Influence of NKG2D Genetic Variants on Response to Anti-TNF Agents in Patients with Rheumatoid Arthritis

Abstract
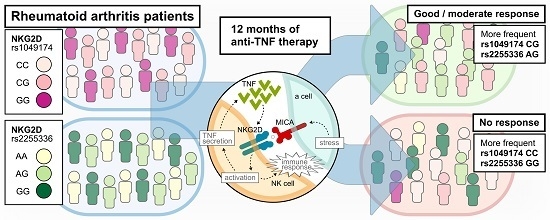
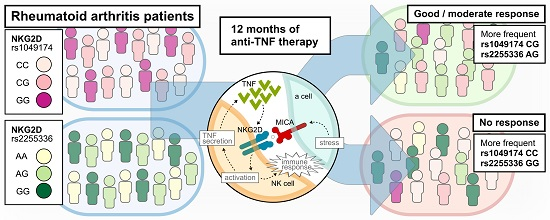
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Treatment Protocol
2.3. Evaluation of Anti-TNF Treatment Outcome
2.4. Single Nucleotide Polymorphisms Selection and Genotyping
2.5. Statistical Analysis
3. Results
3.1. Patients’ Baseline Characteristics and Response to Anti-TNF Treatment
3.2. Linkage Disequilibrium between the NKG2D Polymorphisms
3.3. Effect of NKG2D Genetic Variants on Anti-TNF Treatment Response
3.4. Distribution of the NKG2D Alleles and Genotypes with Regard to Selected Clinical Parameters
3.5. Distribution of the NKG2D Alleles and Genotypes with Regard to EULAR Responses to Adalimumab as well as Etanercept Treatment at 12th Week
3.6. Analysis of NKG2D Haplotypes
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheumatol. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.M.; Huizinga, T.W.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.; de Vries, R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheumatol. 2009, 60, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Haraoui, B. The anti-tumor necrosis factor agents are a major advance in the treatment of rheumatoid arthritis. J. Rheumatol. Suppl. 2005, 72, 46–47. [Google Scholar] [PubMed]
- Hetland, M.L.; Christensen, I.J.; Tarp, U.; Dreyer, L.; Hansen, A.; Hansen, I.T.; Kollerup, G.; Linde, L.; Lindegaard, H.M.; Poulsen, U.E.; et al. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: Results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheumatol. 2010, 62, 22–32. [Google Scholar] [CrossRef]
- Hyrich, K.L.; Watson, K.D.; Silman, A.J.; Symmons, D.P.M. British Society for Rheumatology Biologics Register. Predictors of response to anti-TNF-α therapy among patients with rheumatoid arthritis: Results from the British Society for Rheumatology Biologics Register. Rheumatology 2006, 45, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, M.; Visser, K.; Allaart, C.F.; Huizinga, T.W.J. Personalized medicine: Predicting responses to therapy in patients with RA. Curr. Opin. Pharmacol. 2013, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Verweij, C.L. Pharmacogenetics: Anti-TNF therapy in RA—Towards personalized medicine? Nat. Rev. Rheumatol. 2011, 7, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.P.; Schulze-Koops, H.; Aringer, M. The central role of T cells in rheumatoid arthritis. Clin. Exp. Rheumatol. 2007, 25, S4–S11. [Google Scholar] [PubMed]
- Gizinski, A.M.; Fox, D.A. T cell subsets and their role in the pathogenesis of rheumatic disease. Curr. Opin. Rheumatol. 2014, 26, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Ahern, D.J.; Brennan, F.M. The role of Natural Killer cells in the pathogenesis of rheumatoid arthritis: Major contributors or essential homeostatic modulators? Immunol. Lett. 2011, 136, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, P.; Scrivo, R.; Valesini, G.; Perricone, R. Emerging role for NK cells in the pathogenesis of inflammatory arthropathies. Autoimmun. Rev. 2011, 10, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Glienke, J.; Sobanov, Y.; Brostjan, C.; Steffens, C.; Nguyen, C.; Lehrach, H.; Hofer, E.; Francis, F. The genomic organization of NKG2C, E, F, and D receptor genes in the human natural killer gene complex. Immunogenetics 1998, 48, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.L.; Leibson, P.J. NKG2D-mediated activation of cytotoxic lymphocytes: Unique signaling pathways and distinct functional outcomes. Semin. Immunol. 2006, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef] [PubMed]
- González, S.; Groh, V.; Spies, T. Immunobiology of human NKG2D and its ligands. Curr. Top. Microbiol. Immunol. 2006, 298, 121–138. [Google Scholar] [PubMed]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Diefenbach, A.; Raulet, D.H. Strategies for target cell recognition by natural killer cells. Immunol. Rev. 2001, 181, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.A.; Trowsdale, J. Promiscuity and the single receptor: NKG2D. Nat. Rev. Immunol. 2007, 7, 737–744. [Google Scholar] [CrossRef] [PubMed]
- González, S.; López-Soto, A.; Suarez-Alvarez, B.; López-Vázquez, A.; López-Larrea, C. NKG2D ligands: Key targets of the immune response. Trends Immunol. 2008, 29, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.J.; Maasho, K.; Masilamani, M.; Narayanan, S.; Borrego, F.; Coligan, J.E. The NKG2D receptor: Immunobiology and clinical implications. Immunol. Res. 2008, 40, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Van Belle, T.L.; von Herrath, M.G. The role of the activating receptor NKG2D in autoimmunity. Mol. Immunol. 2009, 47, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bruhl, A.; El-Gabalawy, H.; Nelson, J.L.; Spies, T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2003, 100, 9452–9457. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Sumariwalla, P.F.; McCann, F.E.; Amjadi, P.; Chang, C.; McNamee, K.; Tornehave, D.; Haase, C.; Agersø, H.; Stennicke, V.W.; et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: A potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheumatol. 2011, 63, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kanai, T.; Totsuka, T.; Okamoto, R.; Tsuchiya, K.; Nemoto, Y.; Yoshioka, A.; Tomita, T.; Nagaishi, T.; Sakamoto, N.; et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G199–G207. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, K.; Hamerman, J.A.; Ehrlich, L.R.; Bour-Jordan, H.; Santamaria, P.; Bluestone, J.A.; Lanier, L.L. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity 2004, 20, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Fransen, J.; van Riel, P.L.C.M. The Disease Activity Score and the EULAR response criteria. Clin. Exp. Rheumatol. 2005, 23, S93–S99. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Taylor, J.A. SNPinfo: Integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009, 37, W600–W605. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, M.; Smith, N.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Scheet, P. Accounting for Decay of Linkage Disequilibrium in Haplotype Inference and Missing-Data Imputation. Am. J. Hum. Genet. 2005, 76, 449–462. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing [Internet]. R Foundation for Statistical Computing, Vienna, Austria. 2016. Available online: http://www.R-project.org/ (accessed on 20 June 2017).
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Rhinehart, R.; Randolph-Habecker, J.; Topp, M.S.; Riddell, S.R.; Spies, T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2001, 2, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Maasho, K.; Opoku-Anane, J.; Marusina, A.I.; Coligan, J.E.; Borrego, F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J. Immunol. 2005, 174, 4480–4484. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Castriconi, R.; Espéli, M.; Anfossi, N.; Juarez, T.; Hue, S.; Conway, H.; Romagné, F.; Dondero, A.; Nanni, M.; et al. Comparative analysis of human NK cell activation induced by NKG2D and natural cytotoxicity receptors. Eur. J. Immunol. 2004, 34, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.I.; Lee, L.; Schwarz, E.; Groh, V.; Spies, T.; Ebert, E.C.; Jabri, B. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J. Immunol. 2001, 167, 5527–5530. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, D.E.; Roberts, S.J.; Clarke, S.L.; Filler, R.; Lewis, J.M.; Tigelaar, R.E.; Girardi, M.; Hayday, A.C. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat. Immunol. 2005, 6, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Goronzy, J.J.; Weyand, C.M. CD4+ CD7− CD28− T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Investig. 1996, 97, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, T.; Wagner, U.G.; Goronzy, J.J.; Weyand, C.M. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheumatol. 1998, 41, 2108–2116. [Google Scholar] [CrossRef]
- Vallejo, A.N.; Schirmer, M.; Weyand, C.M.; Goronzy, J.J. Clonality and longevity of CD4+ CD28null T cells are associated with defects in apoptotic pathways. J. Immunol. 2000, 165, 6301–6307. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.J.; Takemura, S.; Goronzy, J.J.; Weyand, C.M. CD4+,CD28− T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheumatol. 2001, 44, 13–20. [Google Scholar] [CrossRef]
- Martens, P.B.; Goronzy, J.J.; Schaid, D.; Weyand, C.M. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheumatol. 1997, 40, 1106–1114. [Google Scholar] [CrossRef]
- Pawlik, A.; Ostanek, L.; Brzosko, I.; Masiuk, M.; Machalinski, B.; Gawronska-Szklarz, B. The expansion of CD4+ CD28− T cells in patients with rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, R210–R213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruck, T.; Bittner, S.; Gross, C.C.; Breuer, J.; Albrecht, S.; Korr, S.; Göbel, K.; Pankratz, S.; Henschel, C.M.; Schwab, N.; et al. CD4+ NKG2D+ T cells exhibit enhanced migratory and encephalitogenic properties in neuroinflammation. PLoS ONE 2013, 8, e81455. [Google Scholar] [CrossRef]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat-Zucman, S.; Paul, P.; Gornet, J.M.; Douay, C.; et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, H.; Ni, B.; He, Y.; Li, J.; Tang, Y.; Fu, X.; Wang, Q.; Xu, G.; Li, K.; et al. Mutual activation of CD4+ T cells and monocytes mediated by NKG2D-MIC interaction requires IFN-gamma production in systemic lupus erythematosus. Mol. Immunol. 2009, 46, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Broux, B.; Markovic-Plese, S.; Stinissen, P.; Hellings, N. Pathogenic features of CD4+ CD28− T cells in immune disorders. Trends Mol. Med. 2012, 18, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Garrity, D.; Call, M.E.; Feng, J.; Wucherpfennig, K.W. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc. Natl. Acad. Sci. USA 2005, 102, 7641–7646. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Imai, K.; Morishita, Y.; Hayashi, I.; Kusunoki, Y.; Nakachi, K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res. 2006, 66, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Roszak, A.; Lianeri, M.; Jagodziński, P.P. Prevalence of the NKG2D Thr72Ala polymorphism in patients with cervical carcinoma. Genet. Test. Mol. Biomark. 2012, 16, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Kabalak, G.; Thomas, R.M.; Martin, J.; Ortego-Centeno, N.; Jimenez-Alonso, J.; de Ramón, E.; Buyny, S.; Hamsen, S.; Gross, W.L.; Schnarr, S.; et al. Association of an NKG2D gene variant with systemic lupus erythematosus in two populations. Hum. Immunol. 2010, 71, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, P.; Lianeri, M.; Olesińska, M.; Jagodziński, P.P. Prevalence of the NKG2D Thr72Ala polymorphism in patients with systemic lupus erythematosus. Mol. Biol. Rep. 2012, 39, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Park, J.H.; Song, Y.W. Inhibitory NKG2A and activating NKG2D and NKG2C natural killer cell receptor genes: Susceptibility for rheumatoid arthritis. Tissue Antigens 2008, 72, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Furue, H.; Matsuo, K.; Kumimoto, H.; Hiraki, A.; Suzuki, T.; Yatabe, Y.; Komori, K.; Kanemitsu, Y.; Hirai, T.; Kato, T.; et al. Decreased risk of colorectal cancer with the high natural killer cell activity NKG2D genotype in Japanese. Carcinogenesis 2008, 29, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Furue, H.; Kumimoto, H.; Matsuo, K.; Suzuki, T.; Hasegawa, Y.; Shinoda, M.; Sugimura, T.; Mitsudo, K.; Tohnai, I.; Ueda, M.; et al. Opposite impact of NKG2D genotype by lifestyle exposure to risk of aerodigestive tract cancer among Japanese. Int. J. Cancer 2008, 123, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Takami, A.; Onizuka, M.; Sao, H.; Akiyama, H.; Miyamura, K.; Okamoto, S.; Inoue, M.; Kanda, Y.; Ohtake, S.; et al. NKG2D gene polymorphism has a significant impact on transplant outcomes after HLA-fully-matched unrelated bone marrow transplantation for standard risk hematologic malignancies. Haematologica 2009, 94, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Hayashi, T.; Yamaoka, M.; Kajimura, J.; Yoshida, K.; Kusunoki, Y.; Nakachi, K. Effects of NKG2D haplotypes on the cell-surface expression of NKG2D protein on natural killer and CD8 T cells of peripheral blood among atomic-bomb survivors. Hum. Immunol. 2012, 73, 686–691. [Google Scholar] [CrossRef] [PubMed]
- McGeough, C.M.; Berrar, D.; Wright, G.; Mathews, C.; Gilmore, P.; Cunningham, R.T.; Bjourson, A.J. Killer immunoglobulin-like receptor and human leukocyte antigen-C genotypes in rheumatoid arthritis primary responders and non-responders to anti-TNF-α therapy. Rheumatol. Int. 2012, 32, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Alasti, F.; Smolen, J.S. Optimisation of a treat-to-target approach in rheumatoid arthritis: Strategies for the 3-month time point. Ann. Rheum. Dis. 2016, 75, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.M.; Thaçi, D.; Gerdes, S.; Arenberger, P.; Pulka, G.; Kingo, K.; EGALITY Study Group; Hattebuhr, N.; Poetzl, J.; et al. The EGALITY study: A confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br. J. Dermatol. 2017, 176, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Burmester, G.R.; Combe, B.; Curtis, J.R.; Hall, S.; Haraoui, B.; van Vollenhoven, R.; Cioffi, C.; Ecoffet, C.; Gervitz, L.; et al. Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet 2016, 388, 2763–2774. [Google Scholar] [CrossRef]
- Ferreiro-Iglesias, A.; Montes, A.; Perez-Pampin, E.; Cañete, J.D.; Raya, E.; Magro-Checa, C.; Vasilopoulos, Y.; Sarafidou, T.; Caliz, R.; Ferrer, M.A.; et al. Replication of PTPRC as genetic biomarker of response to TNF inhibitors in patients with rheumatoid arthritis. Pharmacogenom. J. 2016, 16, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Montes, A.; Perez-Pampin, E.; Narváez, J.; Cañete, J.D.; Navarro-Sarabia, F.; Moreira, V.; Fernández-Nebro, A.; Del Carmen Ordóñez, M.; de la Serna, A.R.; Magallares, B.; et al. Association of FCGR2A with the response to infliximab treatment of patients with rheumatoid arthritis. Pharmacogenet. Genom. 2014, 24, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Morales-Lara, M.J.; Cañete, J.D.; Torres-Moreno, D.; Hernández, M.V.; Pedrero, F.; Celis, R.; García-Simón, M.S.; Conesa-Zamora, P. Effects of polymorphisms in TRAILR1 and TNFR1A on the response to anti-TNF therapies in patients with rheumatoid and psoriatic arthritis. Joint Bone Spine 2012, 79, 591–596. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| RA Patients | N = 280 |
| Demographics | |
| Sex (females/males (% of females)) | 220/60 (78.6%) |
| Age (years) (mean (±SD)) | 51.6 (±12.3) |
| Current smokers (%) | 33.3% |
| Clinical data | |
| Disease duration (years) (mean (±SD)) | 12.6 (±8.1) |
| Disease onset (years) (mean (±SD)) | 39.2 (±12.0) |
| DAS28 at baseline (mean (±SD)) | 6.5 (±0.6) |
| CRP at baseline (mean (±SD)) | 24.4 (±35.7) |
| RF-positive (%) | 65.9% |
| anti-CCP+ (%) | 95.4% |
| Anti-TNF drugs | |
| Etanercept | 54% |
| Adalimumab | 33% |
| Infliximab | 7% |
| Certolizumab pegol | 6% |
| Concomitant treatment | |
| Glucocorticosteroids | 91% |
| Methotrexate | 92% |
| EULAR 12 Weeks | EULAR 24 Weeks | |||
|---|---|---|---|---|
| No Response (Number (%)) | Good/Moderate Response (Number (%)) | No Response (Number (%)) | Good/Moderate Response (Number (%)) | |
| NKG2D rs1049174 | (C|G: general population 51.3%|48.7%; Caucasian population: 68.4%|31.6%) * | |||
| C | 38 (82.6%) a | 342 (67.9%) a | 19 (67.9%) | 348 (68.0%) |
| G | 8 (17.4%) a | 162 (32.1%) a | 9 (32.1%) | 164 (32.0%) |
| CC | 17 (73.9%) b | 106 (41.2%) b | 6 (42.9%) | 113 (44.1%) |
| CG | 4 (17.4%) c | 130 (50.6%) c | 7 (50.0%) | 122 (47.7%) |
| GG | 2 (8.7%) | 21 (8.2%) | 1 (7.1%) | 21 (8.2%) |
| NKG2D rs1154831 | (A|C: general population 10.2%|89.8%; Caucasian population: 21.2%|78.8%) * | |||
| A | 11 (23.9%) | 90 (17.5%) | 6 (21.4%) | 90 (17.6%) |
| C | 35 (76.1%) | 424 (82.5%) | 22 (78.6%) | 422 (82.4%) |
| AA | 1 (4.3%) | 6 (2.3%) | 1 (7.1%) | 6 (2.3%) |
| AC | 9 (39.1%) | 78 (30.4%) | 4 (28.6%) | 78 (30.5%) |
| CC | 13 (56.5%) | 173 (67.3%) | 9 (64.3%) | 172 (67.2%) |
| NKG2D rs2255336 | (A|G: general population 23.8%|76.2%; Caucasian population: 18.6%|81.4%) * | |||
| A | 2 (4.3%) d | 114 (22.2%) d | 4 (14.3%) | 109 (21.3%) |
| G | 44 (95.7%) d | 400 (77.8%) d | 24 (85.7%) | 403 (78.7%) |
| AA | 0 (0.0%) | 12 (4.7%) | 0 (0.0%) | 12 (4.7%) |
| AG | 2 (8.7%) e | 90 (35.0%) e | 4 (28.6%) | 85 (33.2%) |
| GG | 21 (91.3%) f | 155 (60.3%) f | 10 (71.4%) | 159 (62.1%) |
| DAS28 at Baseline (Mean (±SD)) | CRP at Baseline (Mean (±SD)) | RF+ (Number (%)) | CCP+ (Number (%)) | |
|---|---|---|---|---|
| NKG2D rs1049174 | ||||
| CC | 6.58 (±0.64) | 26.36 (±35.52) | 80 (45.5%) | 101 (43.9%) |
| CG | 6.48 (±0.61) | 21.01 (±27.02) | 83 (47.2%) | 108 (47.0%) |
| GG | 6.67 (±0.58) | 33.04 (±64.81) | 13 (7.4%) | 21 (9.1%) |
| NKG2D rs1154831 | ||||
| AA | 6.59 (±0.53) | 24.54 (±18.31) | 4 (2.3%) | 5 (2.2%) |
| AC | 6.57 (±0.65) | 18.14 (±18.19) | 57 (32.4%) | 68 (29.6%) |
| CC | 6.52 (±0.62) | 27.33 (±41.55) | 115 (65.3%) | 157 (68.3%) |
| NKG2D rs2255336 | ||||
| AA | 6.85 (±0.61) | 37.14 (±56.51) | 6 (3.4%) | 7 (3.0%) |
| AG | 6.50 (±0.57) | 24.09 (±36.97) | 60 (34.1%) | 77 (33.5%) |
| GG | 6.53 (±0.65) | 23.69 (±32.91) | 110 (62.5%) | 146 (63.5%) |
| ADA | ETA | |||
|---|---|---|---|---|
| No Response (Number (%)) | Good/Moderate Response (Number (%)) | No Response (Number (%)) | Good/Moderate Response (Number (%)) | |
| NKG2D rs1049174 | ||||
| C | 10 (71.4%) | 103 (64.4%) | 13 (81.3%) | 153 (66.5%) |
| G | 4 (28.6%) | 57 (35.6%) | 3 (18.8%) | 77 (33.5%) |
| CC | 4 (57.1%) | 28 (35.0%) | 6 (75%) | 50 (43.5%) |
| CG | 2 (28.6%) | 47 (58.8%) | 1 (12.5%) | 53 (46.1%) |
| GG | 1 (14.3%) | 5 (6.3%) | 1 (12.5%) | 12 (10.4%) |
| NKG2D rs2255336 | ||||
| A | 2 (14.3%) | 34 (21.3%) | 0 (0%) a | 57 (24.8%) a |
| G | 12 (85.7%) | 126 (78.8%) | 16 (100%) a | 173 (75.2%) a |
| AA | 0 (0%) | 3 (3.8%) | 0 (0%) | 8 (7.0%) |
| AG | 2 (28.6%) | 28 (35.0%) | 0 (0%) | 41 (35.7%) |
| GG | 5 (71.4%) | 49 (61.3%) | 8 (100%) b | 66 (57.4%) b |
| EULAR 12 Weeks | EULAR 24 Weeks | |||
|---|---|---|---|---|
| No Response (Number (%)) | Good/Moderate Response (Number (%)) | No Response (Number (%)) | Good/Moderate Response (Number (%)) | |
| CGA | 10 (3.6%) | 80 (28.6%) | 5 (1.8%) | 80 (28.6%) |
| CGC | 20 (7.1%) | 225 (80.4%) | 12 (4.3%) | 224 (80.0%) |
| GAC | 1 (0.4%) a | 92 (32.9%) a | 4 (1.4%) | 87 (31.1%) |
| GGC | 6 (2.1%) b | 143 (51.1%) b | 8 (2.9%) | 135 (48.2%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwaszko, M.; Świerkot, J.; Kolossa, K.; Jeka, S.; Wiland, P.; Bogunia-Kubik, K. Influence of NKG2D Genetic Variants on Response to Anti-TNF Agents in Patients with Rheumatoid Arthritis. Genes 2018, 9, 64. https://doi.org/10.3390/genes9020064
Iwaszko M, Świerkot J, Kolossa K, Jeka S, Wiland P, Bogunia-Kubik K. Influence of NKG2D Genetic Variants on Response to Anti-TNF Agents in Patients with Rheumatoid Arthritis. Genes. 2018; 9(2):64. https://doi.org/10.3390/genes9020064
Chicago/Turabian StyleIwaszko, Milena, Jerzy Świerkot, Katarzyna Kolossa, Sławomir Jeka, Piotr Wiland, and Katarzyna Bogunia-Kubik. 2018. "Influence of NKG2D Genetic Variants on Response to Anti-TNF Agents in Patients with Rheumatoid Arthritis" Genes 9, no. 2: 64. https://doi.org/10.3390/genes9020064