Differential Enzymatic Activity of Rat ADAR2 Splicing Variants Is Due to Altered Capability to Interact with RNA in the Deaminase Domain

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Cortical Cultures
2.2. RNA Extraction and Retrotranscription
2.3. RNA Editing Quantification
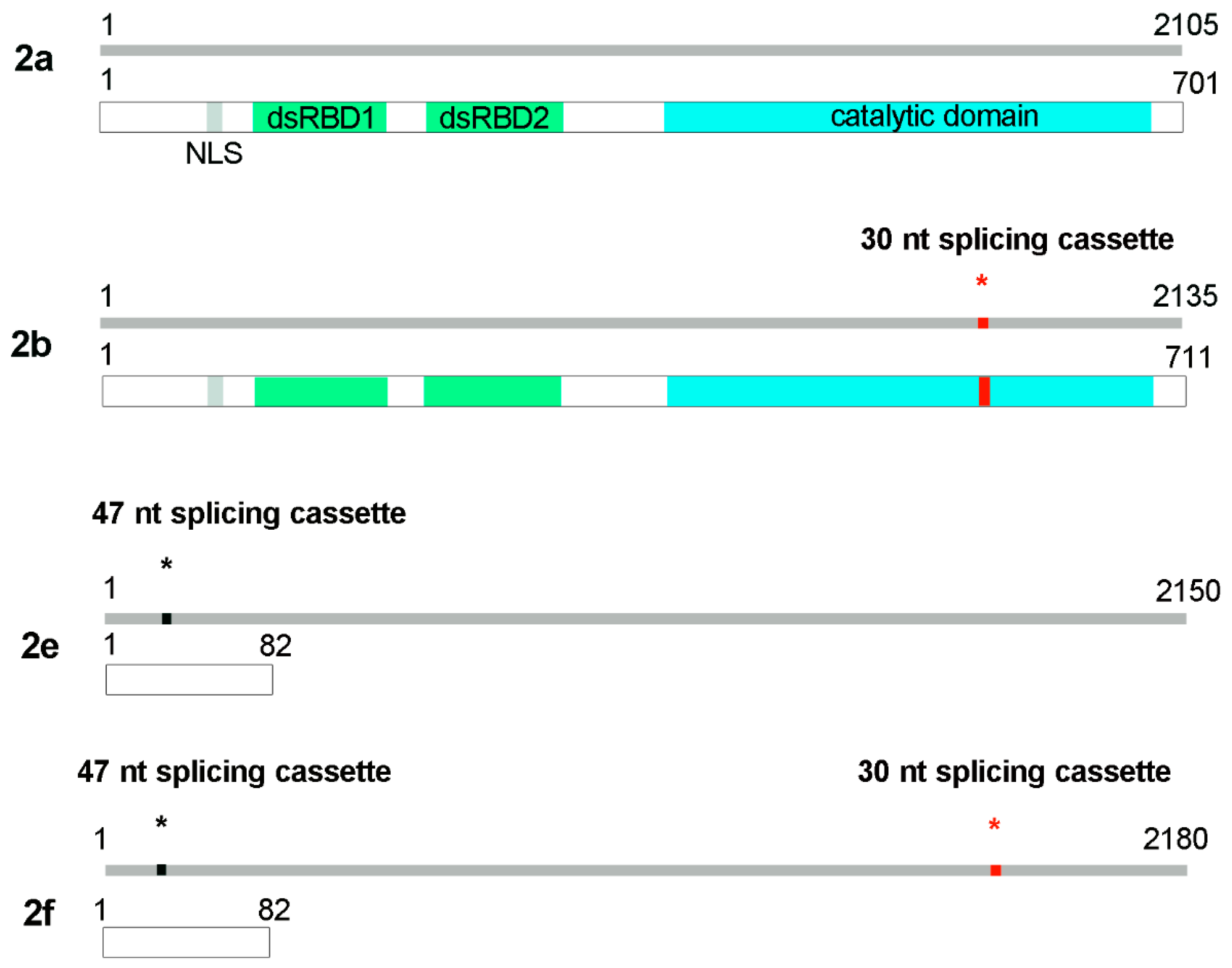
2.4. ADAR2 Splicing Isoforms Quantification
2.5. Lentivirus Preparation
2.5.1. Constructs
2.5.2. Lentivirus Particles Production
2.6. HEK293T Cell Cultures Stably Expressing ADAR2a/ADAR2b Enzymes
Primary Neuronal Cultures Viral Transduction
2.7. Transient Transfection
2.8. In Vitro RNA Editing Assay
2.9. Quantitative Real Time PCR
2.10. Protein Extraction, Quantification and Western Blot
2.11. Proximity Ligation Assay
2.12. Homology Modelling of ADAR2 Splicing Isoforms and Molecular Dynamics
2.13. Statistical Analysis
3. Results
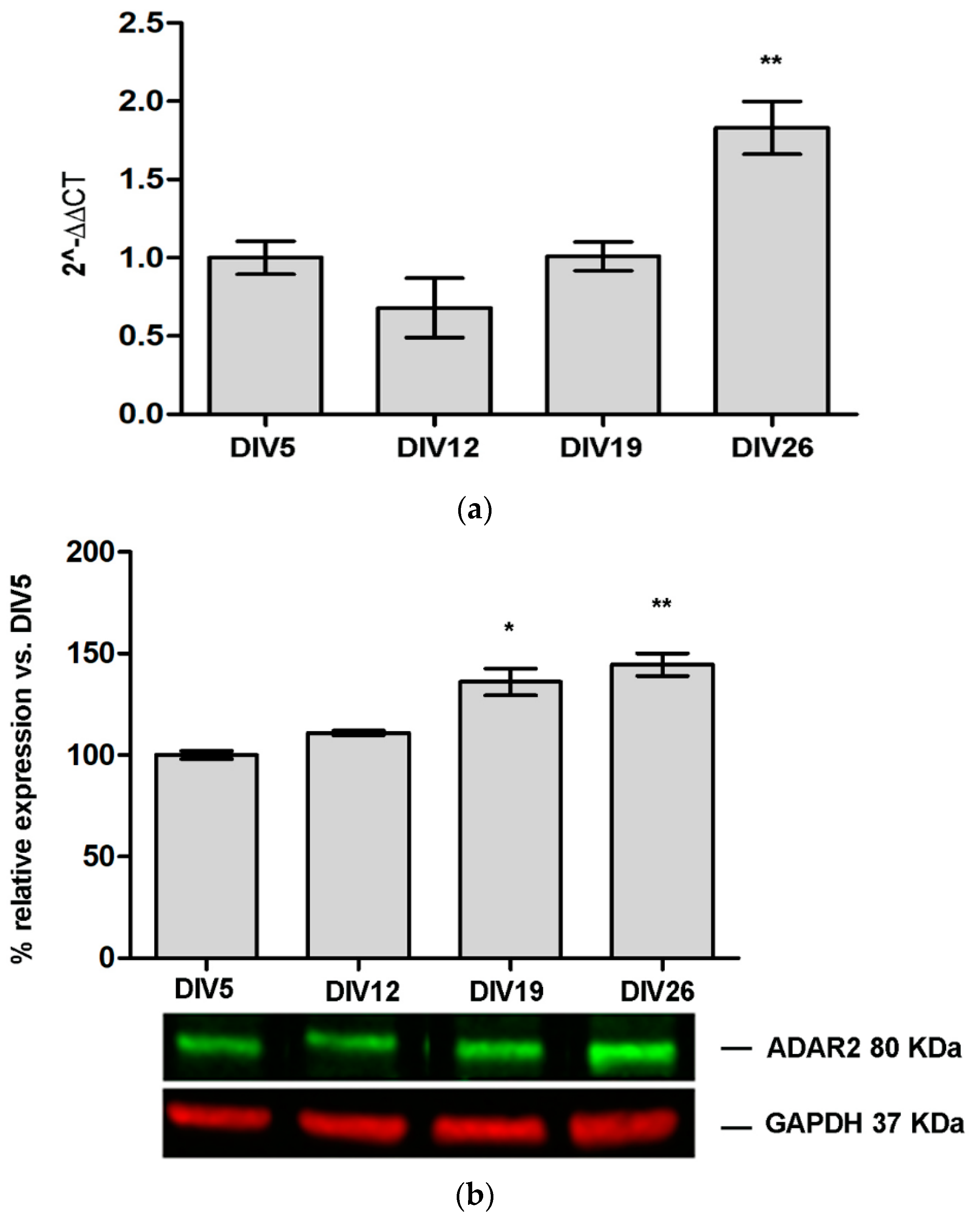
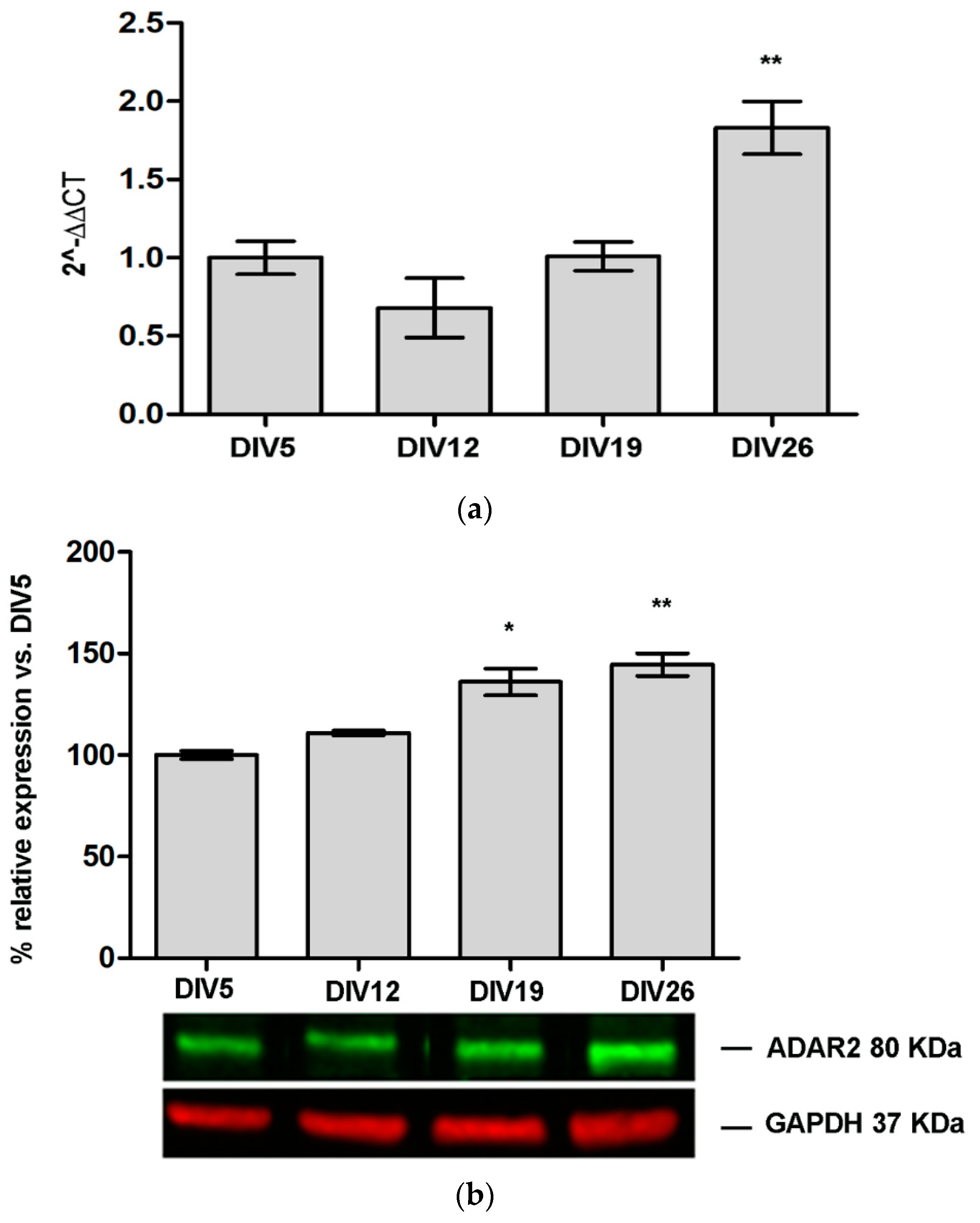
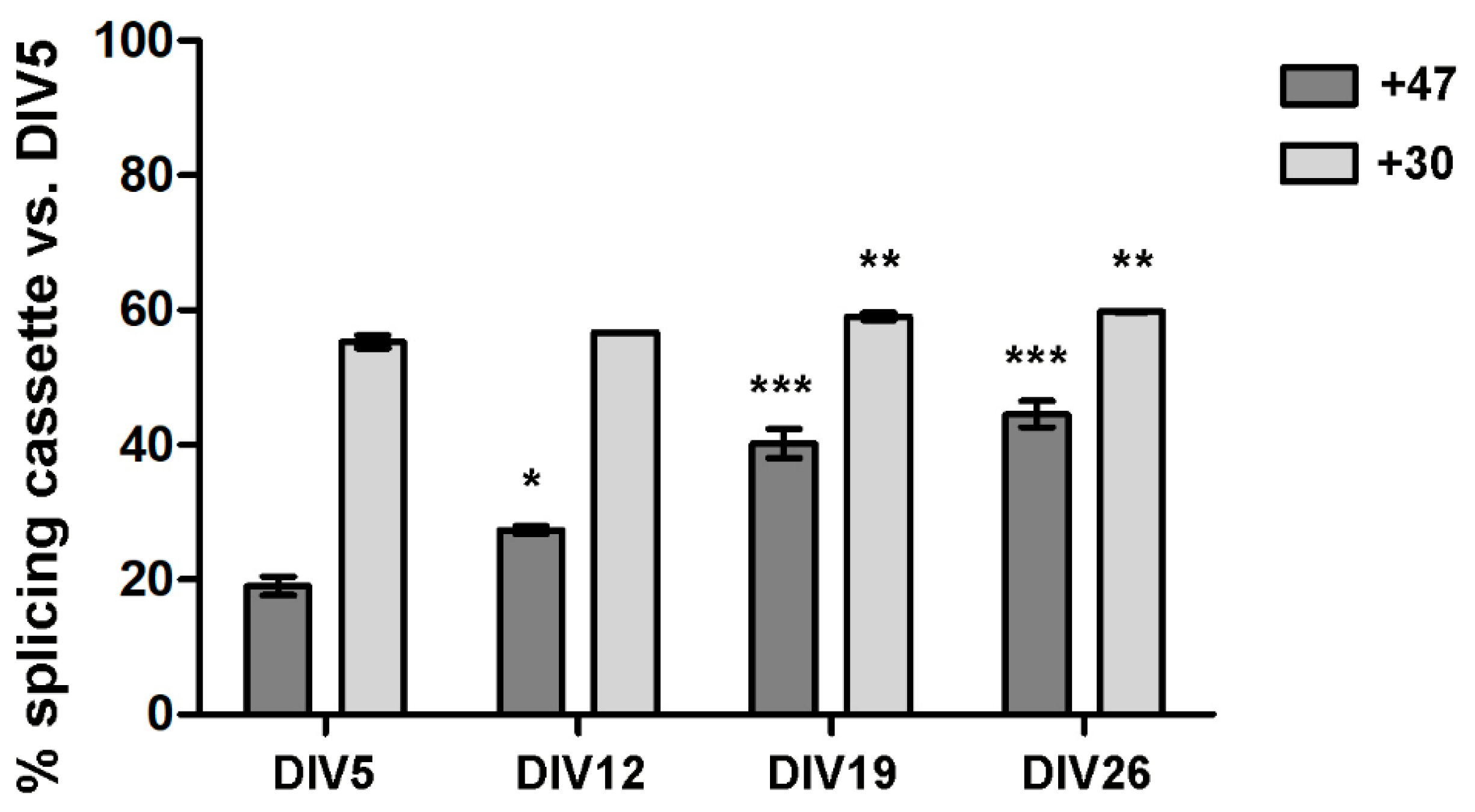
3.1. ADAR2 Expression Increase during Primary Cortical Neuron Culture Maturation
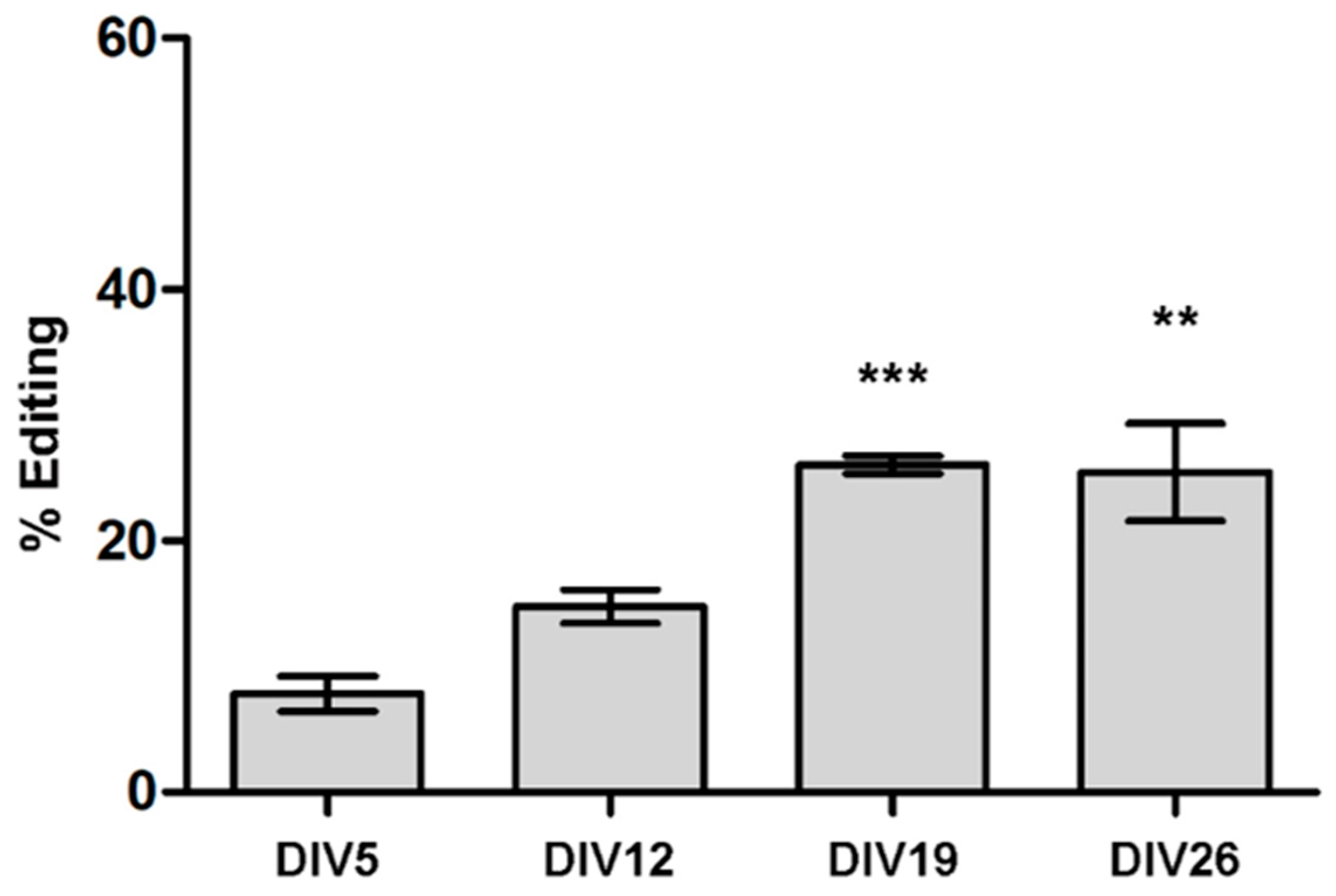
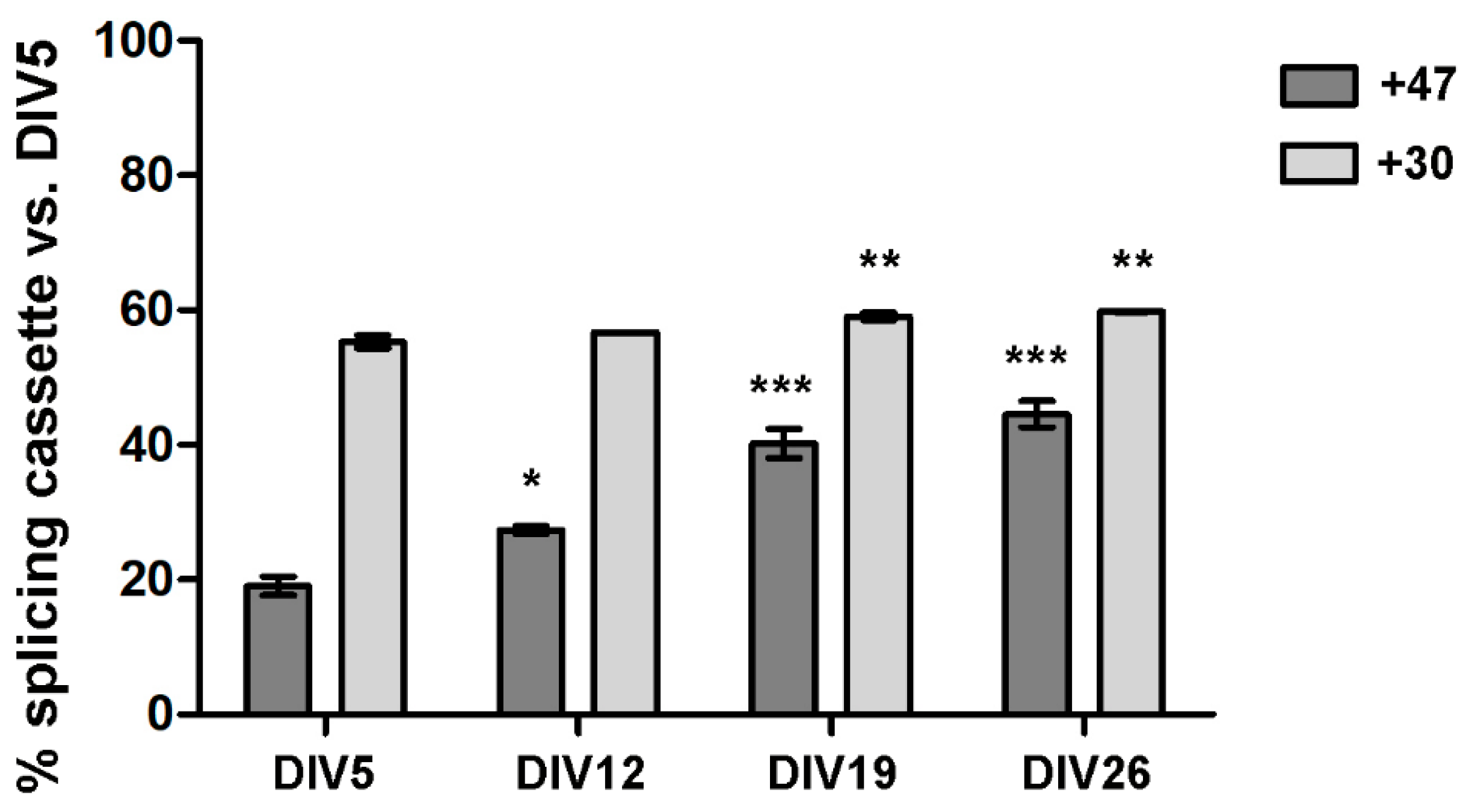
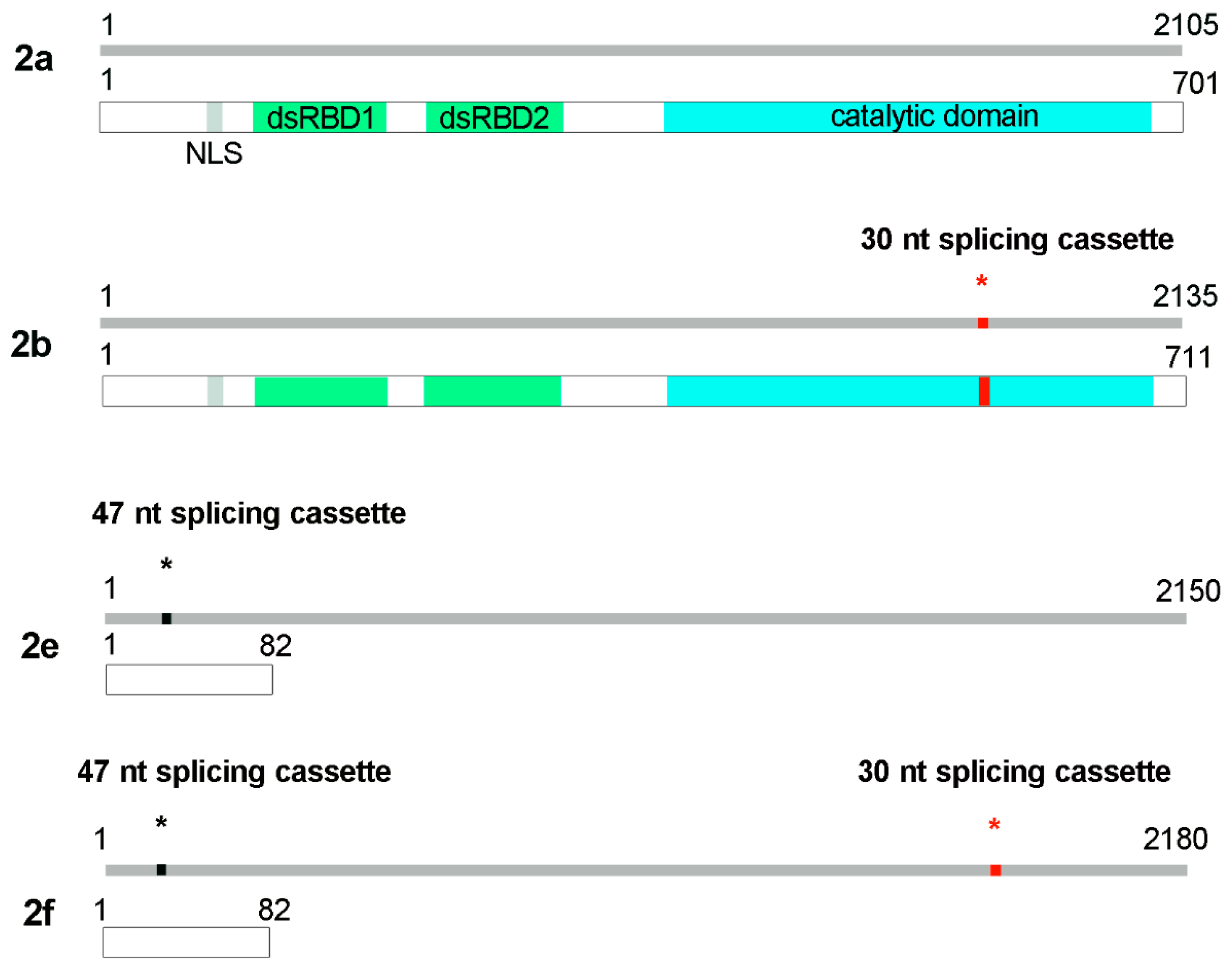
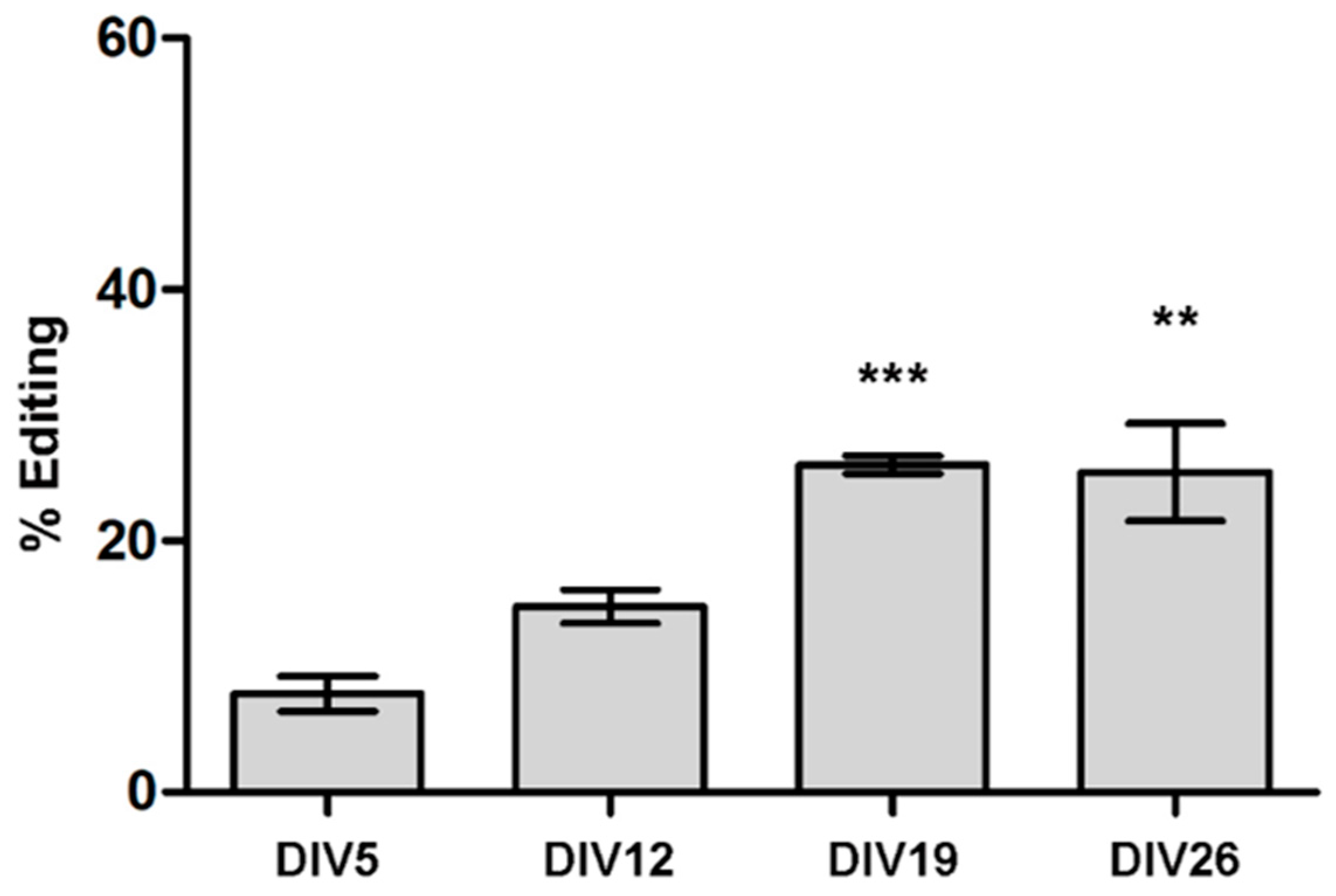
3.2. ADAR2 Self-Editing and Splicing Pattern Is Modified during Cortical Neurons Development
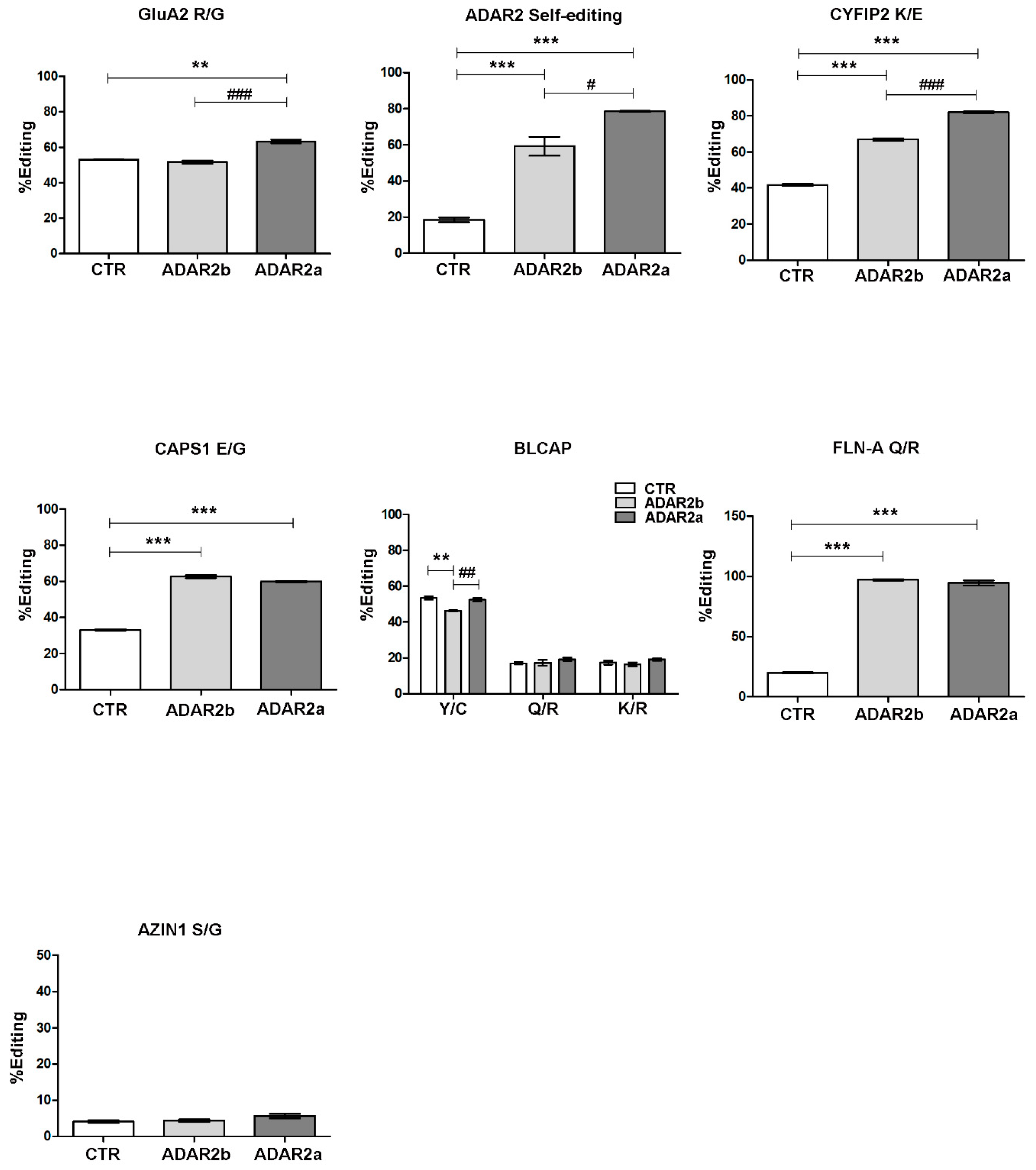
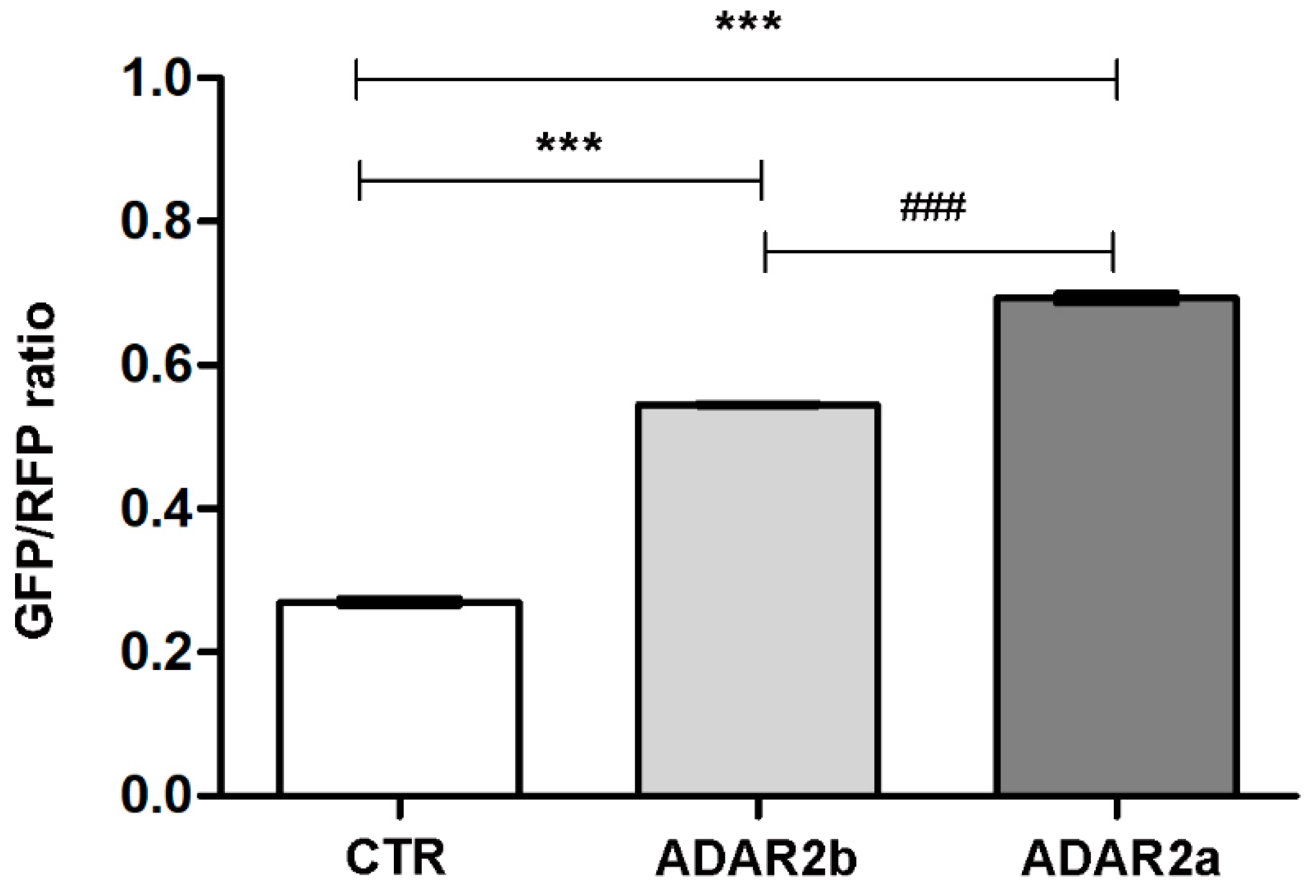
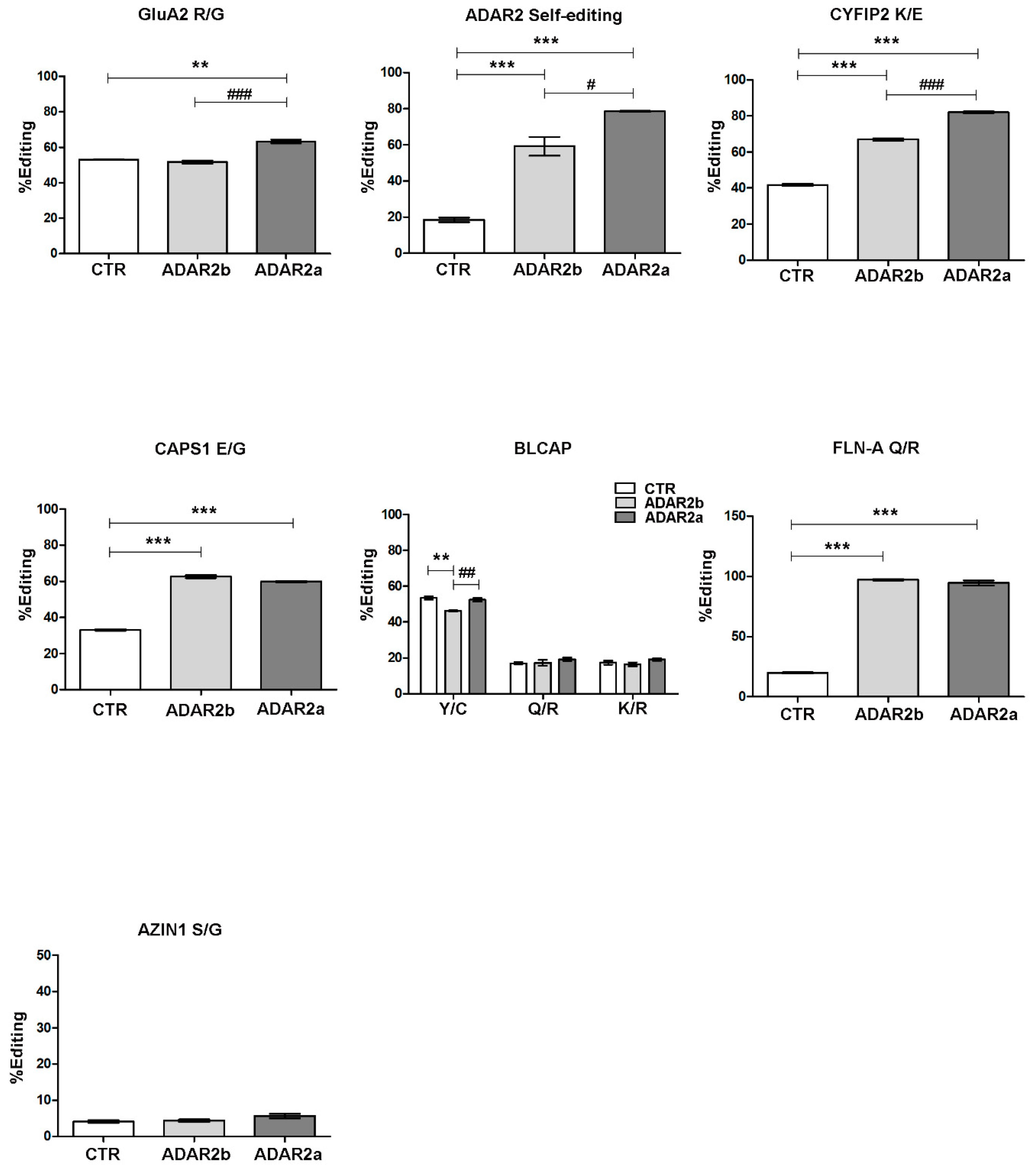
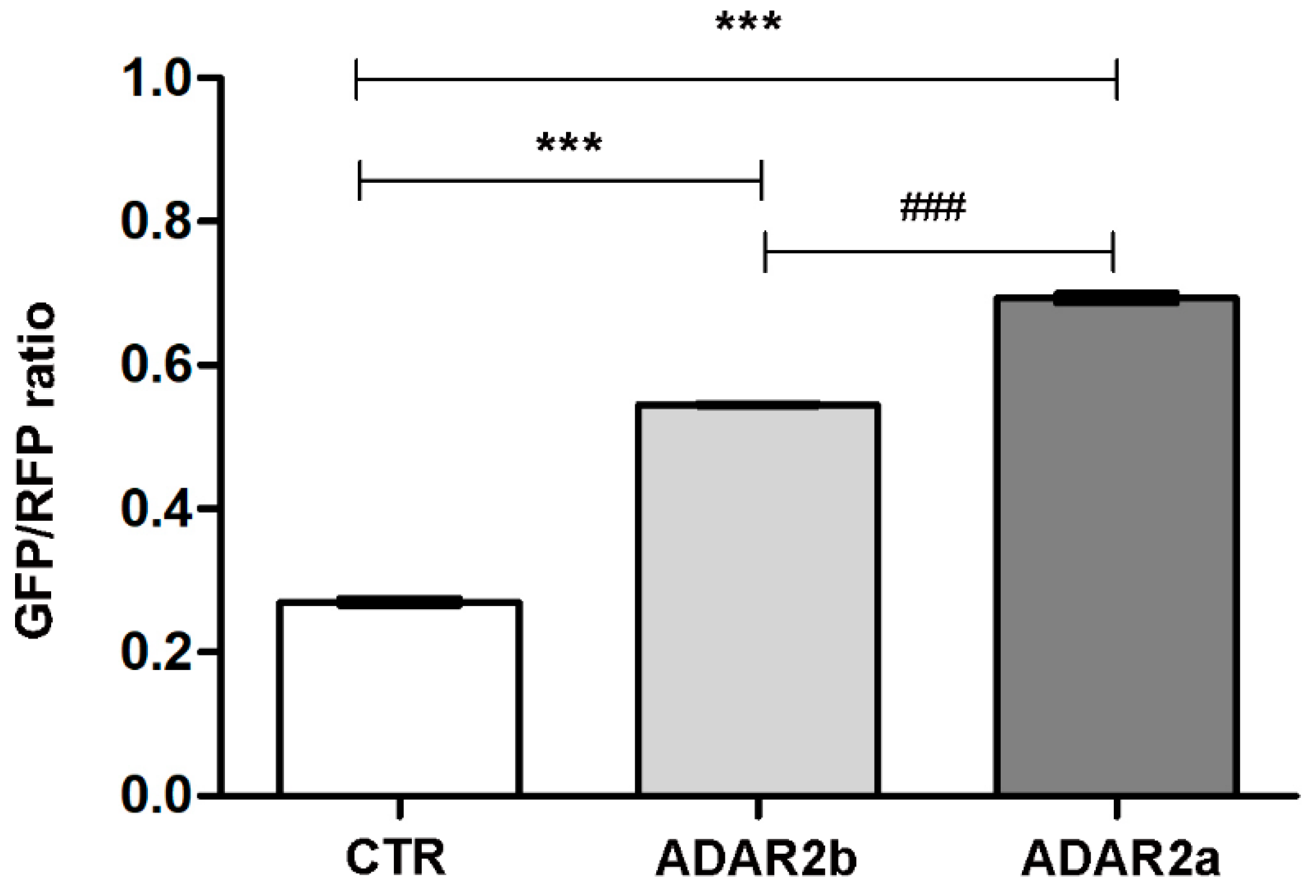
3.3. ADAR2a Splicing Isoform Has the Higher Enzymatic Activity
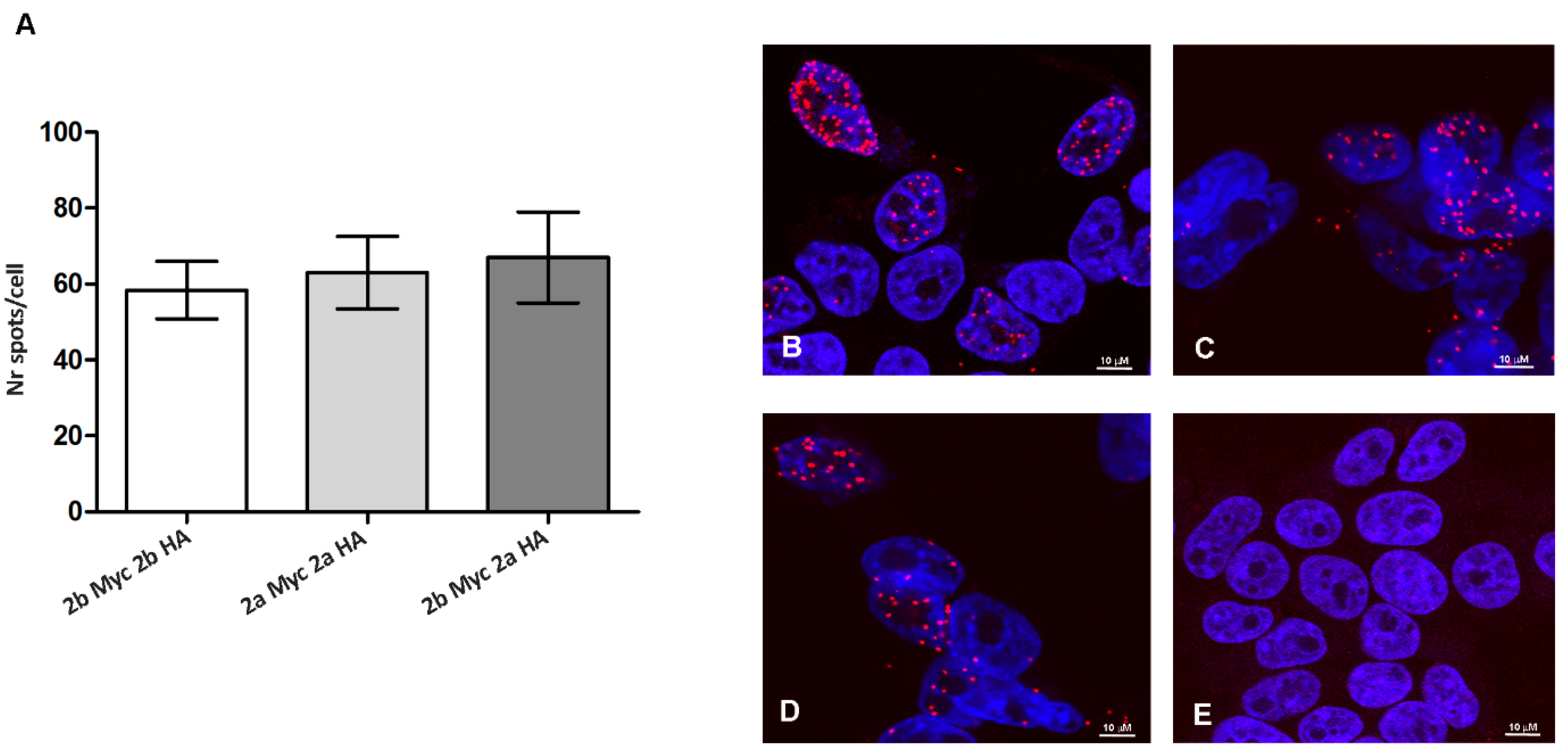
3.4. ADAR2 Splicing Isoforms Do Not Modify the Enzyme Dimerization Properties
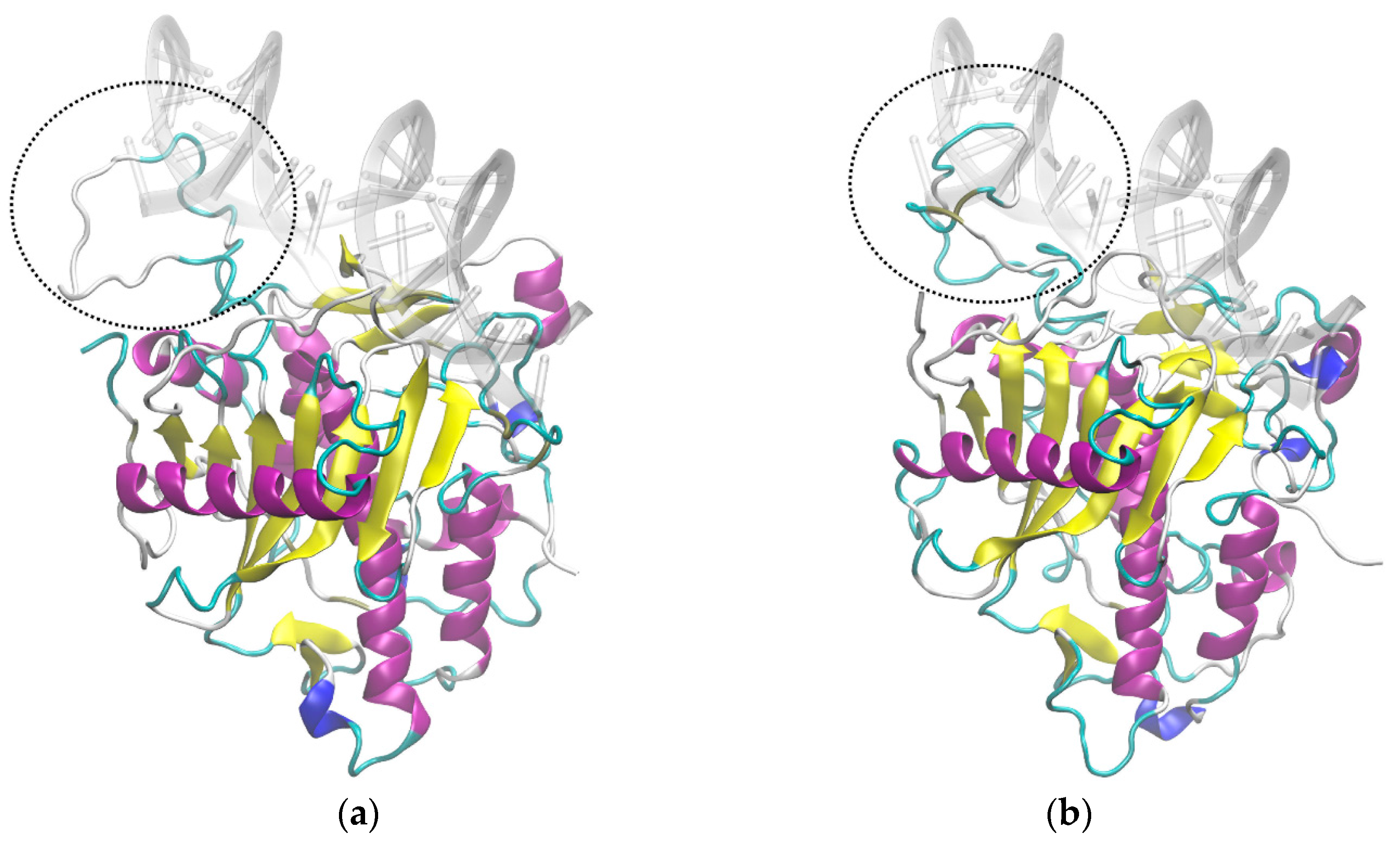
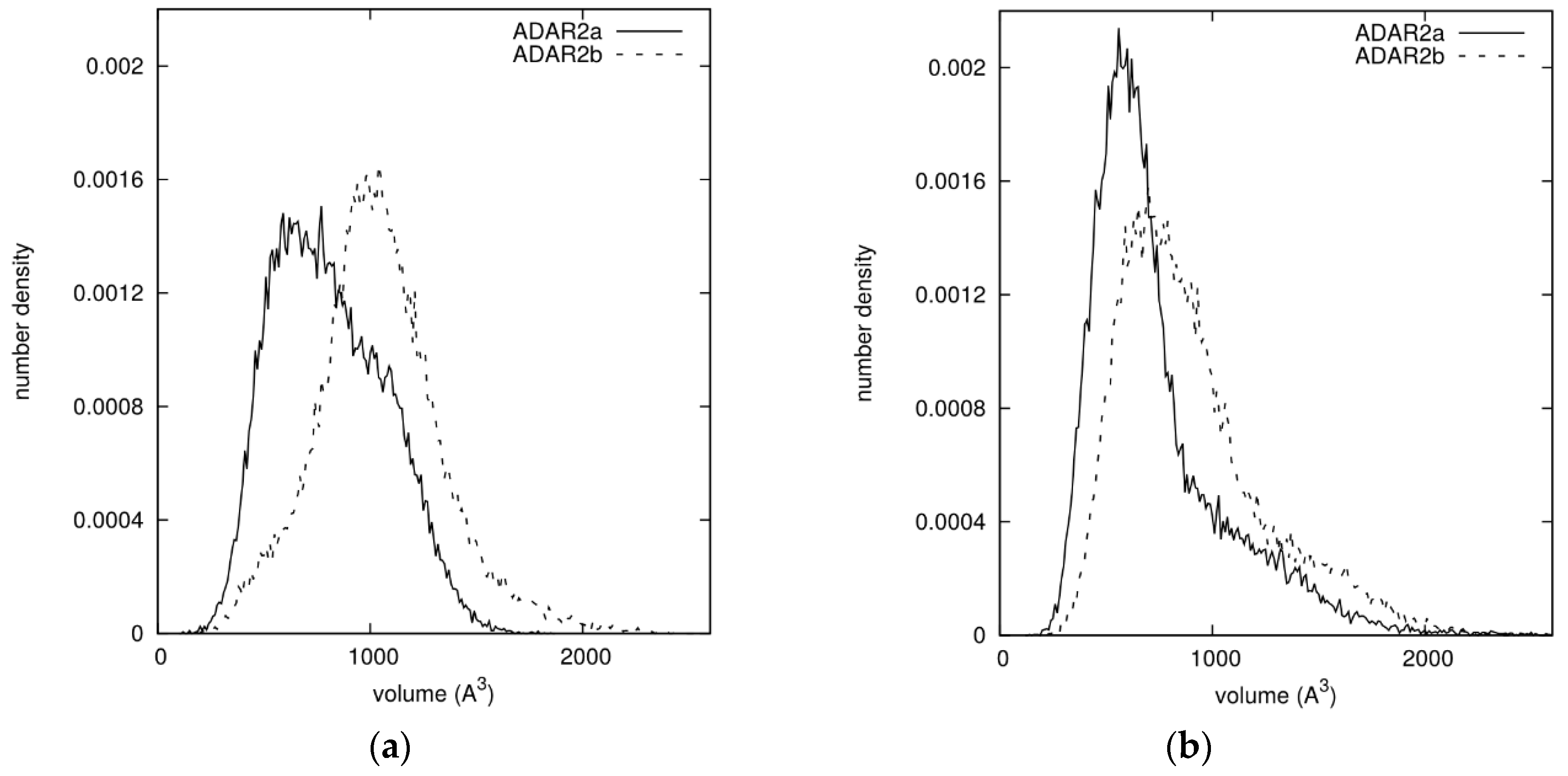
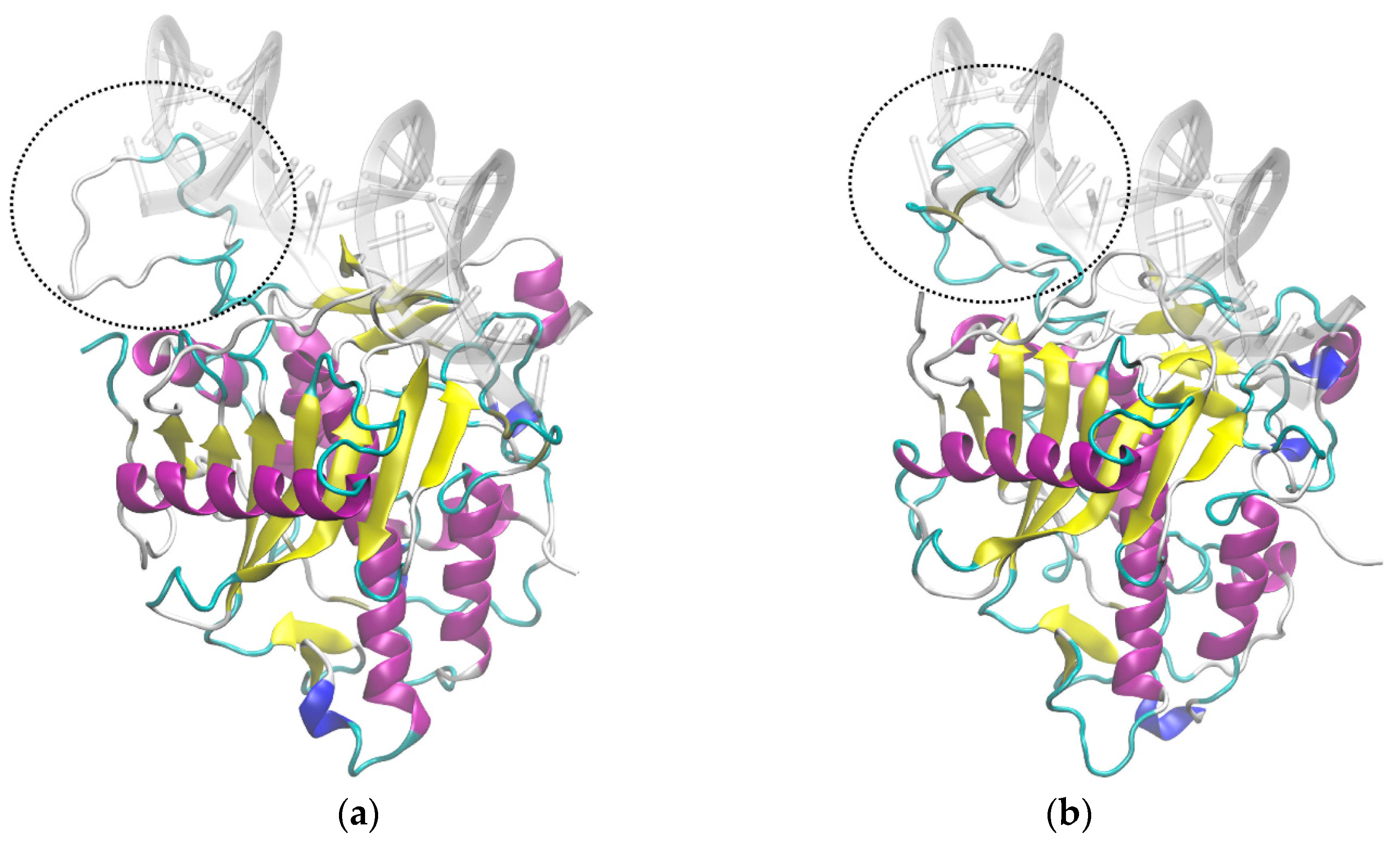
3.5. Molecular Dynamics Suggests Different Interactions with RNA Chain for ADAR2 Isoforms
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tariq, A.; Jantsch, M.F. Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front. Neurosci. 2012, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Keegan, L.P.; Gallo, A.; O’Connell, M.A. The many roles of an RNA editor. Nat. Rev. Genet. 2001, 2, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.; Rich, A.; Nishikura, K. A-to-I RNA editing: Recent news and residual mysteries. J. Biol. Chem. 2003, 278, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Chilibeck, K.A.; Wu, T.; Liang, C.; Schellenberg, M.J.; Gesner, E.M.; Lynch, J.M.; MacMillan, A.M. FRET analysis of in vivo dimerization by RNA-editing enzymes. J. Biol. Chem. 2006, 281, 16530–16535. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Sakurai, M.; Shiromoto, Y.; Nishikura, K. Functions of the RNA editing enzyme ADAR1 and their relevance to human diseases. Genes 2016, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.L.; Zhong, X.; Tu, W.; Soundarapandian, M.M.; Molner, P.; Zhu, D.; Lau, L.; Liu, S.; Liu, F.; Lu, Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 2006, 49, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [PubMed]
- Barbon, A.; Fumagalli, F.; Caracciolo, L.; Madaschi, L.; Lesma, E.; Mora, C.; Carelli, S.; Slotkin, T.A.; Racagni, G.; Di Giulio, A.M.; et al. Acute spinal cord injury persistently reduces R/G RNA editing of AMPA receptors. J. Neurochem. 2010, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Strehblow, A.; Hallegger, M.; Jantsch, M.F. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol. Biol. Cell 2002, 13, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.; Gommans, W.M. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009, 37, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Keegan, L.P.; Lafarga, M.; Berciano, M.T.; O’Connell, M.; Carmo-Fonseca, M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003, 116, 1805–1818. [Google Scholar] [CrossRef] [PubMed]
- Sansam, C.L.; Wells, K.S.; Emeson, R.B. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc. Natl. Acad. Sci. USA 2003, 100, 14018–14023. [Google Scholar] [CrossRef] [PubMed]
- Behm, M.; Wahlstedt, H.; Widmark, A.; Eriksson, M.; Ohman, M. Accumulation of nuclear ADAR2 regulates adenosine-to-inosine RNA editing during neuronal development. J. Cell Sci. 2017, 130, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, C.; Barbon, A.; Barlati, S. Activity regulation of adenosine deaminases acting on RNA (ADARs). Mol. Neurobiol. 2012, 45, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sansam, C.L.; Singh, M.; Emeson, R.B. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol. Cell. Biol. 2006, 26, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.; O’Connell, M.A.; Keller, W. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA 1997, 3, 453–463. [Google Scholar] [PubMed]
- Lai, F.; Chen, C.X.; Carter, K.C.; Nishikura, K. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol. Cell. Biol. 1997, 17, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Slavov, D.; Gardiner, K. Phylogenetic comparison of the pre-mRNA adenosine deaminase ADAR2 genes and transcripts: Conservation and diversity in editing site sequence and alternative splicing patterns. Gene 2002, 299, 83–94. [Google Scholar] [CrossRef]
- Kawahara, Y.; Ito, K.; Ito, M.; Tsuji, S.; Kwak, S. Novel splice variants of human ADAR2 mRNA: Skipping of the exon encoding the dsRNA-binding domains, and multiple C-terminal splice sites. Gene 2005, 363, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.; Gommans, W.M. Novel exon of mammalian ADAR2 extends open rReading frame. PLoS ONE 2009, 4, e4225. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, C.; La Via, L.; Bonini, D.; Mora, C.; Russo, I.; Barbon, A.; Barlati, S. AMPA receptor regulation at the mRNA and protein level in rat primary cortical cultures. PLoS ONE 2011, 6, e25350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbon, A.; Vallini, I.; La Via, L.; Marchina, E.; Barlati, S. Glutamate receptor RNA editing: a molecular analysis of GLUluR2, GLUR5 and GLUR6 in human brain tissues and in NT2 cells following in vitro neural differentiation. Mol. Brain Res. 2003, 117, 168–178. [Google Scholar] [CrossRef]
- La Via, L.; Bonini, D.; Russo, I.; Orlandi, C.; Barlati, S.; Barbon, A. Modulation of dendritic AMPA receptor mRNA trafficking by RNA splicing and editing. Nucleic Acids Res. 2013, 41, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Garncarz, W.; Tariq, A.; Handl, C.; Pusch, O.; Jantsch, M.F. A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol. 2013, 10, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Schoft, V.K.; Schopoff, S.; Jantsch, M.F. Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. 2007, 35, 3723–3732. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.M.; Thomas, J.M.; Zheng, Y.; Tran, K.; Phelps, K.J.; Scott, A.I.; Havel, J.; Fisher, A.J.; Beal, P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016, 23, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hwang, T.; Park, C.K.; Leung, A.K.; Gao, Y.; Hyde, T.M.; Kleinman, J.E.; Rajpurohit, A.; Tao, R.; Shin, J.H.; Weinberger, D.R. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Muller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [PubMed]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Seeburg, P.H.; Higuchi, M. A mammalian RNA editing enzyme. Nature 1996, 379, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Hideyama, T.; Teramoto, S.; Hachiga, K.; Yamashita, T.; Kwak, S. Co-occurrence of TDP-43 mislocalization with reduced activity of an RNA editing enzyme, ADAR2, in aged mouse motor neurons. PLoS ONE 2012, 7, e43469. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, Y.; Farahani, H.S.; Behm, M.; Lagergren, J.; Ohman, M. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res. 2012, 22, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Lesuisse, C.; Martin, L.J. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J. Neurobiol. 2002, 51, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.; Megyesi, J.; Hammond, R. Human central nervous system tissue culture: A historical review and examination of recent advances. Neurobiol. Dis. 2005, 18, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Patrizio, A.; Montalbano, A.; Sciancalepore, M.; Tongiorgi, E. Developmental and maintenance defects in Rett syndrome neurons identified by a new mouse staging system in vitro. Front. Cell. Neurosci. 2014, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Wahlstedt, H.; Daniel, C.; Enstero, M.; Ohman, M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009, 19, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Keegan, L.P.; Jaffray, E.; Hay, R.T.; O’Connell, M.A.; Carmo-Fonseca, M. SUMO-1 Modification Alters ADAR1 Editing Activity. Mol. Biol. Cell 2005, 16, 5115–5126. [Google Scholar] [CrossRef] [PubMed]
- Macbeth, M.R.; Schubert, H.L.; Vandemark, A.P.; Lingam, A.T.; Hill, C.P.; Bass, B.L. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 2005, 309, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, R.; Brindle, J.; Paro, S.; Casadio, A.; Hempel, S.; Morrice, N.; Bisso, A.; Keegan, L.P.; Del Sal, G.; O’Connell, M.A. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011, 30, 4211–4222. [Google Scholar] [CrossRef] [PubMed]
- Tariq, A.; Garncarz, W.; Handl, C.; Balik, A.; Pusch, O.; Jantsch, M.F. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res. 2013, 41, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Bonini, D.; Lacoux, C.; Pacini, L.; Zingariello, M.; Sancillo, L.; Bosisio, D.; Salvi, V.; Mingardi, J.; La Via, L.; et al. Absence of the fragile X mental retardation protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA Biol. 2017, 14, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Hang, P.N.; Tohda, M.; Matsumoto, K. Developmental changes in expression and self-editing of adenosine deaminase type 2 pre-mRNA and mRNA in rat brain and cultured cortical neurons. Neurosci. Res. 2008, 61, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Cenci, C.; Barzotti, R.; Galeano, F.; Corbelli, S.; Rota, R.; Massimi, L.; Di Rocco, C.; O’Connell, M.A.; Gallo, A. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem. 2008, 283, 7251–7260. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5-HT2cR Editing | |||
|---|---|---|---|
| CTR | ADAR2b | ADAR2a | |
| Site A | 79.2 ± 3.83% | 54 ± 0.77% ** | 45.6 ± 1.82% *** |
| Site B | 88.7 ± 0.13% | 65.2 ± 0.45% *** | 57.8 ± 0.59% ***/### |
| Site C′ | 10.9 ± 0.93% | 17.6 ± 1.11% | 20.6 ± 2.38% * |
| Site C | 17 ± 0.46% | 23.4 ± 2.38% * | 26.2 ± 0.49% ** |
| Site D | 71.8 ± 0.3% | 96.3 ± 0.93% *** | 99.1 ± 0.6% *** |
| A | |||||
| Residue | ADAR2a | ADAR2b | Residue | ADAR2a | ADAR2b |
| Val 351 | 0.00 | 0.00 | Lys 475 (485) | 0.20 | 0.69 |
| Thr 375 | 0.58 | 0.40 | Arg 477 (487) | 0.06 | 1.65 |
| Lys 376 | 1.20 | 0.79 | Arg 481 (491) | 1.85 | 1.79 |
| Glu 396 | 0.00 | 0.00 | Ser 486 (496) | 1.07 | 1.28 |
| Cys 451 | 0.00 | 0.00 | Glu 488 (498) | 1.91 | 2.17 |
| Arg 455 | 1.30 | 1.26 | Thr 490 (500) | 0.00 | 0.11 |
| Asn 473 (483) | 0.23 | 0.61 | Ser 495 (505) | 0.00 | 0.35 |
| Arg 474 (484) | 1.09 | 2.44 | Arg 510 (520) | 0.69 | 1.36 |
| B | |||||
| Residue | ADAR2a | ADAR2b | Residue | ADAR2a | ADAR2b |
| Val 351 | 0.00 | 0.00 | Lys 475 (485) | 0.37 | 0.87 |
| Thr 375 | 0.25 | 0.30 | Arg 477 (487) | 0.01 | 1.27 |
| Lys 376 | 0.81 | 0.96 | Arg 481 (491) | 1.28 | 0.97 |
| Glu 396 | 0.00 | 0.00 | Ser 486 (496) | 1.12 | 1.17 |
| Cys 451 | 0.09 | 0.00 | Glu 488 (498) | 1.30 | 1.73 |
| Arg 455 | 0.14 | 0.74 | Thr 490 (500) | 0.03 | 0.12 |
| Asn 473 (483) | 0.42 | 0.62 | Ser 495 (505) | 0.00 | 0.28 |
| Arg 474 (484) | 2.18 | 1.88 | Arg 510 (520) | 0.60 | 0.89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippini, A.; Bonini, D.; Giacopuzzi, E.; La Via, L.; Gangemi, F.; Colombi, M.; Barbon, A. Differential Enzymatic Activity of Rat ADAR2 Splicing Variants Is Due to Altered Capability to Interact with RNA in the Deaminase Domain. Genes 2018, 9, 79. https://doi.org/10.3390/genes9020079
Filippini A, Bonini D, Giacopuzzi E, La Via L, Gangemi F, Colombi M, Barbon A. Differential Enzymatic Activity of Rat ADAR2 Splicing Variants Is Due to Altered Capability to Interact with RNA in the Deaminase Domain. Genes. 2018; 9(2):79. https://doi.org/10.3390/genes9020079
Chicago/Turabian StyleFilippini, Alice, Daniela Bonini, Edoardo Giacopuzzi, Luca La Via, Fabrizio Gangemi, Marina Colombi, and Alessandro Barbon. 2018. "Differential Enzymatic Activity of Rat ADAR2 Splicing Variants Is Due to Altered Capability to Interact with RNA in the Deaminase Domain" Genes 9, no. 2: 79. https://doi.org/10.3390/genes9020079