Proteomic Analysis of Methanonatronarchaeum thermophilum AMET1, a Representative of a Putative New Class of Euryarchaeota, “Methanonatronarchaeia”

 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Cultivation Conditions and Proteomics
2.2. Proteomic Data Normalization and Analysis
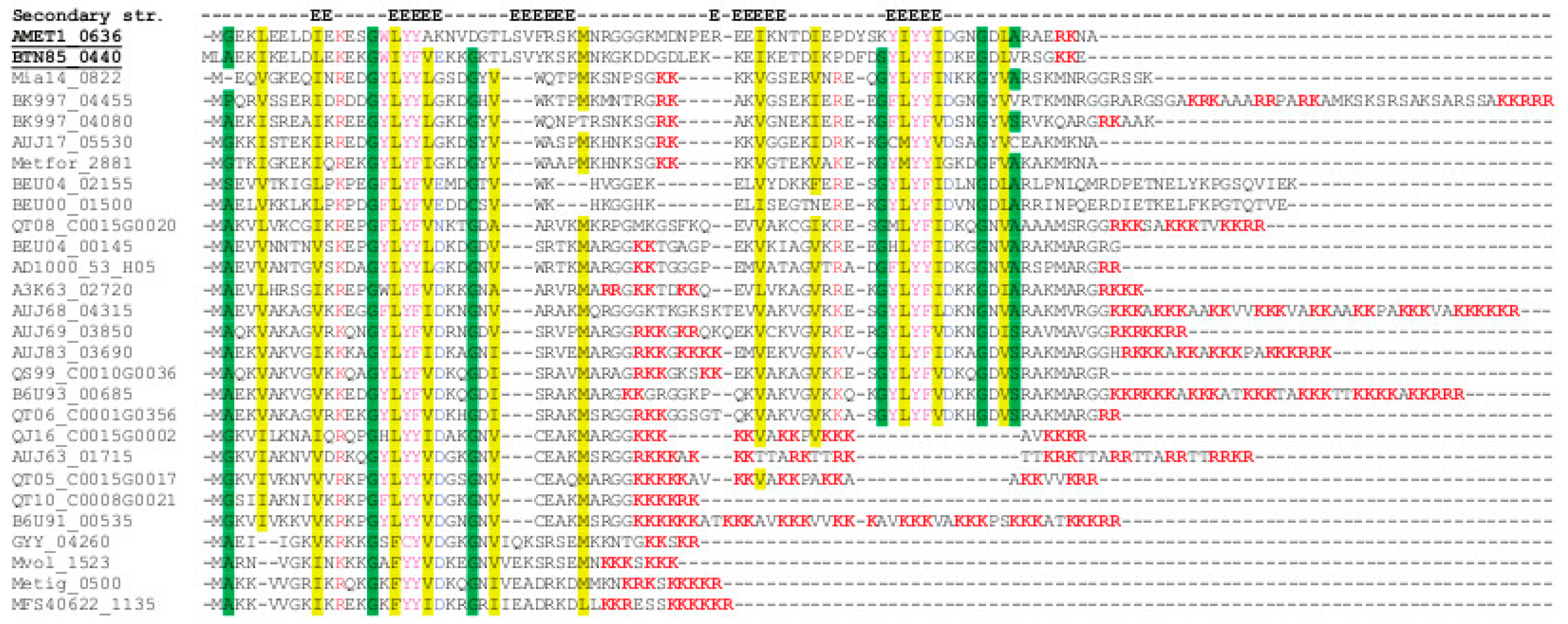
2.3. Sequence Analysis
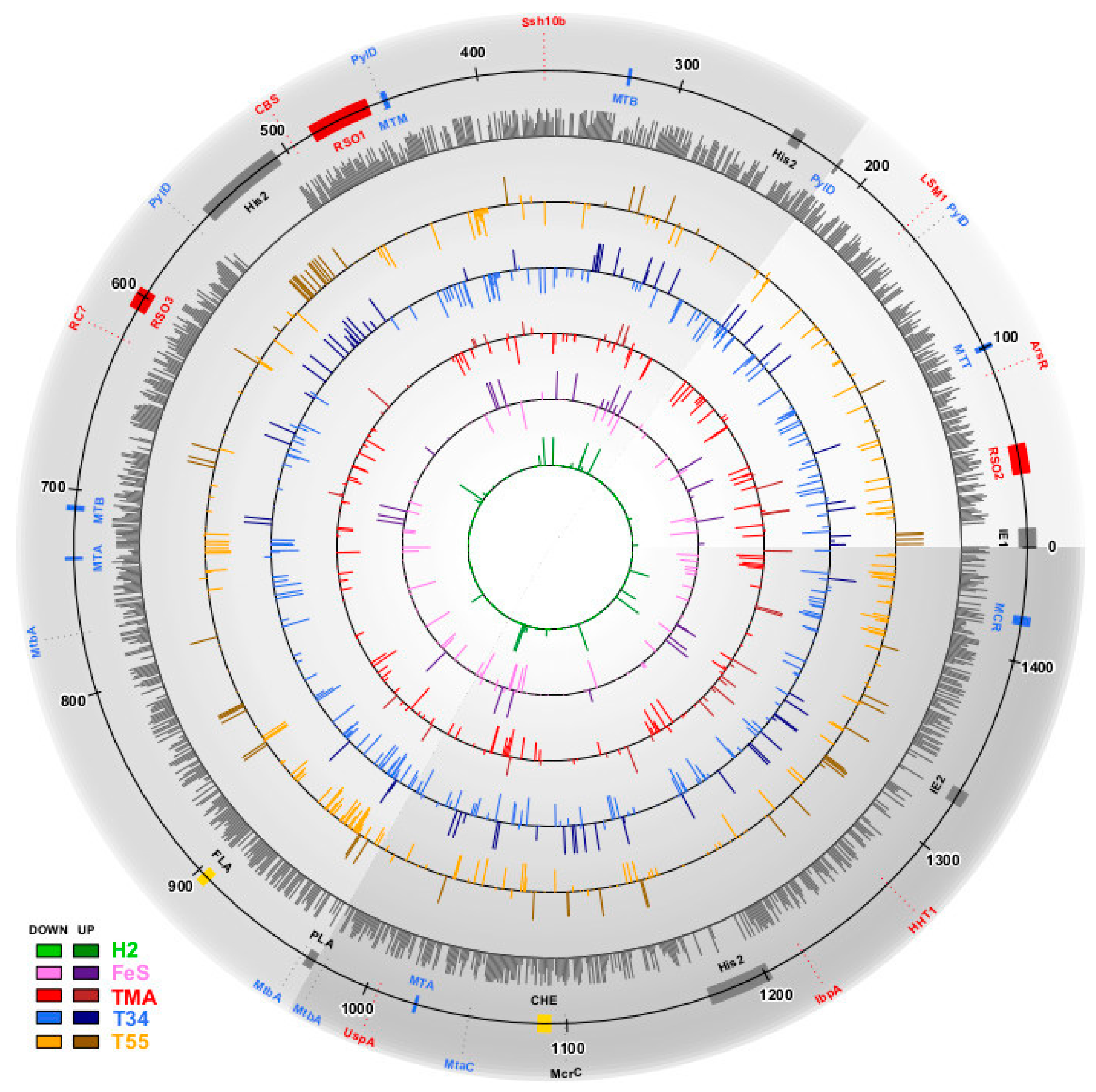
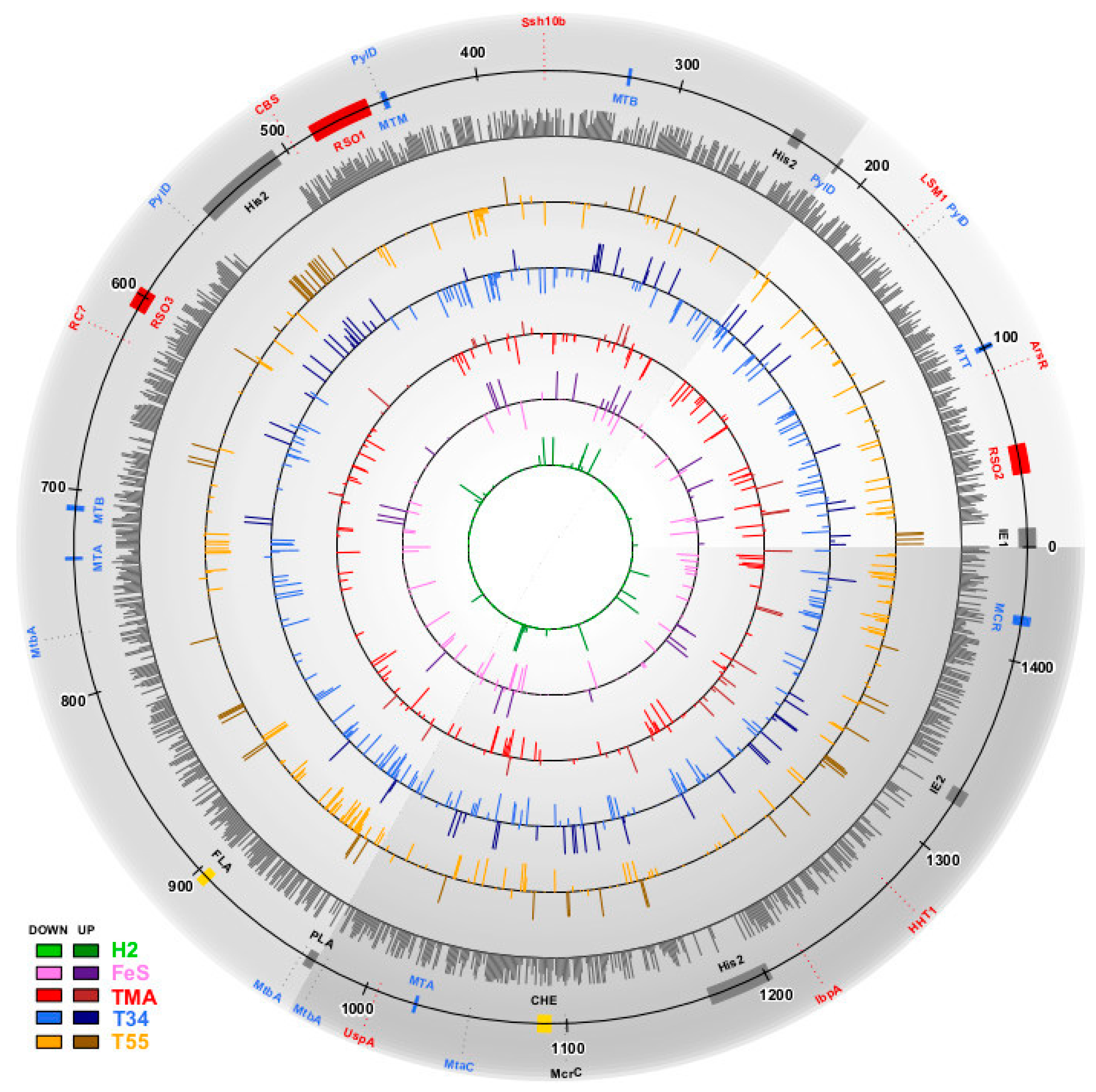
3. Results
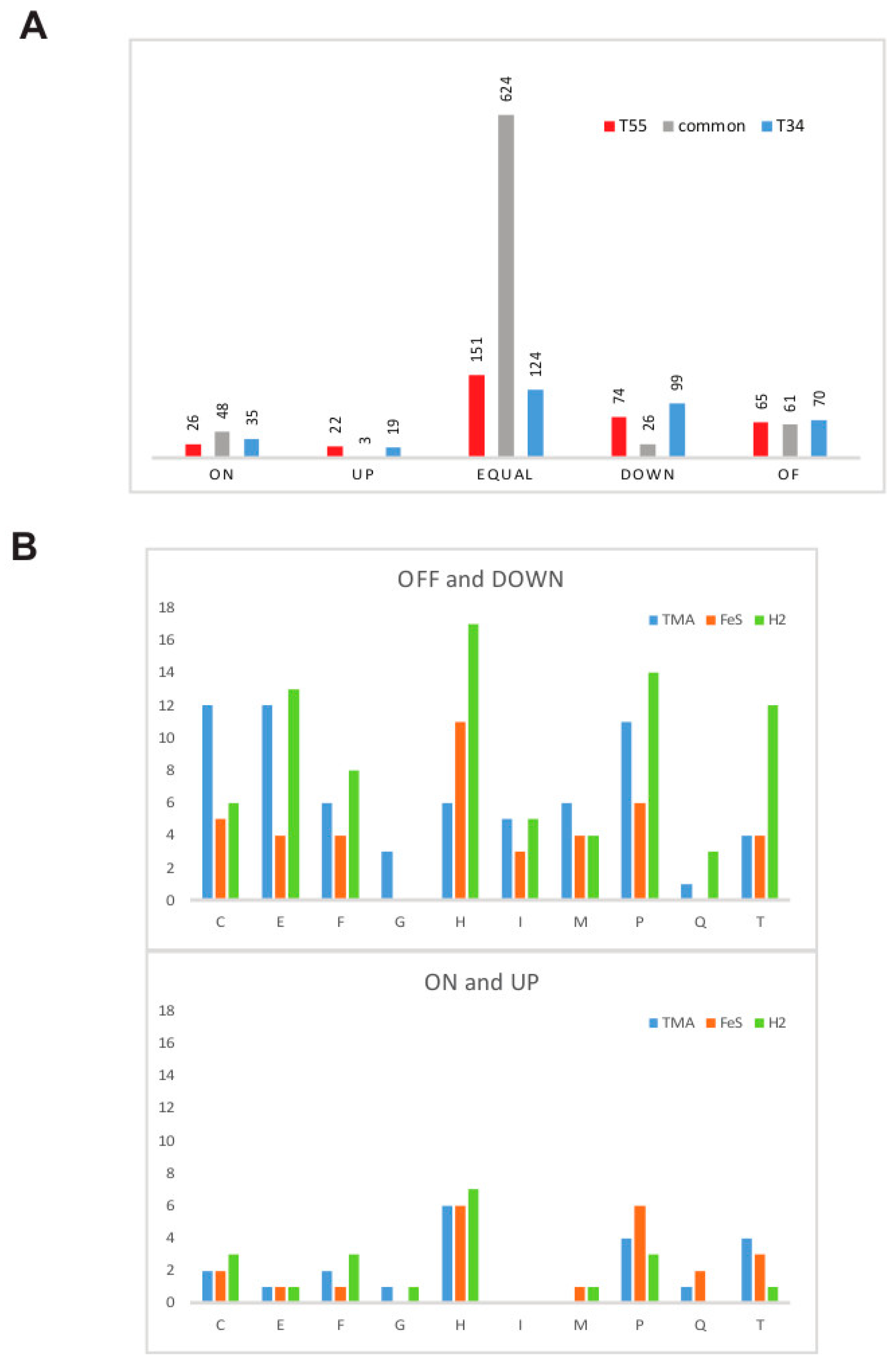
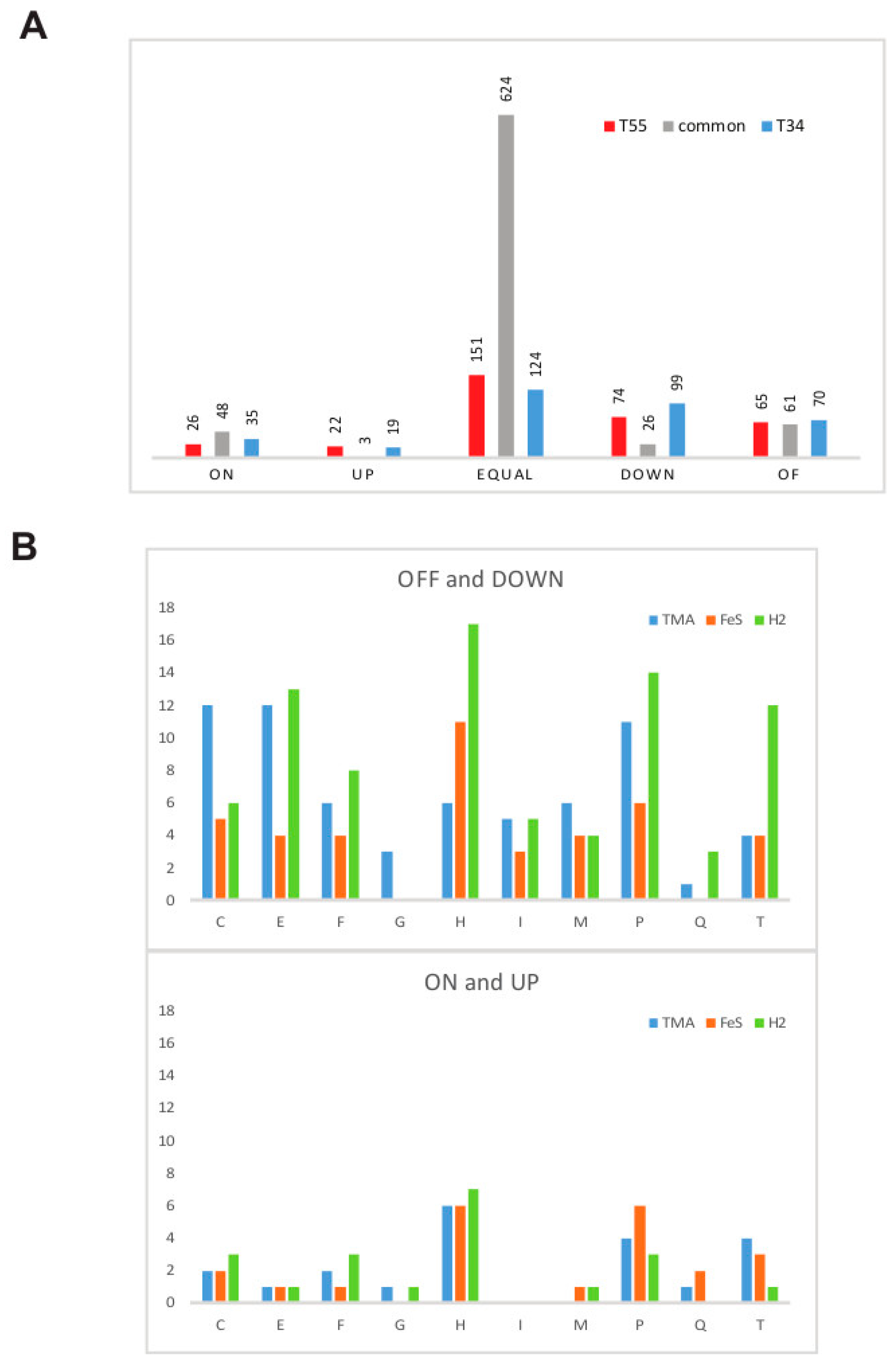
3.1. Estimation of Significant Changes in Protein Abundance
3.2. Methanonatronarchaeum thermophilum AMET1 Protein Abundances under Optimal Growth Conditions
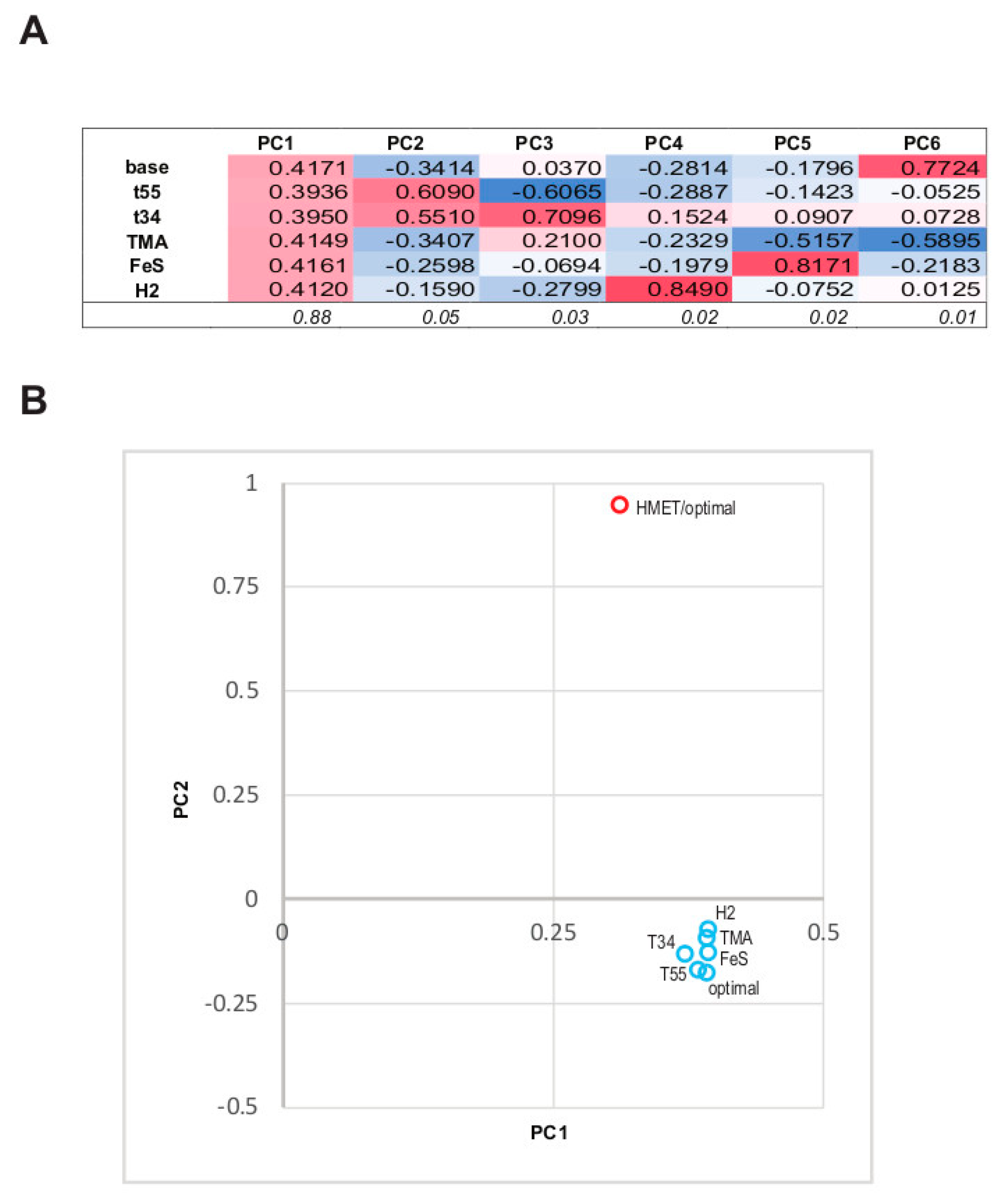
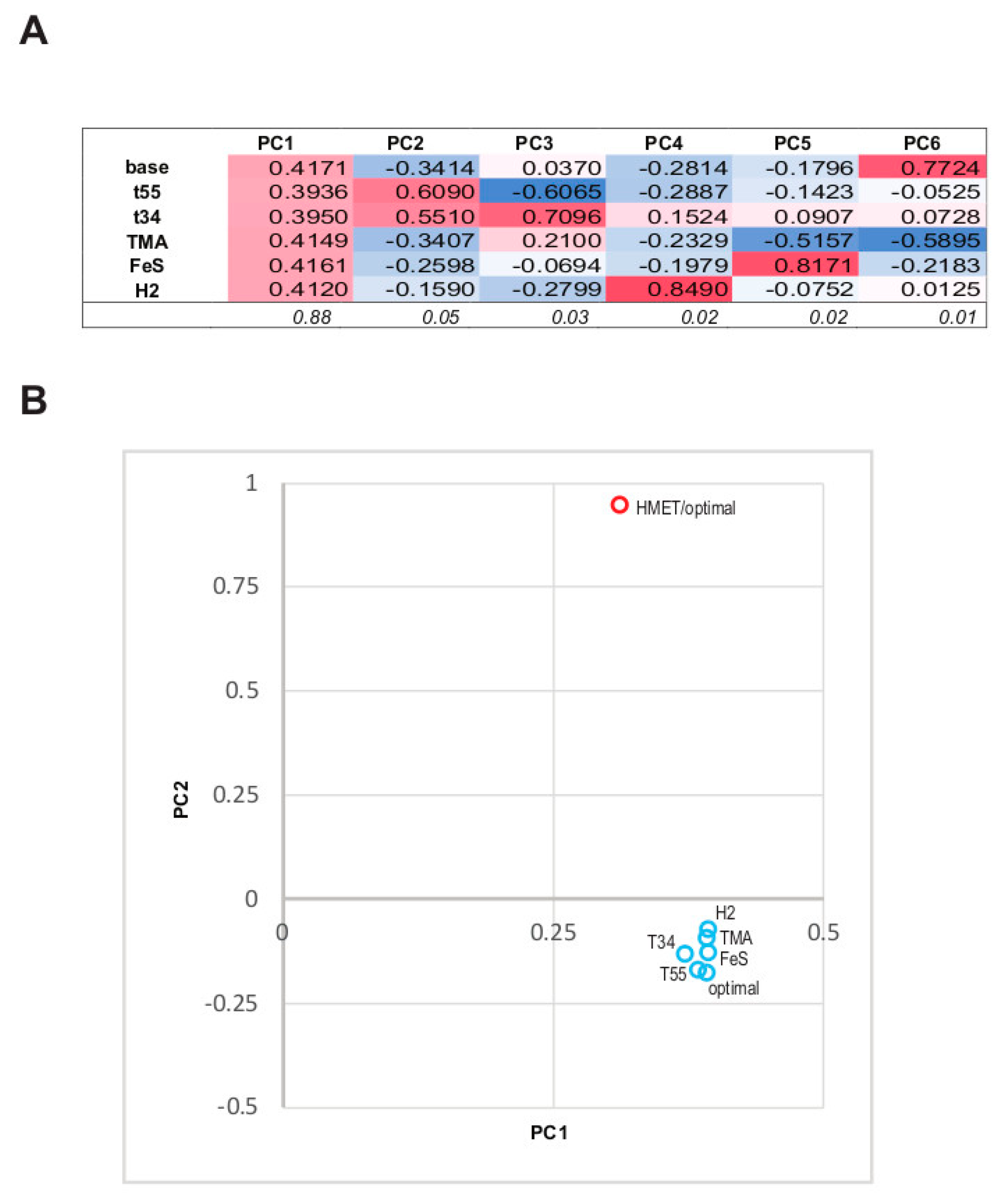
3.3. The Structure of the Protein Abundance Space in Different Conditions
3.4. Supra- and Suboptimal Temperature Effect on Protein Abundance
3.5. Response to Growth Factor Changes
3.6. Positive and Negative Correlations between Abundances of Paralogous Proteins
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Offre, P.; Spang, A.; Schleper, C. Archaea in biogeochemical cycles. Annu. Rev. Microbiol. 2013, 67, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Makarova, K.S.; Abbas, B.; Ferrer, M.; Golyshin, P.N.; Galinski, E.A.; Ciordia, S.; Mena, M.C.; Merkel, A.Y.; Wolf, Y.I.; et al. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2017, 2, 17081. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; O’Toole, P.W.; Harris, H.M.; Peyret, P.; Brugere, J.F.; Gribaldo, S. Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol. Evol. 2013, 5, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Adam, P.S.; Gribaldo, S. Methanogenesis and the wood-ljungdahl pathway: An ancient, versatile and fragile association. Genome Biol. Evol. 2016, 8, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Werner, J.; Chernikova, T.N.; Bargiela, R.; Fernandez, L.; La Cono, V.; Waldmann, J.; Teeling, H.; Golyshina, O.V.; Glockner, F.O.; et al. Unveiling microbial life in the new deep-sea hypersaline lake Thetis. Part ii: A metagenomic study. Environ. Microbiol. 2012, 14, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dong, H.; Yu, B.; Liu, X.; Li, Y.; Ji, S.; Zhang, C.L. Microbial response to salinity change in lake Chaka, a hypersaline lake on tibetan plateau. Environ. Microbiol. 2007, 9, 2603–2621. [Google Scholar] [CrossRef] [PubMed]
- Eder, W.; Schmidt, M.; Koch, M.; Garbe-Schonberg, D.; Huber, R. Prokaryotic phylogenetic diversity and corresponding geochemical data of the brine-seawater interface of the Shaban Deep, Red Sea. Environ. Microbiol. 2002, 4, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Abbas, B.; Geleijnse, M.; Pimenov, N.V.; Sukhacheva, M.V.; van Loosdrecht, M.C. Methanogenesis at extremely haloalkaline conditions in the soda lakes of Kulunda steppe (Altai, Russia). FEMS Microbiol. Ecol. 2015, 91, fiv016. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Abbas, B.; Merkel, A.Y.; Rijpstra, W.I.; Damste, J.S.; Sukhacheva, M.V.; van Loosdrecht, M.C. Methanosalsum natronophilum sp. nov. and Methanocalculus alkaliphilus sp. nov., haloalkaliphilic methanogens from hypersaline soda lakes. Int. J. Syst. Evol. Microbiol. 2015, 65, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Torgerson, W.S. Theory and Methods of Scaling; John Wiley and Sons: New York, NY, USA, 1958. [Google Scholar]
- Arike, L.; Peil, L. Spectral counting label-free proteomics. Methods Mol. Biol. 2014, 1156, 213–222. [Google Scholar] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Petitjean, C.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Extreme deviations from expected evolutionary rates in archaeal protein families. Genome Biol. Evol. 2017, 9, 2791–2811. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. Jpred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a Hidden Markov Model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Bath, C.; Cukalac, T.; Porter, K.; Dyall-Smith, M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, salterprovirus. Virology 2006, 350, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Laurens, N.; Driessen, R.P.; Heller, I.; Vorselen, D.; Noom, M.C.; Hol, F.J.; White, M.F.; Dame, R.T.; Wuite, G.J. Alba shapes the archaeal genome using a delicate balance of bridging and stiffening the DNA. Nat. Commun. 2012, 3, 1328. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.W. Methyl-coenzyme m reductase genes: Unique functional markers for methanogenic and anaerobic methane-oxidizing archaea. Methods Enzymol. 2005, 397, 428–442. [Google Scholar] [PubMed]
- Shima, S.; Krueger, M.; Weinert, T.; Demmer, U.; Kahnt, J.; Thauer, R.K.; Ermler, U. Structure of a methyl-coenzyme m reductase from black sea mats that oxidize methane anaerobically. Nature 2011, 481, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Enoki, M.; Shinzato, N.; Sato, H.; Nakamura, K.; Kamagata, Y. Comparative proteomic analysis of Methanothermobacter themautotrophicus Δh in pure culture and in co-culture with a butyrate-oxidizing bacterium. PLoS ONE 2011, 6, e24309. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Burg, D.W.; Ertan, H.; Raftery, M.J.; Poljak, A.; Guilhaus, M.; Cavicchioli, R. Global proteomic analysis of the insoluble, soluble and supernatant fractions of the psychrophilic archaeon Methanococcoides burtonii. Part ii: The effect of different methylated growth substrates. J. Proteome Res. 2010, 9, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Hendrickson, E.L.; Zhang, Y.; Wang, T.; Taub, F.; Moore, B.C.; Porat, I.; Whitman, W.B.; Hackett, M.; Leigh, J.A. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time pcr. Mol. Cell. Proteom. 2006, 5, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Ereno-Orbea, J.; Oyenarte, I.; Martinez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, M.; Schroeder, R.; Furtig, B. Transient RNA-protein interactions in RNA folding. FEBS J. 2011, 278, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Schey, K.L.; Grey, A.C.; Nicklay, J.J. Mass spectrometry of membrane proteins: A focus on aquaporins. Biochemistry 2013, 52, 3807–3817. [Google Scholar] [CrossRef] [PubMed]
- White, R.H. L-aspartate semialdehyde and a 6-deoxy-5-ketohexose 1-phosphate are the precursors to the aromatic amino acids in Methanocaldococcus jannaschii. Biochemistry 2004, 43, 7618–7627. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.A.; Payne, K.A.P.; Leys, D. The UbiX-UbiD system: The biosynthesis and use of prenylated flavin (prfmn). Arch. Biochem. Biophys. 2017, 632, 209–221. [Google Scholar] [CrossRef] [PubMed]
- White, M.D.; Payne, K.A.; Fisher, K.; Marshall, S.A.; Parker, D.; Rattray, N.J.; Trivedi, D.K.; Goodacre, R.; Rigby, S.E.; Scrutton, N.S.; et al. UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature 2015, 522, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Koonin, E.V. Filling a gap in the central metabolism of archaea: Prediction of a novel aconitase by comparative-genomic analysis. FEMS Microbiol. Lett. 2003, 227, 17–23. [Google Scholar] [CrossRef]
- Elling, F.J.; Becker, K.W.; Konneke, M.; Schroder, J.M.; Kellermann, M.Y.; Thomm, M.; Hinrichs, K.U. Respiratory quinones in archaea: Phylogenetic distribution and application as biomarkers in the marine environment. Environ. Microbiol. 2016, 18, 692–707. [Google Scholar] [CrossRef] [PubMed]
- Tietze, M.; Beuchle, A.; Lamla, I.; Orth, N.; Dehler, M.; Greiner, G.; Beifuss, U. Redox potentials of methanophenazine and CoB-S-S-CoM, factors involved in electron transport in methanogenic archaea. Chembiochem 2003, 4, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Cavicchioli, R.; Thomas, T.; Curmi, P.M. Cold stress response in archaea. Extremophiles 2000, 4, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.B. Genomic analysis of psychrophilic prokaryotes. In Psychrophiles: From Biodiversity to Biotechnology; Springer: Berlin, Germany, 2008; pp. 265–284. [Google Scholar]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; Conway de Macario, E. Molecular chaperones: Multiple functions, pathologies and potential applications. Front. Biosci. J. Virtual Libr. 2007, 12, 2588–2600. [Google Scholar] [CrossRef]
- Williams, T.J.; Liao, Y.; Ye, J.; Kuchel, R.P.; Poljak, A.; Raftery, M.J.; Cavicchioli, R. Cold adaptation of the antarctic haloarchaea Halohasta litchfieldiae and Halorubrum lacusprofundi. Environ. Microbiol. 2017, 19, 2210–2227. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.A.; Jiang, R.; Krzycki, J.A. Functional context, biosynthesis and genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 2011, 14, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Fromont-Racine, M.; Senger, B.; Saveanu, C.; Fasiolo, F. Ribosome assembly in eukaryotes. Gene 2003, 313, 17–42. [Google Scholar] [CrossRef]
- Leidig, C.; Bange, G.; Kopp, J.; Amlacher, S.; Aravind, A.; Wickles, S.; Witte, G.; Hurt, E.; Beckmann, R.; Sinning, I. Structural characterization of a eukaryotic chaperone--the ribosome-associated complex. Nat. Struct. Mol. Biol. 2013, 20, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 a resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Sako, Y.; Prangishvili, D. Provirus induction in hyperthermophilic archaea: Characterization of Aeropyrum pernix spindle-shaped virus 1 and Aeropyrum pernix ovoid virus 1. J. Bacteriol. 2011, 193, 5412–5419. [Google Scholar] [CrossRef] [PubMed]
- Ratner, V.A.; Vasil’eva, L.A. The role of mobile genetic elements (MGE) in microevolution. Genetika 1992, 28, 5–17. [Google Scholar] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Archaeal clusters of orthologous genes (arCOGs): An update and application for analysis of shared features between Thermococcales, Methanococcales and Methanobacteriales. Life 2015, 5, 818–840. [Google Scholar] [CrossRef] [PubMed]
- Vierke, G.; Engelmann, A.; Hebbeln, C.; Thomm, M. A novel archaeal transcriptional regulator of heat shock response. J. Biol. Chem. 2003, 278, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Rohlin, L.; Trent, J.D.; Salmon, K.; Kim, U.; Gunsalus, R.P.; Liao, J.C. Heat shock response of Archaeoglobus fulgidus. J. Bacteriol. 2005, 187, 6046–6057. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Makarova, K.S.; Yutin, N.; Koonin, E.V. Updated clusters of orthologous genes for archaea: A complex ancestor of the archaea and the byways of horizontal gene transfer. Biol. Direct 2012, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Phipps, B.M.; Hoffmann, A.; Stetter, K.O.; Baumeister, W. A novel ATPase complex selectively accumulated upon heat shock is a major cellular component of thermophilic archaebacteria. EMBO J. 1991, 10, 1711–1722. [Google Scholar] [PubMed]
- Shockley, K.R.; Ward, D.E.; Chhabra, S.R.; Conners, S.B.; Montero, C.I.; Kelly, R.M. Heat shock response by the hyperthermophilic archaeon Pyrococcus furiosus. Appl. Environ. Microbiol. 2003, 69, 2365–2371. [Google Scholar] [CrossRef] [PubMed]
- Weng, R.R.; Shu, H.W.; Chin, S.W.; Kao, Y.; Chen, T.W.; Liao, C.C.; Tsay, Y.G.; Ng, W.V. Omics in ecology: Systems level analyses of Halobacterium salinarum reveal large-scale temperature-mediated changes and a requirement of CctA for thermotolerance. Omics J. Integr. Biol. 2014, 18, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, H.K.; Yaoi, T.; Brocchieri, L.; McMillan, R.A.; Alton, T.; Trent, J.D. The composition, structure and stability of a group ii chaperonin are temperature regulated in a hyperthermophilic archaeon. Mol. Microbiol. 2003, 48, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, L.; Curmi, P.M.; Siddiqui, K.S.; Poljak, A.; DeLong, E.; DasSarma, S.; Cavicchioli, R. Structure and function of cold shock proteins in archaea. J. Bacteriol. 2007, 189, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yue, L.; Zhou, L.; Qi, L.; Li, J.; Dong, X. Conserved TRAM domain functions as an archaeal cold shock protein via DNA chaperone activity. Front. Microbiol. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Taha; Siddiqui, K.S.; Campanaro, S.; Najnin, T.; Deshpande, N.; Williams, T.J.; Aldrich-Wright, J.; Wilkins, M.; Curmi, P.M.; Cavicchioli, R. Single TRAM domain RNA-binding proteins in archaea: Functional insight from Ctr3 from the antarctic methanogen Methanococcoides burtonii. Environ. Microbiol. 2016, 18, 2810–2824. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, P.; Neiner, T.; D’Imprima, E.; Banerjee, A.; Reindl, S.; Ghosh, A.; Arvai, A.S.; Mills, D.J.; van der Does, C.; Tainer, J.A.; et al. The nucleotide-dependent interaction of FlaH and FlaI is essential for assembly and function of the archaellum motor. Mol. Microbiol. 2016, 99, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Aylett, C.H.S.; Duggin, I.G. The tubulin superfamily in archaea. In Prokaryotic Cytoskeletons; Löwe, J., Amos, L., Eds.; Springer: Cham, Switzerland, 2017; Volume 84. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genbank Locus ID | Protein Name | Annotation | NemPAI Base Value |
|---|---|---|---|
| Methanogenesis related proteins | |||
| AMET1_1459 | McrB | Methyl coenzyme M reductase, beta subunit | 11.7 |
| AMET1_1461 | McrG | Methyl coenzyme M reductase, gamma subunit | 68.7 * |
| AMET1_1049 | MtaC | Methanogenic corrinoid protein MtaC | 78.6 * |
| AMET1_0104 | MttB1 | Trimethylamine-corrinoid methyltransferase | 68.4 * |
| AMET1_0460 | MtmB | Monomethylamine methyltransferase | 53.6 * |
| AMET1_0747 | MtaB | Methanol-cobalamin methyltransferase B subunit | 9.2 |
| AMET1_0264 | IlvH | Acetolactate synthase, small subunit | 9.0 |
| Translation related proteins | |||
| AMET1_0496 | RplD | Ribosomal protein L4 | 10.3 |
| AMET1_0619 | RPS28A | Ribosomal protein S28E/S33 | 18.6 |
| AMET1_0046 | RPL20A | Ribosomal protein L20A | 10.3 |
| AMET1_0579 | TEF1 | Translation elongation factor EF-1 alpha, GTPase | 9.5 |
| DNA and RNA binding protein | |||
| AMET1_0176 | LSM1 | Small nuclear ribonucleoprotein | 13.2 |
| AMET1_0379 | Ssh10b | Archaeal DNA-binding protein Alba | 161.1 * |
| AMET1_1312 | HHT1 | Histones H3 and H4 | 22.6 |
| Chaperones | |||
| AMET1_1255 | IbpA | Molecular chaperone, HSP20 family, | 9.8 |
| AMET1_1030 | - | Nucleotide-binding protein, UspA family | 43.2* |
| Biological function unknown | |||
| AMET1_0092 | - | Transcriptional regulator, ArsR family | 56.8 * |
| AMET1_0509 | - | CBS domain | 10.0 |
| AMET1_0343 | - | CBS domain | 9.0 |
| AMET1_0636 | - | Uncharacterized protein | 39.0 * |
| Locus # | Description | Change $ | nemPAI or (Fold Change) * |
|---|---|---|---|
| T55 (maximum growth temperature) | |||
| AMET1_1336 | HTH domain containing protein | ON | 55.2 |
| AMET1_1130 | Thermosome subunit, GroEL/HSP60 family | UP | (15.8) |
| AMET1_1197 | Thermosome subunit, GroEL/HSP60 family | UP | (8.5) |
| AMET1_0742 | Cell division GTPase FtsZ | UP | (5.8) |
| AMET1_0032 | Desulfoferredoxin | UP | (9.7) |
| AMET1_0852 | ATPase of the AAA+ class, CDC48 family | UP | (4.0) |
| AMET1_0493 | Ribosomal protein S19 | DOWN | (11) |
| T34 (minimum growth temperature) | |||
| AMET1_0938 | DNA replication ATPase HolB, small subunit | DOWN | (19) |
| AMET1_1237 | Wybutosine biosynthesis enzyme Trm5 | DOWN | (16) |
| AMET1_1255 | Molecular chaperone, HSP20 family | DOWN | (5.3) |
| AMET1_0053 | 16S rRNA N6-dimethyltransferase RsmA/KsgA/DIM1 | OFF | −0.3 |
| AMET1_0781 | SAM-dependent methyltransferase | UP | (10.7) |
| AMET1_0225 | Coproporphyrinogen III oxidase HemN | UP | (5.8) |
| AMET1_0071 | Translation elongation factor EF-1 beta | UP | (5.2) |
| AMET1_1156 | Molecular chaperone, HSP20 family | ON | 0.29 |
| Both T55 and T34 | |||
| AMET1_0168 | (2R,3R)-3-methylornithine synthase PylB | DOWN | (56.5), (5.1) |
| AMET1_0221 | Pyrrolysyl-tRNA-synthetase PylS | DOWN | (36), (6.5) |
| AMET1_0092 | Transcriptional regulator, ArsR family | DOWN | (5.7), (3.2) |
| AMET1_0050 | Ribosomal protein L21E | DOWN | (3.8), (6.2) |
| AMET1_0428 | hypothetical protein | OFF | −1.6 |
| AMET1_0965 | Glycosyltransferase family 1 | OFF | −1.4 |
| AMET1_0970 | Glycosyltransferase family 1 | OFF | −1.0 |
| AMET1_0552 | hypothetical protein | ON | 1.11, 0.47 |
| AMET1_0548 | hypothetical protein | ON | 0.71, 0.80 |
| AMET1_0532 | hypothetical protein | ON | 0.89, 0.3 |
| AMET1_0853 | Transcriptional regulator, ArsR family | UP | (9.6), (4.2) |
| AMET1_0044 | Ribosomal protein L31E | UP | (3.3), (3.7) |
| Locus # | Description | Change $ | nemPAI or (Fold Change) * |
|---|---|---|---|
| TMA | |||
| AMET1_1049 | Methanogenic corrinoid protein MtaC | DOWN | (27.2) |
| AMET1_0372 | Predicted transcriptional regulator | DOWN | (23.2) |
| AMET1_0479 | Ribosomal protein L19E | DOWN | (15.4) |
| AMET1_1538 | Glutamine synthetase | OFF | −1.2 |
| AMET1_0199 | Ribosomal protein L37AE/L43A | OFF | −0.4 |
| AMET1_1486 | Multisubunit Na+/H+ antiporter, MnhG subunit | OFF | −0.26 |
| AMET1_0038 | ACT domain-containing protein | ON | 0.18 |
| AMET1_1329 | Uncharacterized protein | ON | 0.18 |
| AMET1_0105 | Trimethylamine corrinoid protein MtbC1 | UP | (7.1) |
| AMET1_0722 | Dimethylamine methyltransferase MtbB | UP | (4.9) |
| AMET1_0460 | Monomethylamine methyltransferase MtmB | UP | (4.4) |
| FeS | |||
| AMET1_1049 | Methanogenic corrinoid protein MtaC | DOWN | (11.8) |
| AMET1_0222 | (2R,3R)-3-methylornithine synthase PylB | DOWN | (10/8) |
| AMET1_0748 | Methanogenic corrinoid protein MtaC | DOWN | (7.0) |
| AMET1_0462 | Fe-S cluster biogenesis protein NfuA, 4Fe-4S-binding domain | UP | (13.7) |
| AMET1_1183 | Energy-coupling factor transporter ATP-binding protein EcfA2 | UP | (5.5) |
| AMET1_0093 | ABC-type cobalamin/Fe3+-siderophores transport system, ATPase component | UP | (5.4) |
| AMET1_1336 | HTH domain containing protein | ON | 230.9 |
| AMET1_0698 | ABC-type Fe3+-hydroxamate transport system, periplasmic component | ON | 1.0 |
| H2 | |||
| AMET1_0168 | (2R,3R)-3-methylornithine synthase PylB | OFF | −1.3 |
| AMET1_0222 | (2R,3R)-3-methylornithine synthase PylB | OFF | −0.4 |
| AMET1_1113 | Methylase of chemotaxis methyl-accepting protein | OFF | −0.4 |
| AMET1_0575 | Chemotaxis signal transduction protein CheW | OFF | −0.3 |
| AMET1_0021 | Chemotaxis signal transduction protein CheW | OFF | −0.2 |
| AMET1_1117 | Chemotaxis protein histidine kinase CheA | DOWN | (30.7) |
| AMET1_0325 | Chromosome partition protein Smc | DOWN | (28.0) |
| AMET1_1115 | Chemotaxis protein CheC, flagellar motor switch protein | DOWN | (13.1) |
| AMET1_0923 | KaiC family ATPase FlaH involved in archaellum biogenesis | DOWN | (8.5) |
| AMET1_0181 | Nucleoside 2-deoxyribosyltransferase | UP | (6.6) |
| AMET1_0462 | Fe-S cluster biogenesis protein NfuA, 4Fe-4S-binding domain | UP | (6.2) |
| AMET1_0880 | ABC-type metal ion transport system, periplasmic component/surface adhesin LraI | UP | (5.9) |
| AMET1_0879 | ABC-type Mn/Zn transport system, ATPase component ZhuC | UP | (4.8) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer, M.; Sorokin, D.Y.; Wolf, Y.I.; Ciordia, S.; Mena, M.C.; Bargiela, R.; Koonin, E.V.; Makarova, K.S. Proteomic Analysis of Methanonatronarchaeum thermophilum AMET1, a Representative of a Putative New Class of Euryarchaeota, “Methanonatronarchaeia”. Genes 2018, 9, 28. https://doi.org/10.3390/genes9020028
Ferrer M, Sorokin DY, Wolf YI, Ciordia S, Mena MC, Bargiela R, Koonin EV, Makarova KS. Proteomic Analysis of Methanonatronarchaeum thermophilum AMET1, a Representative of a Putative New Class of Euryarchaeota, “Methanonatronarchaeia”. Genes. 2018; 9(2):28. https://doi.org/10.3390/genes9020028
Chicago/Turabian StyleFerrer, Manuel, Dimitry Y. Sorokin, Yuri I. Wolf, Sergio Ciordia, María C. Mena, Rafael Bargiela, Eugene V. Koonin, and Kira S. Makarova. 2018. "Proteomic Analysis of Methanonatronarchaeum thermophilum AMET1, a Representative of a Putative New Class of Euryarchaeota, “Methanonatronarchaeia”" Genes 9, no. 2: 28. https://doi.org/10.3390/genes9020028