Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets


Department of Molecular Science and Technology, Ajou University, Suwon 443-749, Korea
*
Author to whom correspondence should be addressed.
Genes 2017, 8(8), 196; https://doi.org/10.3390/genes8080196
Submission received: 9 May 2017
/
Revised: 19 July 2017
/
Accepted: 31 July 2017
/
Published: 7 August 2017
(This article belongs to the Special Issue Protein-DNA Interactions)
Abstract
:The nucleosome surface is covered with multiple modifications that are perpetuated by eight different classes of enzymes. These enzymes modify specific target sites both on DNA and histone proteins, and these modifications have been well identified and termed “epigenetics”. These modifications play critical roles, either by affecting non-histone protein recruitment to chromatin or by disturbing chromatin contacts. Their presence dictates the condensed packaging of DNA and can coordinate the orderly recruitment of various enzyme complexes for DNA manipulation. This genetic modification machinery involves various writers, readers, and erasers that have unique structures, functions, and modes of action. Regarding human disease, studies have mainly focused on the genetic mechanisms; however, alteration in the balance of epigenetic networks can result in major pathologies including mental retardation, chromosome instability syndromes, and various types of cancers. Owing to its critical influence, great potential lies in developing epigenetic therapies. In this regard, this review has highlighted mechanistic and structural interactions of the main epigenetic families with their targets, which will help to identify more efficient and safe drugs against several diseases.
1. Introduction
Epigenetics provides a partial description for how cloned animals or monozygotic twins show differences in disease susceptibility despite identical DNA sequences [1,2]. Conrad Waddington used the term “epigenetics” for the first time in 1939 to explain “formation of various phenotypes due to interactions between associated genes and their products” [3]. Later, Arthur Riggs defined the term epigenetics as “study of meiotically and/or mitotically heritable changes in gene function unable to be explained by alterations in DNA sequence” [4]. Presently, this term has been widened to encompass both heritable and transient changes in nature [5]. Here, we have used the modern definition of epigenetics, which is described as involving both transient and heritable alterations in gene expression without any change in the primary sequence of DNA.
DNA, being a highly charged polymer, requires intense compaction for its nuclear compartmentalization within eukaryotic cells; this is achieved by its association with a set of basic histone proteins to eventually form a highly organized and compact structure called chromatin. In chromatin, the fundamental repeat unit is the nucleosome, consisting of one octamer comprised of four core histone proteins (H2A, H2B, H3, and H4) and 147 bp of DNA twisted around the outer surface of the octamer in two turns. Less is known regarding the molecular basis of the higher order more folded structure of the nucleosome [6]. The degree of this dynamic folding directly affects some important DNA-related functions such as replication, recombination, and transcription. The formation and maintenance of these differentially folded domains is an important question in the understanding of the regulation of biological processes. Approximately three decades ago, most relevant studies were published explaining post-transcriptional histone modifications and their impact on chromatin folding and activity of relevant genes. In particular, chromatin has been divided into two main types depending upon their folding pattern. (1) The loosely folded part of the chromatin is mostly enriched with acetylation marks and called euchromatin, which is the transcriptionally most active region of the DNA; (2) the tightly folded part of the chromatin is mostly enriched with methylation marks and called heterochromatin, which is a transcriptionally less active region of the DNA. The existing chromatin state can be explained by three biochemical mechanisms: ATP dependent SWI2/SNF2-mediated chromatin remodeling, post-translational modifications of histones, and substitution of histone variant. In comparison to heterochromatin, less is known about the generation, maintenance, and inheritance of euchromatin. Euchromatin is widely considered to be the default state of chromatin; recently silence-antagonizing chromatin modifications have been found that endorse the euchromatic state. These modifications involve the substitution of the H2A histone with H2A.Z [7], methylation of K4H3 and K79H3 [8,9], and acetylation of K16H4 [10].
The acetylated euchromatin was first demonstrated by a chromatin immunoprecipitation assay using hyperacetylated histone-recognizing antibodies, revealing that the global localization of acetylated histones is at DNase I-sensitive regions in correlation with transcriptional activation [11,12]. The N-terminal tails of histones with charged lysine residues provide a promising signaling platform due to their environmental exposure outside of the chromatin polymer, which facilitates various interactions with other proteins and complexes for chromatin remodeling [13,14,15]. These interactions change the charge of the histone tails, which eventually weakens the contact between histone and DNA [16]. Acetylation modification can also change the interactions among histones of adjacent nucleosomes [17], as well as between regulatory proteins and histones [14,15]. These modifications alter the structure and folding of the nucleosome, leading to more permissive and open chromatin for transcription (called euchromatin, as mentioned above); however, there is still confusion regarding whether acetylation is an effect or the cause of increased transcription. Mechanistic studies have identified various enzymes responsible for the removal and addition of these epigenetic marks on DNA and histones.
On histones, over 60 different modified residues have been detected by mass spectrometry or using specific antibodies. Some of these modifications are shown in Figure 1 and Table 1. However, there is a huge underestimation of the total number of histone modifications. This becomes more complex when considering the facts that arginine may exist in either a mono- or di-methyl (symmetric or asymmetric) form and that lysine may exist in a mono-, di-, or tri-methyl form within histones of nucleosomes. This variation in modifications is responsible for different functions; however, none of these modifications present at the same time at the same histone site. Their appearance or removal over time depends on the cellular signaling conditions.
These various modifications can be positively regulated by their own histone marks or marks present on the same transcriptional state and vice versa. This kind of interplay can be between the same or different types of modifications. The best-characterized interplay with reference to alteration in gene expression has been reported for two antagonistic groups of epigenetic proteins, Polycomb (Pc) and Trithorax (Trx) which were first reported for their opposing effects on the Hox gene in Drosophila melanogaster. Various studies confirm that polycomb repressive complex 2 (PRC2) activity can be inhibited by TrxG methyl modifications at H3K4 and H3K36 on the same histone [18,19,20]. Similarly, activity of some Trx proteins can also be inhibited by Pc group of proteins e.g., PRC1-mediated ubiquitin modification at H2AK119 can inhibit the H3K36 methyltransferases [21]. Different modification marks on the same histone can change the expression state of chromatin, e.g., acetylation of H3K27 is the hallmark of active chromatin while its trimethylation causes silencing of the associated gene. The H3K27 acetylation mark is removed by the NURD (Nucleosome Remodeling Deacetylase) complex, which recruits PRC2 for the tri-methylation of H3K27 at the promoter to repress gene expression level [22]. This phenomenon has been observed in the differentiation of ESCs for the silencing of the previously active genes by the association of NURD with CTBP2 [23]. The NURD complex consists of seven subunits: RbAp48 and RbAp46 (histone binding proteins), HDAC1 and HDAC2 (core histone deacetylase proteins), MTA1, MTA2 or MTA3 (metastasis associated proteins), MBD2 or MBD3 (methyl-CpG-binding domain protein) and CHD3 or CHD4 (chromodomain-helicase-DNA-binding protein).
Besides the interaction of Pc and Trx group of proteins, there is another phenomenon that controls the expression of genes, called poly(ADP-ribosyl)ation (PARylation). The PARP family includes 17 enzymes but not all of them are active in transferring ADP-ribose. For example, a PARP1 product, poly(ADP ribose), forms a cloud of negative charges on the surface of the modified protein to affect the functionality of associated proteins by electrostatic interactions [24]. The PARylation of TFIIF (transcription factor II F) and TBP (TATA-binding protein) nullify the PIC (pre-initiation complex) formation [25,26]. Similarly, PARylation of the binding sequence of some other transcription factors (like CREB, NFκB, p53, Sp1, YY1) makes them unable to bind at those regions that eventually stop the transcription of relevant genes [27,28,29,30]. On the other hand, PARP1 has also been reported for transcription activation of particular genes by interacting with some other factors like E2F1 and NFκB [31,32]. This diverse role of PARylation in proteins may be due to the involvement of some other modifiers; it has been reported that the binding of NFκB subunit p50 and PARP1 is due to the acetylation of PARP1 by p300 (histone acetyl transferase) [33].
In this review, we have focused on the acetylation and methylation modification of chromatin. More specifically, structural interactions of proteins associated with these modifications and their targets (DNA and histones) are discussed to better understand the mechanism that will help with the design of therapeutic drugs for various diseases. Moreover, a summary of acetylation and methylation-related players (discussed in this review) and their interaction has been presented in Figure 2.
2. Acetylation-Related Protein Families
2.1. Histone Acetyltransferases (HATs)
Histone acetylation has long been linked with transcriptional activation following its first discovery over 30 years ago [16]. However, no transcription-related activity of HAT was identified until 1996 [51]. Previously, biochemical and genetic studies linked the yeast version of HAT, GCN5, with transcriptional regulation as a coactivator to bridge basal transcription factor and activator protein interactions [52]. This suggested that acetylation of active DNA by DNA-bound activators is due to HATs, and deacetylation of inactive DNA by DNA-bound repressors is due to histone deacetylases [53].
For packaging DNA into chromatin, HATs mainly target histone amino-terminal tails where they acetylate lysine residues at ε-amino groups [54]. DNA of 147 bp spools in two turns around a histone octamer (two molecules of each core histone protein, i.e., H4, H3, H2B, H2A) to form a nucleosome. Each histone molecule consists of the following: (1) an amino-terminal domain (highly charged) responsible for histone modification; and (2) a carboxy-terminal domain responsible for nucleosome assembly. Numerous research groups have tried to elucidate acetyl group-transferring mechanisms from acetyl-CoA (Ac-CoA) to histone acceptors by using partially purified fractions or cell extracts in conventional solution enzymatic assays [55]. Based on cellular origin and function, there are two major classes of HATs: (1) cytoplasmic HAT-B catalyzes the acetylation of freshly synthesized histones to move them from the cytoplasm to the nucleus for their deposition on freshly replicated DNA [56]; and (2) nuclear HAT-A catalyzes acetylation events responsible for the transcription [57].
Depending on the sequence analysis, HATs fall into distinct families with poor to no inter-family but higher intra-family sequence homology [53]. Each HAT family has different substrate-interacting preferences with diverse functional perspectives (see Table 2 and Figure 3a). For instance, the GCN5/PCAF family acetylates H3 at lysine 14 by interacting with transcriptional activators [58]. These proteins also have a bromodomain module at the carboxy-terminal, which acts as a targeting motif for acetyllysine [59]. In contrast, HATs of MYST family (the largest HAT family named after MOZ, Ybf2/Sas3, Sas2 and Tip60) have diverse functions such as dosage compensation in Drosophila melanogaster [60], leukogenesis in Homo sapiens [61], and cell cycle regulation [62] and gene silencing in yeast [63]. Most MYST proteins, except SAS3, have H4 substrate preference and possess a chromodomain responsible for binding RNA [64].
P300/CBP-associated factor (PCAF), HAT1, and general control nonderepressible 5 (GCN5) belong to a functionally diverse N-acetyltransferase superfamily with limited homology within a sequence of four A–D-labeled motifs (15–33 residues), called GNATs (N-acetyltransferases related to GCN5) [65]. HAT domain comparison from HAT1 [66], MYST family member yESA1 [67], and the GCN5/PCAF family [68,69] shows structural homology to GNAT proteins for A and D motifs, which make a three-stranded antiparallel β-sheet with an underlying helix. Including another conserved region (loop-β-strand) in GNAT proteins at the immediate C-terminal of A–D motif helix, these collectively comprise a central core domain that is structurally conserved. Among the three HAT families, structural divergence has been observed at the carboxyl- and amino-termini of their core domains. As compared to H3-specific GCN5/PCAF, the structures of H4-specific yHAT1 and yESA1 are more similar to one another. Despite the structural differences between the C- and N-terminals of these families, the loop-α-helix at the C-terminal and the α-helix loop at the N-terminal to the core region superimpose well onto each other.
Structural comparison among HATs shows a conserved core domain that interacts with coenzyme-A (CoA) facilitated by overall Van der Waals and protein backbone interactions by the motif-A residues of GNAT proteins. Structural and functional HAT correlation signifies a catalysis role of the core domain. No Km change has been identified for either histone or CoA, but a ~360-fold Kcat decrease was observed in a yGCN5 mutant for E173Q, which signifies the importance of Glu173 in catalysis [69]. Moreover, superimposition of the GCN5/PCAF core domain with that of yHAT1 and yESA1 shows that, despite arising from non-analogous structural elements, Glu255 and Glu338 in yHAT1 and yESA1, respectively, superimpose in 3-D space [67]. Mutagenesis studies of E338Q in yESA1 show consistency for its importance in catalysis. Taken together, the core domain of all three discussed HAT proteins shows conservation in its structure as well as function to bind with CoA and perform catalysis.
The ternary complex structure of tGCN5/CoA/histone H3 has enabled visualization of the mode of GCN5/PCAF binding to the histone [68]. The protein structure shows that a random H3 coil structure (formed by 11 H3 residues centered around H3 Lys14) is bound to a distinct protein cleft in tGCN5 and is flanked by N- and C-terminal segments of protein at opposite ends (Figure 3b). Of these interactions, 75% involve Lys14 and its five immediate C-terminal residues at the H3 backbone. Besides Lys14, Gly13 and Pro16 side chains play a discriminative role in recognizing histone. Ternary complex comparison with apo-tGcn5 and binary tGCN5/CoA structures reveals the structural importance of Ac-CoA in the HAT domain configuration for H3 binding. C- and N-terminal segments are bound by Ac-CoA, which enables histone H3 interactions and widens its effective association. This data shows specificity for histone random coil sequences with a small recognition sequence (G-K-X-P), and indicates an important structural role of Ac-CoA in facilitating this association.
There is an impediment in direct visualization of histone and HAT interactions due to the absence of bound peptides in the yESA1/CoA complex (Figure 3c) [67] and the yHAT1/Ac-CoA complex [66]. However, surface-exposed and conserved residues mapping in HAT families signifies a histone-binding region similar to that of GCN5/PCAF [67]. Correlatively, sequence-conserved regions in core domains of the respective HAT families superimpose well with each other. This superimposition gives structural divergence of C- and N-terminal segments of corresponding proteins. Conserved domains in these HAT families depict histone substrate binding, while divergent sequences modulate specific histone target binding.
2.2. Bromodomain-Containing Proteins
HATs are responsible for nucleosomal modifications such as acetylation of histone lysine residues, which are later recognized by protein–protein interacting modules or domains called bromodomains (BRDs). BRDs are evolutionarily conserved domains of 110 amino acids, first discovered in the brahma gene of D. melanogaster [87]. However, histone-acetylated lysine (Kac) motifs can also be recognized by YEATS domains (Yaf9, ENL, AF9, Taf14, and Sas5) [134], which normally bind to crotonylation-modified lysine residues [135].
Sixty-one BRD modules are encoded by the human proteome and present in 42 different proteins that regulate gene expression in a wide range of activities by recognizing Kac. First, BRDs facilitate the assembly of large protein complexes by acting as scaffolds. Secondly, they can act as transcription coregulators and transcription factors. Lastly, they can perform diverse catalytic functions including roles as ATP-dependent chromatin remodeling complexes, helicases, HATS, and methyltransferases (MTases) (Table 3). BRD proteins show variable and broad expression profiles in various tissues [92].
Multidomain proteins contain BRD modules linked to diverse catalytic and interacting domains via flexible sequences [136]. This specific arrangement allows interactions with various sequence motifs due to conformational flexibility. Some BRDs contain diverse domains such as PHD fingers (plant homeodomain), BAH domains (bromo-adjacent homology), and PWWP domains (Pro-Trp-Trp-Pro), which enable them to interact with various proteins to participate in various biological processes as mentioned in Table 3. More than a decade ago, the distinctive architecture of the BRD module was structurally characterized [59,96,97] as having four α-helices (αB, αC, αZ, and αA) that are linked together by two divergent loops (BC and ZA) (Figure 4a). This module contains fold-stabilizing conserved residues including a PxY motif at the C-terminal of the ZA loop and a Tyr residue in the AB loop forming a salt bridge typically to the Asp residue on αB. Kac docking is facilitated by the conserved Asn residue at the N-terminal of the BC loop, while interactions with the acetylated peptide backbone are initiated by a large charged interface provided by the surface surrounding the Kac-binding pocket. Instead of the usually conserved Asn residue, some BRDs contain a Tyr (as in SP100, SP110, and SP140) or Thr residue (as in BRWD3); however, evidence to link these BRDs with Kac-binding and their capability to recognize specific modifications has not been found. Collectively, a neutralized Kac side chain is accommodated within a small hydrophobic pocket of the BRD module formed by its four α-helices, while the charged surface of the BRD surrounding the Kac-binding site facilitates the entire binding of the acetylated peptide. There are eight families of BRD modules in humans determined by sequence similarity and structural topology [136]. There is dramatic charge variation on the surface of BRD modules: some are highly positive, unlike others, and do not target positively charged Lys and Arg residues carrying acetylated histone peptides. Because of surface charge variation and wide expression variation, it can be speculated that BRD modules may also interact with many other acetylated non-histone proteins.
Four α-helices of BRD modules form a hydrophobic cavity, which accommodates the neutralized acetylated lysine side chain of the histone peptide sequence [59,96,97]. The conserved mode of binding has been evaluated by analyzing the crystal structure of BRD modules in complex with histone peptide (acetylated), in which one Kac residue inserts into the Kac-binding cavity in BRD and starts the interaction with water molecules (present in cavity) and conserved Asn residues. The orientation of the bound peptide can show dramatic variation depending upon the adjacency of particular domains to the BRD module. For example, proteins that harbor only a single BRD bind histone peptides in such a way that the peptide N-terminus resides at the back-side of the pocket; the peptide inserts itself between the BC and ZA loops and aligns above the Kac-binding cavity with one exit vector over the ZA loop. Notably, this arrangement is changed by the presence of other modular domains. For example, the chromatin regulator TRIM24 (also recognized as TIF1α) engages N-terminal H3K4 by recognition initiated by a PHD finger, while the adjacent BRD module, linked by a flexible loop to the PHD finger, binds to H3K23ac on the same histone protein (Figure 4b) [100]. Similar PHD-BRD domain organization in nucleosome remodeling factor complex subunit, called BPTF, facilitates its binding to H3K4me2 and H3K4me3 by the PHD domain [137], which then allows specific BRD binding to H4K16ac present in trans within the same nucleosome unlike other H4 acetylations [98]. This multivalency in interactions reveals the peptide orientation: for example, the peptide C-terminus is aligned between BC and ZA loops of BRD, complexed between the TRIM33 PHD-BRD cassette and the H3 peptide containing K18ac and K9me3 [138]. The presence of tandem BRDs bound together by a short rigid linker, as found in TAF1, facilitates H4K12ac and H4K5ac binding, eventually increasing the specificity for the histone H4 tails that have multiple acetylation [96]. Joining of two BRD modules by a long flexible linker, as in the BET family, provides conformational plasticity to the structure, which enables it to simultaneously recognize distant acetylated lysine residues present within the same or different proteins [101,102]. In the absence of these multi-domain arrangements, the mode of binding to single acetylated lysine residues inside histone tails appears to be comparatively well conserved among various BRD modules, with little difference in peptide topology implanted in the grove between the ZA and BC loop regions.
Some BRDs have the ability to recognize double Kac histone marks and to bind H4 peptides, having K8ac and K5ac [99]. This example is related to the BRDT protein, in which H4K5ac directly binds to a conserved Asn residue, while H4K8ac inserts into the binding cavity of Kac, interacts with H4K5ac, and has water-facilitated interactions with the BRDT protein. Recognition of double acetylated lysine marks on histones by a single BRD is a common shared feature of BET family members as the BRD domain at the N-terminal of BRD4 complexed with H4 peptide also shows such an interaction with two Kac residues [136].
2.3. Histone Deacetylases (HDACs)
Many tumor suppressors and oncogenes (e.g., Rb and Mad) are associated with aberrant HDAC activity, resulting in serious consequences [139]. For example, fusion of the retinoic acid receptor α gene and the promyelocytic leukemia (PML) gene in acute promyelocytic leukemia produces an oncoprotein responsible for the recruitment of HDACs to suppress transcription of particular genes, which inhibits cancer cell differentiation and enables their unlimited proliferation [140]. Similar outcomes have been observed for AML1-ETO fusion, PLZF-retinoic acid receptor α fusion, and solid malignancies involving the Myc/Mad/Max signaling pathway [141,142,143,144].
There are two HDAC protein families: the classical HDAC family and the recently discovered SIR2 family (NAD+-dependent). The classical family has two phylogenetic classes, named class I and class II [145]. Class I members (HDAC 1–3 and 8) have resemblance to RPD3 (yeast transcriptional regulator), while class II members (HDAC 4–7, 9, and 10) have resemblance to another yeast deacetylase, HDA1 [145]. HDAC11 is most relevant to class I, but has not yet been placed in any class because of overall sequence variation [146]. Class II HDACs are considered to be involved in developmental processes and cellular differentiation due to their specifically restricted expression behavior, unlike those of class I [147,148]. More details on class members are shown in Table 4.
The action mechanism of HDAC enzymes involves acetyl group removal from nucleosome-forming histones. Hypoacetylation causes tighter association between DNA and nucleosomes due to a decrease in the space between them, which eventually represses transcription due to lower accessibility of transcription factors to that region [156]. The HDAC catalytic domain consists of almost 390 amino acids and has a conserved amino acid set. Its active site has a wider bottom containing a tubular pocket [157]. Two neighboring histidine residues, two aspartate residues (present at around a 30-residue distance from histidines and separated from each other by approx. six residues), and one tyrosine residue (123 residues downstream of aspartate residues) collectively form a charge-relay system that removes an acetyl group from the target site (Figure 4c) [147,157]. Zn2+ is the essential component of this relay system and is bound to the Zn-binding site situated at the bottom of the pocket. Some other cofactors are also required for proper activity of HDAC, thus most of the recombinant enzymes are inactive without such cofactors. Zn2+ displacement is a promising target for most of the HDAC inhibitors (HDACi), while TSA (Trichostatin A) is a good option as a reversible HDACi and has a low nanomolar IC50 value due to its 5-carbon atom phenyl group linker and hydroxamic acid group, which collectively allow it to fit into the HDAC active site [158].
Instead of working alone, HDACs form a repressor complex involving other proteins with facilitating functions such as chromatin remodeling, corepression, and recruitment. DNA itself sends important signals for the initiation of repression. Methylated CpG binding domain-containing proteins, methylated CpG-binding proteins, and MTases recruit HDAC complexes at CpG islands (DNA stretch of methylated cytosine residues at 5′ end of guanosine nucleotides). Epigenetic gene silencing, such as X-chromosome inactivation and imprinting, is mainly based on methyl groups such as those in CpG islands, and it seems that HDACs are the only enzymes responsible for this silencing. However, this is not the case as target gene expression is not always restored by inhibiting HDAC activity [159]. In addition to histones, some other proteins including MyoD, α-tubulin, ESF (pre-rRNA processing protein ESF), and p53 are also deacetylated by HDACs, which indicates their functional complexity in many cellular processes [160,161].
3. Methylation-Related Protein Families
3.1. Methyltransferases
In most vertebrates, including mammals, C5 of the cytosine within CpG dinucleotides is the main site for DNA methylation. This methylation involves the following major steps: methyltransferase (MTase) binds to target DNA; everts target nucleotides from the double helix (base flipping); attacks C6 of cytosine by a conserved cysteine nucleophilic residue; transfers a methyl group to activated cytosine C5 from S-adenosyl methionine (AdoMet); and is finally released from the complex. Histone modifications and methylation modulate the chromatin structure, which eventually controls chromatin-dependent processes such as gene expression [162]. Mammals have two distinct families of DNA nucleotide methyltransferases (DNMTs), which collectively have four members for which the structural and functional information is provided in Figure 5.
The DNA replication fork is hemimethylated (presumably for repairing damaged sites) by DNMT1 when it is targeted by SRA protein/ubiquitin ligase ICBP90 (human)-Np95 (mouse) [163]. It seems that the transition of DNMT1 to the active state involves major conformational changes, which include interactions between catalytic domains and the amino-terminal [164] and/or Ser515 phosphorylation [165].
The germ cell-specific knockouts for DNMT3L and DNMT3 are indistinguishable in terms of DNA methylation pattern in germ cells and retrotransposon dispersion, which indicates the requirement of both for the imprinting of germ cell loci [166,167]. Moreover, DNMT3L enhances de novo methylation by both DNMT3a and DNMT3b due to coimmunoprecipitation and colocalization with these DNMTs [168]. The minimal required region for successful interaction between DNMT3L and DNMT3a or DNMT3b lies in their respective C-terminal domains [169], which show a characteristic fold similar to Class I AdoMet-dependent MTases [170]. However, the methylation reaction product S-adenosyl-L-homocysteine (AdoHcy) was not found in DNMT3L-C, unlike DNMT3a-C, which is consistent with the finding that DNMT3a-C is catalytic in the form of a complex, while DNMT3L alone is not capable of being active and binding to AdoMet [171]. The overall length of the DNMT3L-C/DNMT3a-C complex is approximately 16 nm long (more than the diameter (11 nm) of a core nucleosome). Two monomers of each member comprise this complex and form a tetramer with one 3a–3a interface and two 3L–3a interfaces (3L–3a–3a–3L). Substitution of key residues at these interfaces demolishes the enzymatic activity, which highlights the presence of both interfaces for proper catalysis [172]. The conformation of an active site loop in DNMT3a is stabilized by the interaction of its C-terminal residues (G718–L719–Y720) with DNMT3L. These interactions may explain the role of DNMT3L in DNMT3a activity stimulation [169,173]. Moreover, the intrinsic activity shown by DNMT3a–3L heterodimers is higher than that of DNMT3a–3a homooligomers owing to the positive impact of DNMT3L on the catalytically competent closed conformation of the active-site loop of DNMT3a by reducing the available conformational space for that loop [172].
The smallest DNA-binding domain amongst all known DNA MTases is present in DNMT3a and DNMT3b (it is almost absent in DNMT3L), and consists of ~50 residues as compared to 85 in M.HhaI (bacterial GCGC MTases) [174]. However, the DNA binding surface doubles in size by bringing together two active sites via dimerization of the 3a–3a interface. A contiguous DNA is formed via the connection of two DNA segments in such a way that the two active sites are positioned in the major groove of DNA around 40 Å apart. This model reveals that two CpGs can be methylated simultaneously in one binding event of dimeric DNMT3a if they are present at a separation distance of one helical turn. DNMT3a activity on long DNA substrates shows periodicity, revealing a correlation of methylated CpG sites (8–10 base pairs apart from each other), which could be explained by a structural docked model of oligomeric DNMT3a to DNA [172]. Twelve maternally imprinted genes in the mouse showed similar periodicity for CpG site frequency in differentially methylated regions [172].
So far, the best-studied histone mark associated with DNA methylation is H3K4me0 (unmethylated histone-3 Lys4). Genome-wide analysis showed an inverse correlation between DNA and H3K4 methylation, i.e., DNA can be protected from de novo methylation by H3K4 methylation [175]. Indeed, DNMT3L can only interact with unmethylated H3K4 via its PHD-like domain [176]. DNMT3L recruits DNMT3a2 (Dnmt3a isoform specific to germ cells) to nucleosomes, having unmethylated H3K4 for de novo methylation [172,176]. This hypothesis is supported by an experiment in which mouse KDM1B (H3K4 demethylase) knockout resulted in increased H3K4 methylation and abolished DNA methylation at oocyte imprinted genes [177]. These findings reveal that H3K4 demethylation is acute for DNA de novo methylation of a few imprinted genes of germ cells.
Interestingly, recent structural and biochemical studies have shown that the DNMT3a PHD (also named ADD) domain can directly interact with H3K4me0 without any accessory proteins in vitro (Figure 4d) [178]. Moreover, the PWWP domain of DNMT3a was observed to interact specifically with H3K36me3 (H3 with trimethylated Lys36) in vitro [179]. DNMT3a2 activity at chromatin-bound DNA is increased by both of these interactions [180]. Possibly, DNMT3a identifies particular modifications at histones following methylation of associated DNA, which is consistent with recent genome-wide studies. For example, active gene bodies contain the H3K36me3 modification [181], which has positive correlation with DNA methylation [181]. In somatic cells, strong DNMT3a/3b interaction with nucleosomes has been observed, but it does not involve DNMT3a/3b binding to histone H3, and the presence of any DNMT3a/3b-interacting protein (e.g., HP1α and EZH2) has not been observed [182].
Contrary to the above, lysine, arginine, and histidine histone residues have been reported to be methylated by histone methyltransferases (HMTs); the latter is the least known, unlike the other two. Lysine methylation activity is shown by enzymes as having a conserved 140-amino acid long SET domain (suppressor of variegation, enhancer of zeste, trithorax), except DOT1L. Around 48 SET-containing proteins have been reported to be encoded by the human genome. Recruitment of lysine-methylating enzymes at histones is assisted by specific DNA sequences, e.g., TREs (trithorax group response elements) and PREs (polycomb group response group elements), which recruit the respective proteins for H3K4 and H3K27 methylation, respectively. Arginine residues are methylated by different sets of proteins, called PRMTs (protein arginine methyltransferases), which methylate guanidine nitrogen at arginine residues using S-adenosyl-L-methionine (SAM) as a methyl group donor. PRMTs have a conserved catalytic core and a variable region at their C- and N-terminals. Asymmetric and symmetric dimethylation of arginine residues is carried out by type I and type II HMTs, respectively (reviewed in [183,184]; Table 5).
3.2. Demethylases
There are two mechanisms for DNA demethylation, i.e., passive (no methylation of the newly synthesized DNA strand during replication) and active (replication-independent removal of methylation mark(s)). The former occurs during development in mammals (e.g., during the pre-implant growth period in the maternal genome) [198], and it has been revealed that DNA hypomethylation can be achieved by DNMT1 inhibition [199]. Here, we will mainly be focusing on active DNA demethylation. A considerable body of results supports active genome-wide demethylation in primordial germ cells (PGCs) [200] and zygotes [198], as well as active locus-specific demethylation in somatic cells including T-lymphocytes [201] and neurons [202]. Different mechanisms for the enzyme-mediated removal of 5mC (5′ methyl cytosine) 5-methyl groups, 5mC bases, or 5mC nucleotides are proposed; however, more than one mechanism may be involved in this process. For example, global demethylation may have a different mechanism to that of locus-specific demethylation. Discovery of 5-hydroxymethylcytosine (5hmC) in the mammalian genome has opened new research avenues to understand mechanisms of active demethylation.
Only in plants, direct 5mC base removal by 5mC-specific glycosylases has been observed [203]. Regarding 5mC preference in double-stranded DNA, four members of the 5mC DNA glycosylase family (DME, DML2, DML3, and ROS1) have been identified in Arabidopsis, with strong genetic and biochemical evidence for particular genes in active demethylation [203]. ROS1 is actually a proto-oncogene tyrosine-protein kinase ROS that plays a role in regionalization of the proximal epididymal epithelium and epithelial cell differentiation. For instance, the bifunctional glycosylase ROS1 shows apyrimidinic/apurinic activity, i.e., it eliminates the target base followed by cleavage of the abasic site, which generates a nick (later repaired rapidly) [203]. This process is similar to that present in mammals for mismatch repair with the elimination of alkylated bases, known as base excision repair (BER). Evidence reveals a role of BER in mammals for active demethylation of target, but the initiation mechanism and enzymes may be dissimilar to those of Arabidopsis.
Until now, no homolog of the DME/ROS1 glycosylase family has been identified in mammals; however, fragile glycosylase activity at 5mC has been observed for TDG (thymine DNA glycosylase) and MBD4 (methyl-CpG-binding domain protein 4) [204]. Both of these show 30–40 times lower glycosylase activity on 5mC as compared to T-G mismatch [204], which sheds doubt on their 5mC DNA glycosylase nature [205]. Consistently, zygotic paternal genome global demethylation does not require MBD4, and mbd4 knockout mice show fertility and viability [206]. However, the locus-specific activity of MBD4 as a 5mC DNA glycosylase cannot be ruled out as hormone-induced MBD4 phosphorylation leads to active demethylation at the promoter region of the CYP27B1 gene via stimulation of its glycosylase activity [207].
Other active DNA methylation-related proposed mechanisms also involve BER, but after the 5mC base has been modified. The leading mechanism is conversion of 5mC to thymine by its deamination following the removal of the resulting T-G mismatch via BER machinery. AID (activation induced cytosine deaminase) and APOBEC1 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1) can generate T-G mismatches after 5mC deamination, but both preferentially target single-stranded DNA [208]. Expression of both enzymes has been observed in mouse oocytes, and AID has also been observed in PDCs (plasmacytoid dendritic cells), which suggests a potential role of these enzymes in global DNA methylation [208]. These results led to the hypothesis that mammalian demethylation can occur by deamination of 5mC, with subsequent BER started by T-G mismatch glycosylases, such as TDG and MBD4 [208]. This hypothesis is supported by experiments in zebrafish embryos that overexpress both MBD4 and AID; however, neither alone resulted in DNA demethylation [209]. Moreover, some studies revealed that AID has a role in active mammalian demethylation [210]. Using heterokaryons formed by fusing human fibroblasts with mouse ES cells, Bhutani et al. [211] revealed that AID is required for active demethylation of NANOG and OCT4 promoters during fibroblast genome reprogramming by cell fusion. Another group performed a study in PGCs for wild type and Aid knockout mice and observed wide DNA methylation of the genome [210]. As compared to wild-type PGCs, Aid−/− mice showed a higher methylation level in the whole genome [210]. However, in the absence of AID, significant demethylation still occurred as a low methylation level was detected in Aid−/− PGCs [210] without any developmental defects in mice that were fertile [212], suggesting that there are some other factors in PGCs that participate in global demethylation.
In addition to BER, the involvement of NER (nucleotide excision repair), another DNA repair pathway, has also been examined in relation to active demethylation. Barreto et al. [213] used an expression cloning approach to prove that protein factor GADD45a in mammalian cells can promote active global demethylation by involving the NER pathway owing to DNA synthesis and XPG (NER endonuclease), which binds directly to GADD45a. However, another study could not confirm this finding [214], and neither a global nor a locus-specific increase in methylation was seen in Gadd45a−/− mice [215]. However, a locus-specific DNA demethylation role of GADD45 family proteins has been supported by some studies [202,216]. It has been observed for the rRNA gene promoter that active demethylation happens by NER machinery and GADD45a [216]. Another GADD45 family member, GADD45b, has been observed for locus-specific demethylation at regulatory regions of Fgf1 and Bdnf genes, which are involved in neurogenesis due to neuronal activity in mature hippocampal neurons [202].
The discovery of 5hmC and its associated enzymes in mammalian cells has opened up new avenues for demethylation studies. 5mC was considered to be the only naturally modified mammalian DNA base until the discovery of 5hmC in ES (embryonic stem) cells [217] and mouse Purkinje neurons [218]. By searching for trypanosome thymidine hydroxylase homologs in mammals, researchers found three ten–eleven translocation (TET) family proteins in humans (TET1, TET2, and TET3) and revealed that TET1 can convert 5mC to 5hmC in cultured cells and in vitro [217]. Similar reactions can be performed by all three TET proteins in the mouse; however, TET1 shows a role in self-renewal of ES cells and inner cell mass specification [219]. The crystal structure of TET2 and 5hmC complex is shown in Figure 4e. TET1 retains the hypomethylated state of the Nanog promoter in mouse ES cells, suggesting its role in DNA methylation regulation [219]. One postulated mechanism revealed BER involvement initiated by 5hmC-specific DNA glycosylase [217]. It is notable that the calf thymus has been reported as having 5hmC-associated glycosylase activity [220].
H3K4 and H3K9 methylation marks are removed by lysine-specific demethylases, LSD1 and LSD2, through an FAD-dependent amine oxidation reaction [221]. Meanwhile, H4K20, H3K36, H3K27, H3K9, or H3K4 can be demethylated by another family of histone demethylases, the JMJD (Jumonji C domain-containing) family, which contains a JmjC domain (150 amino acids) [222]. Less is known regarding methylation mark removal from arginine: a new pathway for arginine methylation reversion in mammalian cells has been studied. This involves the conversion of methylarginine to citrulline by removing its methyl group, a process known as deamination, at specific sites on H3 and H4 tails with the help of enzyme PADI4 (peptidyl arginine deiminase 4) [50,223]. Moreover, H3R3me2 and H3R2me2 demethylation have been reported for the first time with JMJD6/PSR/PTDSR (phosphatidylserine receptor) [224]. However, different studies have questioned this activity of JMJD6 [225,226]. Further study is required to validate the arginine demethylase activity of JMJD6.
3.3. Methyl Binding Proteins
Two mechanisms have been identified for the repression of gene expression via DNA methylation. The first direct mechanism involves DNA methylation-mediated alterations in binding sites of transcription factors such as CREB and E2F, which eventually prevents transcription activation [227,228]. Another elaborative mechanism involves the recruitment of methyl-CpG binding proteins (associated with different chromatin modifiers), which creates a repressive chromatin environment [229]. These proteins make a connection between chromatin modification and DNA methylation via reading and interpreting epigenetic signals. Proteins of the methyl-CpG binding domain (MBD) family have been widely studied, and their characterization reveals their various functions (Table 6). Mutations in methyl-CpG binding protein 2 (MeCP2), an MBD family founder, result in X-linked neurodevelopmental disorder and Rett syndrome (RTT) [230]. Other proteins of the MBD family bind to irrationally hypermethylated promoters in different cancer cell lines of human origin [231]. Initially, MBD was identified as the minimal part of MECP2 required for methylated DNA binding [232], and the homology of its amino acid sequence with other proteins led to the discovery of MBD1, MBD2, MBD3, and MBD4 [233]. The solution structure of the methylated CpG binding domain of human MBD1 in complex with methylated DNA is shown in Figure 4f. MBD1, MBD2, and MeCP2 have been reported to contain a non-conserved domain responsible for transcriptional repression. Apart from the MBD domain, MBD1 has a CxxC3 zinc finger domain for DNA binding [234], which has sequence similarity with the CxxC domain of DNMT1 [235]. Preferably, methylated but not unmethylated DNA is recognized by all MBD proteins except mammalian MBD3 [236] and MBD3 LF (amphibian MBD3 long form), which is unable to specifically recognize DNA methylation due to an insertion in the MBD region [237]. Generally, depending on the sequence context, MBD proteins show 3- to 10-fold higher affinities for methylated DNA as compared to unmethylated DNA [238]. In vitro experiments for binding site selection showed that an A/T-rich sequence adjacent to the site of CpG methylation is required by human MeCP2 [239]. Moreover, some other methylated DNA binding proteins aside from the MBD family have also been identified [163,240,241,242,243,244,245].
4. Epigenetics and Human Diseases
Environmental chemical exposure, inherited genetic polymorphisms, and changes in diet lead to fluctuation in patterns of DNA methylation [276]. Diet-acquired methyl groups are transferred to DNA by methionine and folate pathways [277]. Serious clinical consequences including atherosclerosis, cancer, and neural tube defects may occur due to alterations in DNA methylation by consuming a diet with low methionine, folate, or selenium [278,279]. Such nutrient imbalance in the diet cause genetic instability (leading to chromosome rearrangement) and hypomethylation (leading to unfit gene expression) [279]. For example, in vitro models of atherosclerosis revealed global hypomethylation and hyperhomocysteinemia supporting the hypothesis that alterations in patterns of global methylation are attributes of this disease at early stages [278], while hyperproliferation at advanced stages may further enhance hypomethylation of DNA and alterations in gene expression [280].
Environmental agents such as aromatic hydrocarbons (e.g., benzopyrene) and metals (e.g., arsenic) can also cause modification of cellular metabolism or destabilization of the genome, or both [280]. These agents are present in fossil fuel emissions, cigarette smoke, contaminated drinking water, and occupational chemicals [281]. Diet or environmental toxin sensitivity depends on previously existing genetic variants capable of challenging methylation-related metabolism and predisposing a person to changes at the epigenetic level. Some studies have linked the methylenetetrahydrofolate reductase gene (MTHFR) to altered patterns of DNA methylation in response to hormone replacement, alcohol consumption, and diet, which eventually result in a high incidence of colorectal and breast cancer in specific populations. For example, in premenopausal women, a common polymorphism in MTHFR 677CT increases breast cancer by up to 3-fold [282]. Other studies revealed that by hormone replacement therapy in postmenopausal women of the 677TT genotype, a 40% decrease in breast cancer risk was observed, which is probably owing to the limiting nucleic acid precursor availability for hyperproliferating cells [283]. Such examples highlight the complex interplay among disease-enhancing risk factors including epigenetics, genetic individuality (nature), and environment (nurture).
Multicellular organisms require specialized mechanisms for heritable gene silencing patterns, and mutation in these genes alter global epigenetic modification profiles resulting in many somatically acquired or inherited diseases. Interestingly, most of these abnormalities cause learning disabilities and chromosomal alterations. For example, atrx gene mutation changes the methylation pattern of ribosomal DNA, subtelomeric repeats, and Y-specific repeats. Fragile X syndrome occurs by de novo expansion and methylation of the CGG repeat in the 5′-untranslated region of the FMR1 gene, which creates a prominent “fragile” site on the X chromosome by rendering it silent under certain conditions. Globally, mutations in the dnmt3b gene affect establishment of patterns of DNA methylation resulting in immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome [284]. These findings revealed a primary importance of epigenetic modifications in determining chromosomal architecture.
Faulty genome imprinting (described as parent-specific monoallelic gene expression) causes many inherited syndromes such as Beckwith–Wiedemann syndrome (BWS), Prader–Willi syndrome, and Angelman syndrome. In these syndromes, deletion or uniparental disomy (UPD) cause the absence of a maternal or paternal imprinted gene copy or imprinted gene deregulation, which results in abnormal phenotype. For example, an imprinted gene cluster at 11p15.5 participates in BWS pathology: in most cases of BWS, methylation loss in control regions of imprinting causes its deregulation and either silencing (e.g., CDKN1C) or biallelic expression (e.g., IGF2) of associated imprinted regions.
Particular interest has been sparked by the revelation that MeCP2 germ line mutation causes Rett syndrome [230]. Methylcytosine residues are bound by MeCP2 [285], and disease progression is due to derepression of normally DNA methylation-mediated repressed genes. For this, no direct evidence has been found since gene derepression at a global level has not been observed in MeCP2 mutated human cells [285]. Nonetheless, MeCP2 plays a key role in controlling neuronal gene activity, causing Rett syndrome [286].
An interesting recent discovery revealed that antisense RNA transcription causes methylation and silencing of the α-globin gene in thalassemia patients [287]. Conventional diagnostic methods have failed to identify many other such diseases that occur due to improper gene silencing. This thalassemia case might be the tip of the iceberg, indicating that several other diseases might be caused by epigenetic silencing due to inappropriate genomic rearrangements.
Epigenetic changes are also responsible for many kinds of cancers. For example, the MutL homolog-1 (MLH1)-encoding gene is methylated and silenced with a phenotype of microsatellite instability in a high number of sporadic colorectal cancer patients [288]. Thus, genetic instability is directly linked to genetic silencing. Sometimes, promoter-associated MLH1 methylation is found in normal tissues (e.g., spermatozoa) along with tumor cells, and these germline-linked “epimutations” predispose these patients to multiple cancers [289]. De novo methylation of the promoter region of a gene causes disruption of the associated pathways that lead to cancer [290]. Epigenetic silencing is considered the third pathway correlating with Knudson’s hypothesis that tumor-suppressor gene silencing requires two hits [291].
Different chromatin-modifying enzymes are causative agents for various hemopathologies. For example, HMTs and acetyltransferases cause chromosomal translocation and the expression of fusion proteins at target sites in leukemia patients [292]. In acute cases of promyelocytic leukemia, the fused oncogenic protein PML-RARα (promyelocytic leukemia-retinoic acid receptor-α) represses hematopoietic cell differentiation genes via recruiting an HDAC [293]. Similarly, in acute cases of myeloid leukemia, the fusion protein AML1-ETO inhibits myeloid development via recruiting the NCOR–SIN3–HDAC1 complex [294].
The significance of accurate chromatin composition is further supported by the disease involvement of ATP-dependent chromatin remodeling multisubunit complexes, which are capable of transcriptional regulation via shifting and moving nucleosomes. Various cancers involve many members of the SWI–SNF complex, which is a highly conserved chromatin remodeler [295]. For example, SNF5 loss is observed in pediatric cancers, and mutation in BRG1 and BRM ATPase subunits is seen in various primary tumors and cancer cell lines; this is linked with poor prognosis in non-small cell lung cancer patients [295].
5. Conclusions
Many human diseases are linked to inappropriate epigenetic modifications; thus, researchers are attempting to identify relevant drugs to reverse these modifications. For example, various inhibitors against undesired HDACs (HDACi) have been analyzed in animal experiments, normal healthy cells, and clinical trials with no or few side effects within a therapeutic range [296,297,298,299,300,301]. Similar to acetylation, many methylation inhibitors including 5-azacytidine, 5-aza-2-deoxycytidine, zebularine, and procainamide have been discovered to be effective against improper methylation [199,302,303,304]. A short list of drugs under clinical trials against HDACs and methylation is given in Table 7. Each epigenetic family has its own particular structure and functional interacting domain; however, there is still the possibility that some may have the same interacting pocket conformation, and designing a drug against these common pockets, to stop more than one member of different families, would be a worthy strategy. More in-depth structural and mechanistic studies between epigenetic proteins and their target DNA or histones may help to discover such universal drugs, which will be more economical from a commercial point of view.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF-2015R1A2A2A09001059, NRF 2012–0006687) and by a grant from the Korea Health Industry Development Institute (HI14C1992).
Conflicts of Interest
The authors declare no conflict of interests.
References
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Humpherys, D.; Eggan, K.; Akutsu, H.; Hochedlinger, K.; Rideout, W.M.; Biniszkiewicz, D.; Yanagimachi, R.; Jaenisch, R. Epigenetic instability in ES cells and cloned mice. Science 2001, 293, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C. Preliminary notes on the development of the wings in normal and mutant strains of Drosophila. Proc. Natl. Acad. Sci. USA 1939, 25, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.E.; Martienssen, R.A.; Riggs, A.D. Epigenetic Mechanisms of Gene Regulation; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1996. [Google Scholar]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [PubMed]
- Meneghini, M.D.; Wu, M.; Madhani, H.D. Conserved histone variant H2A. Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003, 112, 725–736. [Google Scholar] [CrossRef]
- Ng, H.H.; Ciccone, D.N.; Morshead, K.B.; Oettinger, M.A.; Struhl, K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proc. Natl. Acad. Sci. USA 2003, 100, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Santos-Rosa, H.; Bannister, A.J.; Dehe, P.M.; Géli, V.; Kouzarides, T. Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatin. J. Biol. Chem. 2004, 279, 47506–47512. [Google Scholar] [CrossRef] [PubMed]
- Suka, N.; Luo, K.; Grunstein, M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat. Genet. 2002, 32, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Hebbes, T.R.; Thorne, A.W.; Clayton, A.L.; Crane-Robinson, C. Histone acetylation and globin gene switching. Nucleic Acids Res. 1992, 20, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Hebbes, T.R.; Thorne, A.W.; Crane-Robinson, C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988, 7, 1395. [Google Scholar] [PubMed]
- Logie, C.; Tse, C.; Hansen, J.C.; Peterson, C.L. The core histone N-terminal domains are required for multiple rounds of catalytic chromatin remodeling by the SWI/SNF and RSC complexes. Biochemistry 1999, 38, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.G.; Smith, M.M.; Roth, S.Y. Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev. 1996, 10, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Laroche, T.; Strahl-Bolsinger, S.; Gasser, S.M.; Grunstein, M. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: A molecular model for the formation of heterochromatin in yeast. Cell 1995, 80, 583–592. [Google Scholar] [CrossRef]
- Allfrey, V.G. Structural modifications of histones and their possible role in the regulation of ribonucleic acid synthesis. In Proceedings of the Canadian Cancer Conference, Honey Harbour, ON, Canada, 1 January 1965; pp. 313–335. [Google Scholar]
- Wolffe, A.P.; Hayes, J.J. Chromatin disruption and modification. Nucleic Acids Res. 1999, 27, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stützer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sweet, S.M.; Popovic, R.; Martinez-Garcia, E.; Tipton, J.D.; Thomas, P.M.; Licht, J.D.; Kelleher, N.L. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. USA 2012, 109, 13549–13554. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Ma, B.; Yuan, W.; Zhang, Z.; Chen, P.; Ding, X.; Feng, L.; Shen, X.; Chen, S.; Li, G. Histone H2A ubiquitination inhibits the enzymatic activity of H3 lysine 36 methyltransferases. J. Biol. Chem. 2013, 288, 30832–30842. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kang, B.H.; Jang, H.; Kwak, S.; Shin, J.; Kim, H.; Lee, S.E.; Lee, S.M.; Lee, J.H.; Kim, J.H. Ctbp2 modulates NuRD-mediated deacetylation of H3K27 and facilitates PRC2-mediated H3K27me3 in active embryonic stem cell genes during exit from pluripotency. Stem Cells 2015, 33, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Zahradka, P.; Ebisuzaki, K. A Shuttle Mechanism for DNA-Protein Interactions. FEBS J. 1982, 127, 579–585. [Google Scholar] [CrossRef]
- Oei, S.L.; Griesenbeck, J.; Schweiger, M.; Ziegler, M. Regulation of RNA polymerase II-dependent transcription by poly (ADP-ribosyl) ation of transcription factors. J. Biol. Chem. 1998, 273, 31644–31647. [Google Scholar] [CrossRef] [PubMed]
- Oei, S.L.; Griesenbeck, J.; Ziegler, M.; Schweiger, M. A novel function of poly (ADP-ribosyl) ation: Silencing of RNA polymerase II-dependent transcription. Biochemistry 1998, 37, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Węsierska-Gądek, J.; Schmid, G.; Cerni, C. ADP-Ribosylation of Wild-Type p53in Vitro: Binding of p53 protein to specific p53 consensus sequence prevents its modification. Biochem. Biophys. Res. Commun. 1996, 224, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Oei, S.L.; Griesenbeck, J.; Schweiger, M.; Babich, V.; Kropotov, A.; Tomilin, N. Interaction of the transcription factor YY1 with human poly (ADP-ribosyl) transferase. Biochem. Biophys. Res. Commun. 1997, 240, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-J.; Alvarez-Gonzalez, R. The sequence-specific DNA binding of NF-κB is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J. Biol. Chem. 2001, 276, 47664–47670. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Alvarez, H.; Alvarez-Gonzalez, R. Regulation of p53 sequence-specific DNA-binding by covalent poly (ADP-ribosyl) ation. J. Biol. Chem. 2001, 276, 36425–36430. [Google Scholar] [CrossRef] [PubMed]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Smulson, M.E. Poly (ADP-ribose) polymerase upregulates E2F-1 promoter activity and DNA pol α expression during early S phase. Oncogene 1999, 18, 5015–5023. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-κB coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of poly (ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-κB-dependent transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrozza, M.J.; Li, B.; Florens, L.; Suganuma, T.; Swanson, S.K.; Lee, K.K.; Shia, W.-J.; Anderson, S.; Yates, J.; Washburn, M.P. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005, 123, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.-E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Mowen, K.A.; Tang, J.; Zhu, W.; Schurter, B.T.; Shuai, K.; Herschman, H.R.; David, M. Arginine methylation of STAT1 modulates IFNα/β-induced transcription. Cell 2001, 104, 731–741. [Google Scholar] [CrossRef]
- Murr, R.; Loizou, J.I.; Yun-Gui, Y.; Cuenin, C.; Li, H.; Zhao-Qi, W.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Vogelauer, M.; Rubbi, L.; Lucas, I.; Brewer, B.J.; Grunstein, M. Histone acetylation regulates the time of replication origin firing. Mol. Cell 2002, 10, 1223–1233. [Google Scholar] [CrossRef]
- Basnet, H.; Su, X.B.; Tan, Y.; Meisenhelder, J.; Merkurjev, D.; Ohgi, K.A.; Hunter, T.; Pillus, L.; Rosenfeld, M.G. Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation. Nature 2014, 516, 267. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Best, J.L.; Zon, L.I.; Gill, G. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol. Cell 2002, 10, 831–842. [Google Scholar] [CrossRef]
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Chen, H. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135. [Google Scholar] [CrossRef] [PubMed]
- Olabisi, O.A.; Soto-Nieves, N.; Nieves, E.; Yang, T.T.; Yang, X.; Raymond, Y.; Suk, H.Y.; Macian, F.; Chow, C.-W. Regulation of transcription factor NFAT by ADP-ribosylation. Mol. Cell. Biol. 2008, 28, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Kreimeyer, A.; Wielckens, K.; Adamietz, P.; Hilz, H. DNA repair-associated ADP-ribosylation in vivo. Modification of histone H1 differs from that of the principal acceptor proteins. J. Biol. Chem. 1984, 259, 890–896. [Google Scholar] [PubMed]
- Boulikas, T. Poly (ADP-ribosylated) histones in chromatin replication. J. Biol. Chem. 1990, 265, 14638–14647. [Google Scholar] [PubMed]
- Nelson, C.J.; Santos-Rosa, H.; Kouzarides, T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 2006, 126, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.-H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Wolffe, A. Chromatin: Structure and Function; Academic Press: Cambridge, MA, USA, 1998. [Google Scholar]
- Ait-Si-Ali, S.; Ramirez, S.; Robin, P.; Trouche, D.; Harel-Bellan, A. A rapid and sensitive assay for histone acetyl-transferase activity. Nucleic Acids Res. 1998, 26, 3869–3870. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carrillo, A.; Wangh, L.J.; Allfrey, V.G. Processing of newly synthesized histone molecules. Science 1975, 190, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Allis, C.D. Special HATs for special occasions: Linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 1996, 6, 176–184. [Google Scholar] [CrossRef]
- Kuo, M.-H.; Brownell, J.E.; Sobel, R.E.; Ranalli, T.A. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 1996, 383, 269. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [PubMed]
- Akhtar, A.; Becker, P.B. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 2000, 5, 367–375. [Google Scholar] [CrossRef]
- Borrow, J.; Stanton, V.P.; Andresen, J.M.; Becher, R.; Behm, F.G.; Chaganti, R.S.; Civin, C.I.; Disteche, C.; Dubé, I.; Frischauf, A.M. The translocation t (8; 16)(p11; p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB–binding protein. Nat. Genet. 1996, 14, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.S.; Lowell, J.E.; Jacobson, S.J.; Pillus, L. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol. 1999, 19, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Reifsnyder, C.; Lowell, J.; Clarke, A.; Pillus, L. Yeast SAS silencing genes and human genes associated with AML and HIV–1 Tat interactions are homologous with acetyltransferases. Nat. Genet. 1996, 14, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Zink, D.; Becker, P.B. Chromodomains are protein–RNA interaction modules. Nature 2000, 407, 405–409. [Google Scholar] [PubMed]
- Neuwald, A.F.; Landsman, D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 1997, 22, 154–155. [Google Scholar] [CrossRef]
- Dutnall, R.N.; Tafrov, S.T.; Sternglanz, R.; Ramakrishnan, V. Structure of the histone acetyltransferase Hat1: A paradigm for the GCN5-related N-acetyltransferase superfamily. Cell 1998, 94, 427–438. [Google Scholar] [CrossRef]
- Yan, Y.; Barlev, N.A.; Haley, R.H.; Berger, S.L.; Marmorstein, R. Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol. Cell 2000, 6, 1195–1205. [Google Scholar] [CrossRef]
- Rojas, J.R.; Trievel, R.C.; Zhou, J.; Mo, Y.; Li, X.; Berger, S.L.; Allis, C.D.; Marmorstein, R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 1999, 401, 93–98. [Google Scholar] [PubMed]
- Trievel, R.C.; Rojas, J.R.; Sterner, D.E.; Venkataramani, R.N.; Wang, L.; Zhou, J.; Allis, C.D.; Berger, S.L.; Marmorstein, R. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc. Natl. Acad. Sci. USA 1999, 96, 8931–8936. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.C.; Minucci, S.; Lu, J.; Yang, X.-J.; Walker, K.K.; Chen, H.; Evans, R.M.; Nakatani, Y.; Ozato, K. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 1998, 12, 1638–1651. [Google Scholar] [CrossRef] [PubMed]
- Tanner, K.G.; Trievel, R.C.; Kuo, M.-H.; Howard, R.M.; Berger, S.L.; Allis, C.D.; Marmorstein, R.; Denu, J.M. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 1999, 274, 18157–18160. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.; Muñoz, F.; Schilcher, P.; Imhof, A.; Almouzni, G.; Loyola, A. Sequential establishment of marks on soluble histones H3 and H4. J. Biol. Chem. 2011, 286, 17714–17721. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Che, Y.-Z.; Zou, H.-S.; Cui, Y.-P.; Guo, W.; Zou, L.-F.; Biddle, E.M.; Yang, C.-H.; Chen, G.-Y. Hpa2 required by HrpF to translocate Xanthomonas oryzae transcriptional activator-like effectors into rice for pathogenicity. Appl. Environ. Microbiol. 2011, 77, 3809–3818. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Lu, J.; Duan, J.; Su, D.; Hou, X.; Li, F.; Wang, X.; Huang, B. Gcn5-and Elp3-induced histone H3 acetylation regulates hsp70 gene transcription in yeast. Biochem. J. 2008, 409, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, I.; Aikawa, Y.; Yokoyama, A.; Hosoda, F.; Nagai, M.; Kakazu, N.; Abe, T.; Ohki, M. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with at (8; 22)(p11; q13) chromosome translocation. Leukemia 2001, 15, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Miotto, B.; Struhl, K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol. Cell 2010, 37, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker, A.; Hilfiker-Kleiner, D.; Pannuti, A.; Lucchesi, J.C. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 1997, 16, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Horikoshi, M. Novel substrate specificity of the histone acetyltransferase activity of HIV-1-Tat interactive protein Tip60. J. Biol. Chem. 1997, 272, 30595–30598. [Google Scholar] [CrossRef] [PubMed]
- Katan-Khaykovich, Y.; Struhl, K. Dynamics of global histone acetylation and deacetylation in vivo: Rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 2002, 16, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Mizzen, C.A.; Yang, X.-J.; Kokubo, T.; Brownell, J.E.; Bannister, A.J.; Owen-Hughes, T.; Workman, J.; Wang, L.; Berger, S.L.; Kouzarides, T. The TAF II 250 subunit of TFIID has histone acetyltransferase activity. Cell 1996, 87, 1261–1270. [Google Scholar] [CrossRef]
- Srinivasan, L.; Gopinathan, K.P. Characterization of RNA polymerase III transcription factor TFIIIC from the mulberry silkworm, Bombyx mori. FEBS J. 2002, 269, 1780–1789. [Google Scholar] [CrossRef]
- Liu, Z.; Myers, L.C. Med5 (Nut1) and Med17 (Srb4) are direct targets of mediator histone H4 tail interactions. PLoS ONE 2012, 7, e38416. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, Z.; Xu, J. The cooperative function of nuclear receptor coactivator 1 (NCOA1) and NCOA3 in placental development and embryo survival. Mol. Endocrinol. 2010, 24, 1917–1934. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.-A.; Rose, D.W.; Haque, Z.K.; Kurokawa, R.; McInerney, E.; Westin, S.; Thanos, D.; Rosenfeld, M.G.; Glass, C.K.; Collins, T. Transcriptional activation by NF-κB requires multiple coactivators. Mol. Cell. Biol. 1999, 19, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Kohli, K.; Trivedi, A.; Johnson, D.L.; Stallcup, M.R. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 4948–4952. [Google Scholar] [CrossRef] [PubMed]
- Bhoumik, A.; Singha, N.; O’Connell, M.J.; Ze’ev, A.R. Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem. 2008, 283, 17605–17614. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Alsarraj, J.; Faraji, F.; Geiger, T.R.; Mattaini, K.R.; Williams, M.; Wu, J.; Ha, N.-H.; Merlino, T.; Walker, R.C.; Bosley, A.D. BRD4 short isoform interacts with RRP1B, SIPA1 and components of the LINC complex at the inner face of the nuclear membrane. PLoS ONE 2013, 8, e80746. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sabari, B.R.; Panchenko, T.; Wen, H.; Zhao, D.; Guan, H.; Wan, L.; Huang, H.; Tang, Z.; Zhao, Y. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol. Cell 2016, 62, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell. Biol. 2011, 31, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.E.; Zhou, M.-M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Ornaghi, P.; Yang, J.C.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Ruthenburg, A.J.; Li, H.; Milne, T.A.; Dewell, S.; McGinty, R.K.; Yuen, M.; Ueberheide, B.; Dou, Y.; Muir, T.W.; Patel, D.J. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 2011, 145, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Morinière, J.; Rousseaux, S.; Steuerwald, U.; Soler-López, M.; Curtet, S.; Vitte, A.-L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnölzer, M.; Verdin, E. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Gao, X.; Diaz-Trelles, R.; Ruiz-Lozano, P.; Wang, Z. Coronary development is regulated by ATP-dependent SWI/SNF chromatin remodeling component BAF180. Dev. Biol. 2008, 319, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Reyes, J.; Barra, J.; Muchardt, C.; Camus, A.; Babinet, C.; Yaniv, M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2α). EMBO J. 1998, 17, 6979–6991. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Popowicz, G.M.; Krajewski, M.; Holak, T.A. Structural Ramification for Acetyl-Lysine Recognition by the Bromodomain of Human BRG1 Protein, a Central ATPase of the SWI/SNF Remodeling Complex. ChemBioChem 2007, 8, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Graumann, J.; Wu, G.; Araúzo-Bravo, M.J.; Han, D.W.; Greber, B.; Gentile, L.; Mann, M.; Schöler, H.R. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell 2010, 141, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; Khochbin, S. Histone acylation beyond acetylation: Terra incognita in chromatin biology. Cell J. 2015, 17, 1. [Google Scholar] [PubMed]
- Chen, Y.; Sprung, R.; Tang, Y.; Ball, H.; Sangras, B.; Kim, S.C.; Falck, J.R.; Peng, J.; Gu, W.; Zhao, Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteom. 2007, 6, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, A.; Zhang, D.; Huang, H.; Barral, S.; Kwon, O.K.; Qi, S.; Tang, Z.; Buchou, T.; Vitte, A.-L.; He, T. Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol. Cell 2016, 62, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Bowser, R.; Giambrone, A.; Davies, P. FAC1, a novel gene identified with the monoclonal antibody Alz50, is developmentally regulated in human brain (Part 1 of 2). Dev. Neurosci. 1995, 17, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Banting, G.S.; Barak, O.; Ames, T.M.; Burnham, A.C.; Kardel, M.D.; Cooch, N.S.; Davidson, C.E.; Godbout, R.; McDermid, H.E.; Shiekhattar, R. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum. Mol. Genet. 2005, 14, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Fairbridge, N.A.; Dawe, C.E.; Niri, F.H.; Kooistra, M.K.; King-Jones, K.; McDermid, H.E. Cecr2 mutations causing exencephaly trigger misregulation of mesenchymal/ectodermal transcription factors. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Herrema, H.; Salazar, M.; Cakir, I.; Cabi, S.; Sahin, F.B.; Chiu, Y.-H.; Cantley, L.C.; Ozcan, U. BRD7 regulates XBP1s’ activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab. 2014, 20, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, M.D.; Aslanian, A.; Dong, M.-Q.; Yates, J.R.; Emerson, B.M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 2008, 283, 32254–32263. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, P.M.; Chambers, A.L.; Cloney, R.; Bianchi, A.; Downs, J.A. BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep. 2014, 6, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Saare, M.; Hämarik, U.; Venta, R.; Panarina, M.; Zucchelli, C.; Pihlap, M.; Remm, A.; Kisand, K.; Toots, U.; Möll, K. SP140L, an evolutionarily recent member of the SP100 family, is an autoantigen in primary biliary cirrhosis. J. Immunol. Res. 2015, 2015, 526518. [Google Scholar] [CrossRef] [PubMed]
- Burrows, A.E.; Smogorzewska, A.; Elledge, S.J. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc. Natl. Acad. Sci. USA 2010, 107, 14280–14285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Schmitz, K.-M.; Mayer, C.; Yuan, X.; Akhtar, A.; Grummt, I. Reversible acetylation of the chromatin remodelling complex NoRC is required for non-coding RNA-dependent silencing. Nat. Cell Biol. 2009, 11, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Poot, R.A.; Kukimoto, I.; García-Jiménez, C.; Dellaire, G.; Varga-Weisz, P.D. An ACF1–ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 2002, 32, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K. WSTF regulates the H2A. X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Fukuyama, T.; Biesen, M.A.; Boussouar, F.; Tong, C.; De Pauw, A.; Murray, P.J.; van Deursen, J.M.; Brindle, P.K. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol. Cell. Biol. 2006, 26, 789–809. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Kuttenkeuler, D.; Gesellchen, V.; Zeidler, M.P.; Boutros, M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 2005, 436, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.R.; Karmodiya, K.; Lindahl-Allen, M.; Struhl, K.; Tora, L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol. Cell 2011, 44, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Feng, Y.; Vlassis, A.; Roques, C.; Lalonde, M.E.; González-Aguilera, C.; Lambert, J.P.; Lee, S.B.; Zhao, X.; Alabert, C.; Johansen, J.V. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016, 35, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.J.; Muthurajan, U.M.; Lalonde, M.-E.; Gibson, M.D.; Andrews, F.H.; Hepler, M.; Machida, S.; Yan, K.; Kurumizaka, H.; Poirier, M.G. Bivalent interaction of the PZP domain of BRPF1 with the nucleosome impacts chromatin dynamics and acetylation. Nucleic Acids Res. 2016, 44, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Obri, A.; Ouararhni, K.; Papin, C.; Diebold, M.-L.; Padmanabhan, K.; Marek, M.; Stoll, I.; Roy, L.; Reilly, P.T.; Mak, T.W. ANP32E is a histone chaperone that removes H2A. Z from chromatin. Nature 2014, 505, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.; Li, W. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Andrews, F.H.; Shinsky, S.A.; Shanle, E.K.; Bridgers, J.B.; Gest, A.; Tsun, I.K.; Krajewski, K.; Shi, X.; Strahl, B.D.; Kutateladze, T.G. The Taf14 YEATS domain is a reader of histone crotonylation. Nat. Chem. Biol. 2016, 12, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ilin, S.; Wang, W.; Duncan, E.M.; Wysocka, J.; Allis, C.D.; Patel, D.J. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 2006, 442, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Wang, Z.; Zaromytidou, A.-I.; Zhang, X.H.-F.; Chow-Tsang, L.-F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V. A poised chromatin platform for TGF-β access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell Physiol. 2000, 184, 1–16. [Google Scholar] [CrossRef]
- Zhou, D.-C.; Kim, S.H.; Ding, W.; Schultz, C.; Warrell, R.P.; Gallagher, R.E. Frequent mutations in the ligand-binding domain of PML-RARα after multiple relapses of acute promyelocytic leukemia: Analysis for functional relationship to response to all-transretinoic acid and histone deacetylase inhibitors in vitro and in vivo. Blood 2002, 99, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Nervi, C.; Coco, F.L.; Pelicci, P.G. Histone deacetylases: A common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene 2001, 20, 3110. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.F.; Fazi, F.; Bianchini, A.; Padula, F.; Gelmetti, V.; Minucci, S.; Mancini, M.; Pelicci, P.G.; Coco, F.L.; Nervi, C. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res. 2001, 61, 2–7. [Google Scholar] [PubMed]
- Kitamura, K.; Hoshi, S.; Koike, M.; Kiyoi, H.; Saito, H.; Naoe, T. Histone deacetylase inhibitor but not arsenic trioxide differentiates acute promyelocytic leukaemia cells with t (11; 17) in combination with all-trans retinoic acid. Br. J. Haematol. 2000, 108, 696–702. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Alland, L.; Hong, S.-H.; Wong, C.-W.; DePinho, R.A.; Dejean, A. Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein. Oncogene 1998, 16, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Bjerling, P.; Silverstein, R.A.; Thon, G.; Caudy, A.; Grewal, S.; Ekwall, K. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol. Cell. Biol. 2002, 22, 2170–2181. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Sideris, M.L.; Polly, M.; Lorimer, D.D.; Mcintosh, B.; Clark, J.M. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J. 2000, 350, 199–205. [Google Scholar]
- Galasinski, S.C.; Resing, K.A.; Goodrich, J.A.; Ahn, N.G. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J. Biol. Chem. 2002, 277, 19618–19626. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Lee, C.; Hou, T.; Boldogh, I.; Brasier, A.R. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 2008, 36, 4510–4520. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.J.; Seto, E. Unlocking the mechanisms of transcription factor YY1: Are chromatin modifying enzymes the key? Gene 1999, 236, 197–208. [Google Scholar] [CrossRef]
- Gobinet, J.; Carascossa, S.; Cavaillès, V.; Vignon, F.; Nicolas, J.-C.; Jalaguier, S. SHP represses transcriptional activity via recruitment of histone deacetylases. Biochemistry 2005, 44, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Watamoto, K.; Towatari, M.; Ozawa, Y.; Miyata, Y.; Okamoto, M.; Abe, A.; Naoe, T.; Saito, H. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene 2003, 22, 9176. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Yang, Y.-C. Sumoylation and acetylation play opposite roles in the transactivation of PLAG1 and PLAGL2. J. Biol. Chem. 2005, 280, 40773–40781. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Heldin, C.-H.; Ericsson, J.; Grönroos, E. The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Xie, Y.-B.; Choi, H.-S. Transcriptional corepressor SHP recruits SIRT1 histone deacetylase to inhibit LRH-1 transactivation. Nucleic Acids Res. 2010, 38, 4607–4619. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.A. Transcriptional control at regulatory checkpoints by histone deacetylases: Molecular connections between cancer and chromatin. Hum. Mol. Genet. 2001, 10, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [PubMed]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Juan, L.-J.; Shia, W.-J.; Chen, M.-H.; Yang, W.-M.; Seto, E.; Lin, Y.-S.; Wu, C.-W. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Muto, M.; Takebayashi, S.-I.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, M.; Hermann, A.; Pradhan, S.; Jeltsch, A. The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J. Mol. Biol. 2001, 309, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Rathert, P.; Laser, H.; Gowher, H.; Jeltsch, A. Phosphorylation of serine-515 activates the Mammalian maintenance methyltransferase Dnmt1. Epigenetics 2007, 2, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase DNMT3A in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Kareta, M.S.; Botello, Z.M.; Ennis, J.J.; Chou, C.; Chédin, F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006, 281, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Mann, J.R.; Hsieh, C.L.; Riggs, A.D.; Chédin, F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J. Cell Biochem. 2005, 95, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of DNMT3A and DNMT3B DNA-(cytosine-C5)-methyltransferases by DNMT3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of DNMT3A bound to DNMT3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Chédin, F.; Lieber, M.R.; Hsieh, C.-L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by DNMT3A. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Klimasauskas, S.; Kumar, S.; Roberts, R.J.; Cheng, X. Hhal methyltransferase flips its target base out of the DNA helix. Cell 1994, 76, 357–369. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415. [Google Scholar] [CrossRef] [PubMed]
- Otani, J.; Nankumo, T.; Arita, K.; Inamoto, S.; Ariyoshi, M.; Shirakawa, M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX–DNMT3–DNMT3L domain. EMBO Rep. 2009, 10, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W. Chromatin methylation activity of DNMT3A and DNMT3A/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Smith, A.D.; Kendall, J.; Xuan, Z.; Ravi, K.; Rooks, M.; Zhang, M.Q.; Ye, K.; Bhattacharjee, A.; Brizuela, L. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res. 2009, 19, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Liang, G.; Sharma, S.; Lin, J.C.; Choi, S.H.; Han, H.; Yoo, C.B.; Egger, G.; Yang, A.S.; Jones, P.A. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 2009, 29, 5366–5376. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.; Lee, S.-K.; Lee, J.W. Activating signal cointegrator-2 is an essential adaptor to recruit histone H3 lysine 4 methyltransferases MLL3 and MLL4 to the liver X receptors. Mol. Endocrinol. 2008, 22, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Chun, J.; Wilson, J.R.; Walker, P.A. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Feng, Q.; Ketel, C.S.; Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Simon, J.A.; Zhang, Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 2002, 12, 1086–1099. [Google Scholar] [CrossRef]
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Jung, I.; Lee, H.; Kang, K.; Kim, M.; Jeong, K.; Kwon, C.S.; Han, Y.-M.; Kim, Y.S.; Kim, D. Human histone H3K79 methyltransferase DOT1L methyltransferase binds actively transcribing RNA polymerase II to regulate gene expression. J. Biol. Chem. 2012, 287, 39698–39709. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Branscombe, T.L.; Frankel, A.; Lee, J.-H.; Cook, J.R.; Yang, Z.-H.; Pestka, S.; Clarke, S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 2001, 276, 32971–32976. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.M.; Dowhan, D.H.; Eriksson, N.A.; Muscat, G.E. CARM1/PRMT4 is necessary for the glycogen gene expression programme in skeletal muscle cells. Biochem. J. 2012, 444, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar] [CrossRef] [PubMed]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Embryogenesis: Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed]
- Bruniquel, D.; Schwartz, R.H. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat. Immunol. 2003, 4, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Jang, M.-H.; Guo, J.U.; Kitabatake, Y.; Chang, M.-L.; Pow-Anpongkul, N.; Flavell, R.A.; Lu, B.; Ming, G.-L.; Song, H. Neuronal activity–induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 2009, 323, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Bestor, T.H. The colorful history of active DNA demethylation. Cell 2008, 133, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zheng, Y.; Angliker, H.; Schwarz, S.; Thiry, S.; Siegmann, M.; Jost, J.-P. 5-Methylcytosine DNA glycosylase activity is also present in the human MBD4 (G/T mismatch glycosylase) and in a related avian sequence. Nucleic Acids Res. 2000, 28, 4157–4165. [Google Scholar] [CrossRef] [PubMed]
- Cortázar, D.; Kunz, C.; Saito, Y.; Steinacher, R.; Schär, P. The enigmatic thymine DNA glycosylase. DNA Repair 2007, 6, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Guy, J.; Sansom, O.J.; Selfridge, J.; MacDougall, E.; Hendrich, B.; Keightley, P.D.; Bishop, S.M.; Clarke, A.R.; Bird, A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 2002, 297, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Kondo, T.; Takada, I.; Youn, M.-Y.; Yamamoto, Y.; Takahashi, S.; Matsumoto, T.; Fujiyama, S.; Shirode, Y.; Yamaoka, I. DNA demethylation in hormone-induced transcriptional derepression. Nature 2009, 461, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Dean, W.; Coker, H.A.; Reik, W.; Petersen-Mahrt, S.K. Activation-induced cytidine deaminase deaminates 5-Methylcytosine in DNA and is expressed in pluripotent tissues implications for epigenetic reprogramming. J. Biol. Chem. 2004, 279, 52353–52360. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Huggins, I.J.; James, S.R.; Karpf, A.R.; Jones, D.A.; Cairns, B.R. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 2008, 135, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.; Dean, W.; Feng, S.; Cokus, S.J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 2010, 463, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Barreto, G.; Schäfer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Döderlein, G.; Maltry, N.; Wu, W.; Lyko, F. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-G.; Guo, C.; Pfeifer, G.P. GADD45A does not promote DNA demethylation. PLoS Genet. 2008, 4, e1000013. [Google Scholar] [CrossRef] [PubMed]
- Engel, N.; Tront, J.S.; Erinle, T.; Nguyen, N.; Latham, K.E.; Sapienza, C.; Hoffman, B.; Liebermann, D.A. Conserved DNA methylation in Gadd45a−/− mice. Epigenetics 2009, 4, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.-M.; Schmitt, N.; Hoffmann-Rohrer, U.; Schäfer, A.; Grummt, I.; Mayer, C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol. Cell 2009, 33, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.V.; Cummings, A.; Teebor, G.W. 5-Hydroxymethylcytosine DNA glycosylase activity in mammalian tissue. Biochem. Biophys. Res. Commun. 1988, 151, 1173–1179. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.-H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R. JMJD6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Schmitz, C.; Böttger, A. Changing story of the receptor for phosphatidylserine-dependent clearance of apoptotic cells. EMBO Rep. 2007, 8, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Campanero, M.R.; Armstrong, M.I.; Flemington, E.K. CpG methylation as a mechanism for the regulation of E2F activity. Proc. Natl. Acad. Sci. USA 2000, 97, 6481–6486. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Ariga, S.; Schaffner, W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989, 3, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.C.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Ballestar, E.; Paz, M.F.; Valle, L.; Wei, S.; Fraga, M.F.; Espada, J.; Cigudosa, J.C.; Huang, T.H.M.; Esteller, M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003, 22, 6335–6345. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Meehan, R.R.; Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.F.; Ben-Porath, I.; Bird, A.P. Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol. Cell. Biol. 2004, 24, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, M.; Esteve, P.-O.; Chin, H.G.; Samaranayke, M.; Kim, G.-D.; Pradhan, S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 2008, 47, 10000–10009. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Ishikawa, F. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem. 2002, 277, 35434–35439. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.A.; Gegonne, A.; Jones, P.L.; Ballestar, E.; Aubry, F.; Wolffe, A.P. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet. 1999, 23, 62–66. [Google Scholar] [PubMed]
- Fraga, M.F.; Ballestar, E.; Montoya, G.; Taysavang, P.; Wade, P.A.; Esteller, M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003, 31, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Sarraf, S.A.; Schmiedeberg, L.; McDermott, S.M.; Stancheva, I.; Bird, A.P. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 2005, 19, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Arita, K.; Ariyoshi, M.; Tochio, H.; Nakamura, Y.; Shirakawa, M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 2008, 455, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Li, Y.; Duan, S.; Bronner, C.; Arrowsmith, C.H.; Dhe-Paganon, S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 2008, 455, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.M.; Spring, C.M.; Crawford, H.C.; Reynolds, A.B.; Baig, A. The p120ctn-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 2002, 30, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.; Zhenilo, S.; Salozhin, S.; Yamada, D.; Prokhortchouk, E.; Defossez, P.-A. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 2006, 26, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, A.; Hendrich, B.; Jørgensen, H.; Ruzov, A.; Wilm, M.; Georgiev, G.; Bird, A.; Prokhortchouk, E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001, 15, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Pontes, O.; Pikaard, C.S.; Richards, E.J. VIM1, a methylcytosine-binding protein required for centromeric heterochromatinization. Genes Dev. 2007, 21, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Kaul, S.C.; Wadhwa, R.; Nomura, T.; Khan, M.M.; Shinagawa, T.; Yasukawa, T.; Colmenares, C.; Ishii, S. The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 2001, 276, 34115–34121. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, L.E.; Caldenhoven, E.; Stunnenberg, H.G. In vivo repression of an erythroid-specific gene by distinct corepressor complexes. EMBO J. 2002, 21, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Harikrishnan, K.; Chow, M.Z.; Baker, E.K.; Pal, S.; Bassal, S.; Brasacchio, D.; Wang, L.; Craig, J.M.; Jones, P.L.; Sif, S. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 2005, 37, 254–264. [Google Scholar] [PubMed]
- Agarwal, N.; Hardt, T.; Brero, A.; Nowak, D.; Rothbauer, U.; Becker, A.; Leonhardt, H.; Cardoso, M.C. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 2007, 35, 5402–5408. [Google Scholar] [CrossRef] [PubMed]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Dintilhac, A.; Bernués, J. HMGB1 interacts with many apparently unrelated proteins by recognizing short amino acid sequences. J. Biol. Chem. 2002, 277, 7021–7028. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Watanabe, S.; Ichimura, T.; Tsuruzoe, S.; Shinkai, Y.; Tachibana, M.; Chiba, T.; Nakao, M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 2003, 278, 24132–24138. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ichimura, T.; Fujita, N.; Tsuruzoe, S.; Ohki, I.; Shirakawa, M.; Kawasuji, M.; Nakao, M. Methylated DNA-binding domain 1 and methylpurine–DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc. Natl. Acad. Sci. USA 2003, 100, 12859–12864. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.; Morey, L.; Raker, V.A.; Buschbeck, M.; Gutierrez, A.; De Santis, F.; Corsaro, M.; Varas, F.; Bossi, D.; Minucci, S. The methyl-CpG binding protein MBD1 is required for PML-RARα function. Proc. Natl. Acad. Sci. USA 2006, 103, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Reese, B.E.; Bachman, K.E.; Baylin, S.B.; Rountree, M.R. The methyl-CpG binding protein MBD1 interacts with the p150 subunit of chromatin assembly factor 1. Mol. Cell. Biol. 2003, 23, 3226–3236. [Google Scholar] [CrossRef] [PubMed]
- Fukushige, S.; Kondo, E.; Gu, Z.; Suzuki, H.; Horii, A. RET finger protein enhances MBD2-and MBD4-dependent transcriptional repression. Biochem. Biophys. Res. Commun. 2006, 351, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Le Menuet, D.; Chung, A.C.-K.; Cooney, A.J. Differential recruitment of methylated CpG binding domains by the orphan receptor GCNF initiates the repression and silencing of Oct4 expression. Mol. Cell. Biol. 2006, 26, 9471–9483. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, K.I.; Yamazaki, T.; Ishikawa, F. MBD2-MBD3 complex binds to hemi-methylated DNA and forms a complex containing DNMT1 at the replication foci in late S phase. Genes Cells 2000, 5, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Angrisano, T.; Lembo, F.; Pero, R.; Natale, F.; Fusco, A.; Avvedimento, V.E.; Bruni, C.B.; Chiariotti, L. TACC3 mediates the association of MBD2 with histone acetyltransferases and relieves transcriptional repression of methylated promoters. Nucleic Acids Res. 2006, 34, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Ego, T.; Tanaka, Y.; Shimotohno, K. Interaction of HTLV-1 Tax and methyl-CpG-binding domain 2 positively regulates the gene expression from the hypermethylated LTR. Oncogene 2005, 24, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Boeke, J.; Ammerpohl, O.; Kegel, S.; Moehren, U.; Renkawitz, R. The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J. Biol. Chem. 2000, 275, 34963–34967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ng, H.-H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999, 13, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.M.; Dai, Y.-S.; Kim, Y.; Kim, J.; Kimlin, L.; Gao, K.; Wong, D.T. Cdk2ap1 is required for epigenetic silencing of Oct4 during murine embryonic stem cell differentiation. J. Biol. Chem. 2009, 284, 6043–6047. [Google Scholar] [CrossRef] [PubMed]
- Le Guezennec, X.; Vermeulen, M.; Brinkman, A.B.; Hoeijmakers, W.A.; Cohen, A.; Lasonder, E.; Stunnenberg, H.G. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell. Biol. 2006, 26, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Cicchillitti, L.; Schepis, F.; Riccio, A.; Yeung, A.T.; Matsumoto, Y.; Golemis, E.A.; Genuardi, M.; Neri, G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc. Natl. Acad. Sci. USA 1999, 96, 3969–3974. [Google Scholar] [CrossRef] [PubMed]
- Screaton, R.A.; Kiessling, S.; Sansom, O.J.; Millar, C.B.; Maddison, K.; Bird, A.; Clarke, A.R.; Frisch, S.M. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: A potential link between genome surveillance and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 5211–5216. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Gu, Z.; Horii, A.; Fukushige, S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16INK4a and hMLH1 genes. Mol. Cell. Biol. 2005, 25, 4388–4396. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.-G.; Chan, D.W.; Reynolds, A.B.; Qin, J.; Wong, J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 2003, 12, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.M.; Reynolds, A.B. The catenin p120 ctn interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol. Cell. Biol. 1999, 19, 3614–3623. [Google Scholar] [CrossRef] [PubMed]
- Ruzov, A.; Hackett, J.A.; Prokhortchouk, A.; Reddington, J.P.; Madej, M.J.; Dunican, D.S.; Prokhortchouk, E.; Pennings, S.; Meehan, R.R. The interaction of xKaiso with xTcf3: A revised model for integration of epigenetic and Wnt signalling pathways. Development 2009, 136, 723–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, J.E.; Costa, M. Epigenetics and the environment. Ann. N. Y. Acad. Sci. 2003, 983, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef] [PubMed]
- Zaina, S.; Lindholm, M.W.; Lund, G. Nutrition and aberrant DNA methylation patterns in atherosclerosis: More than just hyperhomocysteinemia? J. Nutr. 2005, 135, 5–8. [Google Scholar] [PubMed]
- Friso, S.; Choi, S.-W. Gene-nutrient interactions and DNA methylation. J. Nutr. 2002, 132, 2382S–2387S. [Google Scholar] [PubMed]
- Hiltunen, M.O.; Turunen, M.P.; Häkkinen, T.P.; Rutanen, J.; Hedman, M.; Mäkinen, K.; Turunen, A.-M.; Aalto-Setalä, K.; Ylä-Herttuala, S. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc. Med. 2002, 7, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Luch, A. Nature and nurture–lessons from chemical carcinogenesis. Nat. Rev. Cancer 2005, 5, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ergul, E.; Sazci, A.; Utkan, Z.; Canturk, N.Z. Polymorphisms in the MTHFR gene are associated with breast cancer. Tumor Biol. 2004, 24, 286–290. [Google Scholar] [CrossRef]
- Le Marchand, L.; Haiman, C.A.; Wilkens, L.R.; Kolonel, L.N.; Henderson, B.E. MTHFR polymorphisms, diet, HRT, and breast cancer risk: The multiethnic cohort study. Cancer Epidemiol. Prev. Biomark. 2004, 13, 2071–2077. [Google Scholar]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Klose, R.; Bird, A. MeCP2 repression goes nonglobal. Science 2003, 302, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Tufarelli, C.; Stanley, J.A.S.; Garrick, D.; Sharpe, J.A.; Ayyub, H.; Wood, W.G.; Higgs, D.R. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat. Genet. 2003, 34, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar] [PubMed]
- Suter, C.M.; Martin, D.I.; Ward, R.L. Germline epimutation of MLH1 in individuals with multiple cancers. Nat. Genet. 2004, 36, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [PubMed]
- Jones, P.A.; Laird, P.W. Cancer-epigenetics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Hake, S.; Xiao, A.; Allis, C. Linking the epigenetic ‘language’of covalent histone modifications to cancer. Br. J. Cancer 2004, 90, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Grignani, F.; De Matteis, S.; Nervi, C.; Tomassoni, L.; Gelmetti, V.; Cioce, M.; Fanelli, M.; Ruthardt, M.; Ferrara, F.F.; Zamir, I. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature 1998, 391, 815–818. [Google Scholar] [PubMed]
- Jones, L.K.; Saha, V. Chromatin modification, leukaemia and implications for therapy. Br. J. Haematol. 2002, 118, 714–727. [Google Scholar] [PubMed]
- Roberts, C.W.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef] [PubMed]
- He, L.-Z.; Tolentino, T.; Grayson, P.; Zhong, S.; Warrell, R.P.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; Pandolfi, P.P. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J. Clin. Investig. 2001, 108, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Bakke, S.; Robey, R.W.; Kang, M.H.; Blagosklonny, M.V.; Bender, J.; Brooks, R.; Piekarz, R.L.; Tucker, E.; Figg, W.D. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin. Cancer Res. 2002, 8, 718–728. [Google Scholar] [PubMed]
- Butler, L.M.; Webb, Y.; Agus, D.B.; Higgins, B.; Tolentino, T.R.; Kutko, M.C.; LaQuaglia, M.P.; Drobnjak, M.; Cordon-Cardo, C.; Scher, H.I. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin. Cancer Res. 2001, 7, 962–970. [Google Scholar] [PubMed]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar] [PubMed]
- Nervi, C.; Borello, U.; Fazi, F.; Buffa, V.; Pelicci, P.G.; Cossu, G. Inhibition of histone deacetylase activity by trichostatin A modulates gene expression during mouse embryogenesis without apparent toxicity. Cancer Res. 2001, 61, 1247–1249. [Google Scholar] [PubMed]
- Coffey, D.C.; Kutko, M.C.; Glick, R.D.; Butler, L.M.; Heller, G.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; La Quaglia, M.P. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001, 61, 3591–3594. [Google Scholar] [PubMed]
- Constantinides, P.G.; Jones, P.A.; Gevers, W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature 1977, 267, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Asgari, K.; Putzi, M.J.; Gage, W.R.; Yu, X.; Cornblatt, B.S.; Kumar, A.; Piantadosi, S.; DeWeese, T.L.; De Marzo, A.M. Reversal of GSTP1 CpG island hypermethylation and reactivation of π-class glutathione S-transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide. Cancer Res. 2001, 61, 8611–8616. [Google Scholar] [PubMed]
- Duvic, M.; Dummer, R.; Becker, J.C.; Poulalhon, N.; Romero, P.O.; Bernengo, M.G.; Lebbé, C.; Assaf, C.; Squier, M.; Williams, D. Panobinostat activity in both bexarotene-exposed and-naive patients with refractory cutaneous T-cell lymphoma: Results of a phase II trial. Eur. J. Cancer 2013, 49, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M.; Vose, J.M.; Harb, W.; Gordon, L.I.; Langdon, R.; Grant, B.; Sprague, J.; Plasencia, C.; Sirisawad, M.; Yue, J. A phase II multicenter study of the histone deacetylase inhibitor (HDACi) abexinostat (PCI-24781) in relapsed/refractory follicular lymphoma (FL) and mantle cell lymphoma (MCL). Am. Soc. Hematol. 2012, 120, 55. [Google Scholar]
- Giaccone, G.; Rajan, A.; Berman, A.; Kelly, R.J.; Szabo, E.; Lopez-Chavez, A.; Trepel, J.; Lee, M.-J.; Cao, L.; Espinoza-Delgado, I. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J. Clin. Oncol. 2011, 29, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Ning, Z.-Q.; Xing, P.-Y.; Xu, J.-L.; Cao, H.-X.; Dou, G.-F.; Meng, Z.-Y.; Shi, Y.-K.; Lu, X.-P.; Feng, F.-Y. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2012, 69, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Gore, L.; Rothenberg, M.L.; O’Bryant, C.L.; Schultz, M.K.; Sandler, A.B.; Coffin, D.; McCoy, C.; Schott, A.; Scholz, C.; Eckhardt, S.G. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin. Cancer Res. 2008, 14, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, A.; Dellacasa, C.M.; Finazzi, G.; Carobbio, A.; Ferrari, M.L.; Guglielmelli, P.; Gattoni, E.; Salmoiraghi, S.; Finazzi, M.C.; Di Tollo, S. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br. J. Haematol. 2010, 150, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Oki, Y.; Bociek, R.G.; Kuruvilla, J.; Fanale, M.; Neelapu, S.; Copeland, A.; Buglio, D.; Galal, A.; Besterman, J. Mocetinostat for relapsed classical Hodgkin's lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011, 12, 1222–1228. [Google Scholar] [CrossRef]
- Razak, A.; Hotte, S.; Siu, L.; Chen, E.; Hirte, H.; Powers, J.; Walsh, W.; Stayner, L.; Laughlin, A.; Novotny-Diermayr, V. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2011, 104, 756. [Google Scholar] [CrossRef] [PubMed]
- Child, F.; Romero, P.O.; Alvarez, R.; Bagot, M.; Stadler, R.; Weichenthal, M.; Alves, R.; Bernengo, M.G.; Beylot-Barry, M.; Cowan, R. Phase 2 Multicenter trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA cutaneous T-cell lymphoma. Br. J. Dermatol. 2016, 175, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Voso, M.T.; Santini, V.; Finelli, C.; Musto, P.; Pogliani, E.; Angelucci, E.; Fioritoni, G.; Alimena, G.; Maurillo, L.; Cortelezzi, A. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin. Cancer Res. 2009, 15, 5002–5007. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.W.; Loaiza-Bonilla, A.; Juckett, M.; DiPersio, J.F.; Roy, V.; Slack, J.; Wu, W.; Laumann, K.; Espinoza-Delgado, I.; Gore, S.D. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica 2009, 94, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, C.C.; Liu, Z.; Bowers, M.A.; Porcu, P.; Flynn, J.M.; Christian, B.; Baiocchi, R.A.; Benson, D.M.; Andritsos, L.A.; Greenfield, C.N. Phase I Study of AR-42 in Relapsed Multiple Myeloma and Lymphoma. Blood 2012, 120, 2955. [Google Scholar]
- Banerji, U.; van Doorn, L.; Papadatos-Pastos, D.; Kristeleit, R.; Debnam, P.; Tall, M.; Stewart, A.; Raynaud, F.; Garrett, M.D.; Toal, M. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin. Cancer Res. 2012, 18, 2687–2694. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Wijermans, P.; Lübbert, M.; Verhoef, G.; Klimek, V.; Bosly, A. An epigenetic approach to the treatment of advanced MDS; the experience with the DNA demethylating agent 5-aza-2′-deoxycytidine (decitabine) in 177 patients. Ann. Hematol. 2005, 84, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Klisovic, R.B.; Stock, W.; Cataland, S.; Klisovic, M.I.; Liu, S.; Blum, W.; Green, M.; Odenike, O.; Godley, L.; Burgt, J.V. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin. Cancer Res. 2008, 14, 2444–2449. [Google Scholar] [CrossRef] [PubMed]
- Burchenal, J.H.; Holmberg, E.A.; Fox, J.J.; Hemphill, S.C.; Reppert, J.A. The effects of 5-fluorodeoxycytidine, 5-fluorodeoxyuridine, and related compounds on transplanted mouse leukemias. Cancer Res. 1959, 19, 494–500. [Google Scholar] [PubMed]
- Yoo, C.; Cheng, J.; Jones, P. Zebularine: A New Drug for Epigenetic Therapy; Portland Press Limited: London, UK, 2004. [Google Scholar]
Figure 1.
Modification marks of various histones (histone residues are represented by a single-letter abbreviation; the numbers mentioned below the residue depict their relevant position from the N-terminus of protein; modification(s) are abbreviated above the relevant residue; complete name of modification is mentioned in box).
Figure 1.
Modification marks of various histones (histone residues are represented by a single-letter abbreviation; the numbers mentioned below the residue depict their relevant position from the N-terminus of protein; modification(s) are abbreviated above the relevant residue; complete name of modification is mentioned in box).
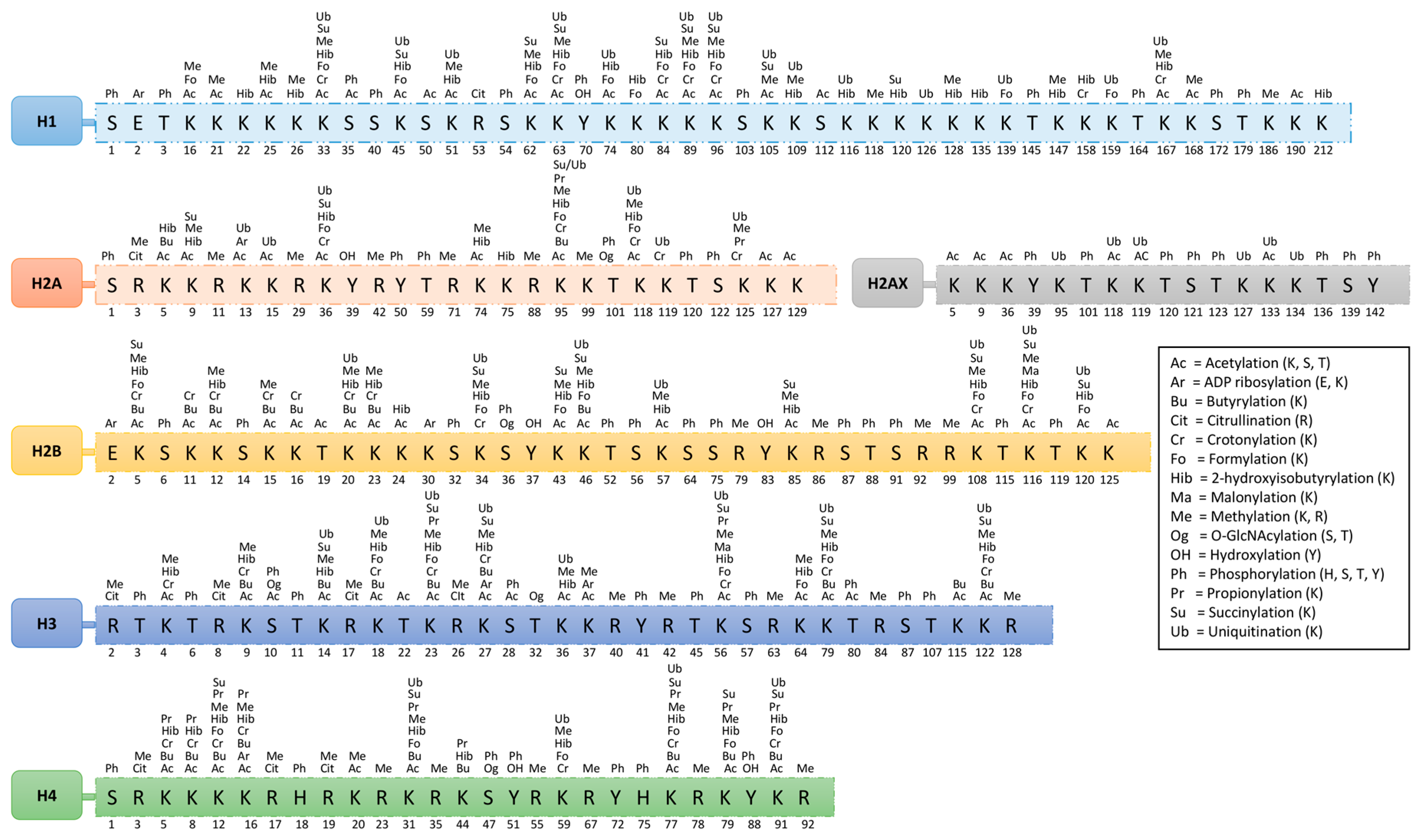
Figure 2.
Acetylation and methylation players and their interaction. (a) Demethylation and histone acetylation: Methyl marks from chromatin DNA are removed by active demethylation or passive demethylation. (I) In active demethylation, (i) 5mC (5-methyl cytosine) is converted to thymine and T/G mismatch is repaired by DNA glycosylases (also called direct removal) and (ii) the TET (10–11 translocation) enzyme makes various modified forms (5hmC, 5fmC, 5caC, etc.) that are repaired by glycosylases via base excision repair (also called indirect removal). (II) In passive demethylation, no methylation of newly replicated DNA happens. Methyl marks from chromatin histones are removed by recruitment of histone demethylases (HDM). Some activators bind to non-methylated chromatin and help in histone acetylation via recruiting histone acetyltransferases (HATs). (b) Deacetylation and methylation: (I) Due to some environmental or intrinsic changes, (i) partial CpG methylation happens and is read by either (α) proteins containing MBD (methyl binding domain) and SET (Su(var)3–9, Enhancer-of-zeste, and Trithorax) domains that recruit histone methyltransferases for direct methylation or (β) MeCP2 (methyl-CpG binding protein2) which recruits HDAC (histone deacetylase), DNMTs (DNA methyltransferases) and HMT (histone methyltransferase) for indirect methylation. (ii) Imbalance between HATs and HDACs happens, which causes hypoacetylation and recruitment of HMTs. Histone methylation marks are read by chromodomain-containing proteins (chromo) that recruit DNMTs for CpG methylation. (II) Acetylated histone marks are read by bromodomain-containing proteins (BRD): some BRD-containing proteins like SMARCA2, SMARCA4 etc. act like activators of transcription, while others like BAZ1A, BAZ2A etc. act as repressors of transcription.
Figure 2.
Acetylation and methylation players and their interaction. (a) Demethylation and histone acetylation: Methyl marks from chromatin DNA are removed by active demethylation or passive demethylation. (I) In active demethylation, (i) 5mC (5-methyl cytosine) is converted to thymine and T/G mismatch is repaired by DNA glycosylases (also called direct removal) and (ii) the TET (10–11 translocation) enzyme makes various modified forms (5hmC, 5fmC, 5caC, etc.) that are repaired by glycosylases via base excision repair (also called indirect removal). (II) In passive demethylation, no methylation of newly replicated DNA happens. Methyl marks from chromatin histones are removed by recruitment of histone demethylases (HDM). Some activators bind to non-methylated chromatin and help in histone acetylation via recruiting histone acetyltransferases (HATs). (b) Deacetylation and methylation: (I) Due to some environmental or intrinsic changes, (i) partial CpG methylation happens and is read by either (α) proteins containing MBD (methyl binding domain) and SET (Su(var)3–9, Enhancer-of-zeste, and Trithorax) domains that recruit histone methyltransferases for direct methylation or (β) MeCP2 (methyl-CpG binding protein2) which recruits HDAC (histone deacetylase), DNMTs (DNA methyltransferases) and HMT (histone methyltransferase) for indirect methylation. (ii) Imbalance between HATs and HDACs happens, which causes hypoacetylation and recruitment of HMTs. Histone methylation marks are read by chromodomain-containing proteins (chromo) that recruit DNMTs for CpG methylation. (II) Acetylated histone marks are read by bromodomain-containing proteins (BRD): some BRD-containing proteins like SMARCA2, SMARCA4 etc. act like activators of transcription, while others like BAZ1A, BAZ2A etc. act as repressors of transcription.

Figure 3.
Histone acetyltransferase (HAT) families and complexes. (a) Bar diagram of different HAT family members (family name in parentheses) with their associated domains; (b) tGCN5/Co-A/histone H3 complex: cyan = tGCN5, purple = histone H3 peptide; (c) yESA1/Co-A complex: cornflower blue = yESA1, elemental structure = Co-A.
Figure 3.
Histone acetyltransferase (HAT) families and complexes. (a) Bar diagram of different HAT family members (family name in parentheses) with their associated domains; (b) tGCN5/Co-A/histone H3 complex: cyan = tGCN5, purple = histone H3 peptide; (c) yESA1/Co-A complex: cornflower blue = yESA1, elemental structure = Co-A.
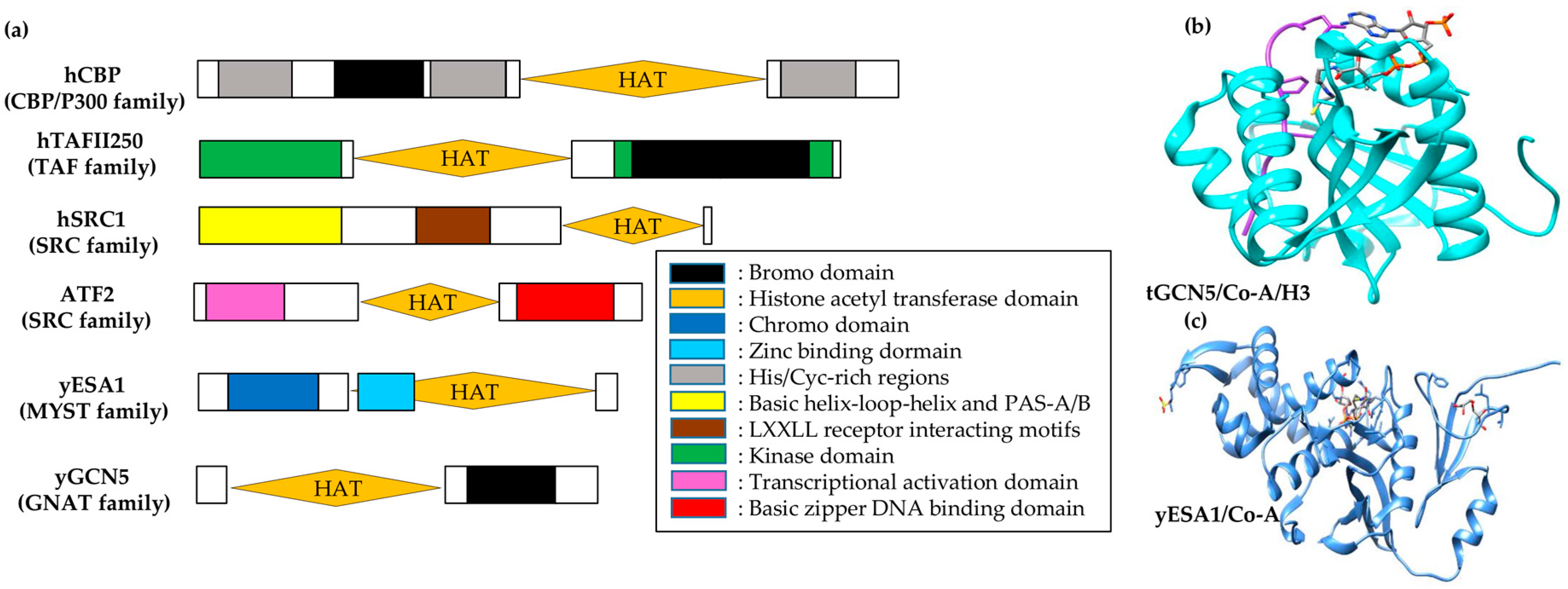
Figure 4.
Structure and complexes of epigenetic proteins. (a) Structure of SP100 (Speckled 100 kDa): the hydrophobic cavity formed by helices can accommodate histone sequences (PDB ID: 4PTB) (orange = helices, gray = loops); (b) Crystal structure of TRIM24 PHD-BRD complexed with H3K27 acetylated peptide (PDB ID: 3O35) (yellow = PHD, red = bromodomain, blue = H3K4ac, cornflower blue = H3K27ac, dark gray = zinc (II) ions (c) Crystal structure of HDAC-like protein (Chain A) bound to SAHA (PDB ID: 1ZZ1) (red = SAHA, dark gray ball = Zinc, purple ball = potassium); (d) Crystal structure of DNMT3A ADD domain (Chain C) bound to H3 peptide (PDB ID: 4QBQ) (cyan = H3 peptide, dark grey = zinc); (e) Crystal structure of TET2 protein in complex with 5hmC (PDB ID: 5DEU) (cyan = TET2, red = DNA, yellow = 2′-deoxy-5-(hydroxymethyl) cytidine 5′-(dihydrogen phosphate), dim gray ball = Fe (III) ion, orange = 2-(n-morpholino)-ethanesulfonic acid, green = n-oxalylglycine, blue = zinc (II) ion (f) Solution structure of the methylated CpG binding domain of human MBD1in complex with methylated DNA (PDB ID: 1IG4) (cyan = binding domain of MBD1, yellow = DNA, red = methylated cytosine).
Figure 4.
Structure and complexes of epigenetic proteins. (a) Structure of SP100 (Speckled 100 kDa): the hydrophobic cavity formed by helices can accommodate histone sequences (PDB ID: 4PTB) (orange = helices, gray = loops); (b) Crystal structure of TRIM24 PHD-BRD complexed with H3K27 acetylated peptide (PDB ID: 3O35) (yellow = PHD, red = bromodomain, blue = H3K4ac, cornflower blue = H3K27ac, dark gray = zinc (II) ions (c) Crystal structure of HDAC-like protein (Chain A) bound to SAHA (PDB ID: 1ZZ1) (red = SAHA, dark gray ball = Zinc, purple ball = potassium); (d) Crystal structure of DNMT3A ADD domain (Chain C) bound to H3 peptide (PDB ID: 4QBQ) (cyan = H3 peptide, dark grey = zinc); (e) Crystal structure of TET2 protein in complex with 5hmC (PDB ID: 5DEU) (cyan = TET2, red = DNA, yellow = 2′-deoxy-5-(hydroxymethyl) cytidine 5′-(dihydrogen phosphate), dim gray ball = Fe (III) ion, orange = 2-(n-morpholino)-ethanesulfonic acid, green = n-oxalylglycine, blue = zinc (II) ion (f) Solution structure of the methylated CpG binding domain of human MBD1in complex with methylated DNA (PDB ID: 1IG4) (cyan = binding domain of MBD1, yellow = DNA, red = methylated cytosine).

Figure 5.
Members of mammalian DNA methyltransferase (DNMT) families. (a) Structural comparison of all mammalian DNMTs; (b) function of each member for DNA methylation: initial CpG methylation (de novo) is established by the DNMT3 family, while it is maintained by another DNMT family (DNMT1). * DNMT2 is an example of divergent evolution and methylates the tRNAAsp anticodon loop at Cys38.
Figure 5.
Members of mammalian DNA methyltransferase (DNMT) families. (a) Structural comparison of all mammalian DNMTs; (b) function of each member for DNA methylation: initial CpG methylation (de novo) is established by the DNMT3 family, while it is maintained by another DNMT family (DNMT1). * DNMT2 is an example of divergent evolution and methylates the tRNAAsp anticodon loop at Cys38.
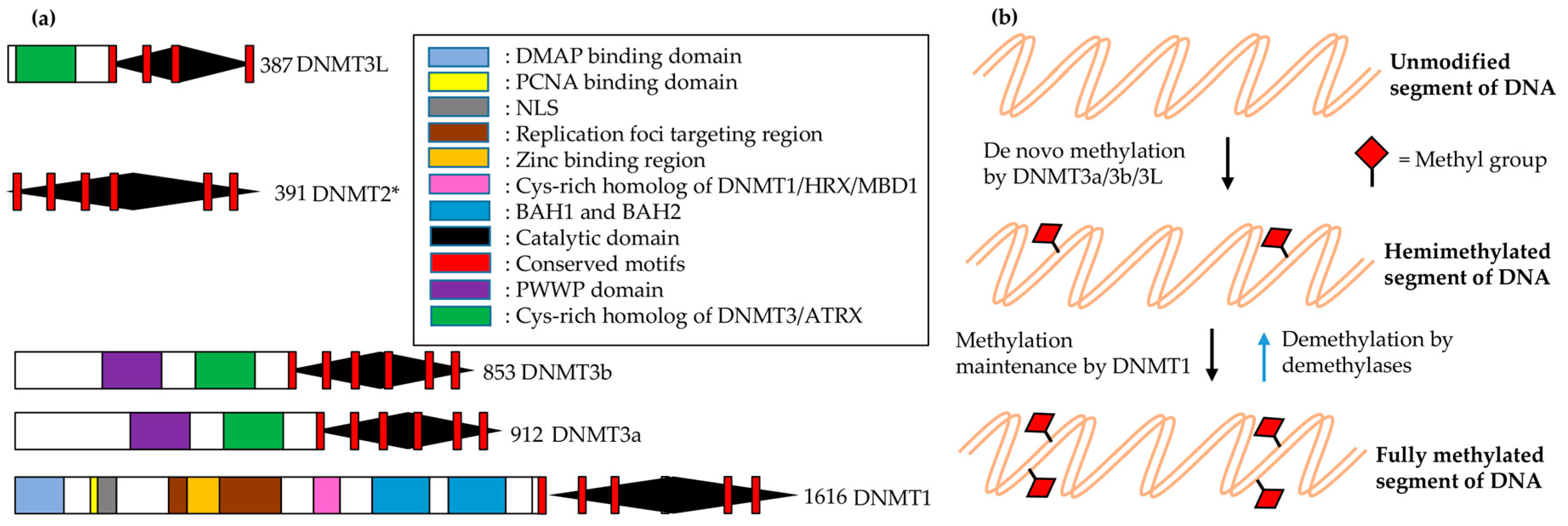

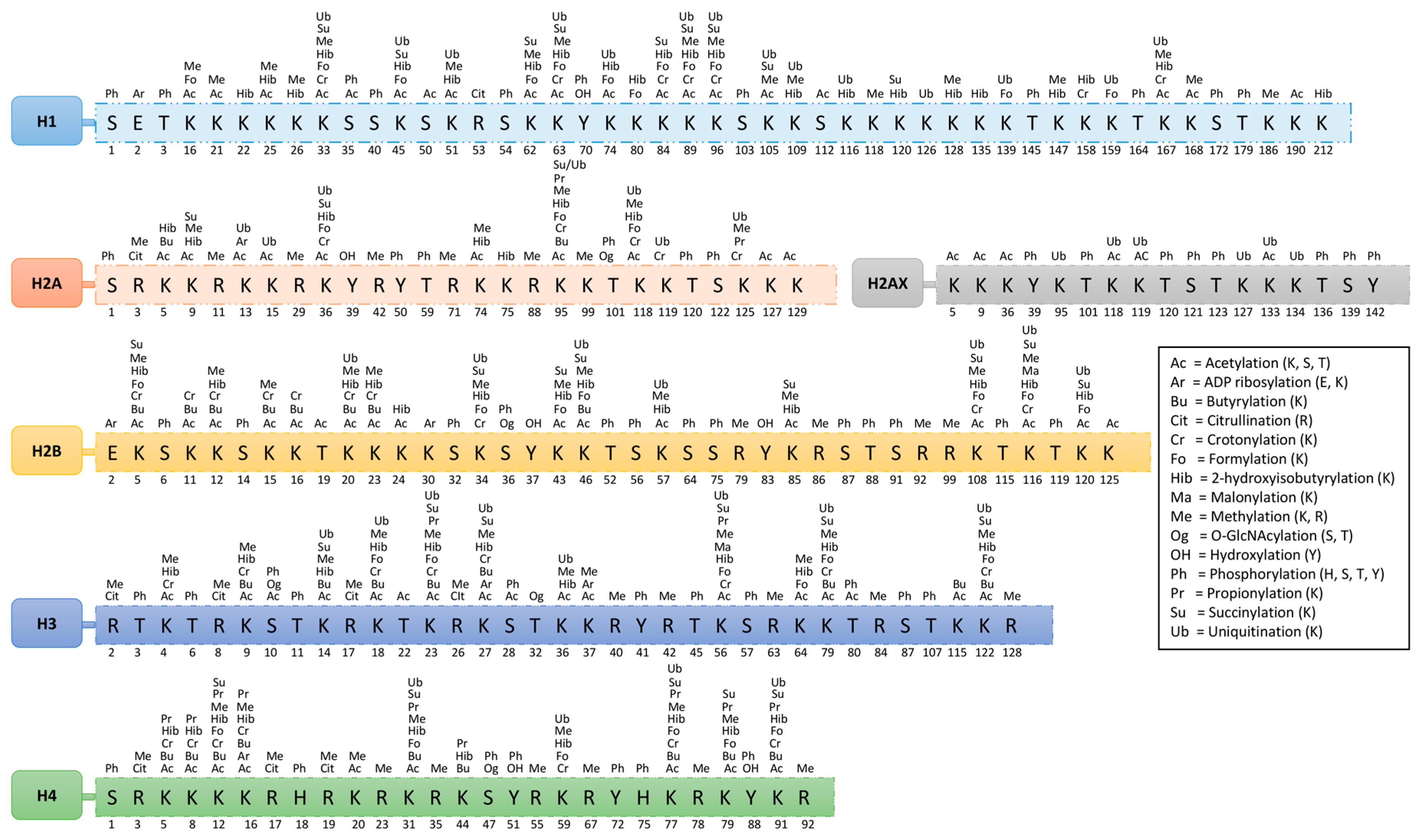{kind=link}
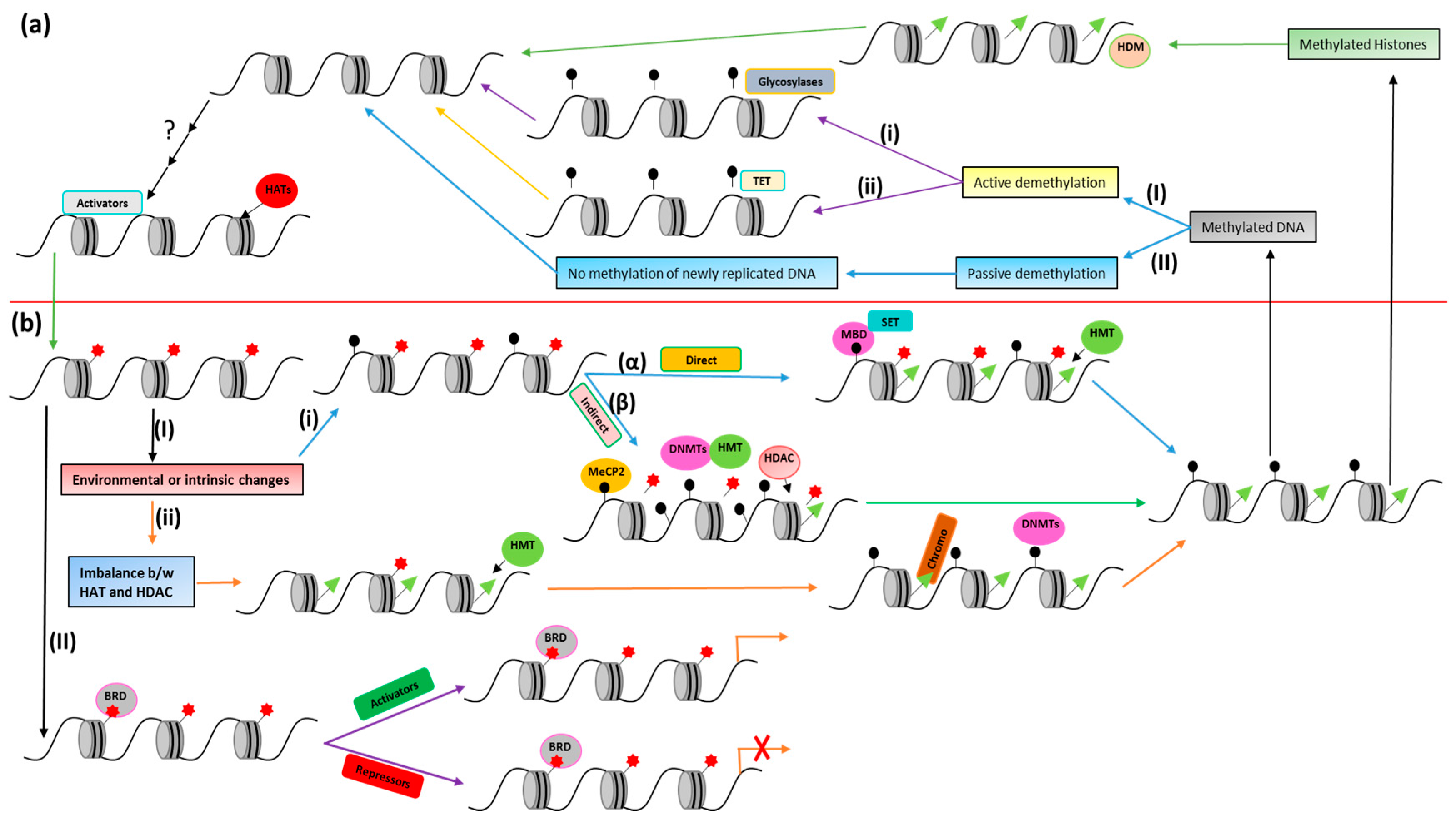{kind=link}
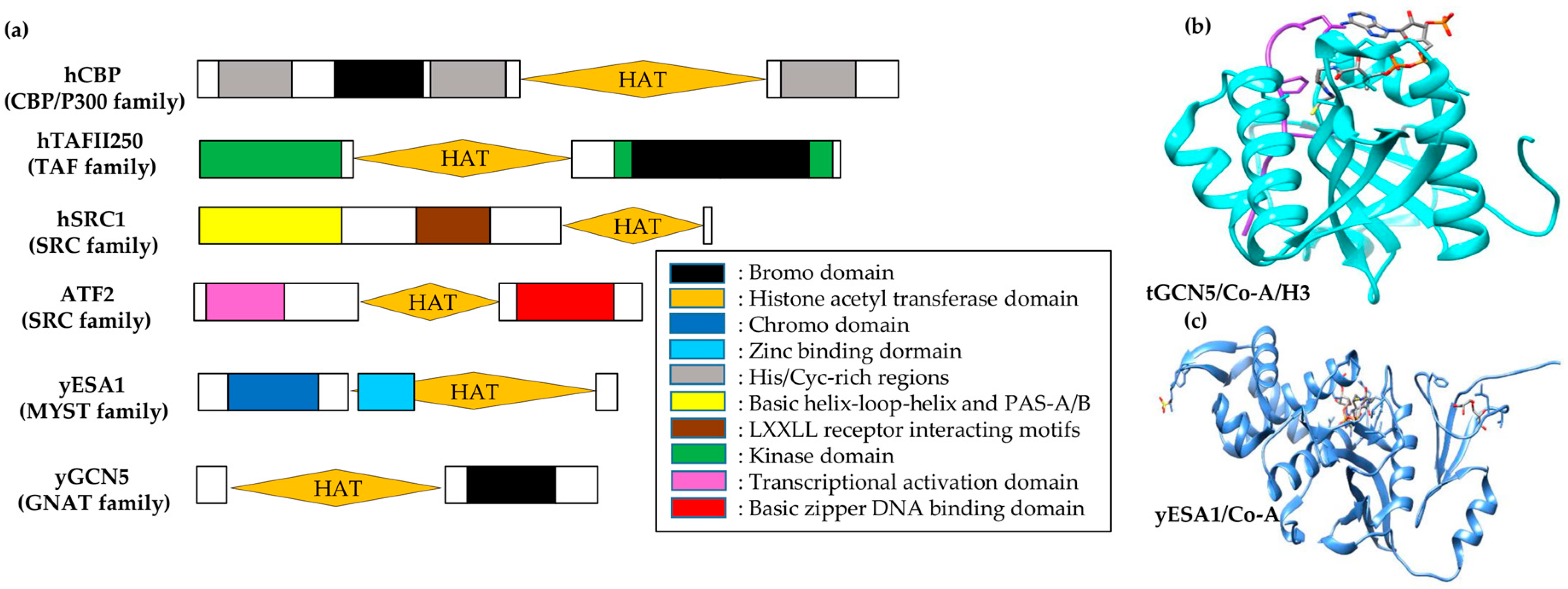{kind=link}
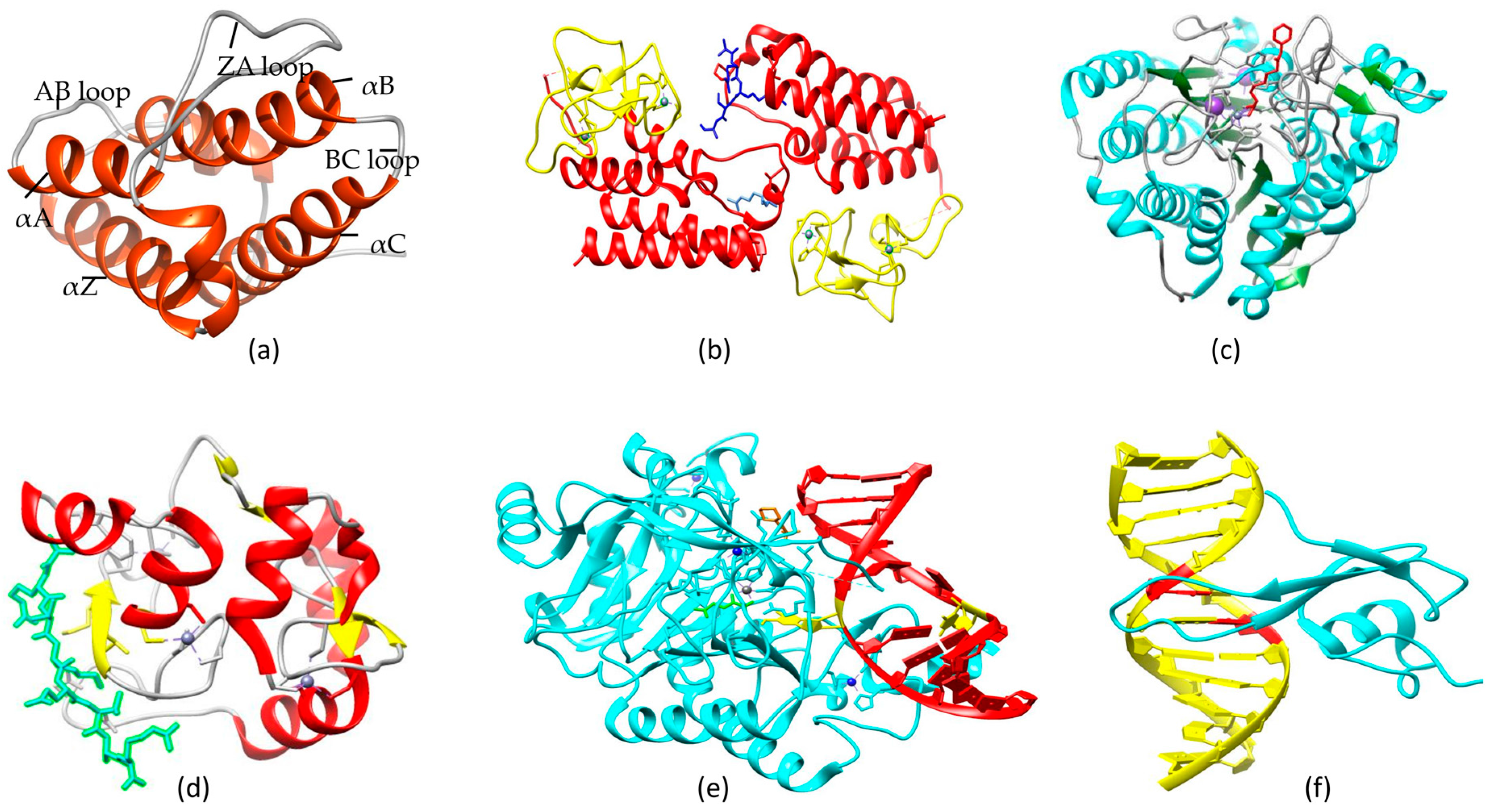{kind=link}
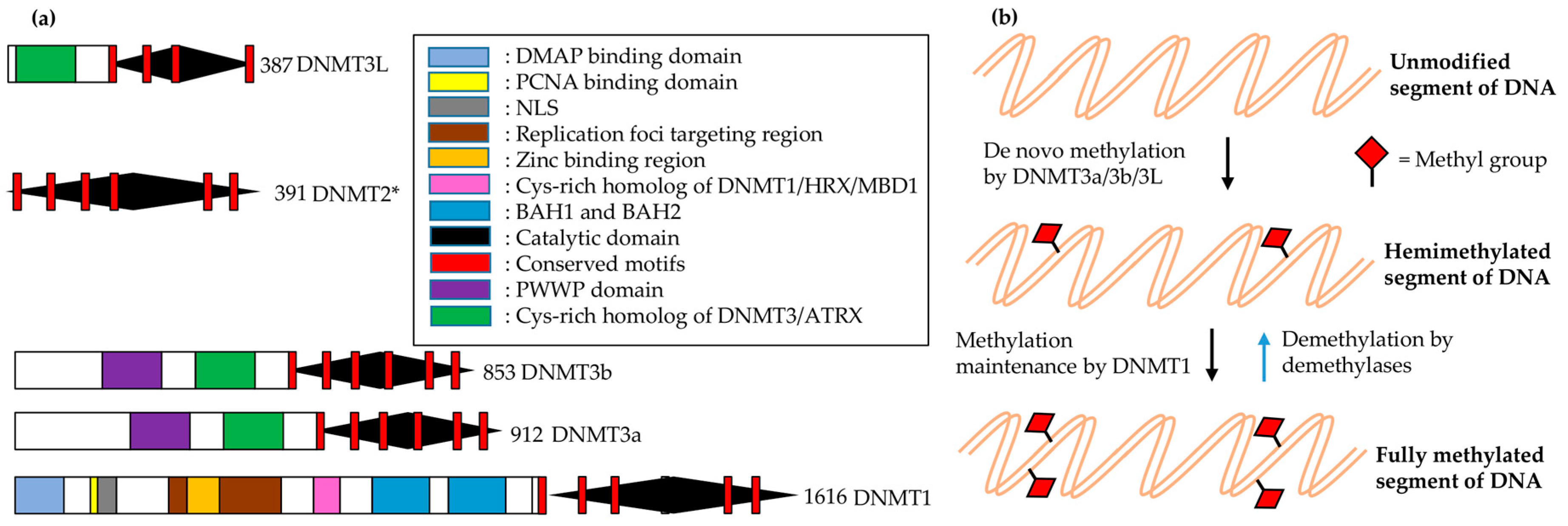{kind=link}
Table 1.
Histone target(s) of various modifications and their associated functions.
| Modification (Short Form) | Function | Target Residue/s | Reference |
|---|---|---|---|
| Methylation (me) | Repair, transcription | K-me1, K-me2, K-me3 | [34,35] |
| Transcription | R-me1, R-me2a, R-me2s | [36] | |
| Acetylation (ac) | Condensation, repair, transcription, replication | K-ac | [37,38,39] |
| Phosphorylation (ph) | Repair, transcription, condensation | T-ph, S-ph | [40,41] |
| Sumoylation (su) | Transcription, repair | K-su | [42,43] |
| Ubiquitylation (ub) | Repair, transcription | K-ub | [44,45] |
| ADP ribosylation (ar) | Transcription, repair, replication | E-ar | [46,47,48] |
| Proline isomerization | Transcription | P-cis > P-trans | [49] |
| Deimination | Transcription | R > Cit | [50] |
Abbreviations: a = asymmetric; Cit = citrulline; E = glutamic acid; K = lysine; P = proline; R = arginine; S = serine; s = symmetric, T = threonine.
Table 2.
HAT family members and their properties.
| HAT Family | Proteins | Enzyme | Organism | Orthologs | Substrate | Function | References |
|---|---|---|---|---|---|---|---|
| GNAT | PCAF | HAT A | Human | Mouse, chicken, lizard, zebrafish | H3 (nu) | Coactivator | [70] |
| GCN5 | HAT A | Human, yeast | Mouse, chicken, lizard, African clawed frog, zebrafish | H2A (nu), H2B (nu/free), H3 (K14), H4 (K8, K16) (nu), Sin1p, All core (nu) | Coactivator | [71] | |
| HAT1 | HAT B | Human | - | H4 (K12, K5) (free) | Acetylation of soluble histones | [72] | |
| HPA2 | Yeast | H3, H4 | Chromatin regulator, transferase | [73] | |||
| ELP3 | - | H4, H3, H2B, H2A | Transcription elongation | [74] | |||
| MYST | MOZ | Human | Mouse, chicken, lizard, zebrafish | Leukemogenesis | [75] | ||
| ESA1 | HAT | Yeast | H2A, H3, H4 (free) | Cell cycle progression | [62] | ||
| SAS3 (NuA3) | Yeast | H3 | Silencing | [63] | |||
| SAS2 | Yeast | Unknown | Silencing | [63] | |||
| HBO1 (KAT7) | Human | Mouse, chicken, lizard, African clawed frog, zebrafish | H3, H4 | Origin recognition interaction | [76] | ||
| MOF (KAT8/MSL) | Fruit fly | Mouse, lizard, African clawed frog, zebrafish | H2A, H3, H4 | Dosage compensation | [77] | ||
| TIP60 (KAT5) | HAT A | Human | Mouse, chicken, lizard, African clawed frog, zebrafish | H2A, H3, H4 | HIV TAT interaction | [78] | |
| P300/CBP | CBP (CREBBP) | HAT A | Humans, worms | - | H4 (K5, 9, 12, 16), all core (nu), p53 (K373, 382, peptide) TFIIF, TFIIEβ | Global coactivator | [79] |
| P300 | HAT A | - | Mouse, chicken, lizard, zebrafish | - | - | [79] | |
| Basal transcription | TAF1 (TAFII-250) | HAT A | Humans | Mouse, chicken, lizard, African clawed frog, zebrafish | TFIIEβ, H4 (free), H3 (K14) | Transcription initiation | [80] |
| TFC3 | HAT A | - | - | H2A, H3, H4 | - | [81] | |
| NUT1 | HAT A | Yeast | H3, H4 | Transcription mediator | [82] | ||
| SRC | NCOA3 (SRC-3) | HAT A | Humans | - | H4 (nu), H3 | Steroid receptor coactivators | [83] |
| NCOA1 (SRC-1) | HAT A | Human | Mouse, chicken, lizard, zebrafish | H4 (nu), H3 (K9, 14, peptide) | Steroid and nuclear hormone coactivator | [83] | |
| NCOA2 (SRC-2) | - | Mouse, chicken, lizard, African clawed frog, zebrafish | - | [84] | |||
| GRIP1 | - | Mouse, chicken, lizard, zebrafish | Trafficking and organization of transmembrane proteins | [85] | |||
| ATF2 (CREB2) | - | Mouse, chicken, lizard, African clawed frog, zebrafish | DNA sequence specific binding activator | [86] |
Abbreviations: ATF2 = activating transcription factor-2; CBP/CREBBP = CREB binding protein; CREB2 = CAMP response element binding protein-2; ELP3 = elongator protein-3; ESA1 = essential Sas2-related acetyltransferase-1; GCN5 = general control nonderepressible-5; GNAT = Gcn5-related N-acetyltransferases; GRIP1 = glutamate receptor interacting protein-1; HAT1 = histone acetyl transferase-1; HBO1 = human acetylase binding to ORC1; HPA2 = histone and other protein acetyltransferase-2; K = lysine; KAT5 = lysine acetyltransferase-5; KAT7 = lysine acetyltransferase-7; KAT8 = lysine acetyltransferase-8; MOF = males absent on the first protein; MOZ = monocytic leukemia zinc finger; MSL = male-specific lethal; MYST = MOZ, YBF2/SAS3, SAS2 and TIP60 protein; NCOA2 = nuclear receptor coactivator-2; ; NCOA3 = nuclear receptor coactivator-3; nu = nucleus; NuA3 = nucleosomal acetyltransferase of histone H3; NUT1 = negative regulation of URS Two-1; PCAF = P300/CBP-associated factor; SAS2 = something about silencing-2; SAS3 = something about silencing-3; SRC-2 = steroid receptor coactivator-2; SRC-3 = steroid receptor coactivator-3; TAF1 = TATA-box binding protein-associated factor-1; TAT = transactivator of transcription; TFIIEβ = transcription factor II E subunit β; TFC3 = transcription factor class-3; TIP60 = 60 kDa Tat-interactive protein.
Table 3.
Bromodomain family members and their properties.
| Subfamily | Proteins | No. of BRDs | Other Domains | Function | Localization | Reference |
|---|---|---|---|---|---|---|
| I | PCAF | 1 | PCAF_nGNAT | HAT | Nu | [87] |
| GCN5L2 | 1 | PCAF_nGNAT | HAT | Nu | [87] | |
| FALZ/BPTF | 1 | WSD, PHD, WHIM1, DDT | Transcription factor | Nu | [88] | |
| CECR2 | 1 | - | Chromatin remodeler | Nu | [89] | |
| II | BAZ1A | 1 | PHD, WSD, WHIM1, DDT, WAC_Acf1_DNA_bd | Chromatin remodeler | Nu | [90] |
| BRDT | 2 | CTM, ET | Transcription regulator, chromatin remodeler, spermatogenesis | Nu | [91] | |
| BRD4 | 2 | CTM, ET | Transcription regulator, chromatin remodeler | Nu | [92] | |
| BRD3 | 2 | ET | Transcription regulator, erythropoiesis | Nu | [93] | |
| BRD2 | 2 | ET | Transcription regulator | Nu | [93] | |
| III | PHIP | 2 | WD40 | Insulin signaling | Nu | [94] |
| BRWD3 | 2 | WD40 | JAK-STAT signaling | Nu, Cyt | [95] | |
| BAZ1B | 1 | PHD, WSD, WHIM1, WAC_Acf1_DNA_bd | Spermatogenesis, tyrosine kinase, transcription regulator, chromatin remodeler | Nu | [59,96,97] | |
| BRD8 | 2 | - | Transcription regulator | Nu | [98,99] | |
| EP300 | 1 | CREB binding, ZZ, HAT, DUF902, KIX, zf-TAZ | HAT | Nu | [100] | |
| CREBBP | 1 | Same as above | HAT | Nu | [100] | |
| WDR9 | 2 | WD40 | Chromatin remodeler | Nu | [101,102] | |
| IV | BRD9 | 1 | DUF3512 | Component of SWI/SNF | Nu, Cyt | [103] |
| BRD7 | 1 | DUF3512 | Transcription regulator | Nu | [104] | |
| BRPF3 | 1 | PWWP, PHD-like, PHD, EPL1 | Same as above | Nu | [105] | |
| BRPF1 | 1 | Same as above | Same as above | Nu, Cyt | [106] | |
| BRD1 | 1 | Same as above | Same as above | Nu, Cyt | [107] | |
| ATAD2B | 1 | AAA | Same as above | Nu | [108] | |
| ATAD2 | 1 | AAA | Same as above | Nu | [109,110,111,112] | |
| V | BAZ2B | 1 | PHD, WSD, WHIM1, DDT, MBD | Unknown | Nu, Cyt | [113] |
| BAZ2A | 1 | Same as above | Transcription repressor | Nu, Cyt | [114] | |
| TRIM66 | 1 | PHD, zf-B_box | Same as above | Nu | [115] | |
| TRIM33 | 1 | PHD, zf-B_box, zf-RING | Transcription elongation, ligase (ubiquitin) | Nu | [116,117] | |
| TRIM24 | 1 | PHD, zf-B_box | Transcription regulator | Nu, Cyt | [118] | |
| SP110 | 1 | SAND, HSR | Same as above | Nu | [119] | |
| SP100 | 1 | SAND, HSR, HMG_box | Same as above | Nu, Cyt | [120] | |
| SP140L | 1 | PHD, SAND, HSR | Unknown | Nu | [121] | |
| SP140 | 1 | SAND, HSR | Transcription regulator | Nu, Cyt | [122] | |
| VI | TRIM28 | 1 | zf-B_box, zf-RING | Transcription regulator, ligase (E3 SUMO) | Nu | [123] |
| MLL | 1 | SET, FYRN, PHD-like, PHD, zf-CXXC | Methyltransferase (histone) | Nu | [124,125] | |
| VII | TAF1L | 2 | zf-CCHC_6, DUF3591, TBP- binding | Transcription initiation | Nu | [126] |
| TAF1 | 2 | Same as above | Transcription initiation, p53 transcription regulation | Nu | [127] | |
| ZMYND11 | 1 | PWWP | Transcription repressor | Nu | [128] | |
| ZMYND8 | 1 | PWWP, DUF3544 | Transcription regulator, DNA damage | Nu | [129] | |
| VIII | SMARCA4 | 1 | SnAC, Helicase_C, SNF2_N, BRK, HAS, QLQ | Chromatin remodeler | Nu | [130] |
| SMARCA2 | 1 | Same as above | Chromatin remodeler, splicing | Nu | [131] | |
| PB1 | 6 | HMG_box, BAH | Chromatin remodeler | Nu, Cyt | [132] | |
| ASH1L | 1 | BAH, SET | Methyl transferase | Nu, Cyt | [133] |
Abbreviations: AAA = ATPases associated with a variety of cellular activities; ACF = ATP-utilizing chromatin assembly and remodeling factor; ASH1L = (absent, small, or homeotic)-1 like protein; ATAD2 = ATPase family, AAA domain containing 2; ATAD2B = ATPase family, AAA domain containing 2B; BAH = bromo-adjacent homology; BAZ1A = BRD adjacent zinc finger-1A; BAZ2A = BRD adjacent zinc finger-2A; BAZ1B = BRD adjacent zinc finger-1B; BAZ2B = BRD adjacent zinc finger-2B; BPTF = bromodomain and PHD domain transcription factor; BRD1 = bromodomain-containing protein 1; BRD2 = bromodomain-containing protein 2; BRD3 = bromodomain-containing protein 3; BRD4 = bromodomain-containing protein 4; BRD7 = bromodomain-containing protein 7; BRD8 = bromodomain-containing protein 8; BRD9 = bromodomain-containing protein 9; BRDT = bromodomain testis associated; BRK = brinker; BRPF3= bromodomain and PHD finger containing 3; BRWD3 = bromodomain and WD repeat domain containing 3; CECR2 = cat eye syndrome chromosome region, candidate 2; CREBBP = CREB binding protein; CTM = carboxy-terminal motif; CXXC = two conserved cysteine-rich clusters; Cyt = cytoplasm; DDT = DNA-binding homeobox and different transcription factors; DUF902 = domain of unknown function-902; EP300 = E1A binding protein P300; EPL1 = enhancer of polycomb-like-1; ET = extra-terminal; FALZ = fetal ALZ-50 clone 1 protein; FYRN = ”FY-rich” domain N-terminal; GCN5L2 = general control of amino acid synthesis protein 5-like 2; HMG box = high mobility group box; HSR = homogeneously-staining region; KIX = interactor of kinase-inducible domain; MBD = methyl-CpG-binding domain; MLL = mixed lineage leukemia; Nu = nucleus; PB1 = polymerase basic protein 1; PCAF = P300/CBP-associated factor; PHD = plant homeodomain; PHIP = PH-interacting protein; PWWP = Pro-Trp-Trp-Pro domain; QLQ = conserved Gln, Leu, Gln containing motif; RING = really interesting new gene; SAND = Sp100, AIRE-1, NucP41/75, DEAF-1; SET = Su(var)3-9, enhancer-of-zeste and trithorax; SMARCA2 = SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 2; SMARCA4 = SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4; SnAC = SNF2 ATP coupling; SP100 = Speckled 100 kDa; SP110 = Speckled 110 kDa; SP140L = SP140 nuclear body protein like; SUMO = small ubiquitin-related modifier; TAF1 = TATA-box binding protein associated factor 1; TAF1L = TATA-box binding protein associated factor 1 like; TRIM24 = tripartite motif containing 24; TRIM28 = tripartite motif containing 28; TRIM33 = tripartite motif containing 33; TRIM66 = tripartite motif containing 66; WAC =WSTF/Acf1/Cbp146; WD40 = 40 amino acids motif terminating in tryptophan-aspartic acid (W-D) dipeptide; WDR9 = WD Repeat domain 9; WHIM1 = WSTF, HB1, Itc1p, MBD9 motif 1; WSD = Williams-Beuren Syndrome DDT; zf-CCHC_6 = cysteine- and histidine-rich zinc finger domain; zf-TAZ = transcription adaptor putative zinc finger; ZMYND8 = zinc finger MYND-type containing 8; ZMYND11 = zinc finger MYND-type containing 11; ZZ = two zinc ion binding domain.
Table 4.
HDAC family members and their properties.
| Family | Class | Members | Tissue Distribution | Subcellular Localization | Catalytic Site | Substrates | References |
|---|---|---|---|---|---|---|---|
| Classic (Zn-dependent) | I | HDAC1 | Pervasive | Nucleus | 1 | STAT3, E2F1, MyoD, p53, SHP, androgen receptor | [149] |
| HDAC2 | Pervasive | Nucleus | 1 | STAT3, BCL6, YY1, glucocorticoid receptor | [150] | ||
| HDAC3 | Pervasive | Nucleus | 1 | MEF2D, STAT3, RELA, GATA1, YY1, SHP | [151] | ||
| HDAC8 | Pervasive? | Cytoplasm/nucleus | 1 | - | [151] | ||
| IIA | HDAC4 | Brain, skeletal muscle, heart | Cytoplasm/nucleus | 1 | HP1, GATA1, GCMA | [152] | |
| HDAC5 | Brain, skeletal muscle, heart | Cytoplasm/nucleus | 1 | HP1, SMAD7, GCMA | [152] | ||
| HDAC7 | Placenta, skeletal muscle, heart, pancreas | Cytoplasm/nucleus/mitochondria | 1 | PLAG2, PLAG1 | [153] | ||
| HDAC9 | Skeletal muscle, brain | Cytoplasm/nucleus | 1 | - | [153] | ||
| IIB | HDAC6 | Placenta, kidney, liver, heart | Cytoplasm (mostly) | 2 | SMAD7, SHP, HSP90, α-tubulin | [154] | |
| HDAC10 | Kidney, spleen, liver | Cytoplasm (mostly) | 1 | - | [151] | ||
| IV | HDAC11 | Kidney, skeletal muscle, heart, brain | Cytoplasm/nucleus | 2 | - | [151] | |
| Modern (NAD+-dependent) | III | Mammalian sirtuins (SIRT 1–7) | - | - | - | - | [155] |
| Yeast Sir2 | - | - | - | - | [155] |
Abbreviations: BCL6 = B-cell CLL/lymphoma 6; E2F1 = E2F transcription factor 1; GATA1 = GATA binding protein 1; GCMA = glial cells missing homolog A; HP1 = heterochromatin protein 1; HSP90 = heat shock protein 90 kDa; MEF2D = myocyte enhancer factor 2D; MyoD = myogenic differentiation; PLAG1 = pleomorphic adenoma gene-1; PLAG2 = pleomorphic adenoma gene-2; RELA = V-Rel avian reticuloendotheliosis viral oncogene homolog A; SHP = small heterodimer partner; SIR2 = silent information regulator-2.; SIRT = Sirtuin; SMAD7 = SMAD family member-7; STAT3 = signal transducer and activator of transcription 3; YY1 = Yin and Yang 1.
Table 5.
Histone methyltransferases (HMTs) and their properties.
| Family Name | Complex Member | Substrate | Function | References |
|---|---|---|---|---|
| Lysine-associated HMTs | ||||
| hSET1 | HCF1/ASH2/SET1 | H3 (K4) | Transcription activation | [185] |
| MLL4 | MENIN, SET1 | Same as above | Same as above | [186] |
| MLL1 | Same as above | Same as above | Cell proliferation, transcription activation | [185] |
| SET7/9 | Same as above | Silencing, transcription activation | [187] | |
| SMYD3 | Same as above | Same as above | [188] | |
| SET8 | H4 (K20) | Heterochromatin, cell cycle | [189] | |
| SUV39H1/2 | E2F1/4 | H3 (K9) | Heterochromatin, transcription repression | [190] |
| DOT1L | H3 (K79) | Silencing, transcription activation | [191] | |
| EZH2 | EDD-EZH2 | H3 (K9, 27) | Silencing, transcription repression | [192] |
| G9a | Same as above | Same as above | [193] | |
| SETDB1 | He (K9) | Silencing, heterochromatin | [194] | |
| Arginine-associated HMTs | ||||
| PRMT5 | Methylosome | SMN, H4, H2A | Heterochromatin, cell cycle | [195] |
| PRMT4 | NUMAC, P300, NCOA2, PCAF, AR | H3 (R1T, 26), PAB1, CBP, TARP | Transcription coactivation | [196] |
| PRMT1 | H4 (R3), HNRPA2B1, ETOILE, ILE3 | Transcription activation | [197] | |
Abbreviations: AR = androgen receptor; ASH2 = absent, small, or homeotic 2; CBP = CREB binding protein; DOT1L = disruptor of telomeric silencing 1-like; EDD = enriched domain detector; ETIOLE = mouse homolog of T-STAR; EZH2 = enhancer of zeste homolog-2; HCF1 = host cell factor 1; HNRPA2B1 = heterogeneous nuclear ribonucleoproteins A2/B1; MENIN = protein encoded by men1 gene (multiple endocrine neoplasia type-1); MLL4 = myeloid/lymphoid or mixed-lineage leukemia-4; NCOA2 = nuclear receptor coactivator-2; NUMAC = nucleosomal methylation activator complex; PAB1 = polyadenylate-binding protein-1; PCAF = P300/CBP-associated factor; PRMT = protein arginine N-methyltransferase 5; SET = Su(var)3-9, enhancer-of-zeste and Trithorax; SMYD3 = SET and MYND domain containing-3, TARP = TCR gamma alternate reading frame protein.
Table 6.
Methyl binding proteins and their properties.
| Protein Name | Interaction Partner | Function | References |
|---|---|---|---|
| MeCP2 | HDACs, SIN3a | Transcription repression | [229] |
| NCOR, c-SKI | Same as above | [246] | |
| HDAC2, Sin3B | Same as above | [247] | |
| Methyltransferase (H3K9) | Same as above | [248] | |
| BRM (SWI/SNF complex) | Same as above | [249] | |
| HP1 | Transcription repression during myogenic differentiation | [250] | |
| CoREST complex | Neural genes repression | [251] | |
| HMGB1 | Unknown | [252] | |
| DNMT1 | DNA methylation maintenance targeting | [253] | |
| ATRX | Neural development epigenetic regulation | [254] | |
| YB-1 | Alternative splicing | [255] | |
| CREB1 | Transcription activation | [256] | |
| MBD1 | SUV39H1-HP1 | Transcription repression | [257] |
| MPG | DNA repairing | [258] | |
| HDAC3, PML-RARα | Silencing | [259] | |
| CAF-1 p150, SETDB1, MCAF1, MCAF2 | Epigenetic inheritance, transcription repression | [260] | |
| MBD2 | RFP | Transcription repression enhancement | [261] |
| GCNF | Silencing of Oct-4 | [262] | |
| DNMT1 | DNA methylation maintenance | [263] | |
| pCAF, HATs, TACC3 | Transcription activation | [264] | |
| TAX | Same as above | [265] | |
| SIN3A | Transcription repression | [266] | |
| MEP50, PRMT5, DOC-1, RbAp46/48, HDAC1/2, P66α/β, MTA1-3, Mi-2 (NuRD complex) | Same as above | [267] | |
| MBD3 | GCNF, CDK2AP1 | Silencing of Oct-4 | [268] |
| Dnmt1 | DNA methylation maintenance | [263] | |
| MEP50, PRMT5, DOC-1, RbAp46/48, HDAC1/2, P66α/β, MTA1-3, Mi-2 (NuRD complex) | Transcription repression | [237,269] | |
| MBD4 | RFP | Transcription repression enhancement | [261] |
| MLH1 | DNA repair | [270] | |
| FADD | Apoptosis | [271] | |
| HDAC1, SIN3A | Transcription repression | [272] | |
| Kaiso | NCOR | Transcription repression | [273] |
| P120 | Wnt signaling | [274] | |
| TCF3 | Wnt signaling suppression | [275] |
Abbreviations: ATRX = alpha thalassemia/mental retardation syndrome X-linked; BRM = brahma; CAF1 = chromatin assembly factor-1; CoREST = cofactor of repressor element-1 silencing transcription factor; CREB1 = CAMP responsive element binding protein-1; c-SKI = SKI proto-oncogene; DNMT1 = DNA methyl transferase-1; DOC1 = destruction of cyclin B protein-1; FADD = Fas-associated protein with death domain; GCNF = germ cell nuclear factor; HMGB1 = high mobility group box-1; HP1 = heterochromatin protein 1; MCAF1 = MBD1-containing chromatin-associated factor-1; MCAF2 = MBD1-containing chromatin-associated factor-2; MEP50 = methylosome protein-50; MLH1 = MutL homolog-1; MPG = N-methylpurine DNA glycosylase; MTA = metastasis associated; NCOR = nuclear receptor corepressor; NuRD = nucleosome remodeling deacetylase; PML = promyelocytic leukemia; PRMT5 = protein arginine N-methyltransferase-5; RARα = retinoic acid receptor alpha; RbAP 48 = retinoblastoma-binding protein p48; RFP = RET finger protein; SETDB1 = SET domain bifurcated-1; SIN3A = SIN3 transcription regulator homolog A; SIN3B = SIN3 transcription regulator homolog B; SUV39H1 = suppressor of variegation 3–9 homolog-1; TACC3 = transforming acidic coiled-coil containing protein-3; TAX = transactivator from the X-gene region; TCF3 = transcription factor-3; YB1 = Y-box binding protein-1.
Table 7.
Some drugs against HDAC and DNA methylation that are in clinical trials.
| Name of Drug | Cancer Type | Phase | Reference |
|---|---|---|---|
| HDAC inhibitors | |||
| Panobinostat | Cutaneous T cell lymphoma | III | [305] |
| Abexinostat | Follicular lymphoma | II | [306] |
| Belinostat | Thymic malignancies | II | [307] |
| Chidamide | Solid lymphomas and tumors | II | [308] |
| Entinostat | Melanoma | II | [309] |
| Givinostat | Myeloproliferative neoplasms | II | [310] |
| Mocetinostat | B-cell malignancies | II | [311] |
| Practinostat | Prostate cancer | II | [312] |
| Quisinostat | Cutaneous T cell lymphoma | II | [313] |
| Valporate | Myelodysplasia | II | [314] |
| Vorinostat | Acute myeloid leukemia | II | [315] |
| AR-42 | Hematological malignancies | I | [316] |
| CHR-3996 | Solid tumors | I | [317] |
| Methylation inhibitors | |||
| 5-azacytidine | Myelodysplasia | III | [318] |
| 5-aza-2′-deoxycytidine (decitabine) | Myelodysplasia | III | [319] |
| MG98 | Leukemia | II | [320] |
| 5-Fluorodeoxycytidine | Leukemia | I | [321] |
| Zebularine | Acute myeloid leukemia | Preclinical | [322] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Javaid, N.; Choi, S. Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets. Genes 2017, 8, 196. https://doi.org/10.3390/genes8080196
AMA Style
Javaid N, Choi S. Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets. Genes. 2017; 8(8):196. https://doi.org/10.3390/genes8080196
Chicago/Turabian StyleJavaid, Nasir, and Sangdun Choi. 2017. "Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets" Genes 8, no. 8: 196. https://doi.org/10.3390/genes8080196
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.