Analysis of Argonaute 4-Associated Long Non-Coding RNA in Arabidopsis thaliana Sheds Novel Insights into Gene Regulation through RNA-Directed DNA Methylation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. RNA Extraction and cDNA Synthesis
2.3. Expression Analysis by Semi-Quantitative RT-PCR and RT-qPCR
2.4. Sequence-Specific RT-PCR
2.5. Nuclear RNA-Immunoprecipitation
2.6. Chromatin-Immunoprecipitation Assay
2.7. 5’ End Labeling and SDS-PAGE
2.8. Template-Switch cDNA Library Preparation and Expression Analysis of AGO4- and AGO1-Associated RNA by PCR
2.9. Deep Sequencing
2.10. Bioinformatics
2.11. Western Blot Analysis
2.12. Bisulphite Sequencing
2.13. 5’ Rapid Amplification of cDNA Ends
2.14. Infection of Arabidopsis Plants with Fusarium Oxysporum
2.15. Data Access
3. Results
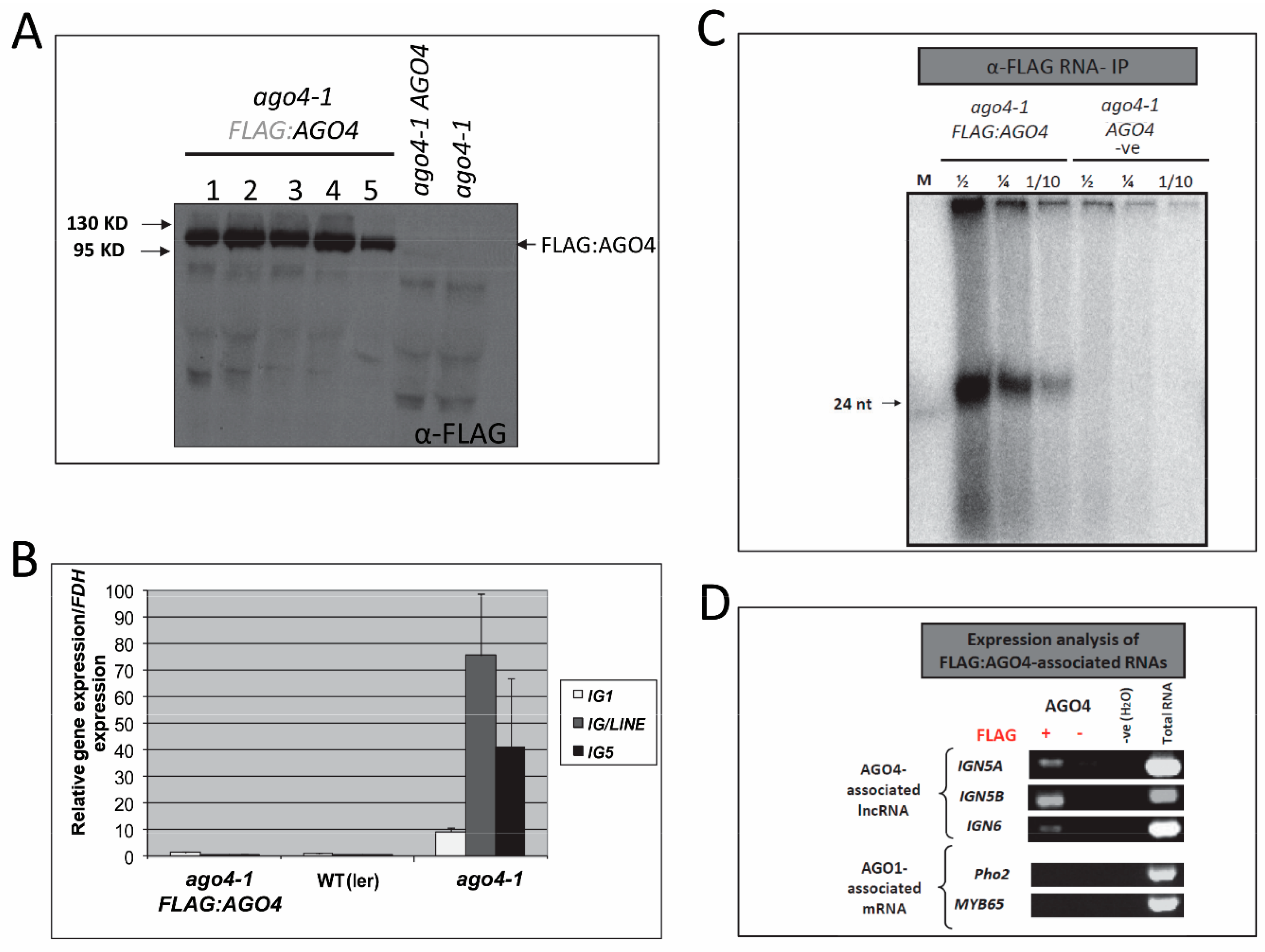
3.1. Transgenic FLAG:AGO4 Functions Like Endogenous AGO4
3.2. Anti-FLAG:AGO4 RNA Immunoprecipitation Gives Highly Enriched AGO4-Specific RNA Species
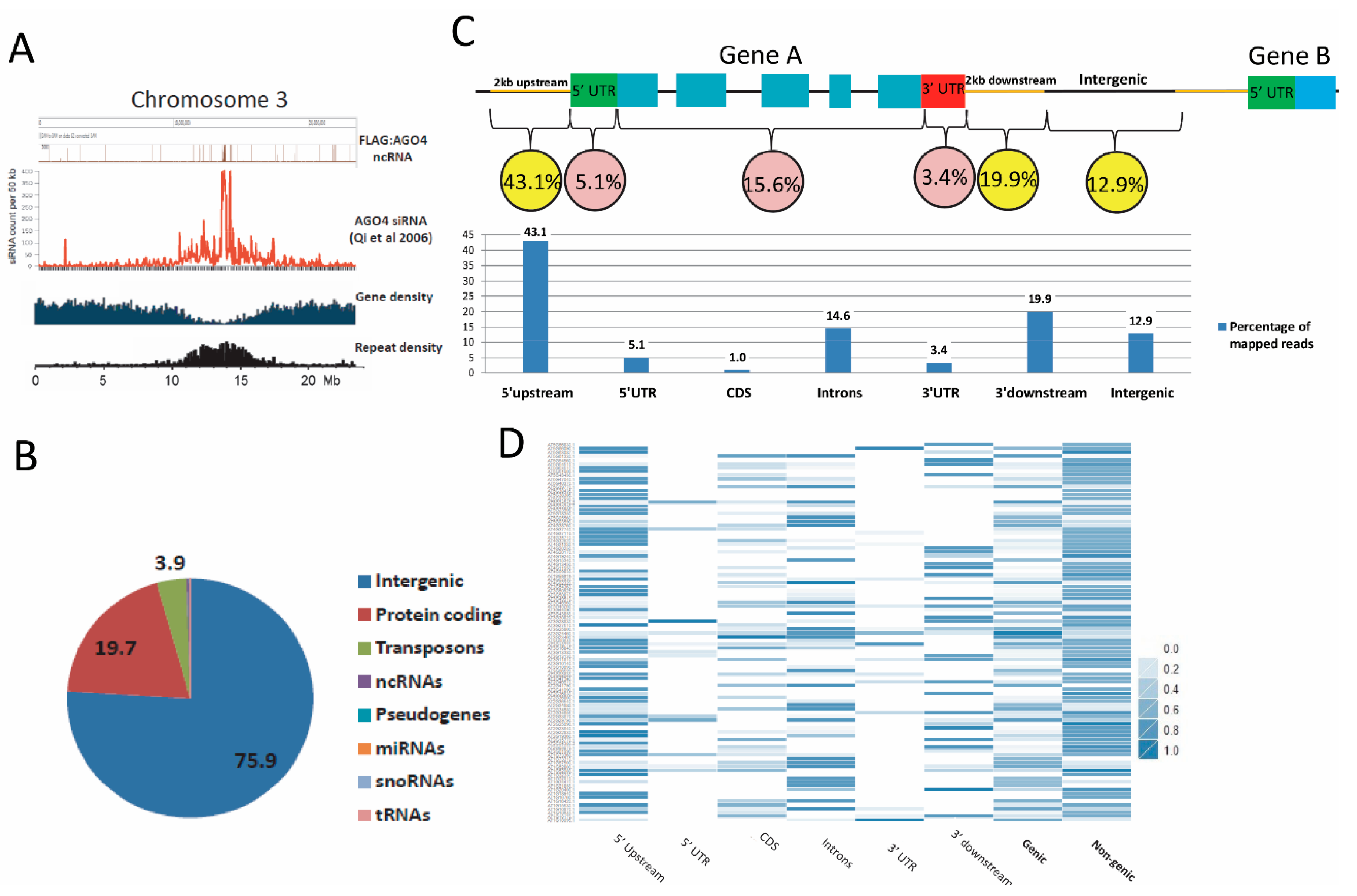
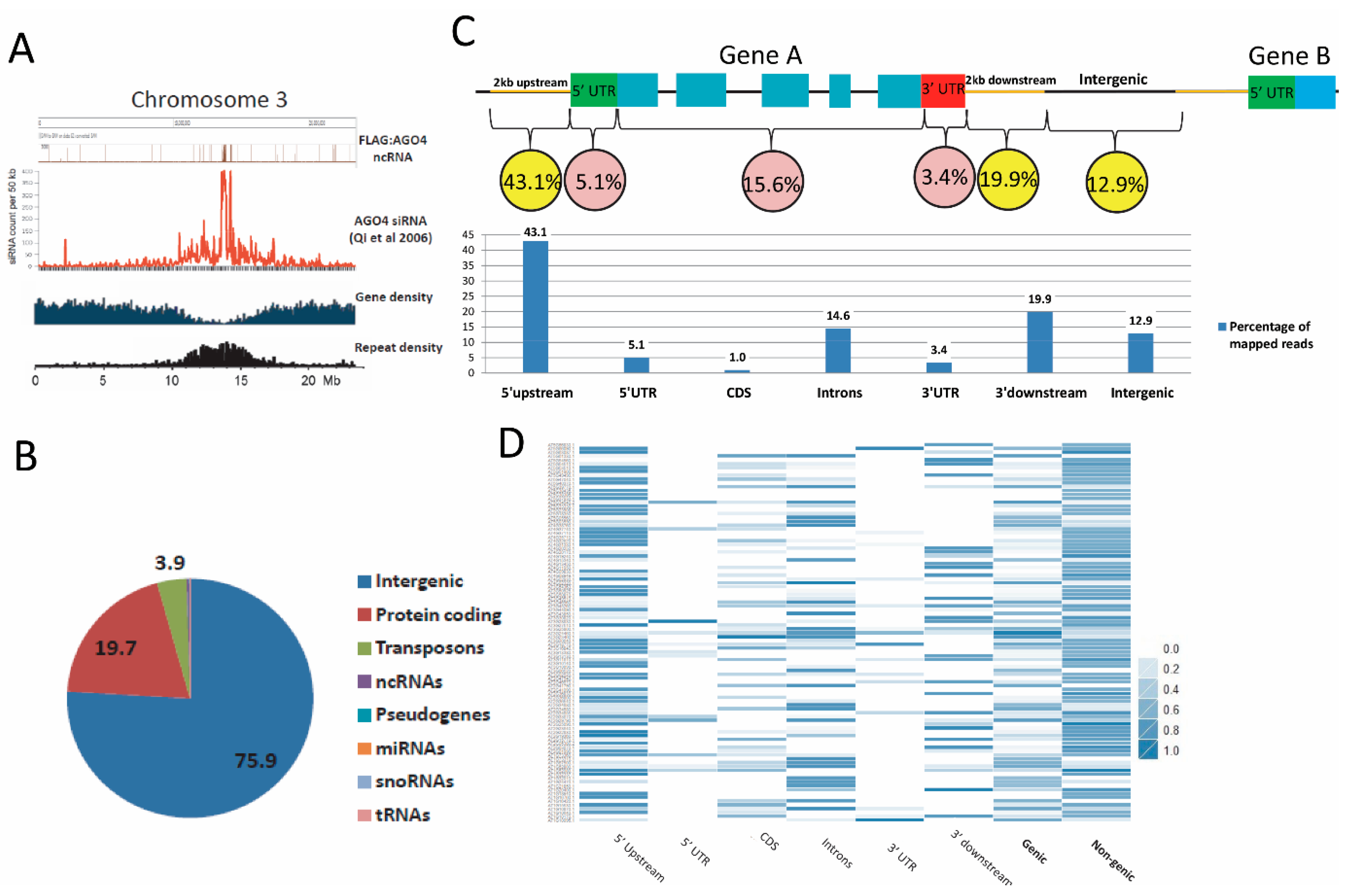
3.3. Establishment of an AGO4-Associated Non-Coding RNA Landscape by RNA-IP Seq
3.4. FLAG:AGO4 RNA-IP Seq Data Show Enrichment in RNA Reads from Noncoding Regions of Genes
3.5. AGO4-Associated lncRNAs Overlap with AGO4 and POL V ChIP-Seq Data, TEs and AGO4-IP sRNAs
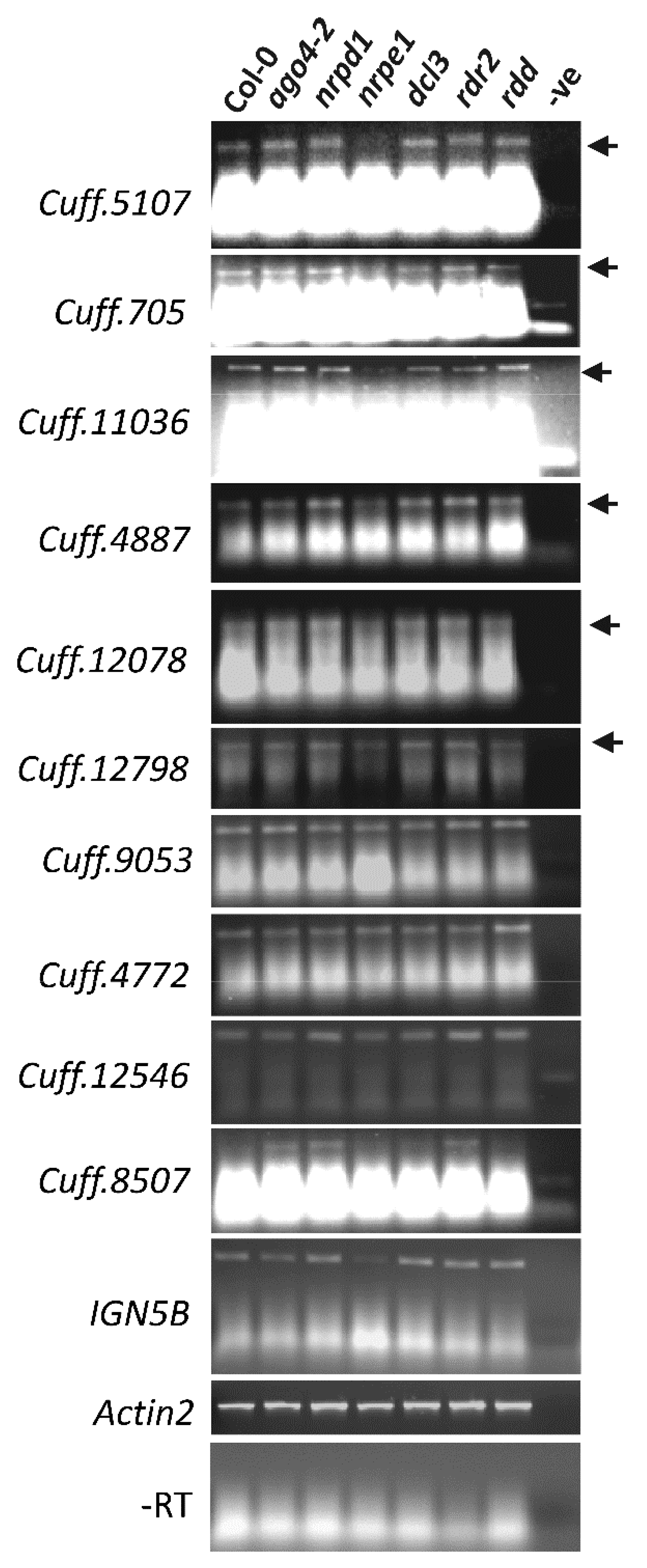
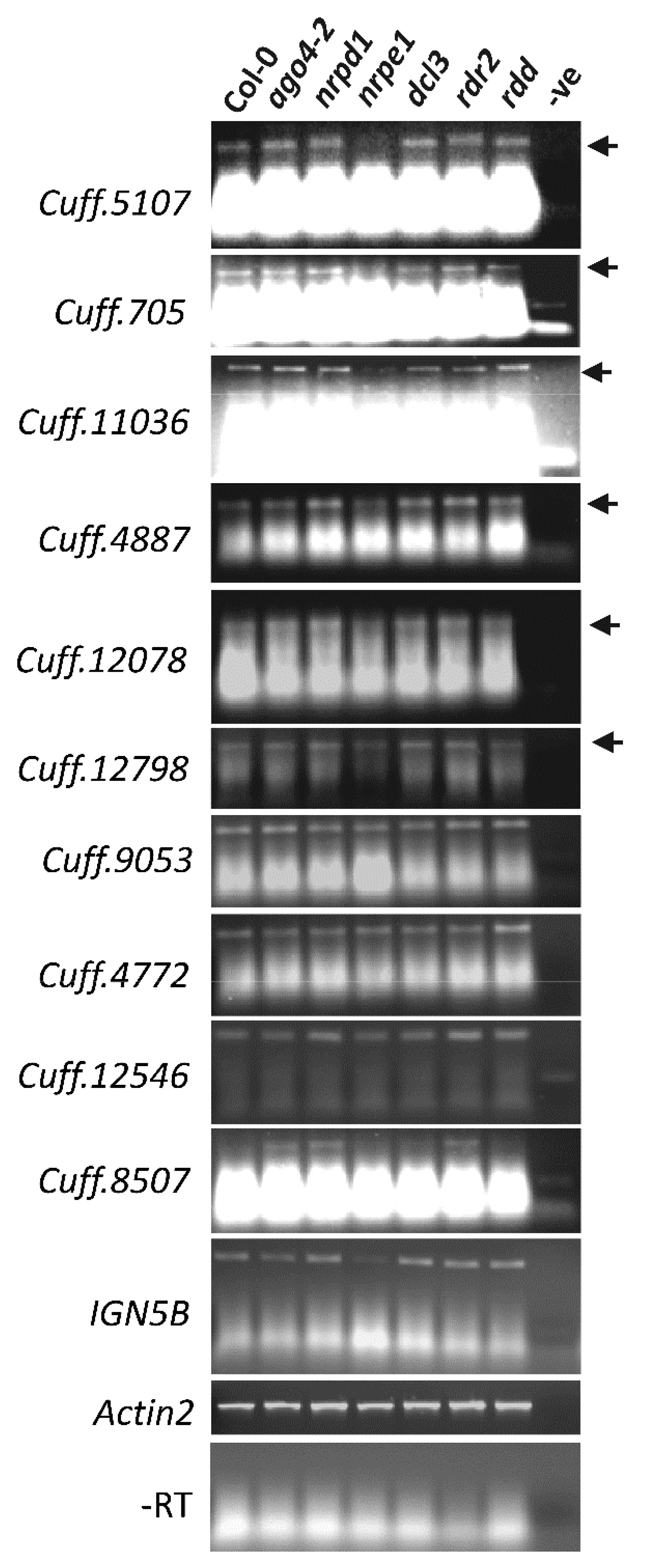
3.6. AGO4-Associated lncRNAs Show Partial Dependence on POL V for Transcription
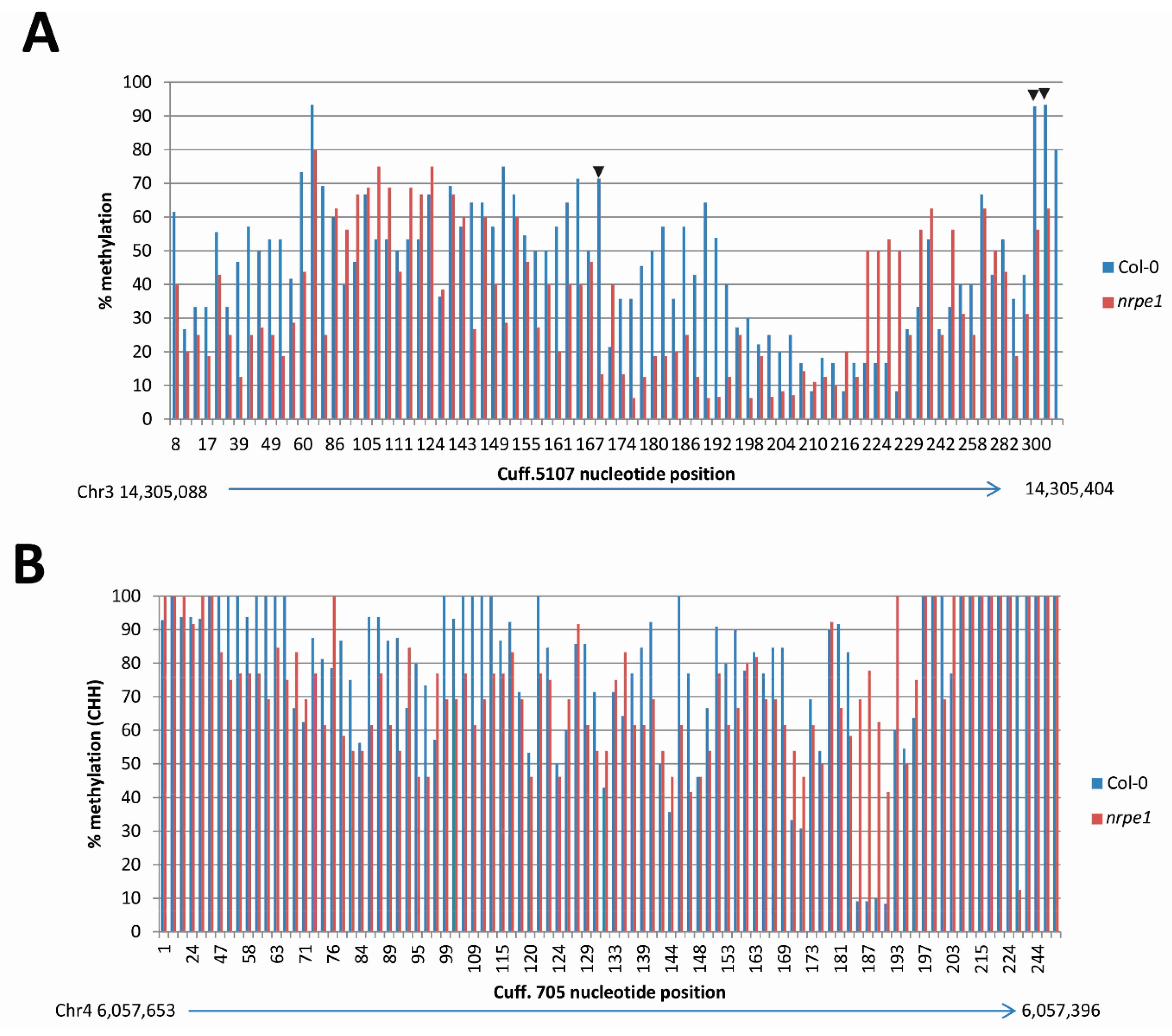
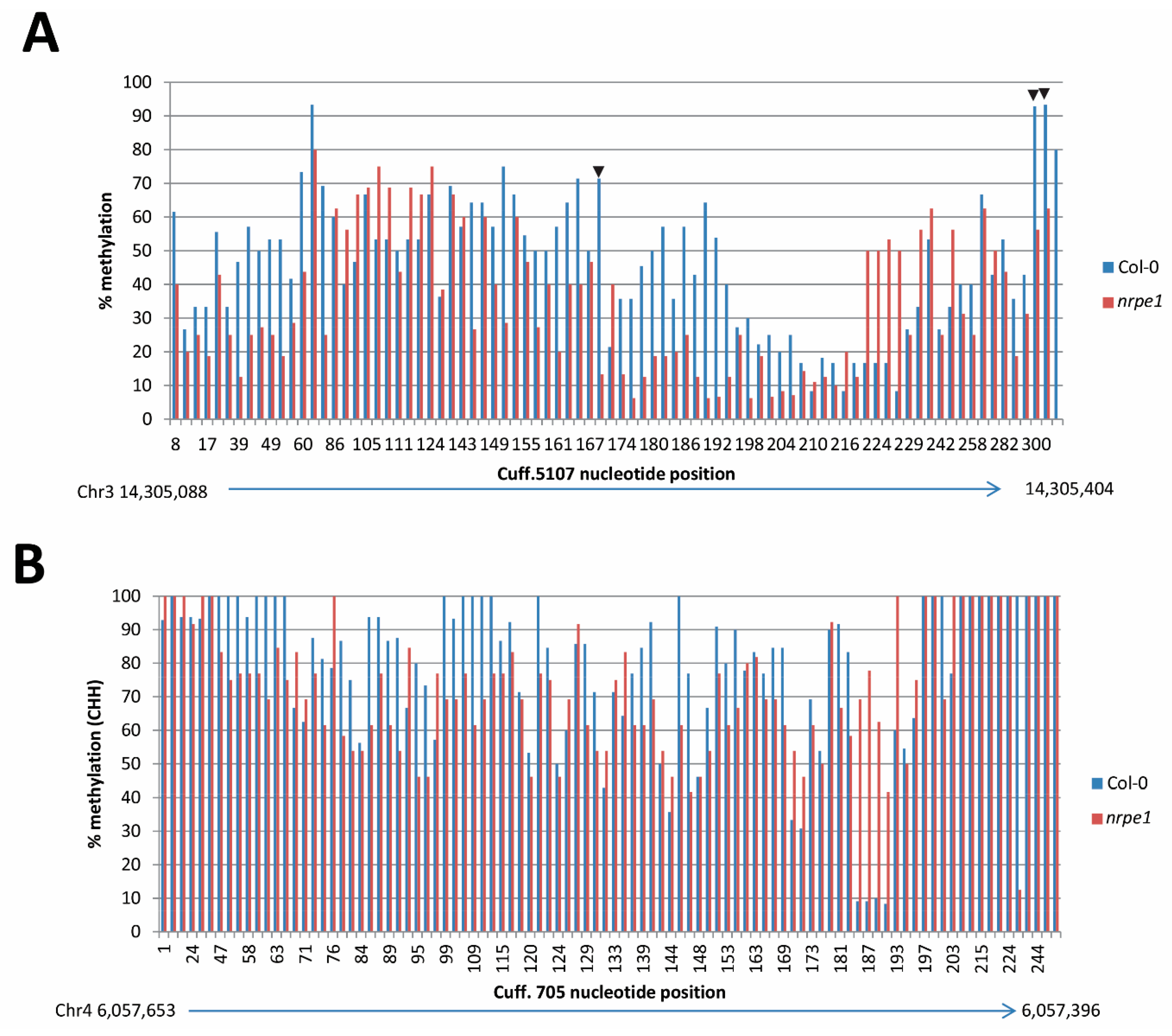
3.7. nrpe1 Plants Show CHH Hypomethylation in Genomic Loci that Generate AGO4-Associated and POL V Dependent lncRNAs
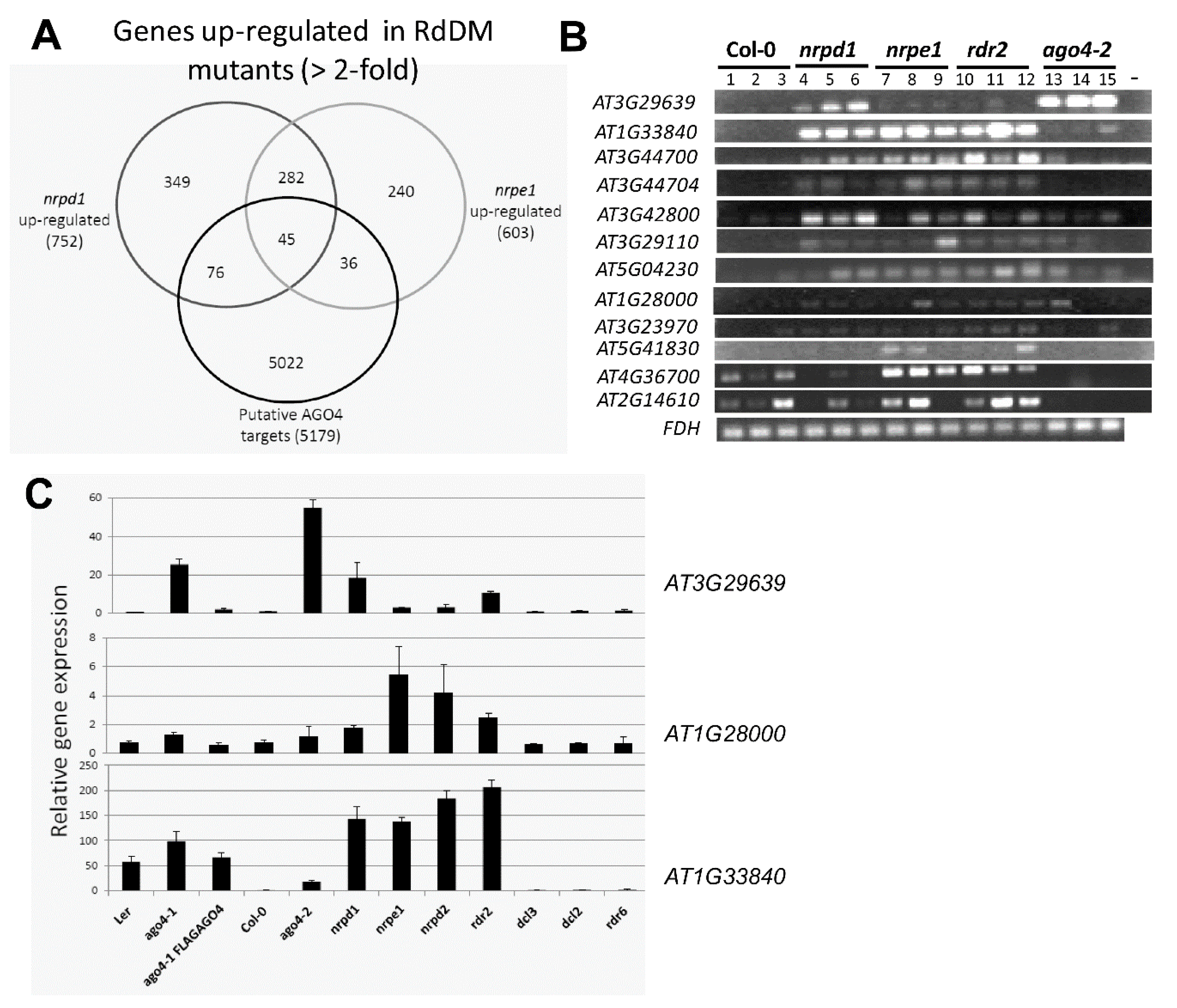
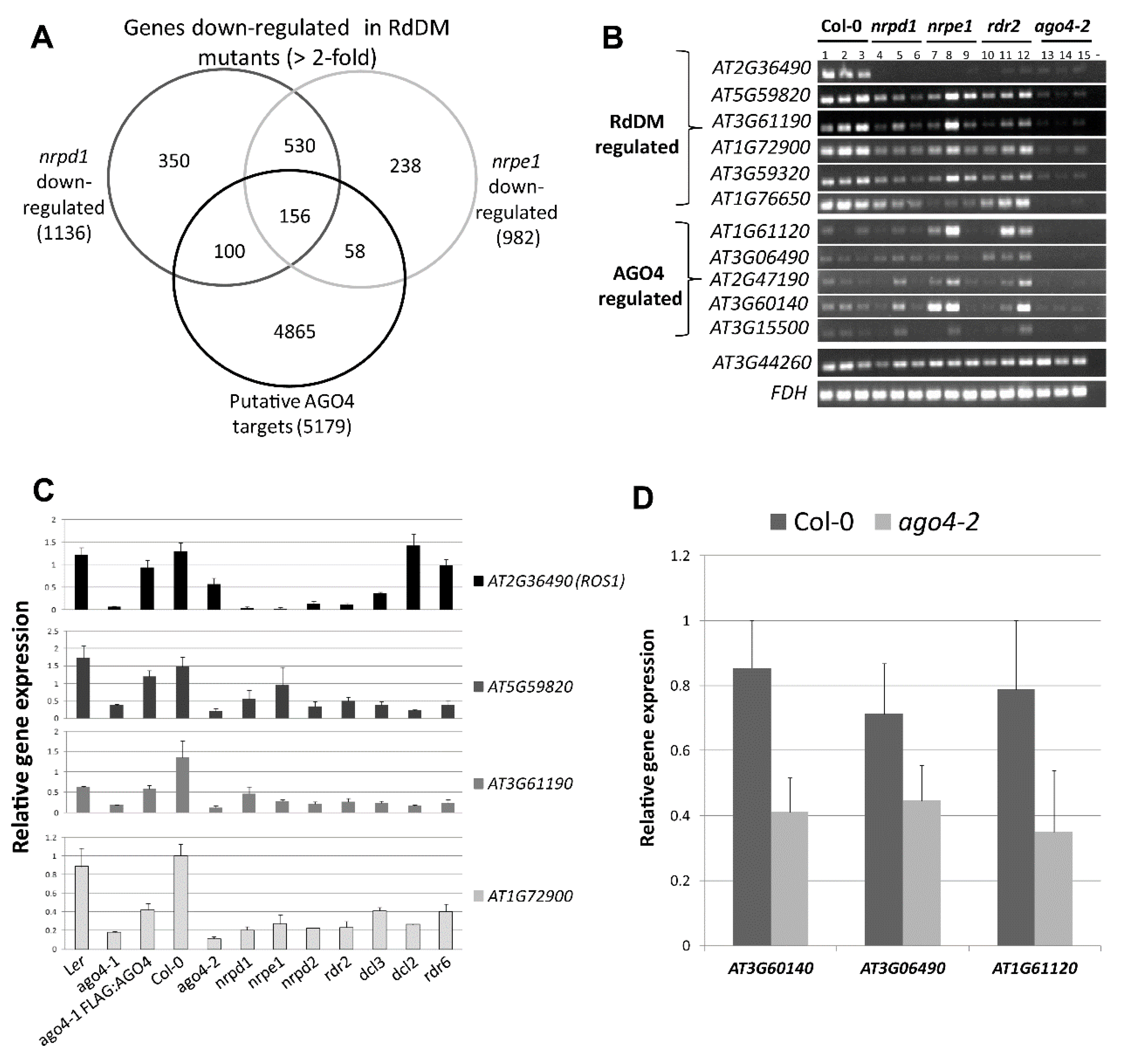
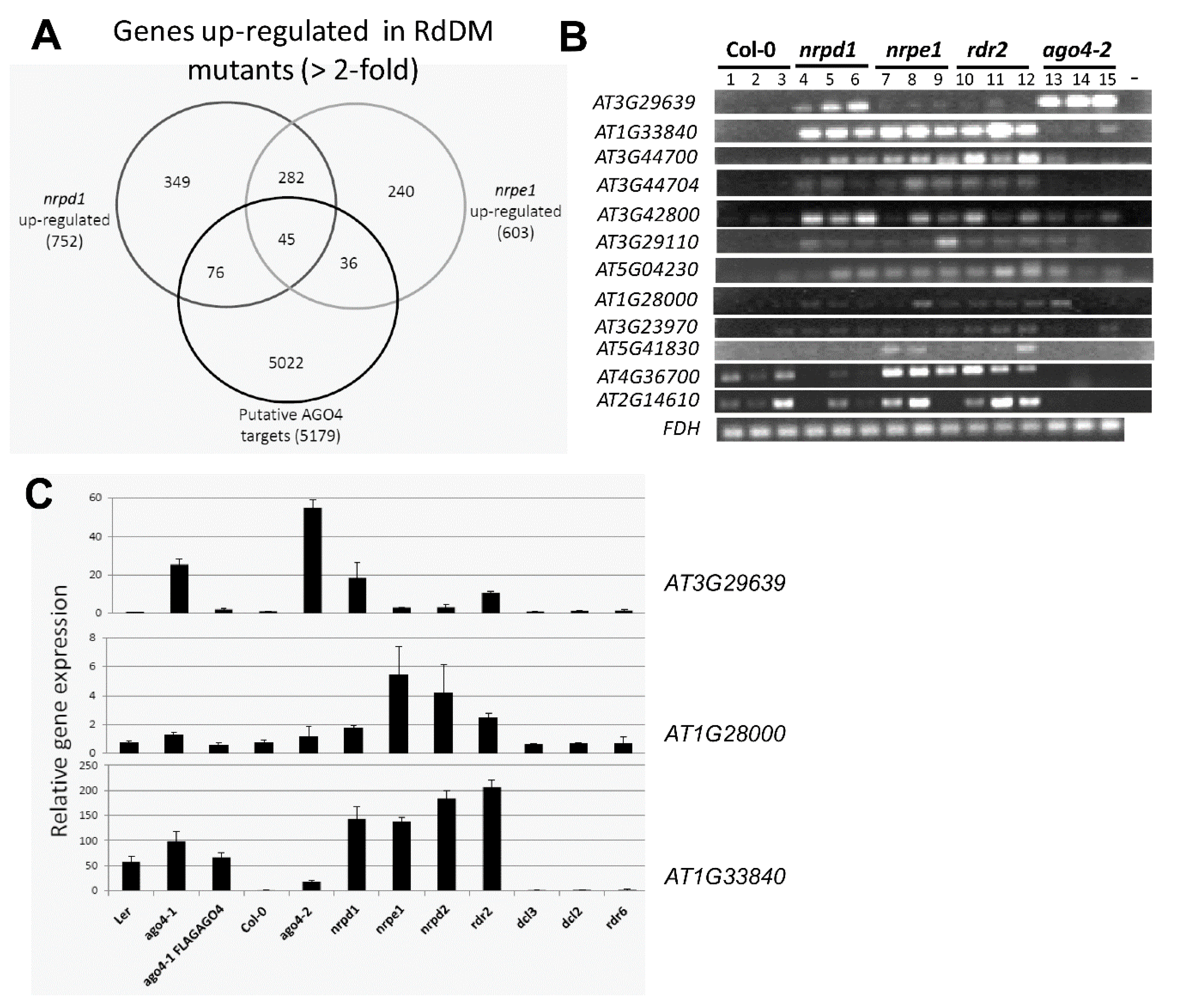
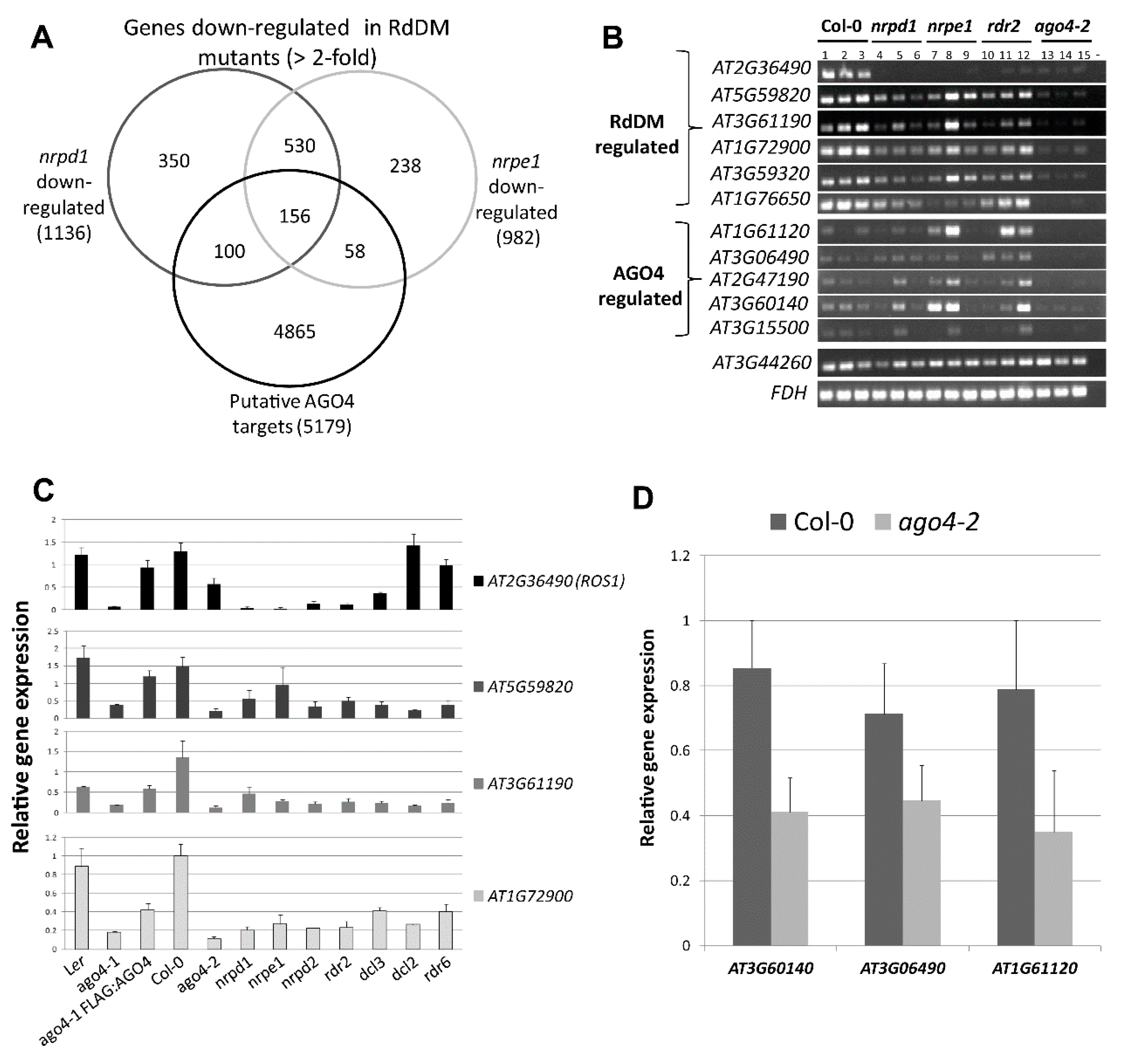
3.8. The FLAG:AGO4 RNA-IP Seq Data Reveal Novel RdDM Target Genes
3.9. RdDM Is Involved in the Activation of Stress-Responsive Genes
3.10. RdDM/AGO4-Activated Stress-Response Genes Are Induced upon Fusarium Oxysporum Infection
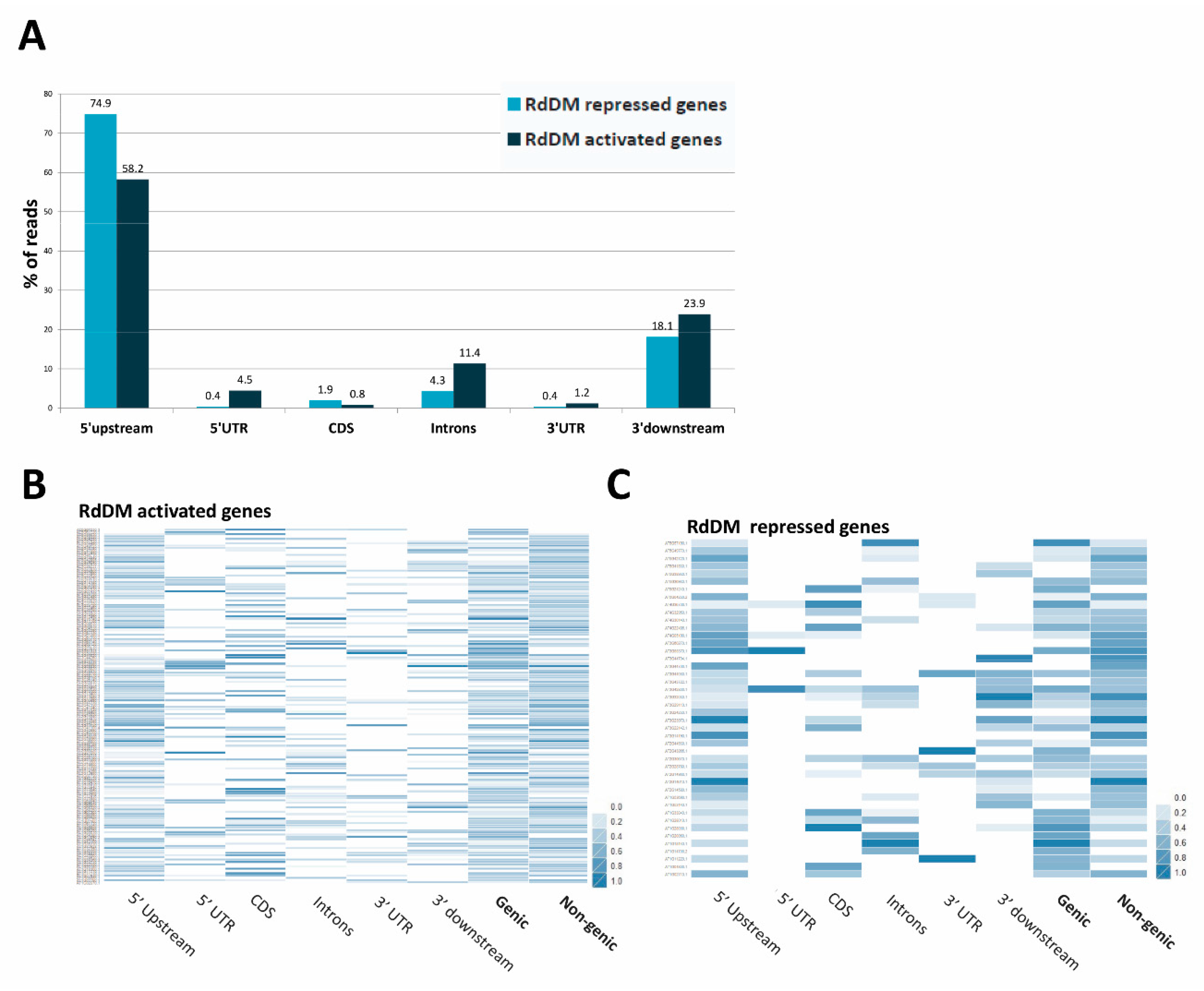
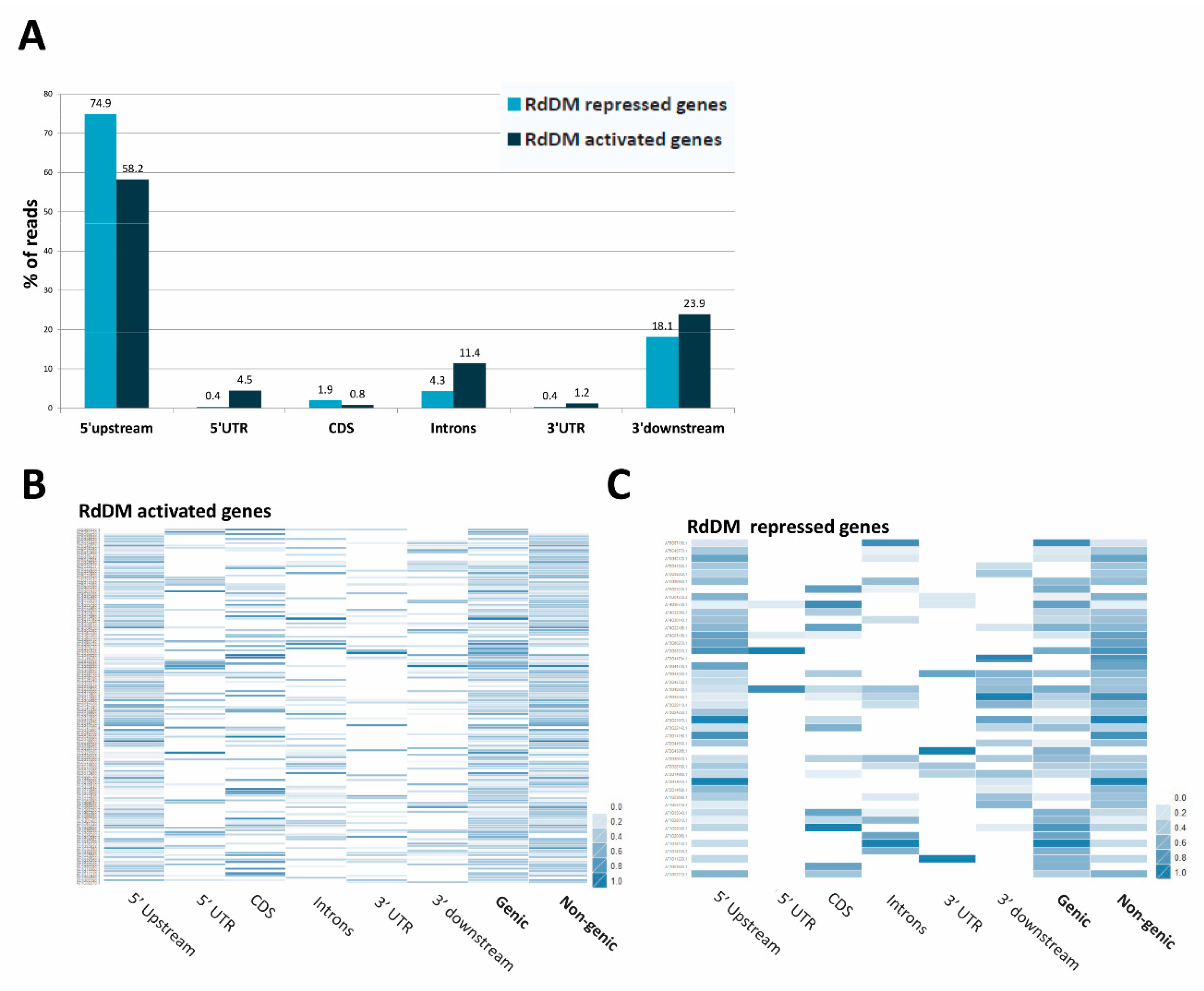
3.11. Repression or Activation of Gene Expression by RdDM May Depend on the Location of the Target Site in the Gene
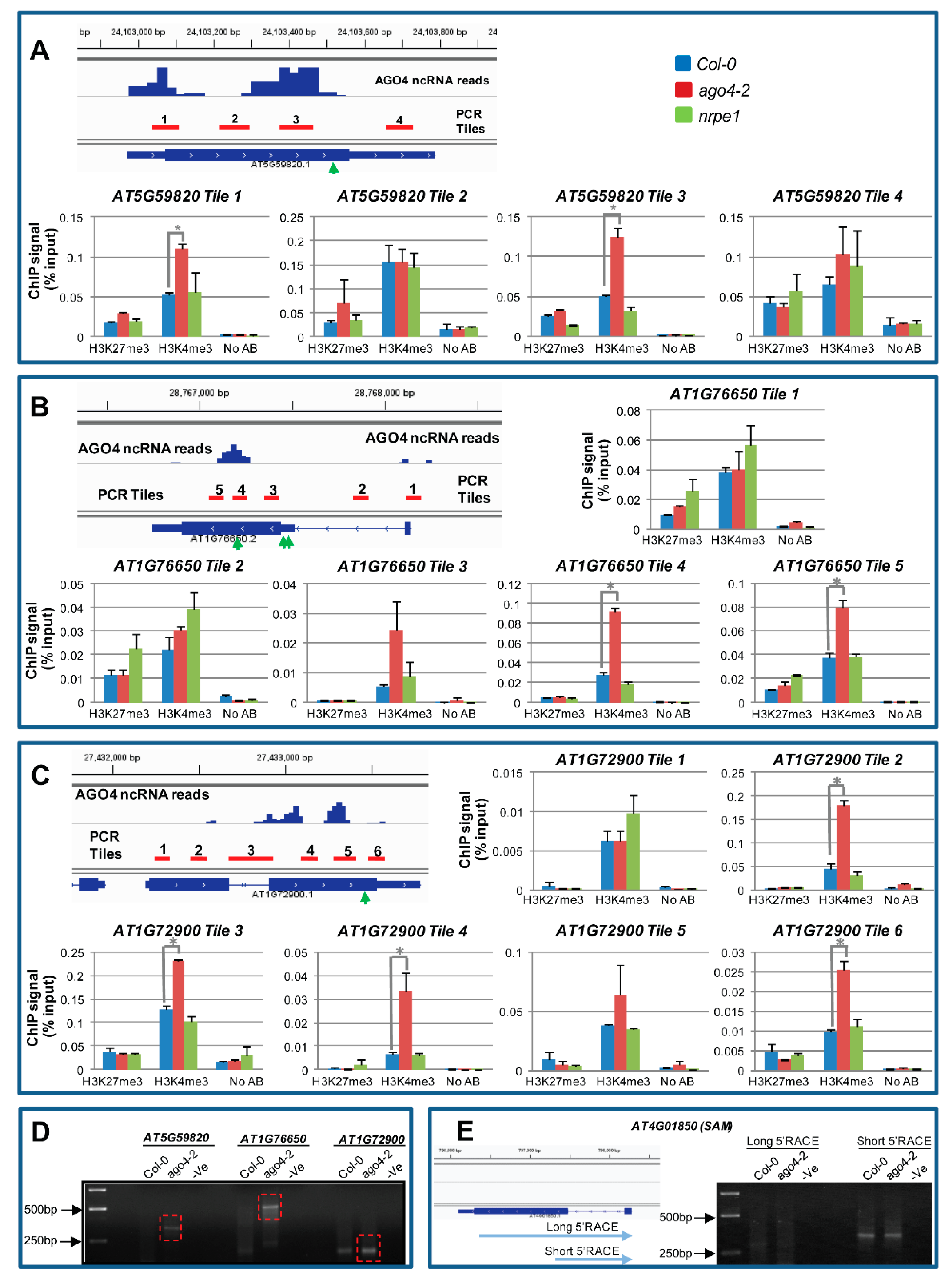
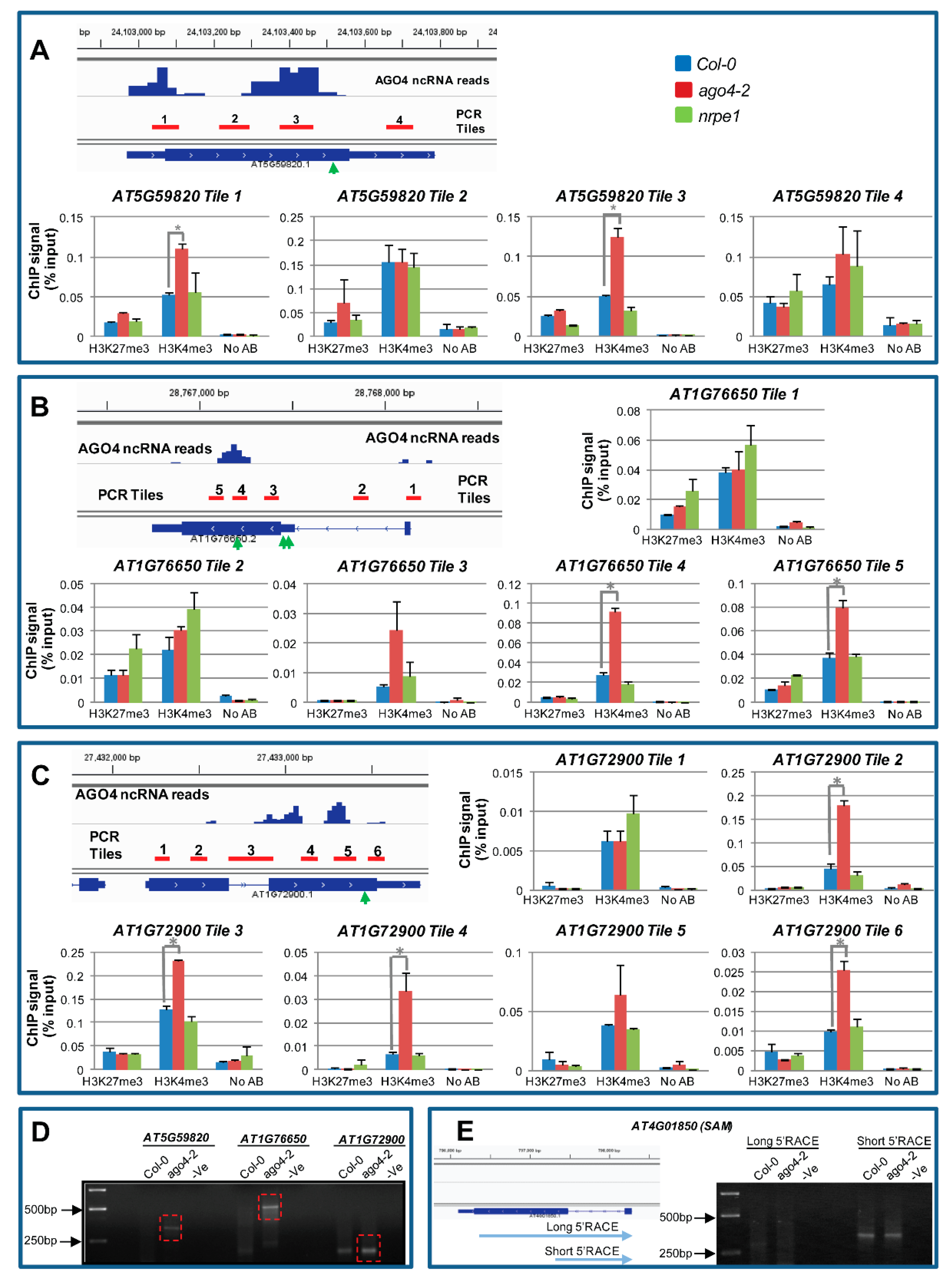
3.12. AGO4-Associated lncRNAs Derived from Gene Body Are Associated with H3K4me3 Marks
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Haag, J.R.; Pikaard, C.S. Multisubunit RNA polymerases IV and V: Purveyors of non-coding RNA for plant gene silencing. Nat. Rev. Mol. Cell Biol. 2011, 12, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.B.; Dennis, E.S. SPT5-like, a new component in plant RdDM. EMBO Rep. 2009, 10, 573–575. [Google Scholar] [CrossRef] [PubMed]
- Huettel, B.; Kanno, T.; Daxinger, L.; Aufsatz, W.; Matzke, A.J.; Matzke, M. Endogenous targets of RNA-directed DNA methylation and Pol IV in Arabidopsis. EMBO J. 2006, 25, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Mosher, R.A.; Schwach, F.; Studholme, D.; Baulcombe, D.C. PolIVb influences RNA-directed DNA methylation independently of its role in siRNA biogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3145–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, P.; Wolf, J.N.; Bisaro, D.M. RNA silencing directed against geminiviruses: Post-transcriptional and epigenetic components. Biochim. Biophys. Acta 2010, 1799, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Henderson, I.R.; Lu, C.; Green, P.J.; Jacobsen, S.E. Role of RNA polymerase IV in plant small RNA metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Kasschau, K.D.; Fahlgren, N.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Carrington, J.C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007, 5, e57. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Matsui, A.; Kawashima, M.; Kaminuma, E.; Ishida, J.; Morosawa, T.; Mochizuki, Y.; Kobayashi, N.; Toyoda, T.; Shinozaki, K.; et al. Identification of the candidate genes regulated by RNA-directed DNA methylation in Arabidopsis. Biochem. Biophys. Res. Commun. 2008, 376, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Cao, X.; Jacobsen, S.E. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 2003, 299, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, E104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blevins, T.; Podicheti, R.; Mishra, V.; Marasco, M.; Wang, J.; Rusch, D.; Tang, H.; Pikaard, C.S. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. Elife 2015, 4, e09591. [Google Scholar] [CrossRef] [PubMed]
- Havecker, E.R.; Wallbridge, L.M.; Hardcastle, T.J.; Bush, M.S.; Kelly, K.A.; Dunn, R.M.; Schwach, F.; Doonan, J.H.; Baulcombe, D.C. The Arabidopsis RNA-directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell 2010, 22, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C.; et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5’ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; He, X.; Wang, X.J.; Kohany, O.; Jurka, J.; Hannon, G.J. Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature 2006, 443, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Ream, T.S.; Haag, J.R.; Pikaard, C.S. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat. Genet. 2009, 41, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Agorio, A.; Vera, P. ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 2007, 19, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Ramirez, V.; Garcia-Andrade, J.; Flors, V.; Vera, P. The RNA silencing enzyme RNA polymerase V is required for plant immunity. PLoS Genet. 2011, 7, e1002434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glazebrook, J. Genes controlling expression of defense responses in Arabidopsis—2001 Status. Curr. Opin. Plant Biol. 2001, 4, 301–308. [Google Scholar] [CrossRef]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Smith, N.A.; Zhang, L.; Dennis, E.S.; Waterhouse, P.M.; Unrau, P.J.; Wang, M.B. Synthesis of complementary RNA by RNA-dependent RNA polymerases in plant extracts is independent of an RNA primer. Funct. Plant Biol. 2008, 35, 1091–1099. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Goecks, J.; Nekrutenko, A.; Taylor, J. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.B.; Helliwell, C.A.; Wu, L.M.; Waterhouse, P.M.; Peacock, W.J.; Dennis, E.S. Hairpin RNAs derived from RNA polymerase II and polymerase III promoter-directed transgenes are processed differently in plants. RNA 2008, 14, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Hetzl, J.; Foerster, A.M.; Raidl, G.; Mittelsten Scheid, O. CyMATE: A new tool for methylation analysis of plant genomic DNA after bisulphite sequencing. Plant J. 2007, 51, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, L.F.; Manners, J.M.; Kazan, K. Fusarium oxysporum hijacks COI1-mediated jasmonate signaling to promote disease development in Arabidopsis. Plant J. 2009, 58, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Mosher, R.A.; Melnyk, C.W.; Kelly, K.A.; Dunn, R.M.; Studholme, D.J.; Baulcombe, D.C. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 2009, 460, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Liu, J.; Kiba, T.; Woo, J.; Ojo, T.; Hafner, M.; Tuschl, T.; Chua, N.H.; Wang, X.J. Deep sequencing of small RNAs specifically associated with Arabidopsis AGO1 and AGO4 uncovers new AGO functions. Plant J. 2011, 67, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Li, C.F.; Pontes, O.; El-Shami, M.; Henderson, I.R.; Bernatavichute, Y.V.; Chan, S.W.; Lagrange, T.; Pikaard, C.S.; Jacobsen, S.E. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 2006, 126, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Pontes, O.; Li, C.F.; Costa Nunes, P.; Haag, J.; Ream, T.; Vitins, A.; Jacobsen, S.E.; Pikaard, C.S. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell 2006, 126, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Au, P.C.; Helliwell, C.; Wang, M.B. Characterizing RNA-protein interaction using cross-linking and metabolite supplemented nuclear RNA-immunoprecipitation. Mol. Biol. Rep. 2014, 41, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Rowley, M.J.; Bohmdorfer, G.; Sandhu, D.; Gregory, B.D.; Wierzbicki, A.T. RNA polymerase V targets transcriptional silencing components to promoters of protein-coding genes. Plant J. 2013, 73, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Cocklin, R.; Mayampurath, A.; Lister, R.; Rowley, M.J.; Gregory, B.D.; Ecker, J.R.; Tang, H.; Pikaard, C.S. Spatial and functional relationships among Pol V-associated loci, Pol IV-dependent siRNAs, and cytosine methylation in the Arabidopsis epigenome. Genes Dev. 2010, 26, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.; Zhu, Q.H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R.; et al. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Davison, T.S.; Henz, S.R.; Pape, U.J.; Demar, M.; Vingron, M.; Scholkopf, B.; Weigel, D.; Lohmann, J.U. A gene expression map of Arabidopsis thaliana development. Nat. Genet. 2005, 37, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, W.; Zhao, Y.; Wang, C.; Shen, J.; Zhu, J.K.; Gong, Z. Antisilencing role of the RNA-directed DNA methylation pathway and a histone acetyltransferase in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 11425–11430. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mao, L.; Qi, Y. Roles of dicer-like and argonaute proteins in TAS-derived small interfering RNA-triggered DNA methylation. Plant Physiol. 2012, 160, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Stephen, S.; Kazan, K.; Jin, G.; Fan, L.; Taylor, J.; Dennis, E.S.; Helliwell, C.A.; Wang, M.B. Characterization of the defense transcriptome responsive to Fusarium oxysporum-infection in Arabidopsis using RNA-seq. Gene 2013, 512, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Fukushima, S.; Takatsuji, H. RNA-directed DNA methylation induces transcriptional activation in plants. Proc. Natl. Acad. Sci. USA 2009, 106, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Lauria, M.; Rossi, V. Epigenetic control of gene regulation in plants. Biochim. Biophys. Acta 2011, 1809, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, Z.; Li, S.; Yu, B.; Liu, J.Y.; Chen, X. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev. 2009, 23, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y.; et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, S.; Kakutani, T. Control of genic DNA methylation in Arabidopsis. J. Plant Res. 2010, 123, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Miyazaki, Y.; Takuno, S.; Saze, H. Epigenetic regulation of intragenic transposable elements impacts gene transcription in Arabidopsis thaliana. Nucleic Acids Res. 2015, 43, 3911–3921. [Google Scholar] [CrossRef] [PubMed]
- Nuthikattu, S.; McCue, A.D.; Panda, K.; Fultz, D.; DeFraia, C.; Thomas, E.N.; Slotkin, R.K. The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21–22 nucleotide small interfering RNAs. Plant Physiol. 2013, 162, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Pontier, D.; Picart, C.; Roudier, F.; Garcia, D.; Lahmy, S.; Azevedo, J.; Alart, E.; Laudie, M.; Karlowski, W.M.; Cooke, R.; et al. NERD, a plant-specific GW protein, defines an additional RNAi-dependent chromatin-based pathway in Arabidopsis. Mol. Cell 2012, 48, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FLAG:AGO4 RNA-IP | Negative RNA-IP (+15 Cycles) | Nuclear RNA | |
|---|---|---|---|
| # reads after adaptor trim | 18,878,339 | 10,799,912 | 17,537,595 |
| # reads map to Chr 1-5 | 15,905,349 (84%) | 1,071,294 (13%) | 6,307,354 (36%) |
| # reads map to Chr C & M | 50,238 (0.3%) | 8,087,305 (75%) | 3,467,242 (20%) |
| % total mapped reads | 84.3% | 88% | 56% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Au, P.C.K.; Dennis, E.S.; Wang, M.-B. Analysis of Argonaute 4-Associated Long Non-Coding RNA in Arabidopsis thaliana Sheds Novel Insights into Gene Regulation through RNA-Directed DNA Methylation. Genes 2017, 8, 198. https://doi.org/10.3390/genes8080198
Au PCK, Dennis ES, Wang M-B. Analysis of Argonaute 4-Associated Long Non-Coding RNA in Arabidopsis thaliana Sheds Novel Insights into Gene Regulation through RNA-Directed DNA Methylation. Genes. 2017; 8(8):198. https://doi.org/10.3390/genes8080198
Chicago/Turabian StyleAu, Phil Chi Khang, Elizabeth S. Dennis, and Ming-Bo Wang. 2017. "Analysis of Argonaute 4-Associated Long Non-Coding RNA in Arabidopsis thaliana Sheds Novel Insights into Gene Regulation through RNA-Directed DNA Methylation" Genes 8, no. 8: 198. https://doi.org/10.3390/genes8080198