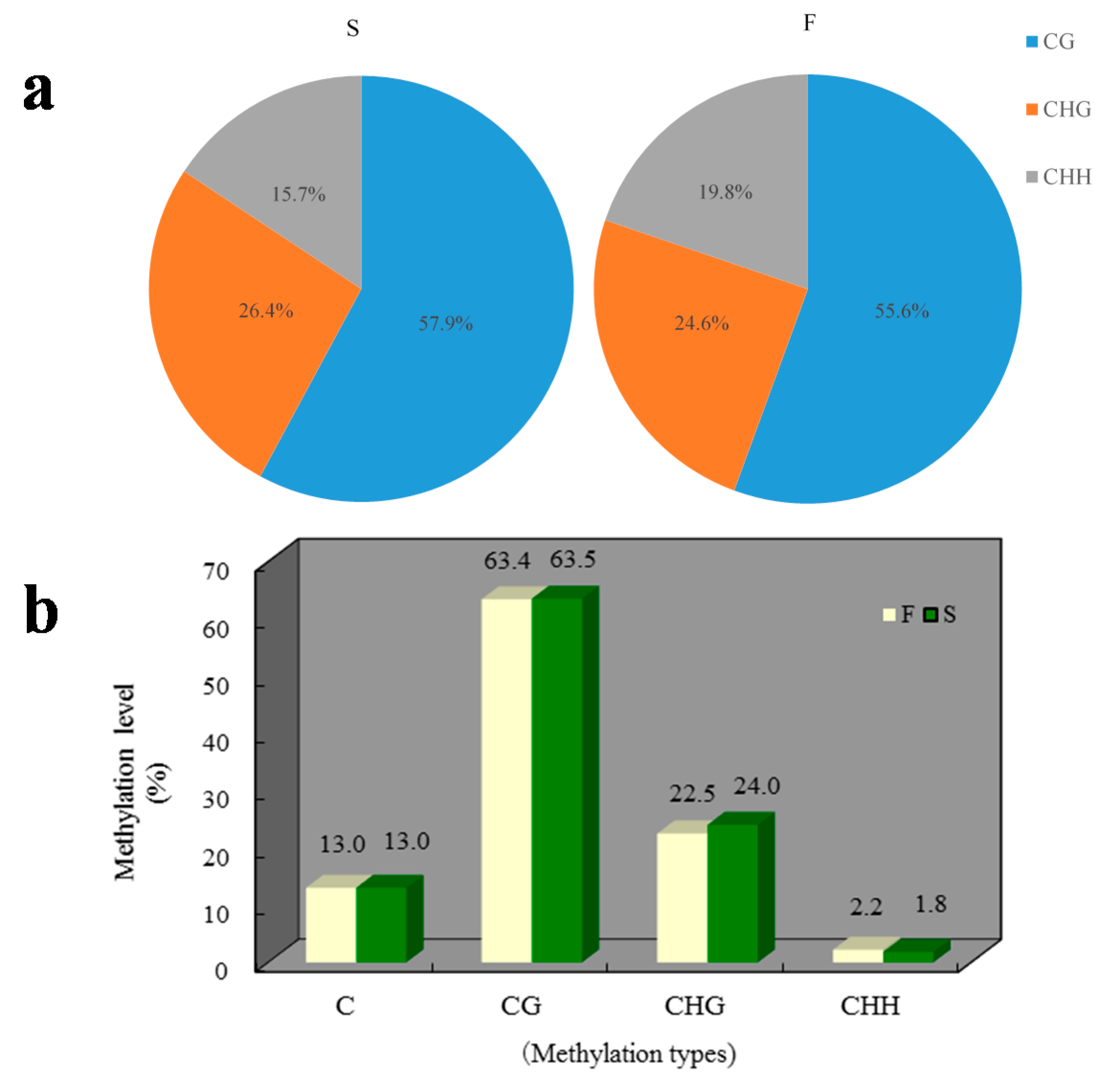
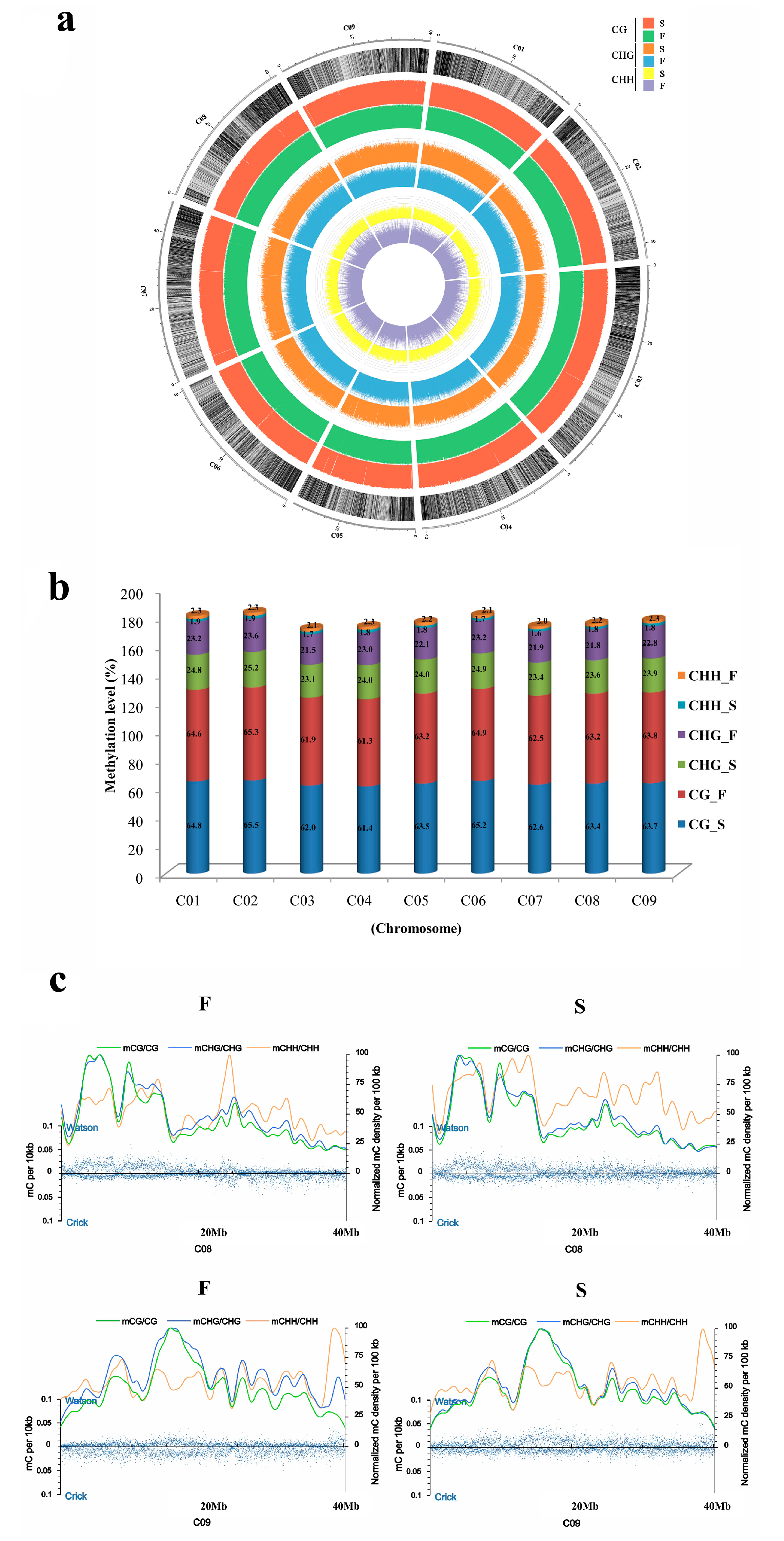
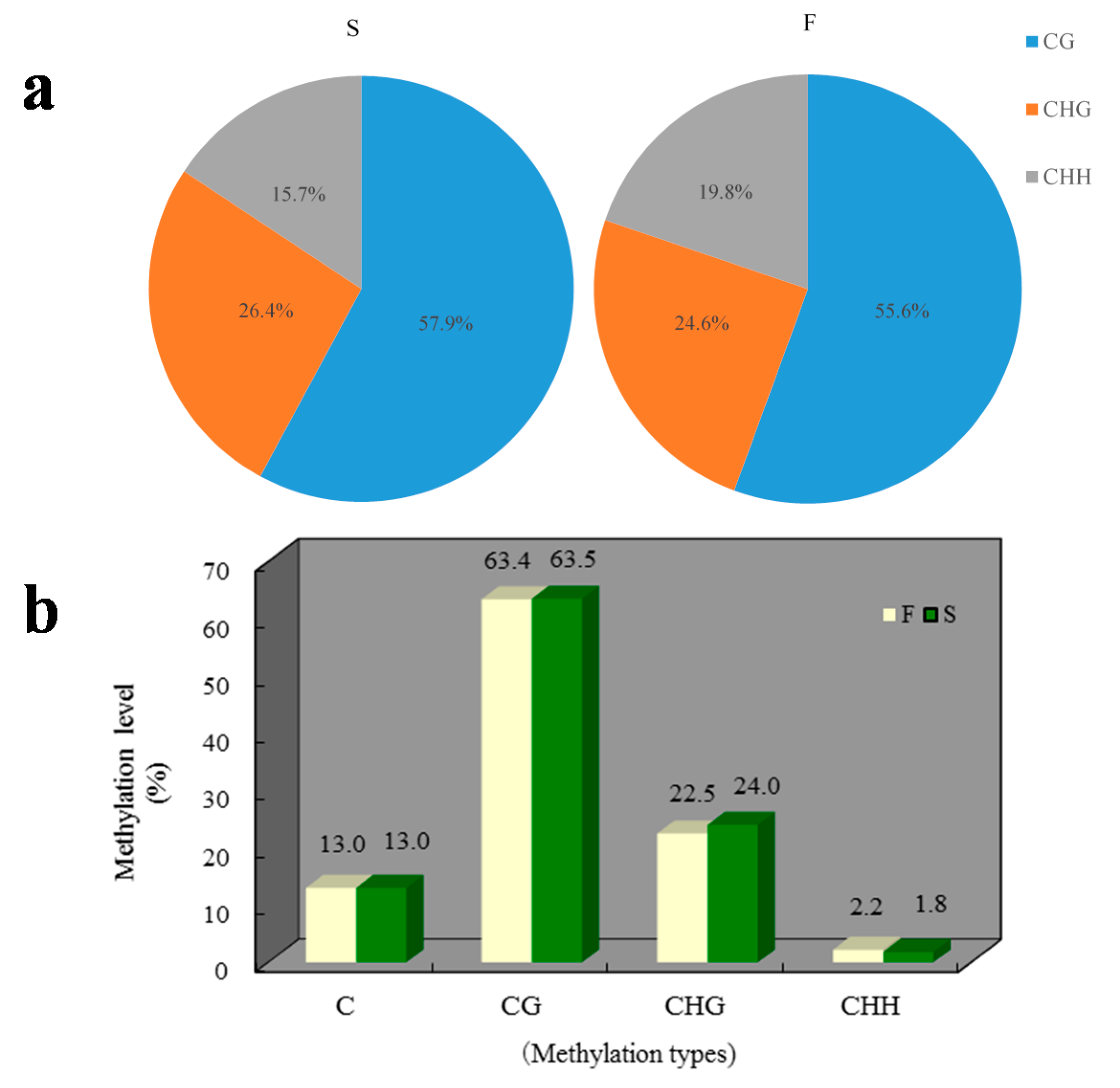
2.1. Methylation Landscape of F and S
2.2. DNA Methylation Patterns of Genes
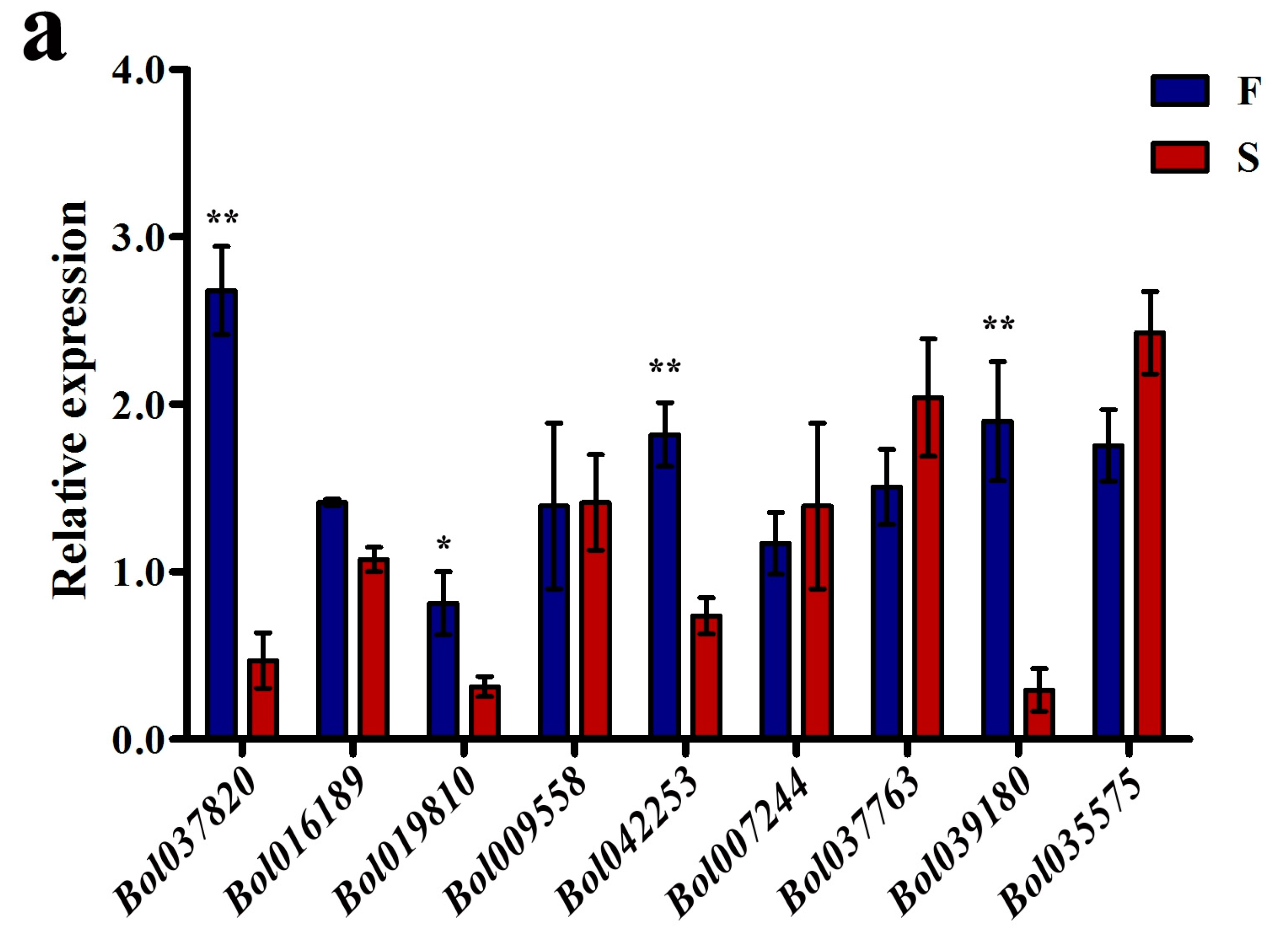
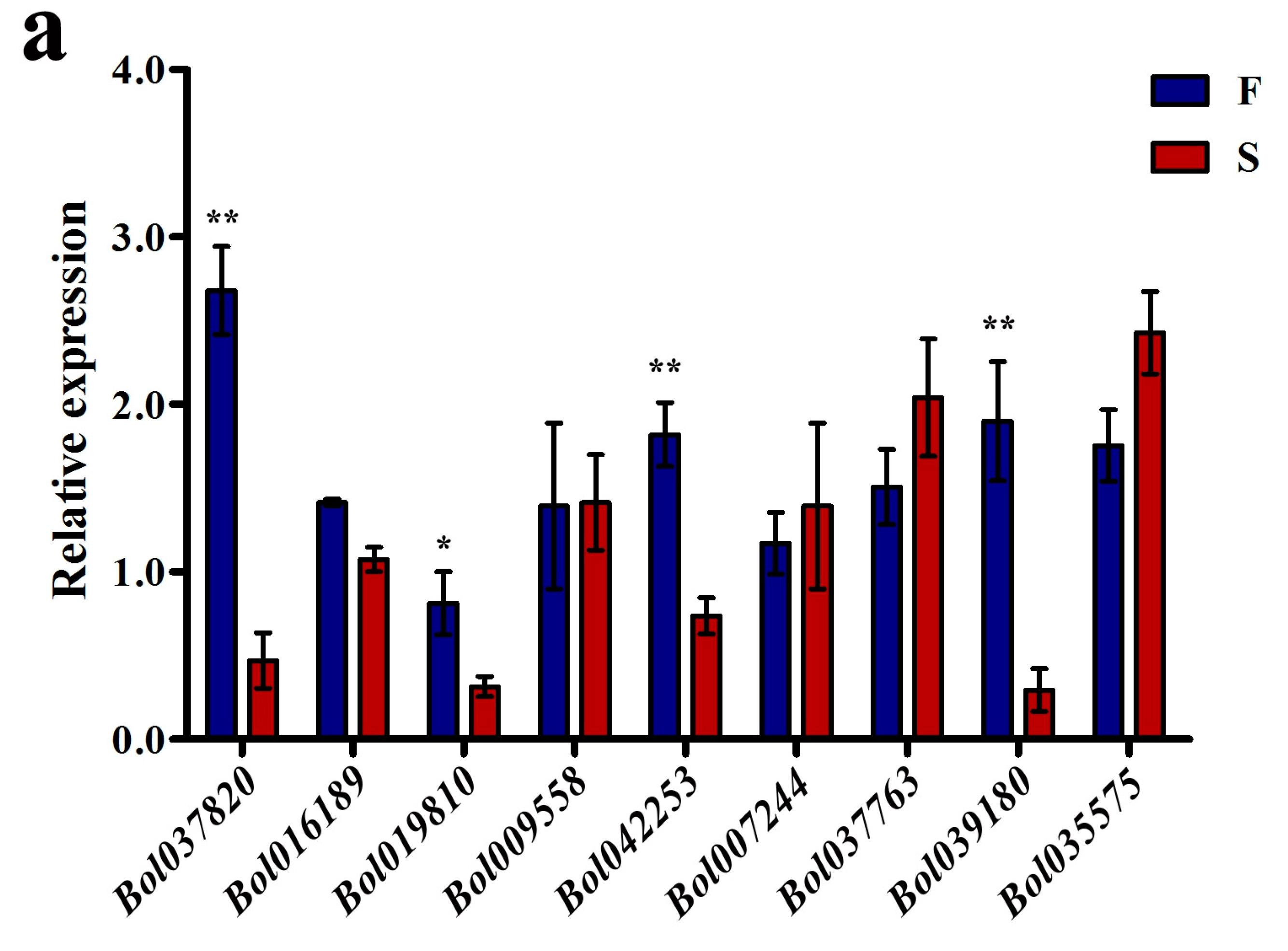
2.3. The S Background Altered the Expression Levels of Some DMR-Associated Genes Involved in Pollen Development
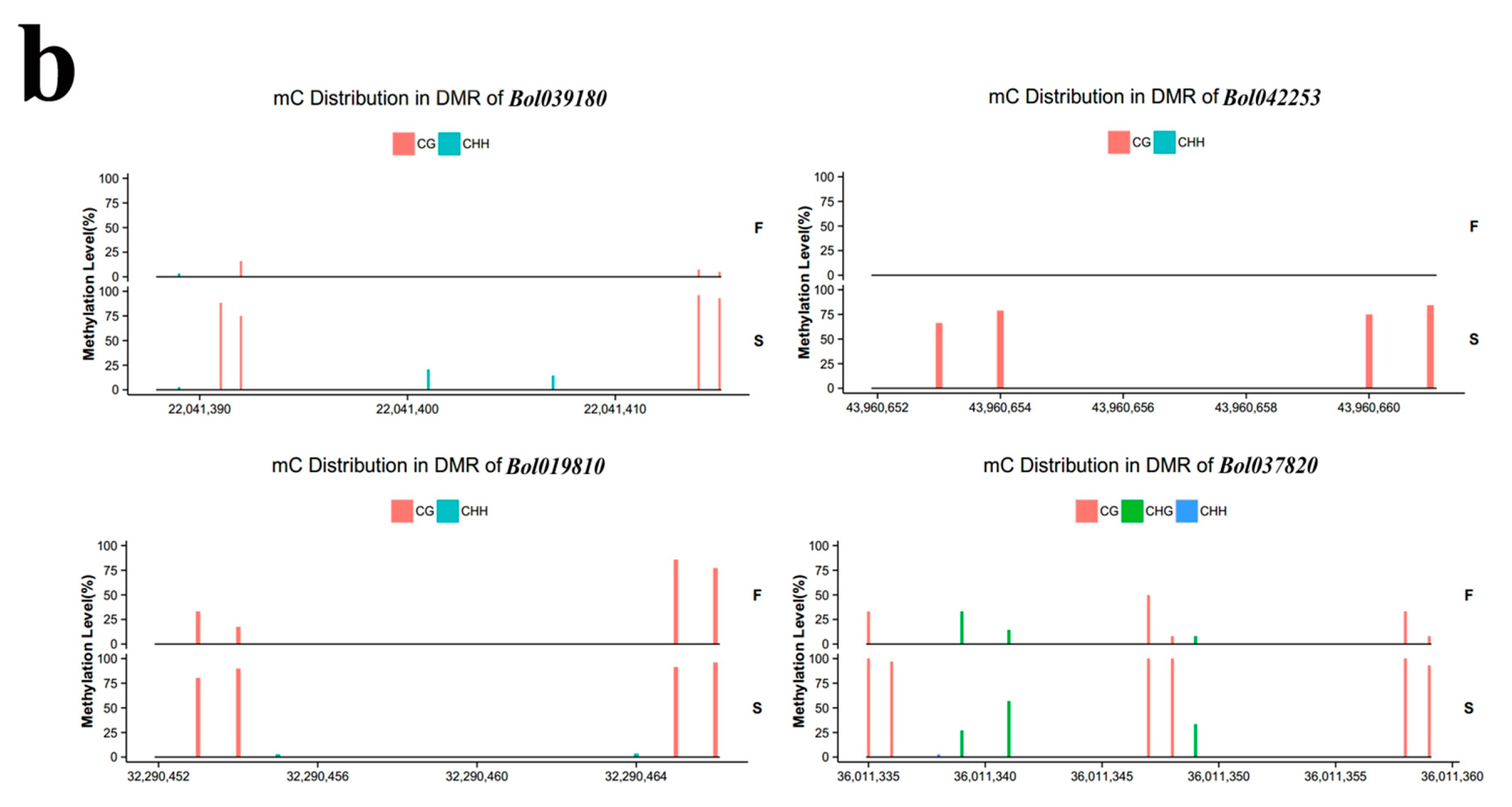
2.4. Validation of BS-Seq by Bisulfite PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Library Construction and BS-Seq
4.3. Bioinformatic Analysis
4.4. Bisulphite Sequencing PCR
4.5. Real-Time RT-PCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BS-Seq | Single-base-resolution bisulfite sequencing |
| DMRs | Differentially methylated genomic regions |
| qRT-PCR | Quantitative reverse-transcription polymerase chain reaction |
| MET1 | Methyltransferase 1 |
| CMT3 | Chromomethylase 3 |
| DDM1 | Decrease in DNA methylation 1 |
| DME | DNA demethylases DEMETER |
| ROS1 | Repressor of silencing 1 |
| TEs | Transposable elements |
| PMC | Pollen mother cell |
| EGMS | Environmentally sensitive genic male sterile |
| LDMAR | Long-day-specific male-fertility-associated RNA |
| MSAP | Methylation-sensitive amplified polymorphism |
| MeDIP-seq | DNA immunoprecipitation sequencing |
| CMS | Cytoplasmic male sterility |
| DGMS | Dominant male sterility |
| BRAD | Brassica Database |
| PMEI | Pectin methylesterase inhibitor family protein |
| NIL | Near-isogenic line |
| GDSL | Consensus amino acid sequence of Gly, Asp, Ser, and Leu around the active site Ser |
| IVFCAAS | Flowers and Vegetables of the Chinese Academy of Agriculture Sciences |
References
- Chan, S.W.L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Grafi, G. Epigenetics in plant development and response to stress. Biochim. Biophys. Acta 2011, 1809, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J. Inherited epigenetic variation–revisiting soft inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Ecker, J.R. Epigenetic and epigenomic variation in Arabidopsis thaliana. Trends Plant Sci. 2012, 17, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Kovac, K.A. Plant DNA methyltransferases. Plant Mol. Biol. 2000, 43, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [PubMed]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Penterman, J.; Zilberman, D.; Huh, J.H.; Ballinger, T.; Henikoff, S.; Fischer, R.L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. USA 2007, 104, 6752–6757. [Google Scholar] [CrossRef] [PubMed]
- Saze, H.; Kakutani, T. Differentiation of epigenetic modifications between transposons and genes. Curr. Opin. Plant Biol. 2011, 14, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.P.; Chaille, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijó, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzic, M.; Becker, J.D.; Feijo, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Schoft, V.K.; Chumak, N.; Choi, Y.; Hannon, M.; Garcia-Aguilar, M.; Machlicova, A.; Slusarz, L.; Mosiolek, M.; Park, J.S.; Park, G.T.; et al. Function of the DEMETER DNA glycosylase in the Arabidopsis thaliana male gametophyte. Proc. Natl. Acad. Sci. USA 2011, 108, 8042–8047. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Oses-Prieto, J.A.; Li, K.H.; Fernandes, J.F.; Burlingame, A.L.; Walbot, V. The male sterile 8 mutation of maize disrupts the temporal progression of the transcriptome and results in the mis-regulation of metabolic functions. Plant J. 2010, 63, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Shen, J.; Mao, H.; Xie, W.; Li, X.; Zhang, Q. RNA-directed DNA methylation is involved in regulating photoperiod-sensitive male sterility in rice. Mol. Plant 2012, 5, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, J.; Zhang, H.; Ding, Y. DNA methylation changes in photoperiod-thermo-sensitive male sterile rice PA64S under two different conditions. Gene 2014, 537, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, X.; Zhang, H.; Ding, Y. Genome-wide analysis of DNA methylation in photoperiod- and thermo-sensitive male sterile rice Peiai 64S. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Yan, W.; He, J.; Li, Y.; Zhang, H.; Peng, H.; Wu, X. DNA methylation affected by male sterile cytoplasm in rice (Oryza sativa L.). Mol. Breed. 2013, 31, 719–727. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, Y.; Lu, Y.; Wang, J.; Zhang, S.; Lan, H.; Rong, T.; Cao, M. DNA methylation analysis of sterile and fertile CMS-C hybrids and their parents in maize. J. Plant Biochem. Biotechnol. 2016, 25, 3–11. [Google Scholar] [CrossRef]
- Omidvar, V.; Fellner, M. DNA methylation and transcriptomic changes in response to different lights and stresses in 7B-1 male-sterile tomato. PLoS ONE 2015, 10, e0121864. [Google Scholar] [CrossRef] [PubMed]
- Ba, Q.; Zhang, G.; Wang, J.; Niu, N.; Ma, S.; Wang, J. Gene expression and DNA methylation alterations in chemically induced male sterility anthers in wheat (Triticum aestivum L.). Acta Physiol. Plant. 2014, 36, 503–512. [Google Scholar] [CrossRef]
- Fang, Z.; Sun, P.; Liu, Y.; Yang, L.; Wang, X.; Hou, A.; Bian, C. A male sterile line with dominant gene (Ms) in cabbage (Brassica oleracea var. capitata) and its utilization for hybrid seed production. Euphytica 1997, 97, 265–268. [Google Scholar]
- Lou, P.; Kang, J.; Zhang, G.; Bonnema, G.; Fang, Z.; Wang, X. Transcript profiling of a dominant male sterile mutant (Ms-cd1) in cabbage during flower bud development. Plant Sci. 2007, 172, 111–119. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, G.; Bonnema, G.; Fang, Z.; Wang, X. Global analysis of gene expression in flower buds of Ms-cd1 Brassica oleracea conferring male sterility by using an Arabidopsis microarray. Plant Mol. Biol. 2008, 66, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ma, Y.; Wu, J.; Cheng, F.; Liu, B.; Wang, X. Map-based cloning of the dominant genic male sterile Ms-cd1 gene in cabbage (Brassica oleracea). Theor. Appl. Genet. 2017, 130, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Parkin, I.A.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.; et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.J.; Liu, X.S.; Tao, H.; Tan, S.K.; Chu, S.S.; Oono, Y.; Zhang, X.D.; Chen, J.; Yang, Z.M. Variation of DNA methylation patterns associated with gene expression in rice (Oryza sativa) exposed to cadmium. Plant Cell Environ. 2016, 39, 2629–2649. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.D.; Boavida, L.C.; Carneiro, J.; Haury, M.; Feijó, J.A. Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiol. 2003, 133, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.M.; Quan, L.; Cannon, A.E.; Wen, J.; Blancaflor, E.B. AGD1, a class 1 ARF-GAP, acts in common signaling pathways with phosphoinositide metabolism and the actin cytoskeleton in controlling Arabidopsis root hair polarity. Plant J. 2012, 69, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Katsumata, H.; Abe, M.; Yabe, N.; Komeda, Y.; Yamamoto, K.T.; Takahashi, T. Characterization of the class IV homeodomain-leucine zipper gene family in Arabidopsis. Plant Physiol. 2006, 141, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Oono, Y.; Narumi, I. Arabidopsis pab1, a mutant with reduced anthocyanins in immature seeds from banyuls, harbors a mutation in the MATE transporter FFT. Plant Mol. Boil. 2016, 90, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.P.; Wilkins, C.; Demidchik, V.; Davies, J.M.; Glover, B.J. An Arabidopsis flavonoid transporter is required for anther dehiscence and pollen development. J. Exp. Bot. 2010, 61, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, T.; Hatakeyama, K.; Hinata, K.; Inatsugi, R.; Nishida, I.; Sato, S.; Kato, T.; Tabata, S.; Toriyama, K. Disruption of the novel plant protein NEF1 affects lipid accumulation in the plastids of the tapetum and exine formation of pollen, resulting in male sterility in Arabidopsis thaliana. Plant J. 2004, 39, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, A.; Mayfield, J.A.; Miley, N.L.; Chau, S.; Fischer, R.L.; Preuss, D. Alterations in CER6, a gene identical to CUT1, differentially affect long-chain lipid content on the surface of pollen and stems. Plant Cell 2000, 12, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Nagel, C.; Taylor, L.P. Biochemical complementation of chalcone synthase mutants defines a role for flavonols in functional pollen. Proc. Natl. Acad. Sci. USA 1992, 89, 7213–7217. [Google Scholar] [CrossRef] [PubMed]
- Buer, C.S.; Imin, N.; Djordjevic, M.A. Flavonoids: New roles for old molecules. J. Integr. Plant Biol. 2010, 52, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Z.; Guo, Z.; Song, G.; Cheng, Q.; Jiang, D.; Zhu, Y.; Yang, D. Comparative transcriptomes profiling of photoperiod sensitive male sterile rice Nongken 58S during the male sterility transition between short-day and long-day. BMC Genom. 2011, 12, 462. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Boil. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Garrett-Bakelman, F.E.; Akalin, A.; Zumbo, P.; Levine, R.; To, B.L.; Lewis, I.D.; Brown, A.L.; D’Andrea, R.J.; Melnick, A.; et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Reither, S.; Mikeska, T.; Paulsen, M.; Walter, J.; Lengauer, T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 2005, 21, 4067–4068. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Different Methylation Region | Hyper or Hypo in S | p-Value | Homologous Gene in A. thaliana | E Value | Predicted Functional Description |
|---|---|---|---|---|---|---|
| Bol009558 | Intron | hyper | 1.74 × 10−3 | AT5G49720 | 1.00 × 10−16 | Endoglucanase 21 |
| Bol037820 | Upstream | hyper | 5.09 × 10−12 | AT2G35210 | 1.00 × 10−117 | ADP-ribosylation factor GTPase-activating protein AGD10 |
| Bol016189 | Upstream | hyper | 1.84 × 10−7 | AT1G70780 | 1.00 × 10−55 | Unknown protein; expressed in sperm cell, male gametophyte, pollen tube |
| Bol042253 | Intron | hyper | 2.76 × 10−8 | AT4G25640 | 0.00 | ABC transporter; multidrug and toxin efflux family transporter DTX35 |
| Bol007244 | Intron | hyper | 1.98 × 10−7 | AT4G08160 | 0.00 | Endo-1,4-beta-xylanase A |
| Bol019810 | Upstream | hyper | 6.36 × 10−5 | AT1G34650 | 0.00 | Homeobox-leucine zipper protein HDG10 |
| Bol035575 | downstream | hypo | 2.62 × 10−5 | AT1G53840 | 0.00 | Pectinesterase, active site |
| Bol037763 | downstream | hyper | 1.16 × 10−5 | AT2G34880 | 0.00 | Transcription factor jumonji |
| Bol039180 | downstream | hyper | 1.93 × 10−11 | AT2G47340 | 0.38 | invertase/pectin methylesterase inhibitor family protein |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, F.; Zhang, X.; Liu, X.; Su, H.; Kong, C.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Liu, Y.; et al. Comparative Analysis of Genome Wide DNA Methylation Profiles for the Genic Male Sterile Cabbage Line 01-20S and Its Maintainer Line. Genes 2017, 8, 159. https://doi.org/10.3390/genes8060159
Han F, Zhang X, Liu X, Su H, Kong C, Fang Z, Yang L, Zhuang M, Zhang Y, Liu Y, et al. Comparative Analysis of Genome Wide DNA Methylation Profiles for the Genic Male Sterile Cabbage Line 01-20S and Its Maintainer Line. Genes. 2017; 8(6):159. https://doi.org/10.3390/genes8060159
Chicago/Turabian StyleHan, Fengqing, Xiaoli Zhang, Xing Liu, Henan Su, Congcong Kong, Zhiyuan Fang, Limei Yang, Mu Zhuang, Yangyong Zhang, Yumei Liu, and et al. 2017. "Comparative Analysis of Genome Wide DNA Methylation Profiles for the Genic Male Sterile Cabbage Line 01-20S and Its Maintainer Line" Genes 8, no. 6: 159. https://doi.org/10.3390/genes8060159