DNA Methylation and All-Cause Mortality in Middle-Aged and Elderly Danish Twins

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Methylation Data
2.3. Validation Studies
2.4. Statistical Analysis
2.4.1. Univariate Analysis
2.4.2. Construction of the Predictor
2.4.3. Heritability
2.4.4. Validation
2.4.5. Comparison to the Literature
3. Results
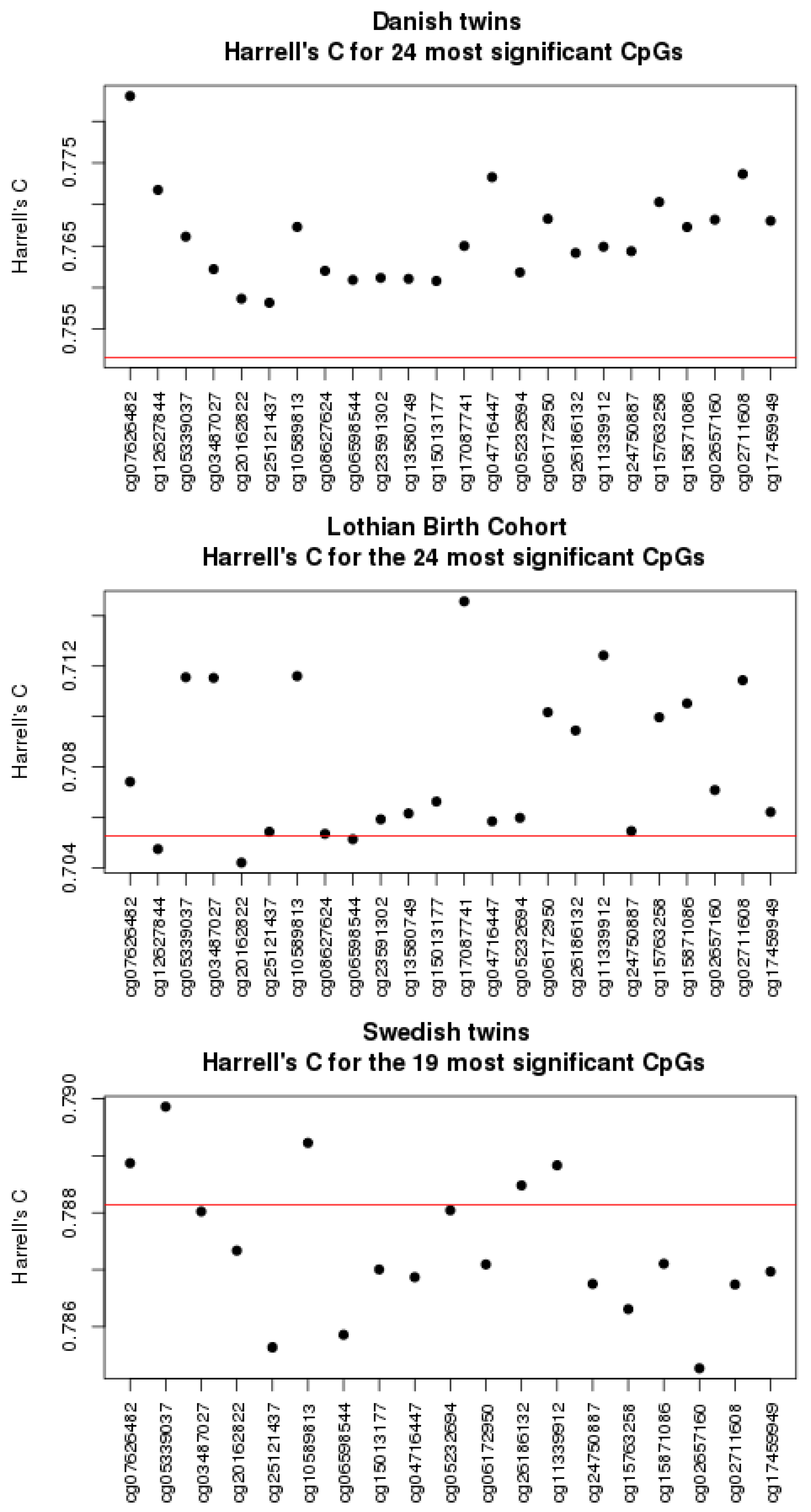
3.1. Univariate Analyses
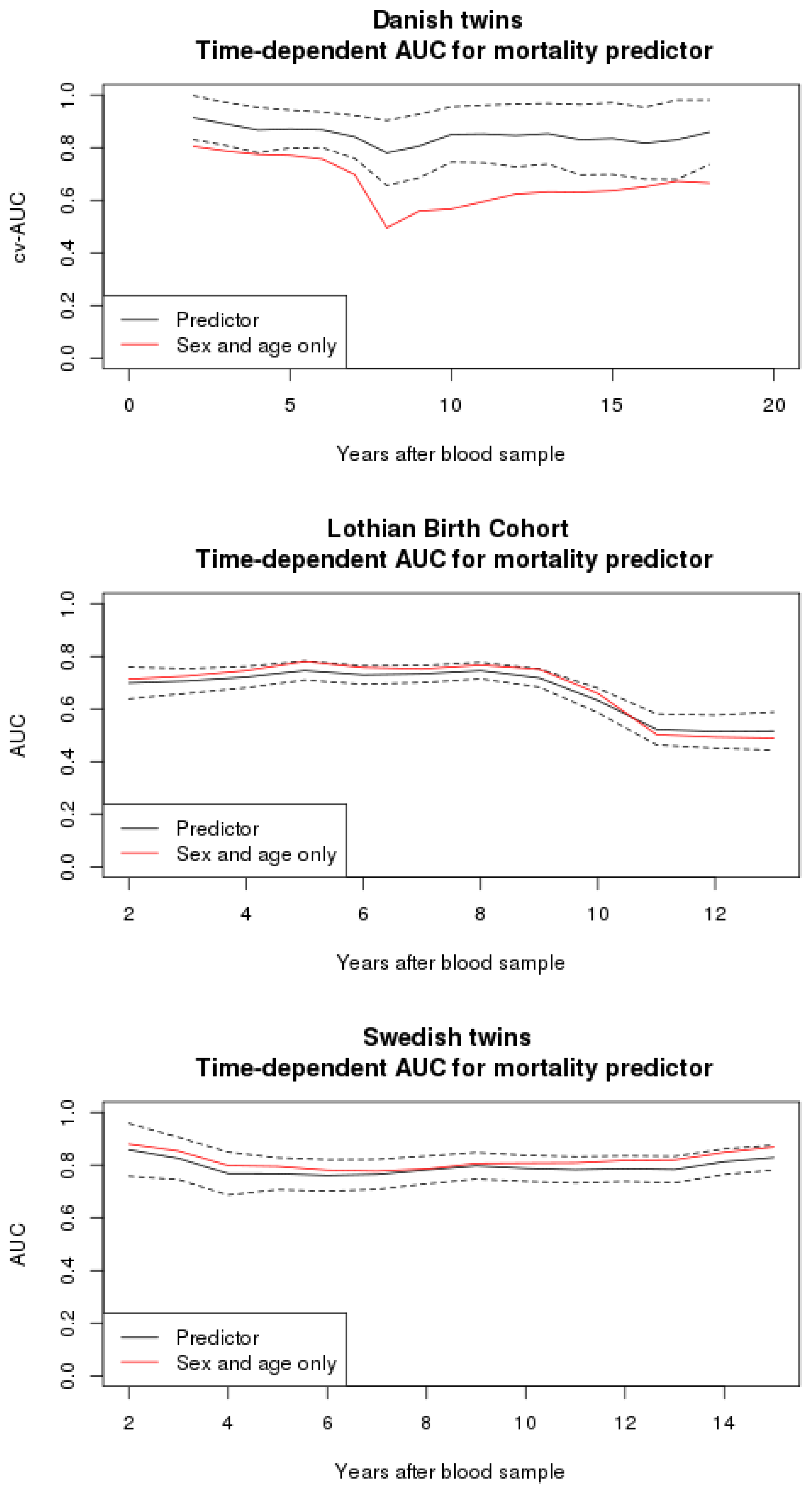
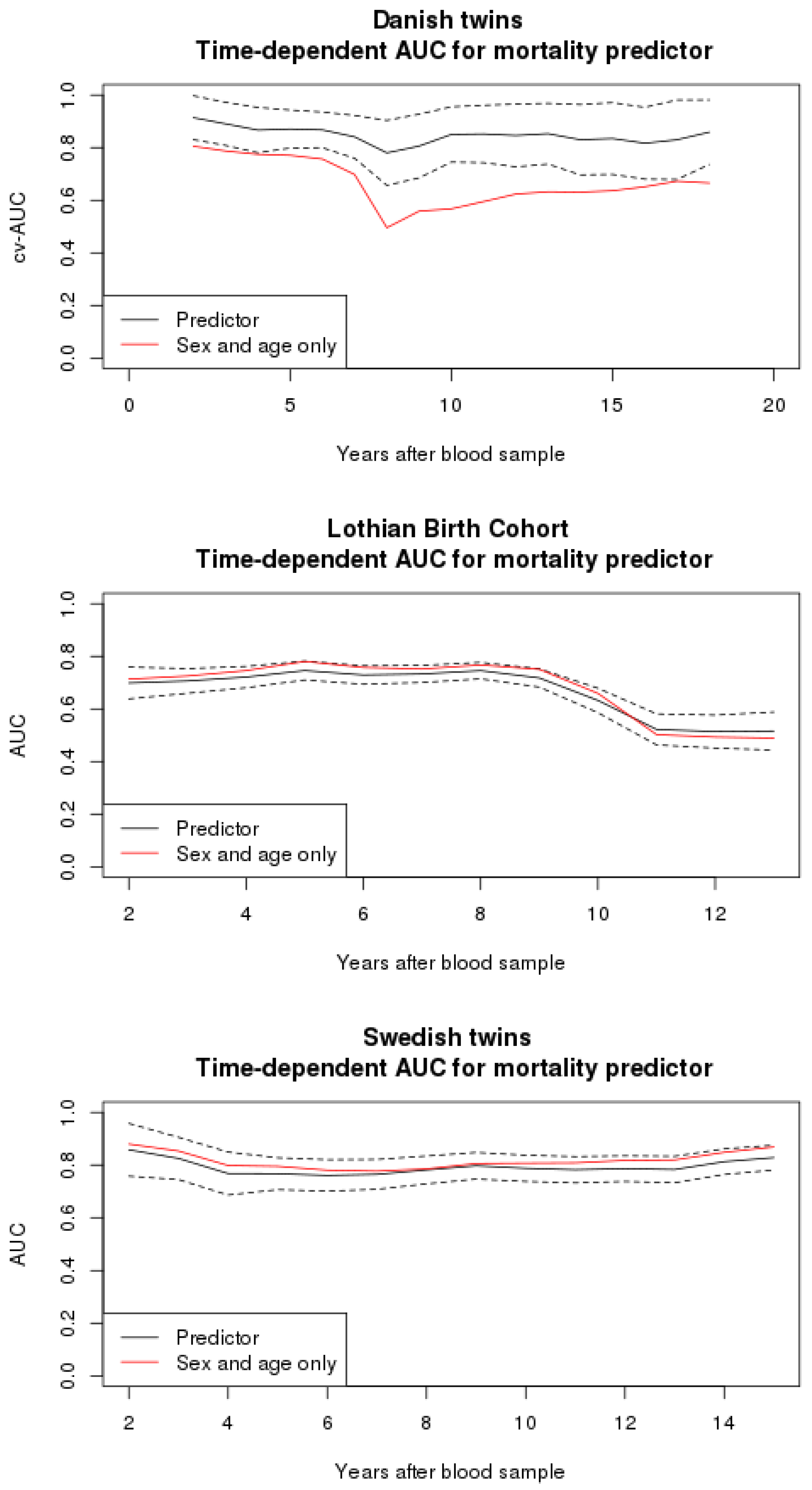
3.2. Mortality Predictor
3.3. Heritability
3.4. Comparison to Other Studies
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Volkmar, M.; Dedeurwaerder, S.; Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 31, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.L.; Yan, M.S.; Marsden, P.A. Epigenetics and cardiovascular disease. Can. J. Cardiol. 2013, 29, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.M. Epigenetic regulation of memory: implications in human cognitive disorders. Biomol. Concepts 2012, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, L.; Lenart, A.; Tan, Q.; Vaupel, J.W.; Aviv, A.; McGue, M.; Christensen, K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 2016, 15, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wilson, R.; Heiss, J.; Breitling, L.P.; Saum, K.-U.; Schöttker, B.; Holleczek, B.; Waldenberger, M.; Peters, A.; Brenner, H. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun. 2017, 8, 14617. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Kananen, L.; Raitanen, J.; Marttila, S.; Nevalainen, T.; Hervonen, A.; Jylhä, M.; Hurme, M. Methylomic predictors demonstrate the role of NF-κB in old-age mortality and are unrelated to the aging-associated epigenetic drift. Oncotarget 2016, 7, 19228–19241. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.Z.; Hernandez, D.G.; Tanaka, T.; Pilling, L.C.; Nalls, M.A.; Bandinelli, S.; Singleton, A.B.; Ferrucci, L. Change in Epigenome-Wide DNA Methylation Over 9 Years and Subsequent Mortality: Results From the InCHIANTI Study. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.; Petersen, I.; Brixen, K.; Beck-Nielsen, H.; Holst, J.J.; Christiansen, L.; Højlund, K.; Christensen, K. Adult glucose metabolism in extremely birthweight-discordant monozygotic twins. Diabetologia 2012, 55, 3204–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, K.; Holm, N.V.; Mcgue, M.; Corder, L.; Vaupel, J.W. A Danish Population-Based Twin Study on General Health in the Elderly. J. Aging Health 1999, 11, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Gaist, D.; Bathum, L.; Skytthe, A.; Jensen, T.K.; McGue, M.; Vaupel, J.W.; Christensen, K. Strength and anthropometric measures in identical and fraternal twins: no evidence of masculinization of females with male co-twins. Epidemiology 2000, 11, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Slieker, R.C.; Stein, A.D.; Suchiman, H.E.D.; Slagboom, P.E.; van Zwet, E.W.; Heijmans, B.T.; Lumey, L.H. Early gestation as the critical time-window for changes in the prenatal environment to affect the adult human blood methylome. Int. J. Epidemiol. 2015, 44, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.T.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed]
- Debrabant, B.; Sørensen, M.; Christiansen, L.; Tan, Q.; McGue, M.; Christensen, K.; Hjelmborg, J. DNA methylation age and perceived age in elderly Danish twins. Mech. Ageing Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Baumbach, J.; Li, S.; Svane, A.M.; Hjelmborg, J.; Christiansen, L.; Christensen, K.; Redmond, P.; Marioni, R.E.; Deary, I.J.; et al. DNA methylation linked to all-cause mortality in the elderly: An epigenome-wide association study. 2018. in progress. [Google Scholar]
- Deary, I.J.; Gow, A.J.; Pattie, A.; Starr, J.M. Cohort Profile: The Lothian Birth Cohorts of 1921 and 1936. Int. J. Epidemiol. 2012, 41, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Finkel, D.; Pedersen, N.L. Processing Speed and Longitudinal Trajectories of Change for Cognitive Abilities: The Swedish Adoption/Twin Study of Aging. Aging Neuropsychol. Cognit. 2004, 11, 325–345. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Meinshausen, N.; Bühlmann, P. Stability selection. J. R. Stat. Soc. Ser. B 2010, 72, 417–473. [Google Scholar] [CrossRef]
- Mogensen, S.W.; Petersen, A.H.; Buchardt, A.S.; Hansen, N.R. Survival prognosis and variable selection: A case study for metastatic castrate resistant prostate cancer patients. F1000Research 2016, 5, 2680. [Google Scholar] [CrossRef] [PubMed]
- Harrell, F.E.; Califf, R.M.; Pryor, D.B.; Lee, K.L.; Rosati, R.A. Evaluating the yield of medical tests. J. Am. Med. Assoc. 1982, 247, 2543–2546. [Google Scholar] [CrossRef]
- Harrell, F.E.; Lee, K.L.; Califf, R.M.; Pryor, D.B.; Lee, K.L.; Rosati, R.A. Regression modeling strategies for improved prognostic prediction. Stat. Med. 1984, 3, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Larriba, S.; Sumoy, L.; Ramos, M.D.; Giménez, J.; Estivill, X.; Casals, T.; Nunes, V. ATB0/SLC1A5 gene. Fine localisation and exclusion of association with the intestinal phenotype of cystic fibrosis. Eur. J. Hum. Genet. 2001, 9, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid Transporters in Cancer and Their Relevance to ‘Glutamine Addiction’: Novel Targets for the Design of a New Class of Anticancer Drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, L.; An, H.; Chang, Y.; Zhang, W.; Zhu, Y.; Xu, L.; Xu, J. High expression of Solute Carrier Family 1, member 5 (SLC1A5) is associated with poor prognosis in clear-cell renal cell carcinoma. Sci. Rep. 2015, 5, 16954. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunose, Y.; Arakawa, K.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; Nagamori, S.; Kanai, Y.; Oyama, T.; et al. Clinicopathological significance of ASC amino acid transporter-2 expression in pancreatic ductal carcinoma. Histopathology 2015, 66, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Kriebel, J.; Herder, C.; Rathmann, W.; Wahl, S.; Kunze, S.; Molnos, S.; Volkova, N.; Schramm, K.; Carstensen-Kirberg, M.; Waldenberger, M.; et al. Association between DNA methylation in whole blood and measures of glucose metabolism: KORA F4 Study. PLoS ONE 2016, 11, e0152314. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Akira, S.; Kishimoto, T. A member of the C/EBP family, NF-IL6β, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc. Natl. Acad. Sci. USA 1992, 89, 1473–1476. [Google Scholar] [CrossRef] [PubMed]
- Poli, V. The Role of C/EBP Isoforms in the Control of Inflammatory and Native Immunity Functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef] [PubMed]
- Pless, O.; Kowenz-Leutz, E.; Knoblich, M.; Lausen, J.; Beyermann, M.; Walsh, M.J.; Leutz, A. G9a-mediated Lysine Methylation Alters the Function of CCAAT/Enhancer-binding Protein-β. J. Biol. Chem. 2008, 283, 26357–26363. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kang, Y.K.; Park, Z.Y.; Kim, Y.H.; Hong, S.W.; Oh, S.J.; Sohn, H.A.; Yang, S.-J.; Jang, Y.J.; Lee, D.C.; et al. SH3RF2 functions as an oncogene by mediating PAK4 protein stability. Carcinogenesis 2014, 35, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Heijmans, B.T.; Hjelmborg, J.V.B.H.; Soerensen, M.; Christensen, K.; Christiansen, L. Epigenetic drift in the aging genome: A ten-year follow-up in an elderly twin cohort. Int. J. Epidemiol. 2016, 45, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Hjelmborg, J.; Iachine, I.; Skytthe, A.; Vaupel, J.W.; McGue, M.; Koskenvuo, M.; Kaprio, J.; Pedersen, N.L.; Christensen, K. Genetic influence on human lifespan and longevity. Hum. Genet. 2006, 119, 312–321. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Dataset | BWD | LSADT | MADT | Total |
|---|---|---|---|---|
| N | 150 | 238 | 482 | 870 |
| Women, N (%) | 74 (49%) | 156 (76%) | 218 (46%) | 448 |
| Deaths, N (%) | 11 (7.3%) | 213 (89%) | 34 (7.0%) | 258 |
| Age at blood sample | 57–74 | 73–90 | 55–79 | 55–90 |
| Year of blood sample | 2009–2010 | 1996–1997 | 2008–2011 | |
| Follow-up year | 2016 | 2016 | 2016 | |
| Cause of death: | ||||
| Cancer | 1 | 35 | 4 | 40 |
| Cardiovascular disease | 0 | 72 | 2 | 74 |
| Respiratory causes | 1 | 18 | 1 | 20 |
| CpG Site | HR | p | FDR | Chromosome | Gene Name | MZ Correlation |
|---|---|---|---|---|---|---|
| cg07626482 | 0.64 | 3.21l | 1.42 | 19 | SLC1A5 | 0.43 *** |
| cg12627844 | 0.69 | 5.20 | 1.15 | 2 | VPS54 | 0.44 *** |
| cg05339037 | 0.77 | 1.18 | 1.31 | 19 | 0.44 *** | |
| cg03487027 | 0.73 | 1.40 | 1.31 | 10 | ZNF503 | 0.51 *** |
| cg20162822 | 0.78 | 1.81 | 1.31 | 17 | SERPINF2 | −0.05 |
| cg25121437 | 0.73 | 2.04 | 1.31 | 11 | FEZ1 | 0.29 *** |
| cg10589813 | 0.71 | 2.76 | 1.31 | 20 | 0.41 *** | |
| cg08627624 | 0.69 | 2.85 | 1.31 | 10 | −0.04 | |
| cg06598544 | 0.78 | 3.13 | 1.31 | 20 | COL9A3 | 0.31 *** |
| cg23591302 ** | 0.74 | 3.14 | 1.31 | 12 | PRICKLE1 | 0.15 *** |
| cg13580749 | 0.72 | 3.25 | 1.31 | 9 | 0.18 *** | |
| cg15013177 *** | 0.77 | 3.98 | 1.31 | 3 | CNTN6 | 0.12 *** |
| cg17087741 ** | 0.71 | 4.16 | 1.31 | 2 | 0.45 *** | |
| cg04716447 ** | 1.42 | 4.16 | 1.31 | 12 | 0.18 *** | |
| cg05232694 | 0.72 | 4.64 | 1.33 | 20 | 0.31 *** | |
| cg06172950 | 0.74 | 4.81 | 1.33 | 13 | COG3 | 0.27 *** |
| cg26186132 | 0.71 | 5.66 | 1.41 | 6 | C6orf147 | −0.07 |
| cg11339912 | 0.72 | 5.74 | 1.41 | 5 | SH3RF2 | 0.39 *** |
| cg24750887 | 1.42 | 6.22 | 1.44 | 4 | HERC3 | 0.05 |
| cg15763258 | 0.73 | 6.52 | 1.44 | 11 | FLI1 | 0.36 *** |
| cg15871086 | 0.74 | 7.65 | 1.61 | 18 | 0.36 *** | |
| cg02657160 | 0.70 | 8.81 | 1.72 | 3 | CPOX | 0.18 *** |
| cg02711608 ** | 0.69 | 8.99 | 1.72 | 19 | SLC1A5 | 0.48 *** |
| cg17459949 | 0.73 | 9.79 | 1.80 | 10 | −0.06 |
| CpG Site | Danish Twins | Lothian Birth Cohort | Swedish Twins | |||
|---|---|---|---|---|---|---|
| HR | p | HR | p | HR | p | |
| cg07626482 | 0.64 | 3.21 | 0.55 | 3.18 | 0.88 | 0.09 |
| cg12627844 | 0.69 | 5.20 | 0.94 | 0.53 | - | - |
| cg05339037 | 0.77 | 1.18 | 0.42 | 1.70 | 0.85 | 0.02 |
| cg03487027 | 0.73 | 1.40 | 1.09 | 4.81 | 0.99 | 0.87 |
| cg20162822 | 0.78 | 1.81 | 0.96 | 0.64 | 1.08 | 0.31 |
| cg25121437 | 0.73 | 2.04 | 0.91 | 0.48 | 1.07 | 0.30 |
| cg10589813 | 0.71 | 2.76 | 0.56 | 2.71 | 0.85 | 0.03 |
| cg08627624 | 0.69 | 2.85 | 1.08 | 0.48 | - | - |
| cg06598544 | 0.78 | 3.13 | 1.06 | 0.13 | 1.04 | 0.57 |
| cg23591302 | 0.74 | 3.14 | 1.05 | 0.11 | - | - |
| cg13580749 | 0.72 | 3.25 | 1.00 | 1.00 | - | - |
| cg15013177 | 0.77 | 3.98 | 1.09 | 0.42 | 0.97 | 0.66 |
| cg17087741 | 0.71 | 4.16 | 0.57 | 3.22 | - | - |
| cg04716447 | 1.42 | 4.16 | 1.37 | 0.15 | 1.06 | 0.47 |
| cg05232694 | 0.72 | 4.64 | 0.86 | 0.07 | 0.90 | 0.17 |
| cg06172950 | 0.74 | 4.81 | 0.77 | 0.034 | 0.98 | 0.75 |
| cg26186132 | 0.71 | 5.66 | 1.18 | 0.33 | 1.10 | 0.17 |
| cg11339912 | 0.72 | 5.74 | 0.65 | 2.27 | 0.91 | 0.18 |
| cg24750887 | 1.42 | 6.22 | 1.05 | 0.52 | 1.02 | 0.82 |
| cg15763258 | 0.73 | 6.52 | 0.70 | 0.03 | 0.97 | 0.69 |
| cg15871086 | 0.74 | 7.65 | 0.66 | 6.82 | 0.97 | 0.71 |
| cg02657160 | 0.70 | 8.81 | 1.01 | 0.93 | 0.90 | 0.13 |
| cg02711608 | 0.69 | 8.99 | 0.53 | 5.30 | 0.99 | 0.94 |
| cg17459949 | 0.73 | 9.79 | 1.07 | 0.11 | 0.97 | 0.68 |
| Covariate | Coefficient ** | SE ** | p ** | Chromosome | Gene Name | MZ Correlation |
|---|---|---|---|---|---|---|
| cg02537149 | 0.158 | 0.0664 | 0.0173 | 1 | LRRC41, UQCRH | 0.00 |
| cg02691019 | 0.0906 | 0.0564 | 0.108 | 16 | −0.07 | |
| cg04716447 * | 0.308 | 0.0623 | 7.36 | 12 | 0.18 *** | |
| cg05232694 | −0.155 | 0.0686 | 0.0234 | 20 | 0.31 *** | |
| cg07626482 | −0.160 | 0.0820 | 0.0508 | 19 | SLC1A5 | 0.43 *** |
| cg12880095 | 0.115 | 0.0621 | 0.0650 | 17 | 0.53 *** | |
| cg14304264 | 0.140 | 0.0705 | 0.0467 | 15 | MCTP2 | 0.00 |
| cg17459949 | −0.176 | 0.0640 | 5.95 | 10 | −0.06 | |
| cg20164226 * | 0.0579 | 0.0579 | 0.317 | 7 | 0.27 *** | |
| cg21381949 | −0.112 | 0.0592 | 0.0585 | 3 | LEPREL1 | 0.12 *** |
| cg22304262 | −0.273 | 0.0822 | 8.97 | 19 | SLC1A5 | 0.38 *** |
| cg24750887 | 0.205 | 0.0663 | 1.96 | 4 | HERC3 | 0.05 |
| cg24967142 | 0.219 | 0.0669 | 1.04 | 12 | C12orf47, MAPKAPK5 | −0.08 |
| cg26186132 | −0.228 | 0.0688 | 9.11 | 6 | C6orf147 | −0.07 |
| Sex (female) | −0.702 | 0.140 | 5.71 | |||
| Age (in years) | 0.139 | 0.0165 | <2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svane, A.M.; Soerensen, M.; Lund, J.; Tan, Q.; Jylhävä, J.; Wang, Y.; Pedersen, N.L.; Hägg, S.; Debrabant, B.; Deary, I.J.; et al. DNA Methylation and All-Cause Mortality in Middle-Aged and Elderly Danish Twins. Genes 2018, 9, 78. https://doi.org/10.3390/genes9020078
Svane AM, Soerensen M, Lund J, Tan Q, Jylhävä J, Wang Y, Pedersen NL, Hägg S, Debrabant B, Deary IJ, et al. DNA Methylation and All-Cause Mortality in Middle-Aged and Elderly Danish Twins. Genes. 2018; 9(2):78. https://doi.org/10.3390/genes9020078
Chicago/Turabian StyleSvane, Anne Marie, Mette Soerensen, Jesper Lund, Qihua Tan, Juulia Jylhävä, Yunzhang Wang, Nancy L. Pedersen, Sara Hägg, Birgit Debrabant, Ian J. Deary, and et al. 2018. "DNA Methylation and All-Cause Mortality in Middle-Aged and Elderly Danish Twins" Genes 9, no. 2: 78. https://doi.org/10.3390/genes9020078