Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sampling and RNA Preparation
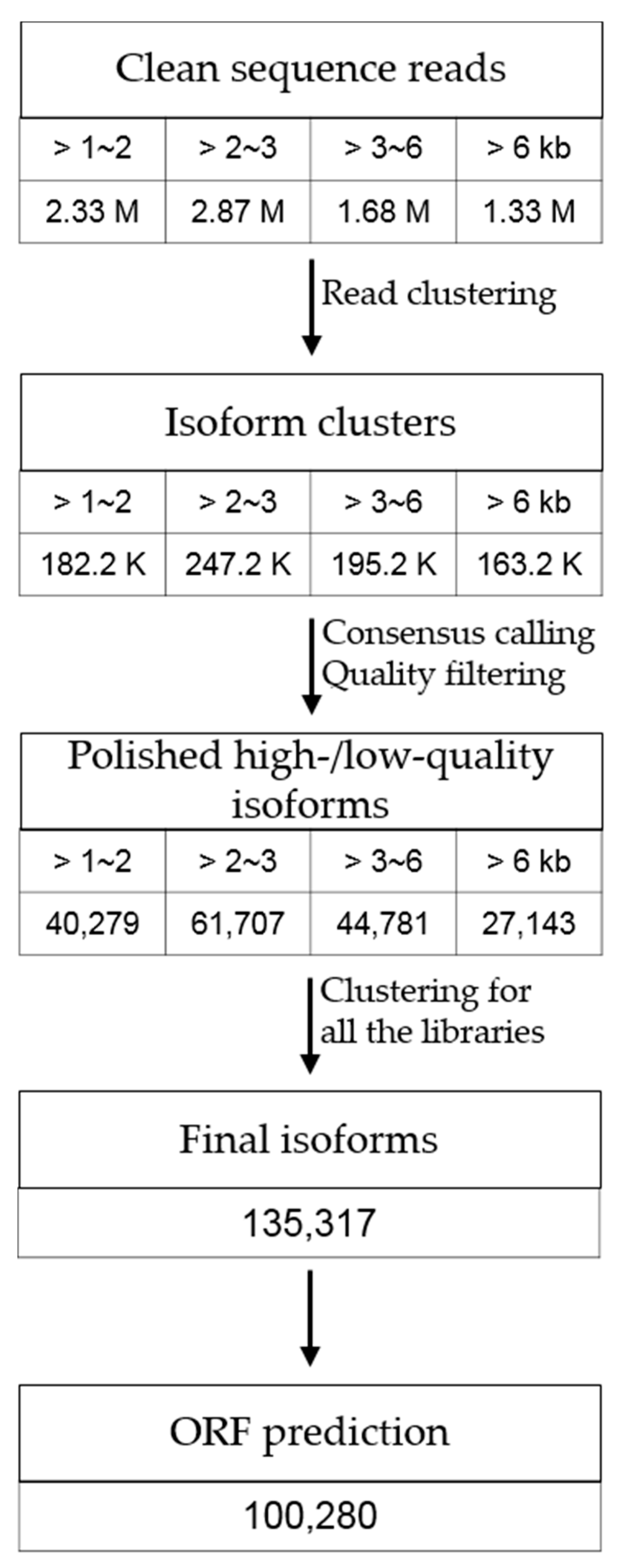
2.2. PacBio Iso-Seq
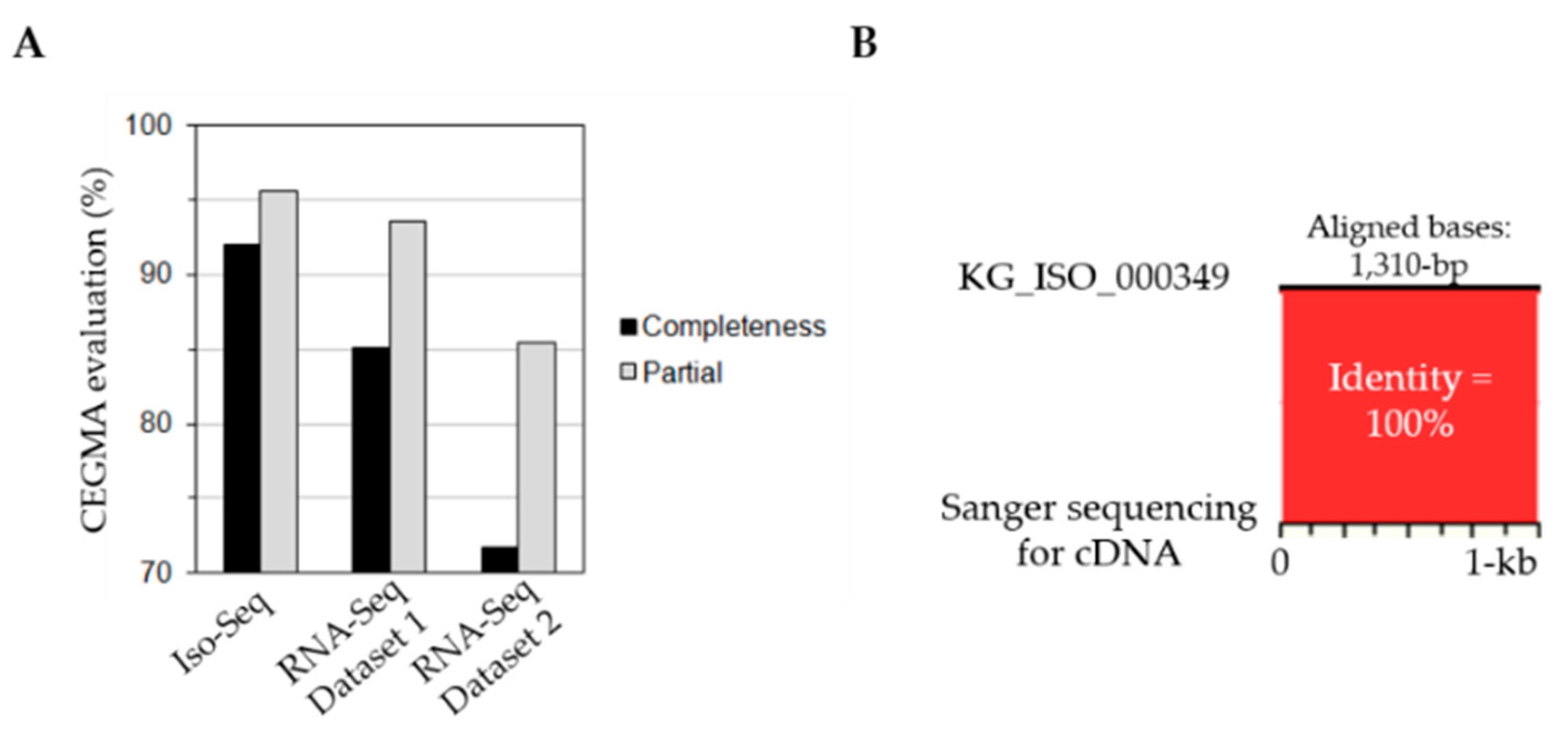
2.3. Iso-Seq Assembly and Quality Assessment
2.4. Unigene Annotation
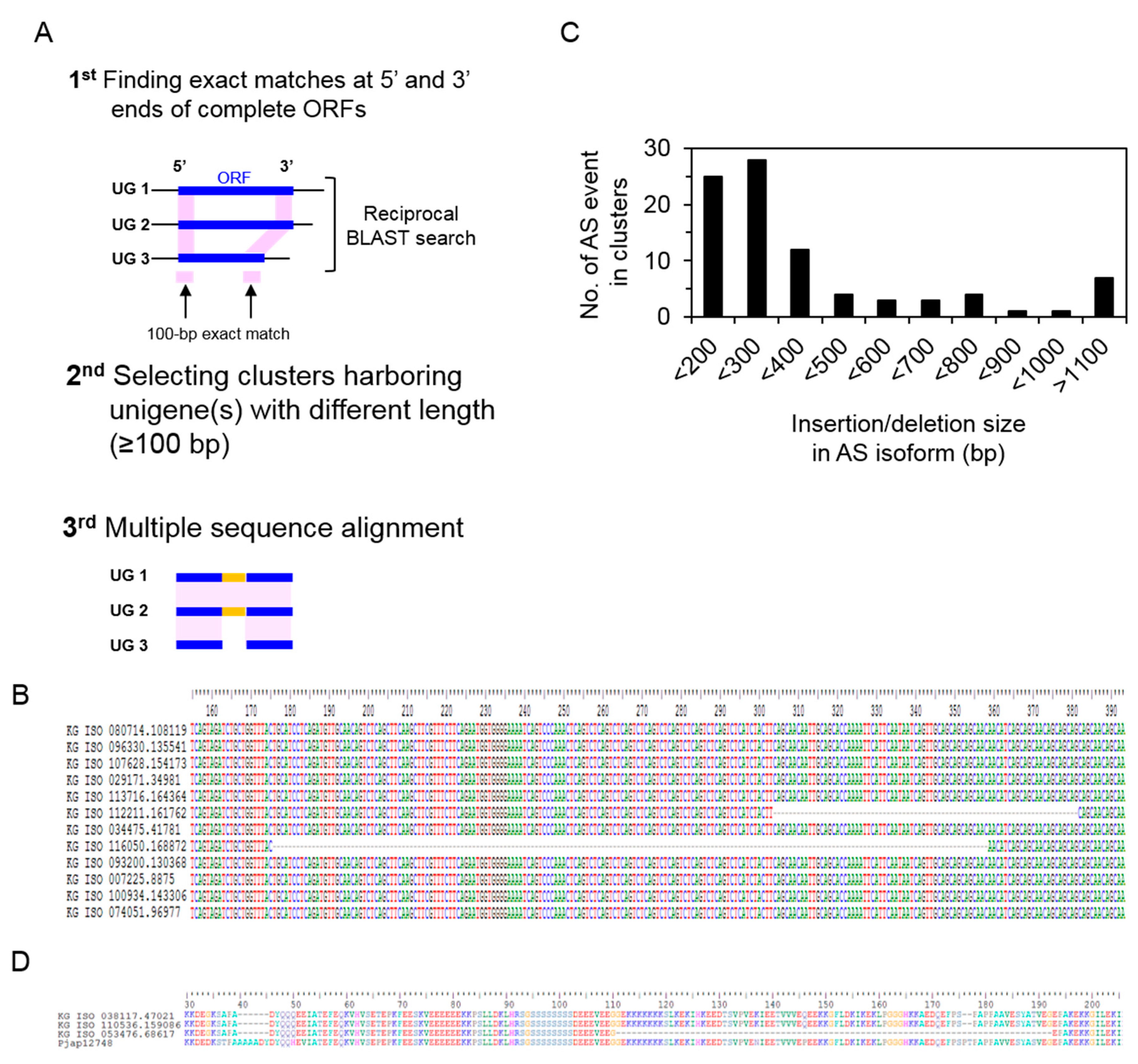
2.5. Identification of Alternative Splicing Isoforms
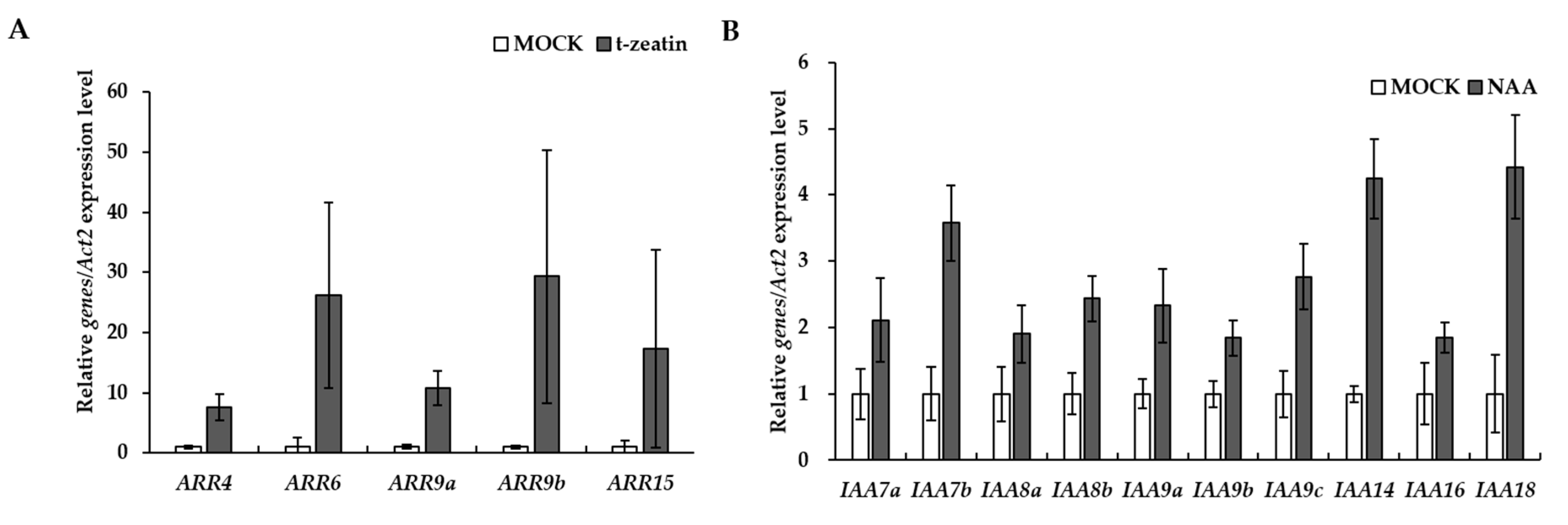
2.6. Plant Hormone Treatments and Real Time qRT-PCR
2.7. GenBank Accession Code
3. Results
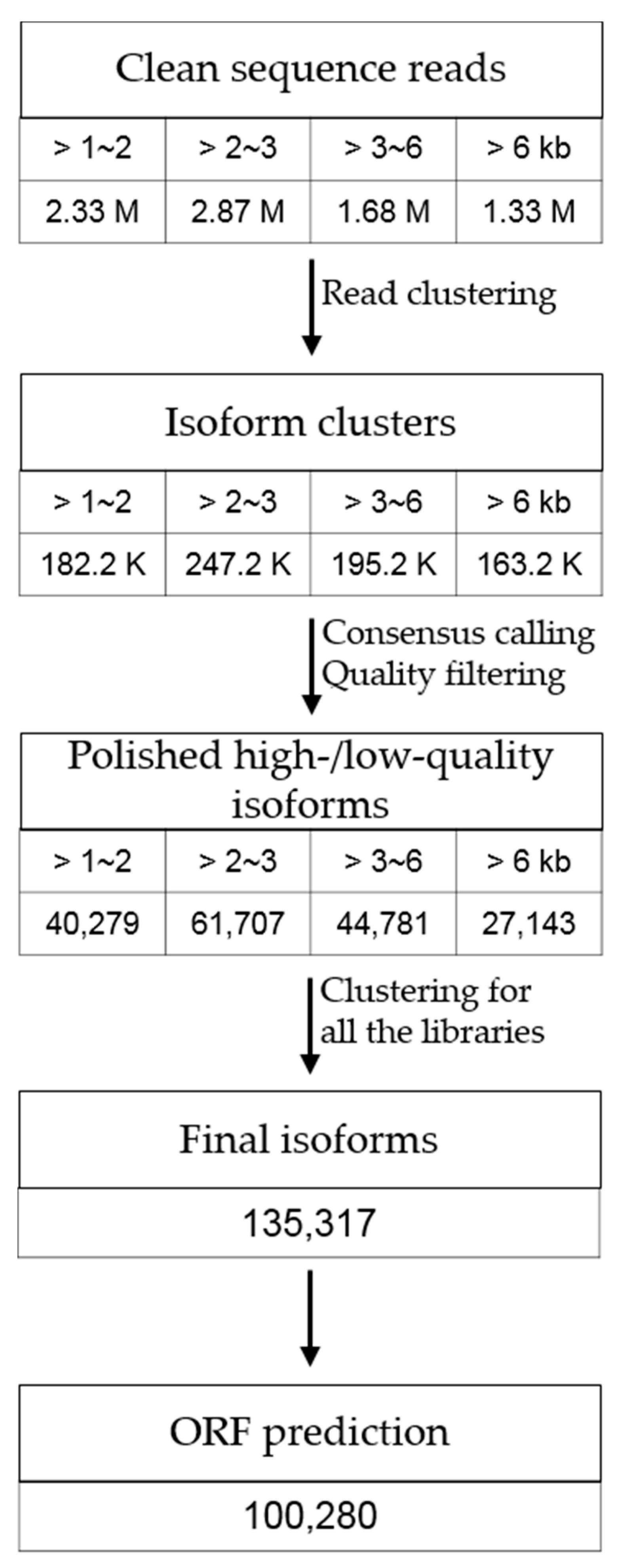
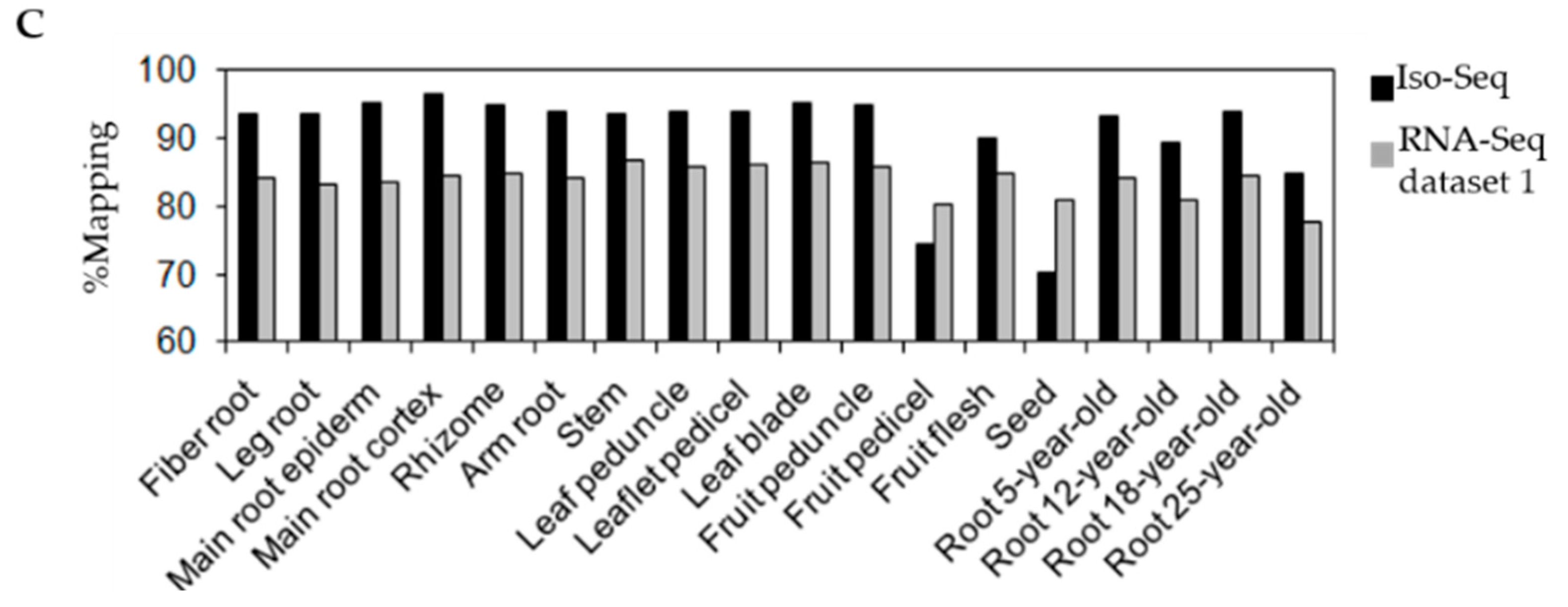
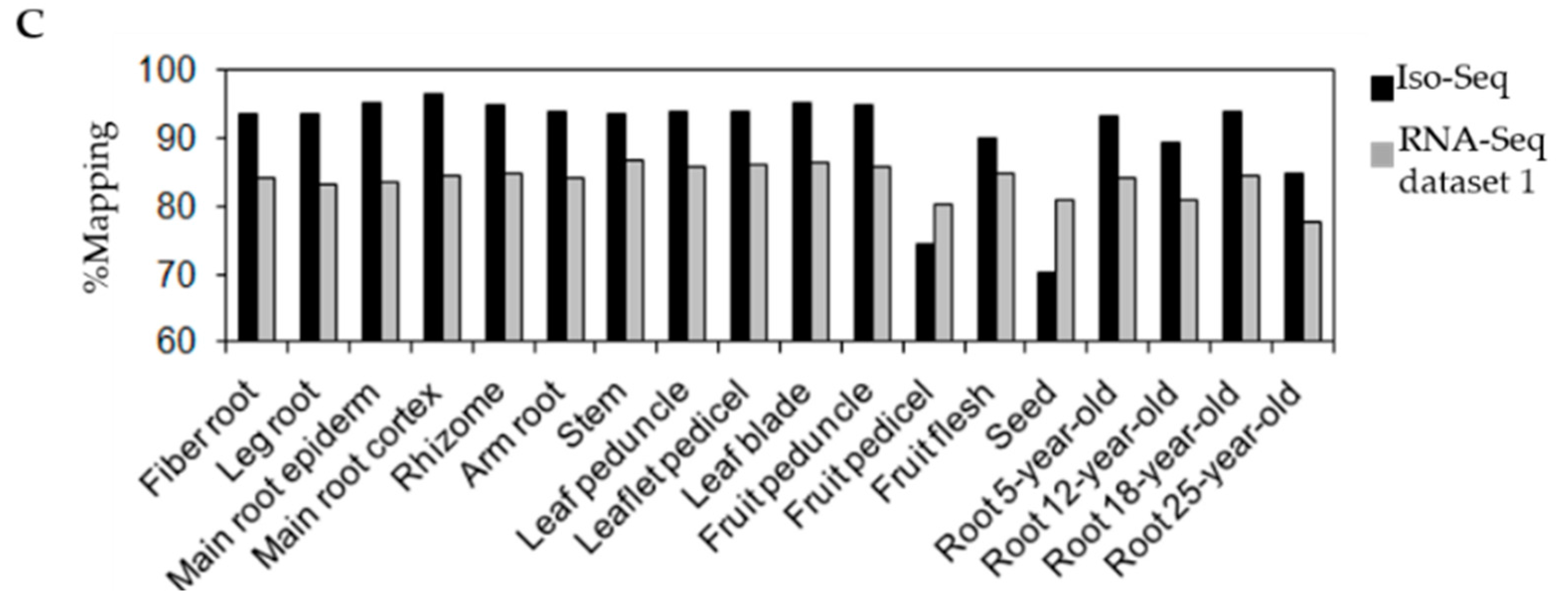
3.1. Transcriptome Sequencing of P. ginseng Using PacBio Iso-Seq
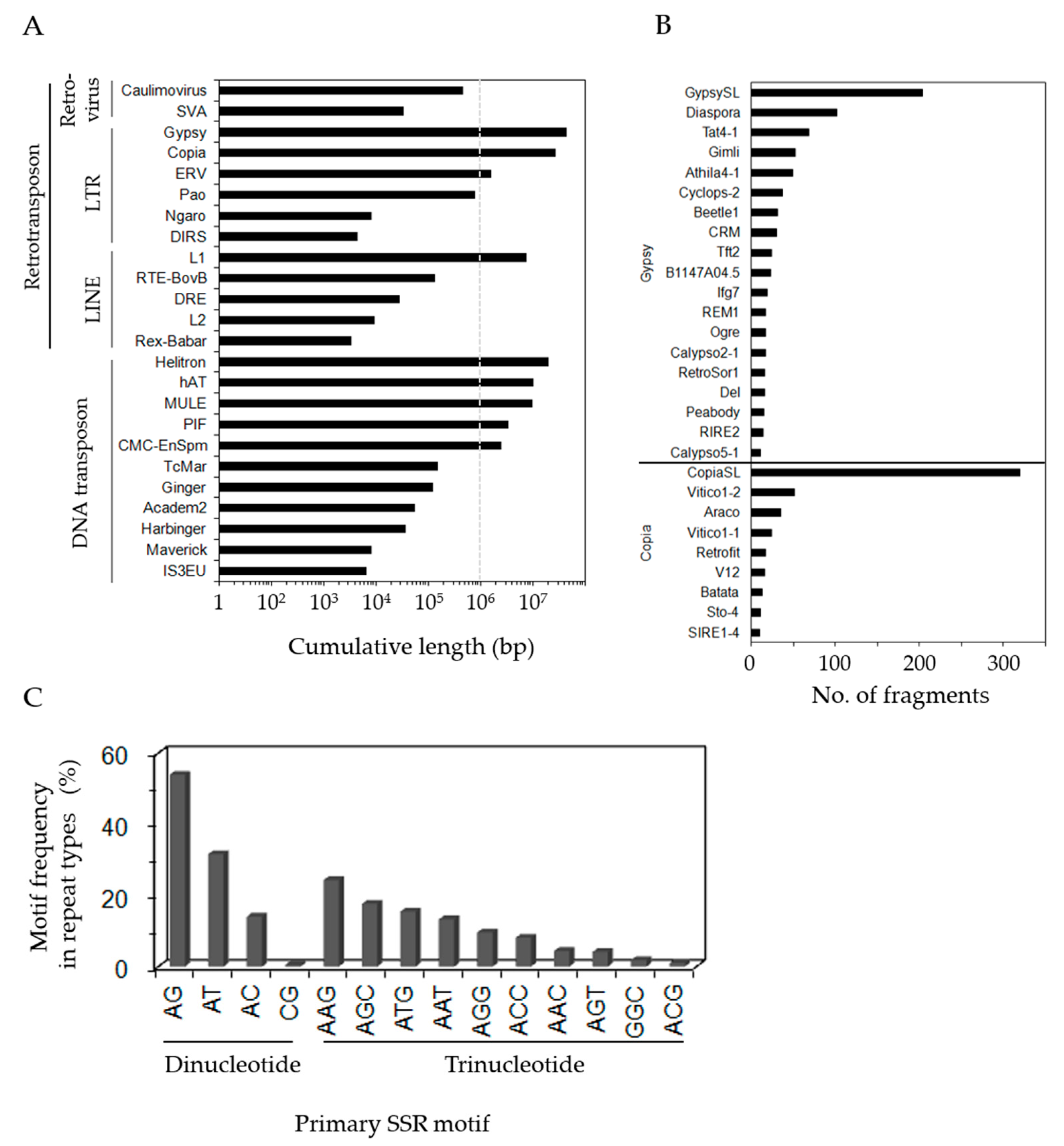
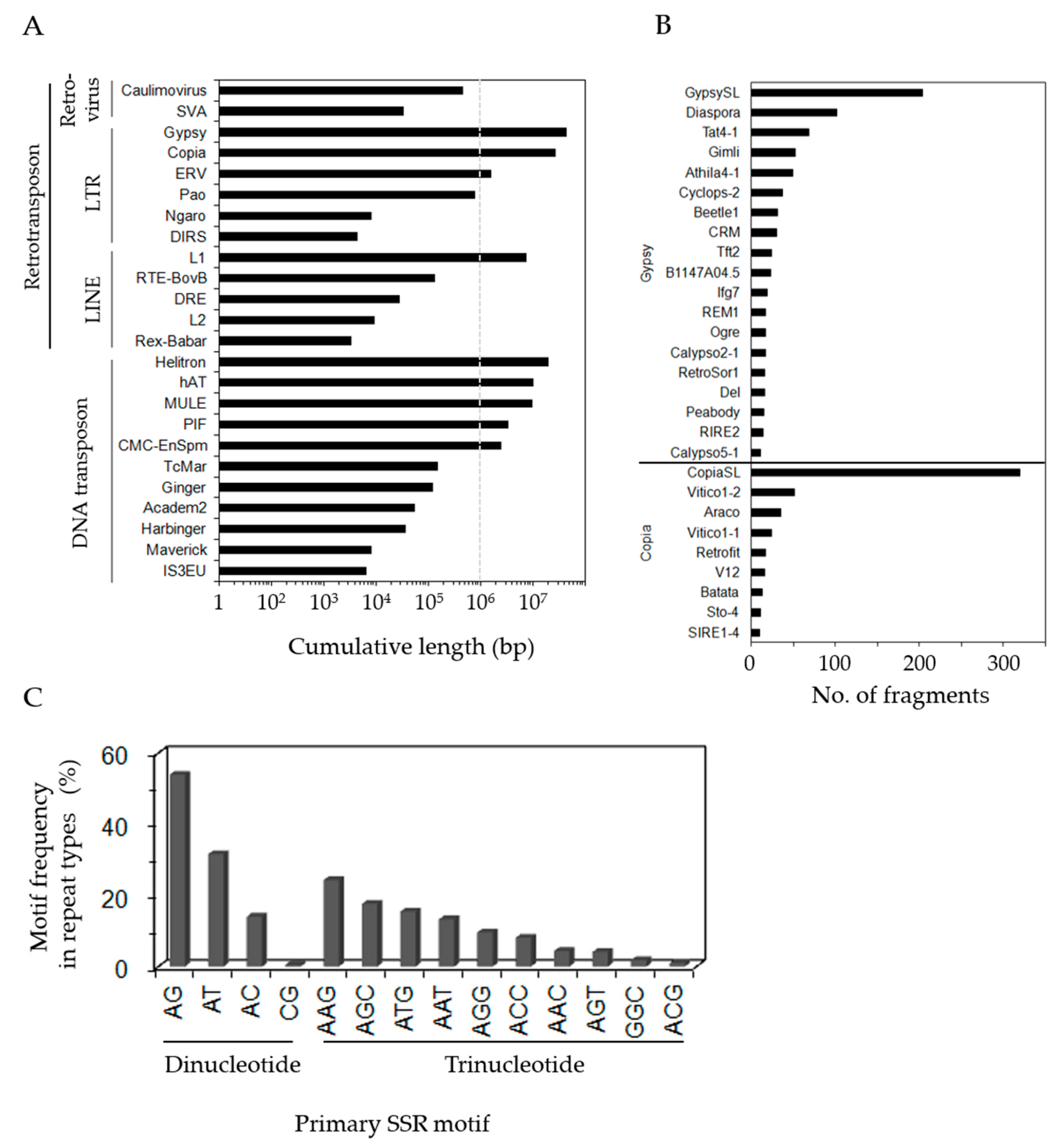
3.2. Identification of Repeat Sequences in P. ginseng Unigenes
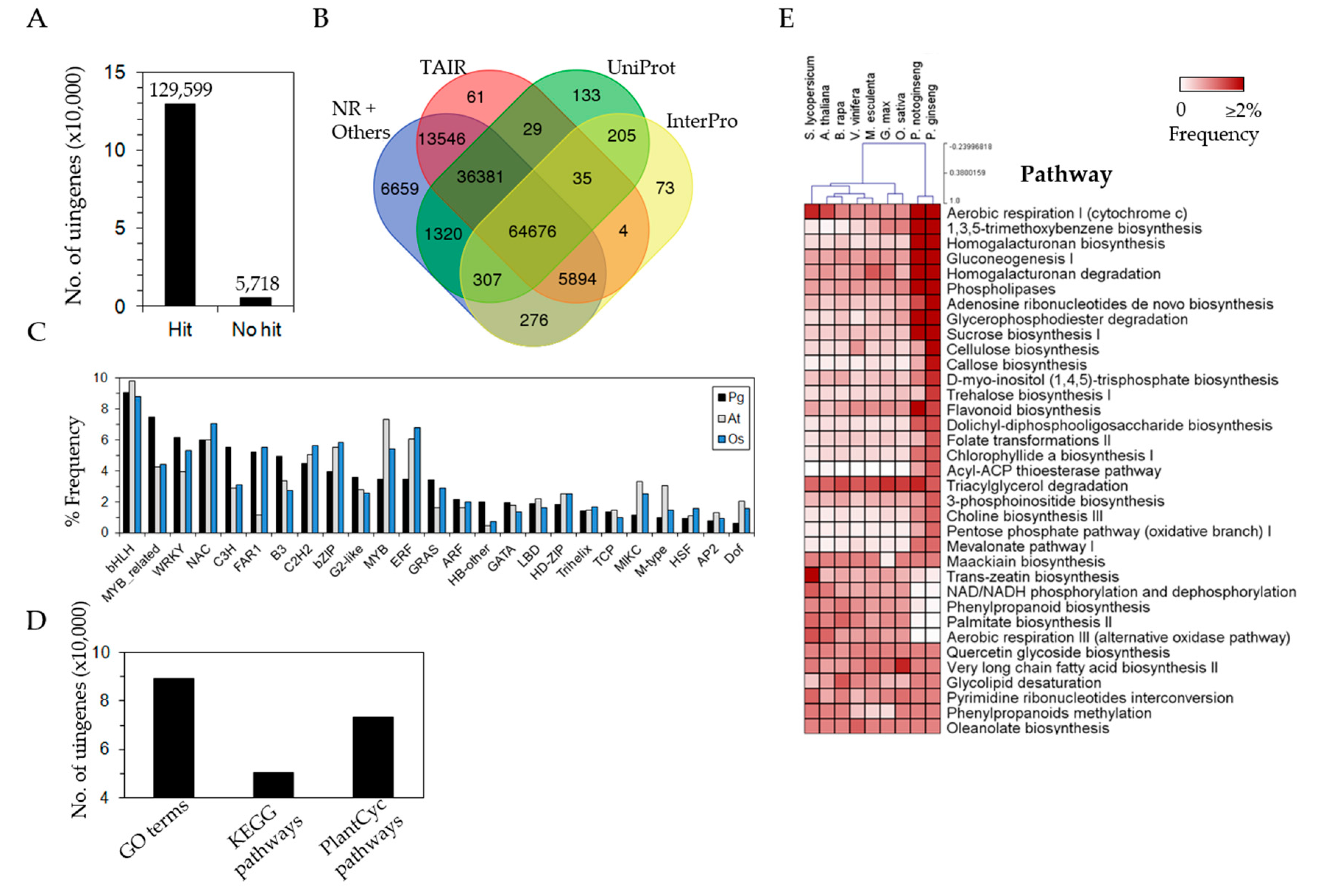
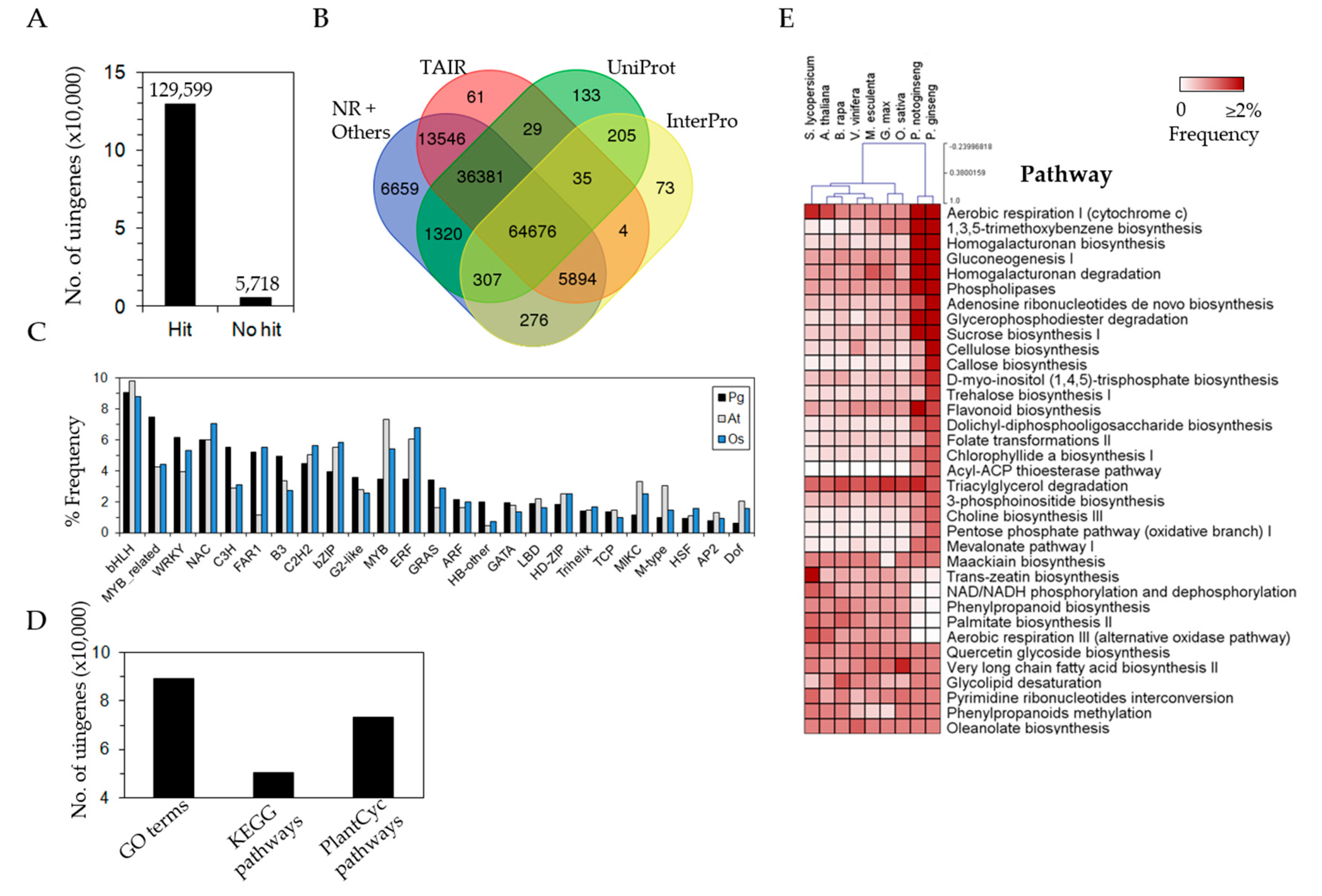
3.3. Efficient Gene Annotation of P. ginseng
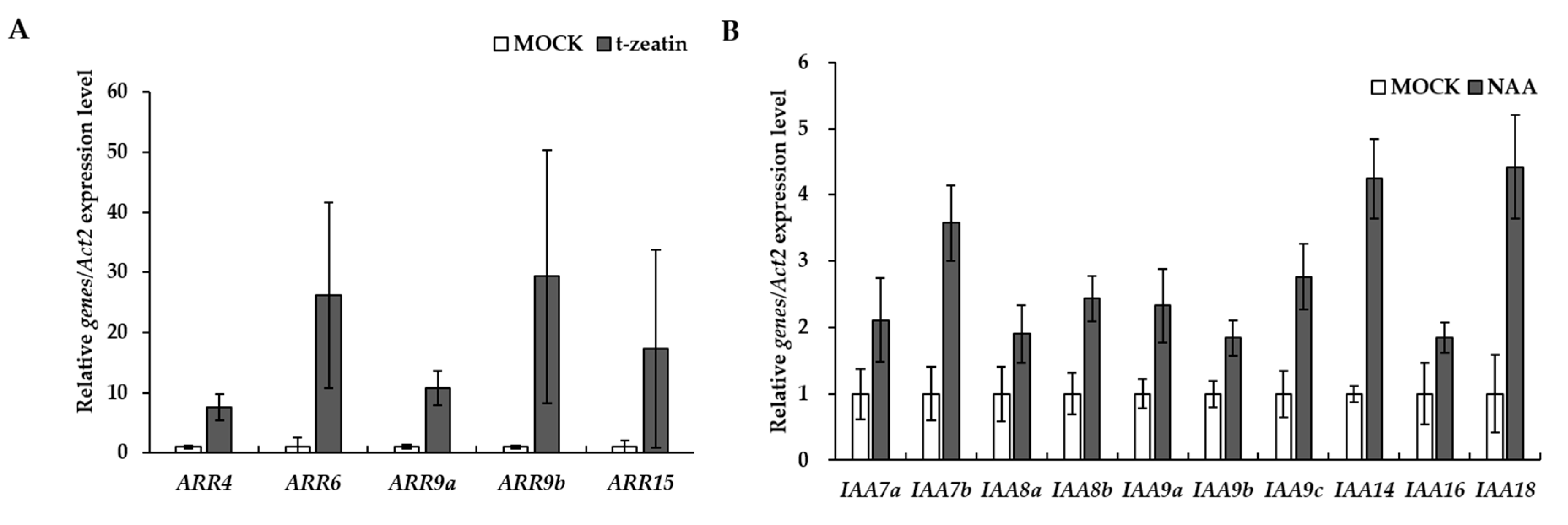
3.4. Identification and Functional Analysis of Auxin and Cytokinin Signaling Pathways in P. ginseng
3.5. Identification of Alternative Splicing Isoforms
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hu, S.Y. The genus Panax (ginseng) in chinese medicine. Econ. Bot. 1976, 30, 11–28. [Google Scholar] [CrossRef]
- Kang, S.; Min, H. Ginseng, the ‘immunity boost’: The effects of Panax ginseng on immune system. J. Ginseng Res. 2012, 36, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.-M.; Lim, J.-J.; Ahn, M.-S.; Jeong, H.-N.; An, T.-J.; Kim, S.-H. Comparative phenolic compound profiles and antioxidative activity of the fruit, leaves, and roots of Korean ginseng (Panax ginseng Meyer) according to cultivation years. J. Ginseng Res. 2016, 40, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-J.; Han, I.-H.; Kim, Y.-J.; Yamabe, N.; Lee, D.; Hwang, G.S.; Oh, M.; Choi, K.-C.; Kim, S.-N.; Ham, J.; et al. Anticarcinogenic effects of products of heat-processed ginsenoside Re, a major constituent of ginseng berry, on human gastric cancer cells. J. Agric. Food Chem. 2014, 62, 2830–2836. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Kim, S.; Lee, S.J.; Oh, B.-C.; Jun, H.-S. Ginseng berry extract supplementation improves age-related decline of insulin signaling in mice. Nutrients 2015, 7, 3038–3053. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, K.; Whang, W.-K.; Ko, S.-K. Inhibitory effects of ginseng (Panax ginseng) berry on tumour promotion and inflammatory ear oedema induced by TPA. J. Nutr. Ther. 2016, 4, 143–148. [Google Scholar] [CrossRef]
- Luo, H.; Sun, C.; Sun, Y.; Wu, Q.; Li, Y.; Song, J. Analysis of the transcriptome of Panax notoginseng root uncovers putative triterpene saponin-biosynthetic genes and genetic markers. BMC Genom. 2011, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-I.; Waminal, N.E.; Park, H.M.; Kim, N.-H.; Choi, B.S.; Park, M.; Choi, D.; Lim, Y.P.; Kwon, S.-J.; Park, B.-S.; et al. Major repeat components covering one-third of the ginseng (Panax ginseng C.A. Meyer) genome and evidence for allotetraploidy. Plant J. 2014, 77, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Lee, S.-C.; Park, H.-S.; Jang, W.; Lee, Y.S.; Choi, B.-S.; Nah, G.J.; Kim, D.-S.; Natesan, S.; Sun, C.; et al. Transcriptome profiling and comparative analysis of Panax ginseng adventitious roots. J. Ginseng Res. 2014, 38, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.P.; Lee, S.J.; Park, J.Y.; Plaha, P.; Park, Y.S.; Lee, Y.K.; Choi, J.E.; Kim, K.Y.; Lee, J.H.; Lee, J.; et al. Construction of a BAC library of Korean ginseng and initial analysis of BAC-end sequences. Mol. Genet. Genomics 2004, 271, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.; Kim, N.-H.; Lee, J.; Waminal, N.E.; Lee, S.-C.; Jayakodi, M.; Choi, H.-I.; Park, J.Y.; Lee, J.-E.; Yang, T.-J. A glimpse of Panax ginseng genome structure revealed from ten BAC clone sequences obtained by SMRT sequencing platform. Plant Breed. Biotechnol. 2017, 5, 25–35. [Google Scholar] [CrossRef]
- Haralampidis, K.; Trojanowska, M.; Osbourn, A.E. Biosynthesis of triterpenoid saponins in plants. In History and Trends in Bioprocessing and Biotransformation; Dutta, N.N., Hammar, F., Haralampidis, K., Karanth, N.G., König, A., Krishna, S.H., Kunze, G., Nagy, E., Orlich, B., Osbourn, A.E., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 31–49. [Google Scholar]
- Wang, K.; Jiang, S.; Sun, C.; Lin, Y.; Yin, R.; Wang, Y.; Zhang, M. The spatial and temporal transcriptomic landscapes of ginseng, Panax ginseng C. A. Meyer. Sci. Rep. 2015, 5, 18283. [Google Scholar] [CrossRef] [PubMed]
- Jo, I.-H.; Lee, S.-H.; Kim, Y.-C.; Kim, D.-H.; Kim, H.-S.; Kim, K.-H.; Chung, J.-W.; Bang, K.-H. De novo transcriptome assembly and the identification of gene-associated single-nucleotide polymorphism markers in asian and american ginseng roots. Mol. Genet. Genom. 2015, 290, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wang, M.; Ma, Y.; Yuan, L.; Lu, S. High-throughput sequencing and characterization of the small RNA transcriptome reveal features of novel and conserved microRNAs in Panax ginseng. PLoS ONE 2012, 7, e44385. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-S.; Raveendar, S.; Suresh, S.; Lee, G.-A.; Lee, J.-R.; Cho, J.-H.; Lee, S.-Y.; Ma, K.-H.; Cho, G.-T.; Chung, J.-W. Transcriptome analysis of two Vicia sativa subspecies: Mining molecular markers to enhance genomic resources for vetch improvement. Genes 2015, 6, 1164–1182. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.Q.; Tanzi, S.A.; Ho, K.W.; Malla, S.; Blythe, M.; Karunaratne, A.; Massawe, F.; Mayes, S. Development of gene-based SSR markers in winged bean (Psophocarpus tetragonolobus (L.) DC.) for diversity assessment. Genes 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. Cd-hit suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Parra, G.; Guigó, R. Using geneID to identify genes. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. Interproscan: Protein domains identifier. Nucl. Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Repeatmasker. Available online: http://repeatmasker.org (accessed on 3 July 2017).
- Zhang, D.; Li, W.; Xia, E.H.; Zhang, Q.J.; Liu, Y.; Zhang, Y.; Tong, Y.; Zhao, Y.; Niu, Y.C.; Xu, J.H.; et al. The medicinal herb Panax notoginseng genome provides insights into ginsenoside biosynthesis and genome evolution. Mol. Plant 2017, 10, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Repbase. Available online: http://girinst.org/repbase/ (accessed on 3 July 2017).
- Gydb. Available online: http://gydb.org/index.php/Main_Page (accessed on 3 July 2017).
- SSR Finder. Available online: ftp://ftp.gramene.org/pub/gramene/archives/software/scripts/ssr.pl (accessed on 3 July 2017).
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Lee, S.-C.; Lee, Y.S.; Park, H.-S.; Kim, N.-H.; Jang, W.; Lee, H.O.; Joh, H.J.; Yang, T.-J. Comprehensive analysis of Panax ginseng root transcriptomes. BMC Plant Biol. 2015, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sugimoto, K.; Otsuki, H.; Hirochika, H. Transcriptional activation of the tobacco retrotransposon tto1 by wounding and methyl jasmonate. Plant Mol. Biol. 1998, 36, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, Y.; Guo, X.; Sun, C.; Luo, H.; Song, J.; Li, Y.; Wang, L.; Qian, J.; Chen, S. Transcriptome analysis reveals ginsenosides biosynthetic genes, microRNAs and simple sequence repeats in Panax ginsengc. A. Meyer. BMC Genom. 2013, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-H.; Ma, C.-H.; Zhang, J.-J.; Chen, J.-W.; Tang, Q.-Y.; He, M.-H.; Xu, X.-Z.; Jiang, N.-H.; Yang, S.-C. Transcriptome analysis of Panax vietnamensis var. Fuscidicus discovers putative ocotillol-type ginsenosides biosynthesis genes and genetic markers. BMC Genom. 2015, 16, 159. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, K.T.; Mohanta, N.; Bae, H. Identification and expression analysis of pin-like (PILS) gene family of rice treated with auxin and cytokinin. Genes 2015, 6, 622–640. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-H.; Liu, Y.-B.; Zhang, X.-S. Auxin–cytokinin interaction regulates meristem development. Mol. Plant 2011, 4, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Su, H.; Zhang, L.; Liao, B.-S.; Xiao, S.-M.; Dong, L.-L.; Hu, Z.-G.; Wang, P.; Li, X.-W.; Huang, Z.-H.; et al. Comprehensive characterization for ginsenosides biosynthesis in ginseng root by integration analysis of chemical and transcriptome. Molecules 2017, 22, 889. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Nuruzzaman, M.; Xiu, H.; Huang, J.; Wu, K.; Chen, X.; Li, J.; Wang, L.; Jeong, J.-H.; Park, S.-J.; et al. Transcriptome analysis of methyl jasmonate-elicited Panax ginseng adventitious roots to discover putative ginsenoside biosynthesis and transport genes. Int. J. Mol. Sci. 2015, 16, 3035–3057. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Zhu, J.; Cao, H.; Huang, J.; Xiu, H.; Luo, T.; Luo, Z. Molecular cloning and expression profile of an abiotic stress and hormone responsive MYB transcription factor gene from Panax ginseng. Acta Biochim. Biophys. Sin. 2015, 47, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Cao, H.; Xiu, H.; Luo, T.; Li, J.; Chen, X.; Luo, J.; Luo, Z. Transcriptomics-based identification of WRKY genes and characterization of a salt and hormone-responsive pgWRKY1 gene in Panax ginseng. Acta Biochim. Biophys. Sin. 2016, 48, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhong, Y.; Guo, A.; Zhu, Q.; Tang, W.; Zheng, W.; Gu, X.; Wei, L.; Luo, J. Drtf: A database of rice transcription factors. Bioinformatics 2006, 22, 1286–1287. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Nishiyama, Y.; Hirai, M.Y.; Yano, M.; Nakajima, J.-I.; Awazuhara, M.; Inoue, E.; Takahashi, H.; Goodenowe, D.B.; Kitayama, M.; et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005, 42, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The molecular genetics of crop domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name & Description | EC Number | Number of Unigene | Number of Unigene with Complete ORF |
|---|---|---|---|
| AACT, acetyl-CoA acetyltransferase | 2.3.1.9 | 18 | 12 |
| HMGS, hydroxymethylglutaryl-CoA synthase | 2.3.3.10 | 22 | 18 |
| HMGR, hydroxymethylglutaryl-CoA reductase | 1.1.1.34 | 35 | 26 |
| MVK, mevalonate kinase | 2.7.1.36 | 13 | 10 |
| PMK, phosphomevalonate kinase | 2.7.4.2 | 17 | 10 |
| MVD, mevalonate diphosphate decarboxylase | 4.1.1.33 | 8 | 5 |
| GGPPS, geranylgeranyl pyrophosphate synthase | 2.5.1.29 | 17 | 14 |
| FPPS, farnesyl diphosphate synthase | 2.5.1.10 | 36 | 23 |
| IPPI, isopentenyl diphospate isomerase | 5.3.3.2 | 7 | 2 |
| SS, squalene synthase | 2.5.1.21 | 15 | 14 |
| SE, squalene epoxidase | 1.14.99.7 | 64 | 47 |
| DS, dammarenediol-II synthase | 4.2.1.125 | 33 | 29 |
| β-AS, β-amyrin synthase | 5.4.99.39 | 5 | 5 |
| β-A28O, β-amyrin 28-oxidase | 1.14.13 | 2 | 2 |
| D12H, dammarenediol 12-hydroxylase | 1.14.13.183 | 13 | 9 |
| P6H, protopanaxadiol 6-hydroxylase | 1.14.13.184 | 9 | 7 |
| Gene Name | [TAIR] Description | [TAIR] AGI Number | Number of Unigenes | Number of Unigene with Complete ORF |
|---|---|---|---|---|
| Aux/IAA | ||||
| IAA7 | Indole-3-acetic acid 7 | AT3G23050.1 | 3 | 2 |
| IAA8 | Indoleacetic acid-induced protein 8 | AT2G22670.4 | 18 | 10 |
| IAA9 | Indole-3-actic acid inducible 9 | AT5G65670.1 | 21 | 17 |
| IAA14 | Indole-3-acetic acid inducible 14 | AT4G14550.1 | 1 | 1 |
| IAA16 | Indoleacetic acid-induced protein 16 | AT3G04730.1 | 2 | 1 |
| IAA17 | Indole-3-acetic acid inducible 17 | AT1G04250.1 | 2 | 1 |
| IAA18 | Indole-3-acetic acid inducible 18 | AT1G51950.1 | 2 | 1 |
| SCF complex | ||||
| SKP1 | S phase kinase-associated protein 1 | AT1G75950.1 | 11 | 7 |
| CUL1 | Cullin 1 | AT4G02570.1 | 55 | 38 |
| AFB1 | Auxin signaling F box protein 1 | AT4G03190.1 | 5 | 2 |
| Auxin Response Factor | ||||
| ARF3 | Auxin response factor 3 | AT2G33860.1 | 17 | 15 |
| ARF5 | Auxin response factor 5 | AT1G19850.1 | 16 | 10 |
| ARF6 | Auxin response factor 6 | AT1G30330.2 | 146 | 127 |
| ARF7 | Auxin response factor 7 | AT5G20730.1 | 19 | 9 |
| ARF8 | Auxin response factor 8 | AT5G37020.1 | 41 | 38 |
| ARF16 | Auxin response factor 16 | AT4G30080.1 | 30 | 27 |
| ARF17 | Auxin response factor 17 | AT1G77850.1 | 4 | 3 |
| ARF19 | Auxin response factor 19 | AT1G19220.1 | 56 | 45 |
| Gene Name | [TAIR] Description | [TAIR] AGI Number | Number of Unigenes | Number of Unigene with Complete ORF |
|---|---|---|---|---|
| Histidine Kinase | ||||
| AHK2 | Arabidopsis histidine kinase 2 | AT5G35750.1 | 3 | 2 |
| AHK3 | Arabidopsis histidine kinase 3 | AT1G27320.1 | 5 | 5 |
| AHK4 | Arabidopsis histidine kinase 4 | AT2G01830.2 | 5 | 5 |
| Histidine-Containing Phosphotransmitter | ||||
| AHP1 | HP 1 | AT3G21510.1 | 2 | 2 |
| AHP5 | HP 5 | AT1G03430.1 | 1 | 1 |
| Type A-Response Regulator | ||||
| ARR4 | Response regulator 4 | AT1G10470.1 | 1 | 1 |
| ARR6 | Response regulator 6 | AT5G62920.1 | 1 | 0 |
| ARR9 | Response regulator 9 | AT3G57040.1 | 2 | 2 |
| ARR15 | Response regulator 15 | AT1G74890.1 | 1 | 0 |
| Type B-Response Regulator | ||||
| ARR1 | Response regulator 1 | AT3G16857.2 | 4 | 4 |
| ARR2 | Response regulator 2 | AT4G16110.1 | 19 | 19 |
| ARR10 | Response regulator 10 | AT4G31920.1 | 2 | 2 |
| ARR11 | Response regulator 11 | AT1G67710.1 | 3 | 3 |
| ARR12 | Response regulator 12 | AT2G25180.1 | 5 | 5 |
| ARR18 | Response regulator 18 | AT5G58080.1 | 2 | 2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, I.-H.; Lee, J.; Hong, C.E.; Lee, D.J.; Bae, W.; Park, S.-G.; Ahn, Y.J.; Kim, Y.C.; Kim, J.U.; Lee, J.W.; et al. Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome. Genes 2017, 8, 228. https://doi.org/10.3390/genes8090228
Jo I-H, Lee J, Hong CE, Lee DJ, Bae W, Park S-G, Ahn YJ, Kim YC, Kim JU, Lee JW, et al. Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome. Genes. 2017; 8(9):228. https://doi.org/10.3390/genes8090228
Chicago/Turabian StyleJo, Ick-Hyun, Jinsu Lee, Chi Eun Hong, Dong Jin Lee, Wonsil Bae, Sin-Gi Park, Yong Ju Ahn, Young Chang Kim, Jang Uk Kim, Jung Woo Lee, and et al. 2017. "Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome" Genes 8, no. 9: 228. https://doi.org/10.3390/genes8090228