Characterizing the DNA Methyltransferases of Haloferax volcanii via Bioinformatics, Gene Deletion, and SMRT Sequencing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions
2.2. Deletion of Annotated Restriction Modification Genes
2.3. DNA Purification for Single-Molecule Real-Time Sequencing
2.4. Single-Molecule Real-Time Sequencing
2.5. Bioinformatics Analysis
2.6. Haloferax volcanii Growth Experiments
3. Results
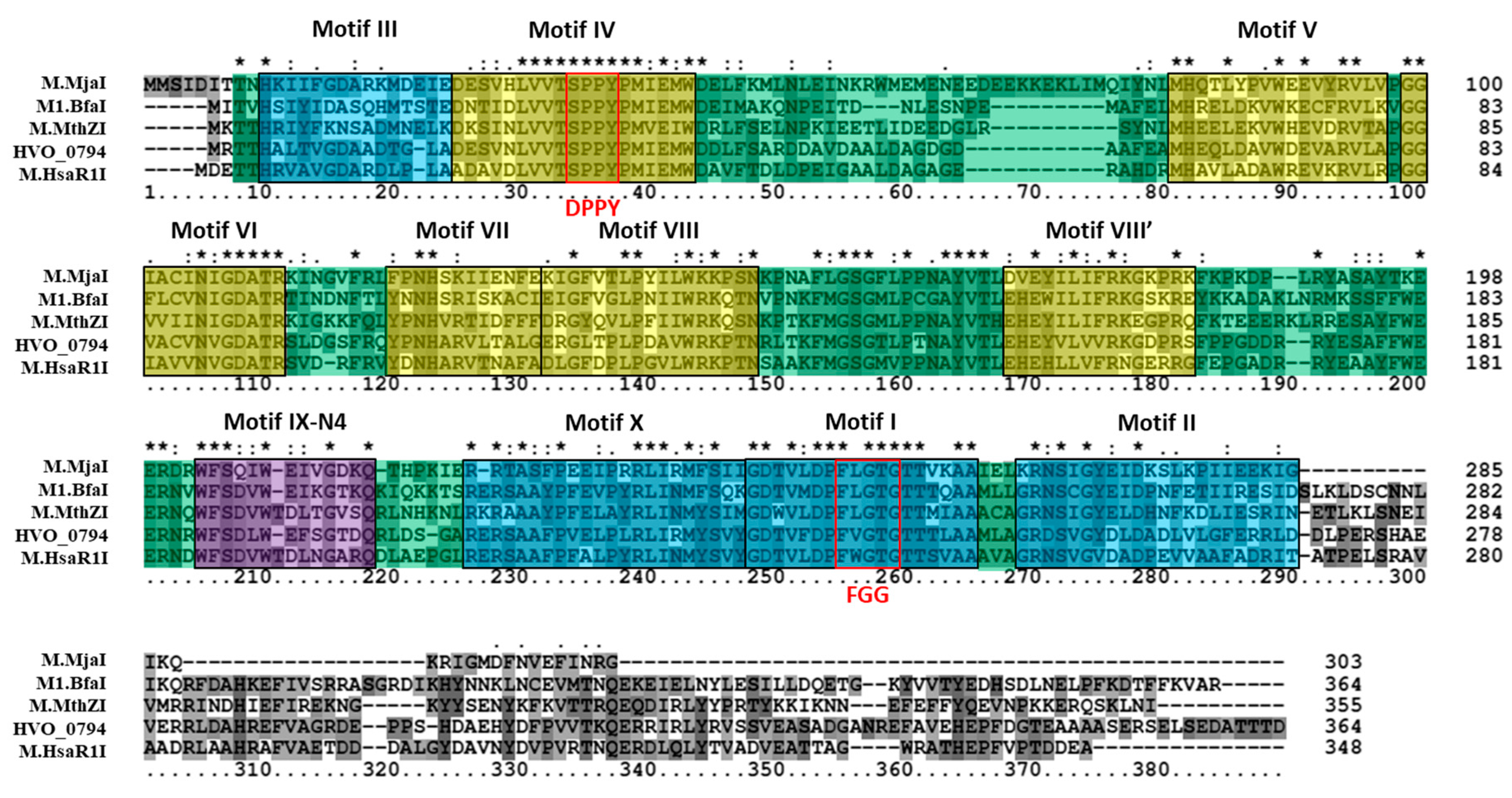
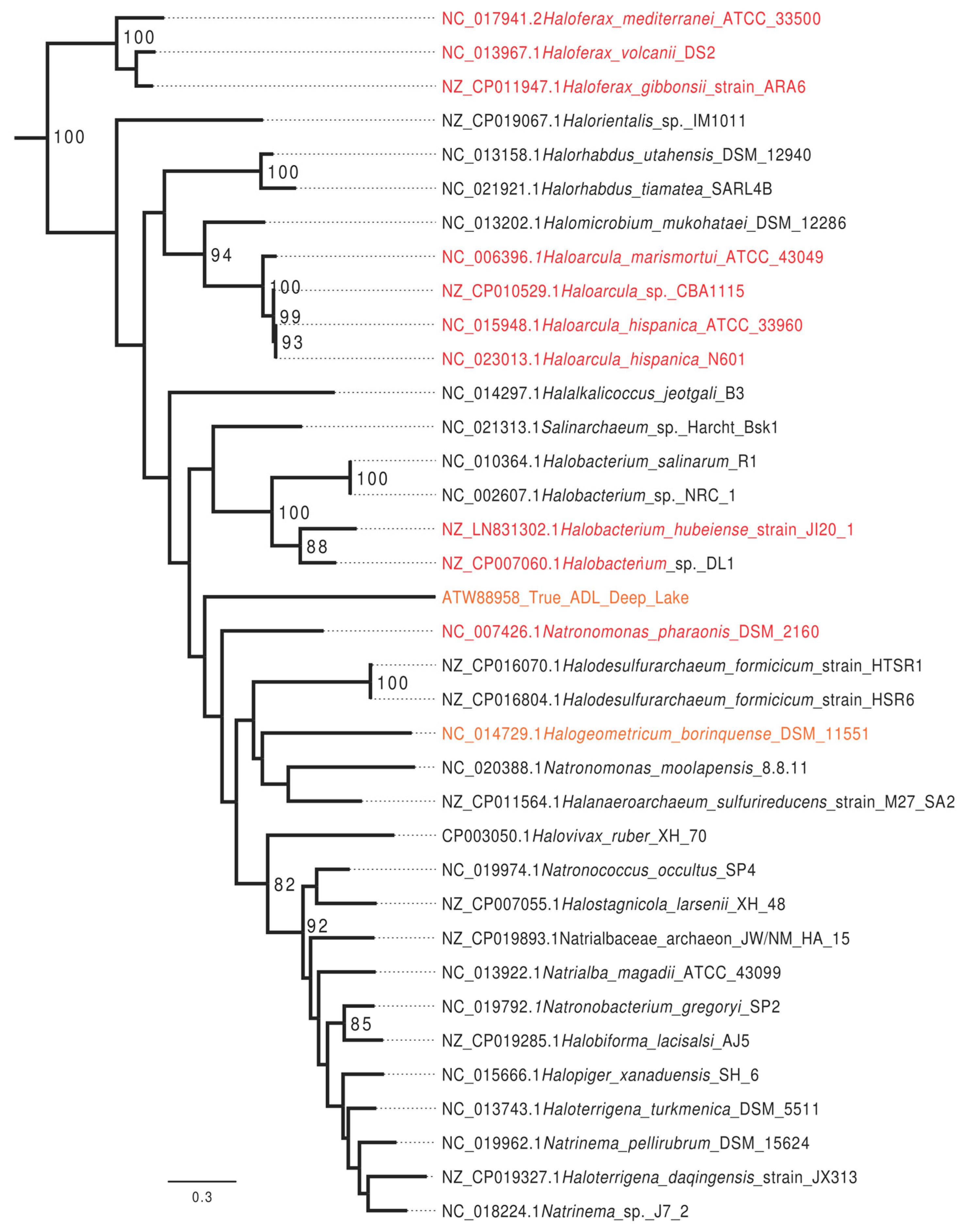
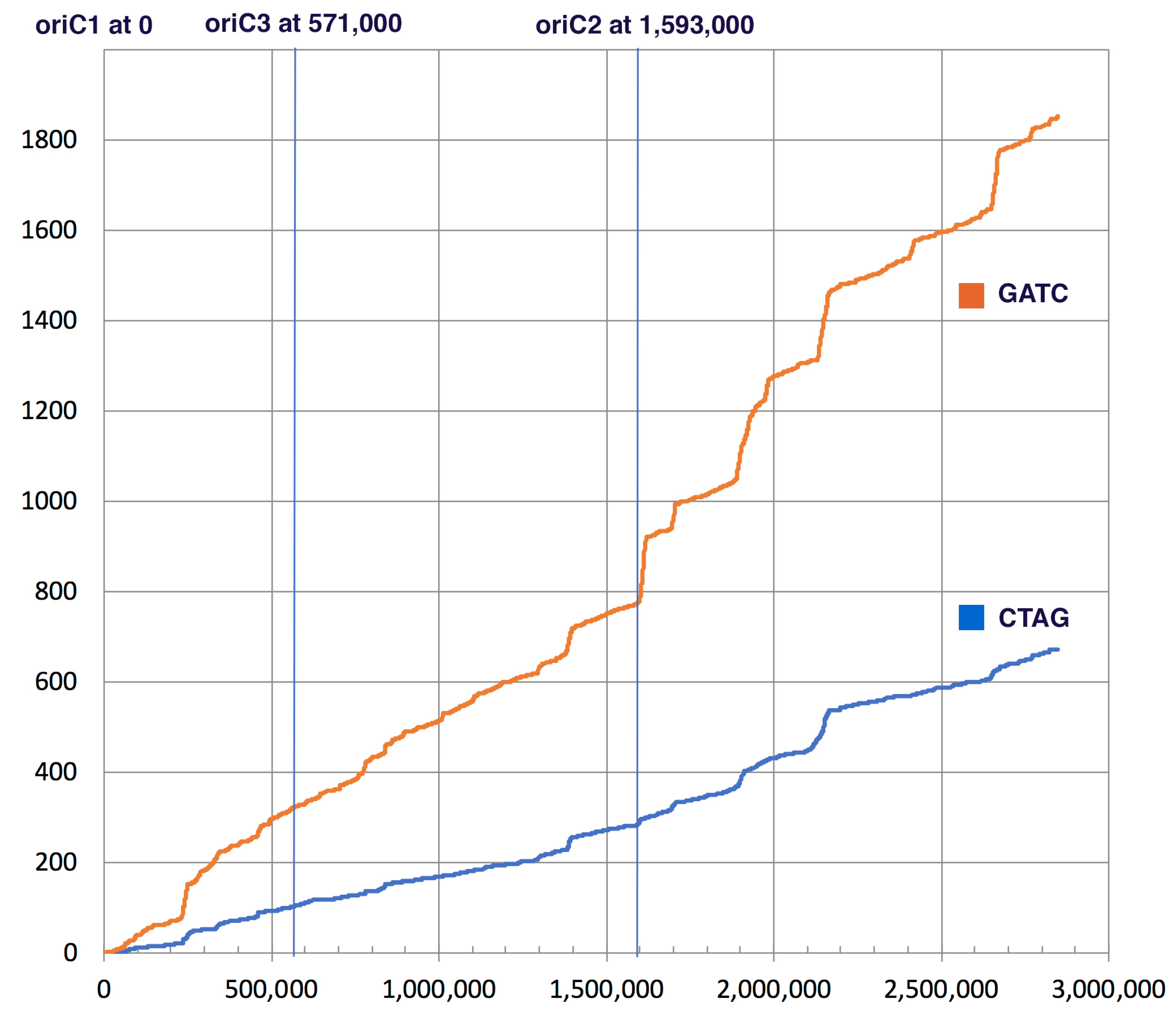
3.1. Bioinformatics Analysis Supports Identification of HVO_0794 as a Chromosomal 4mC CTAG MTase
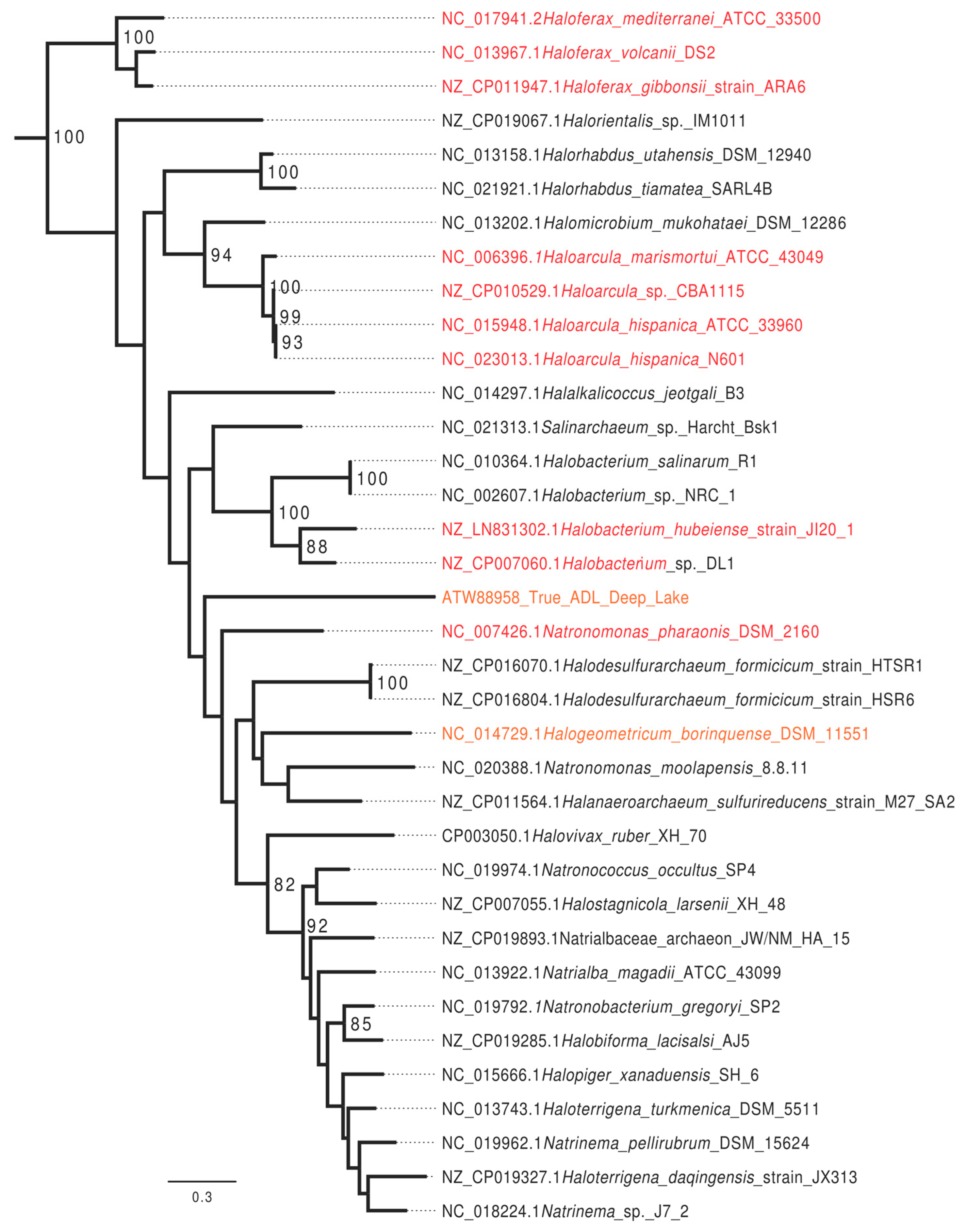
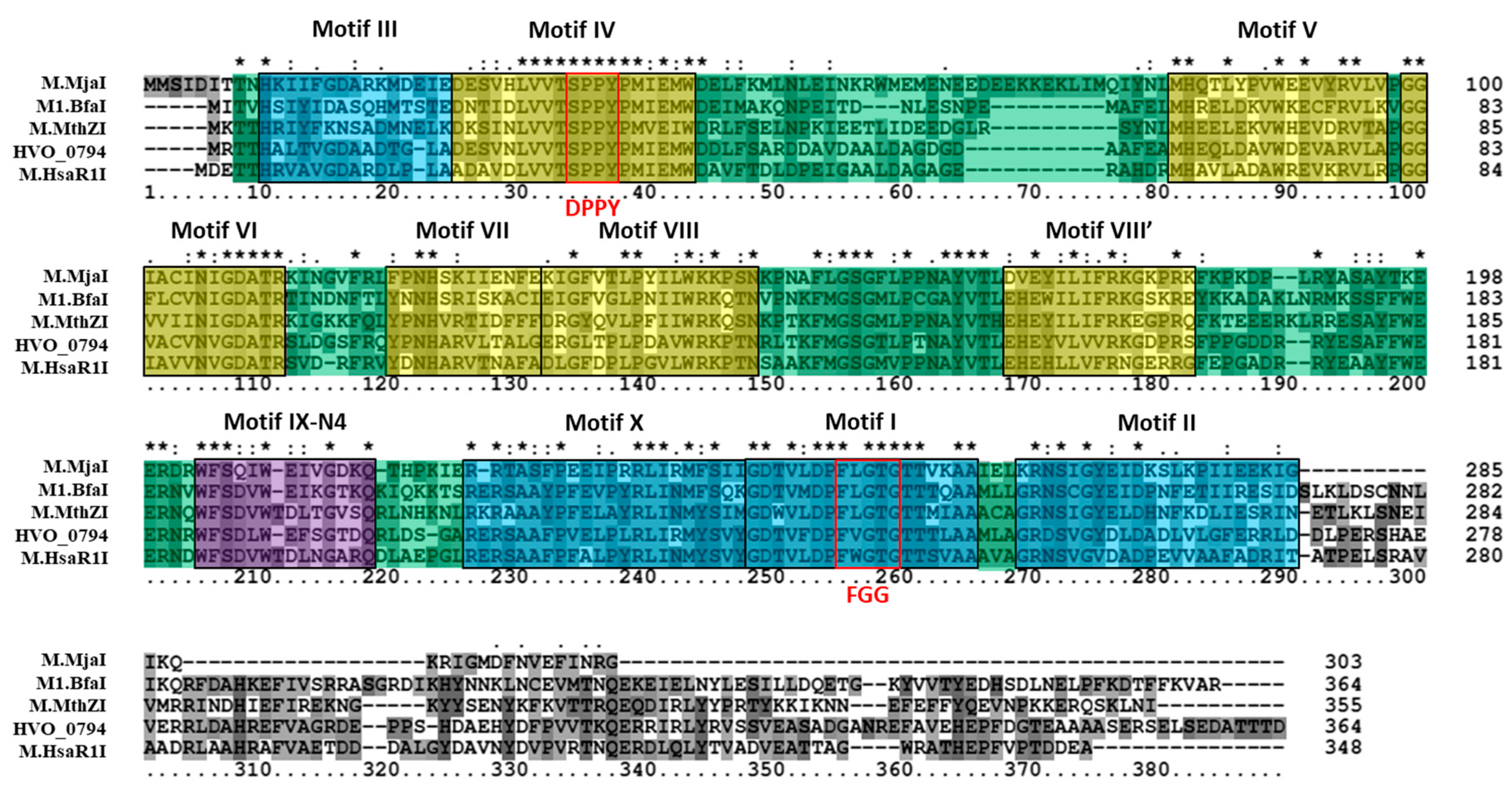
3.2. Bioinformatics Analysis Supports Identification of RmeM as a Type I 6mA MTase and RmeS as a Type I Specificity Subunit on the Chromosome
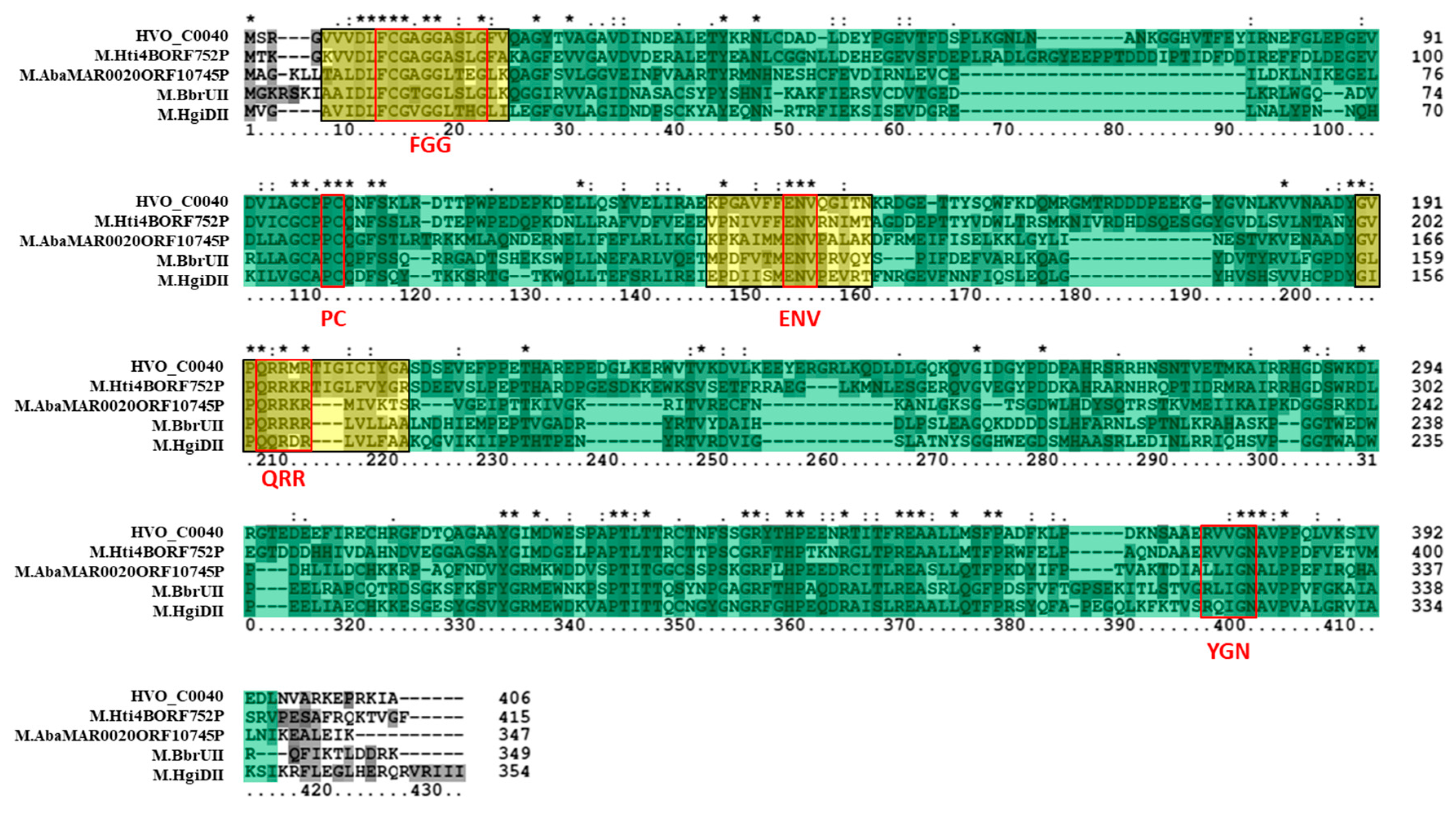
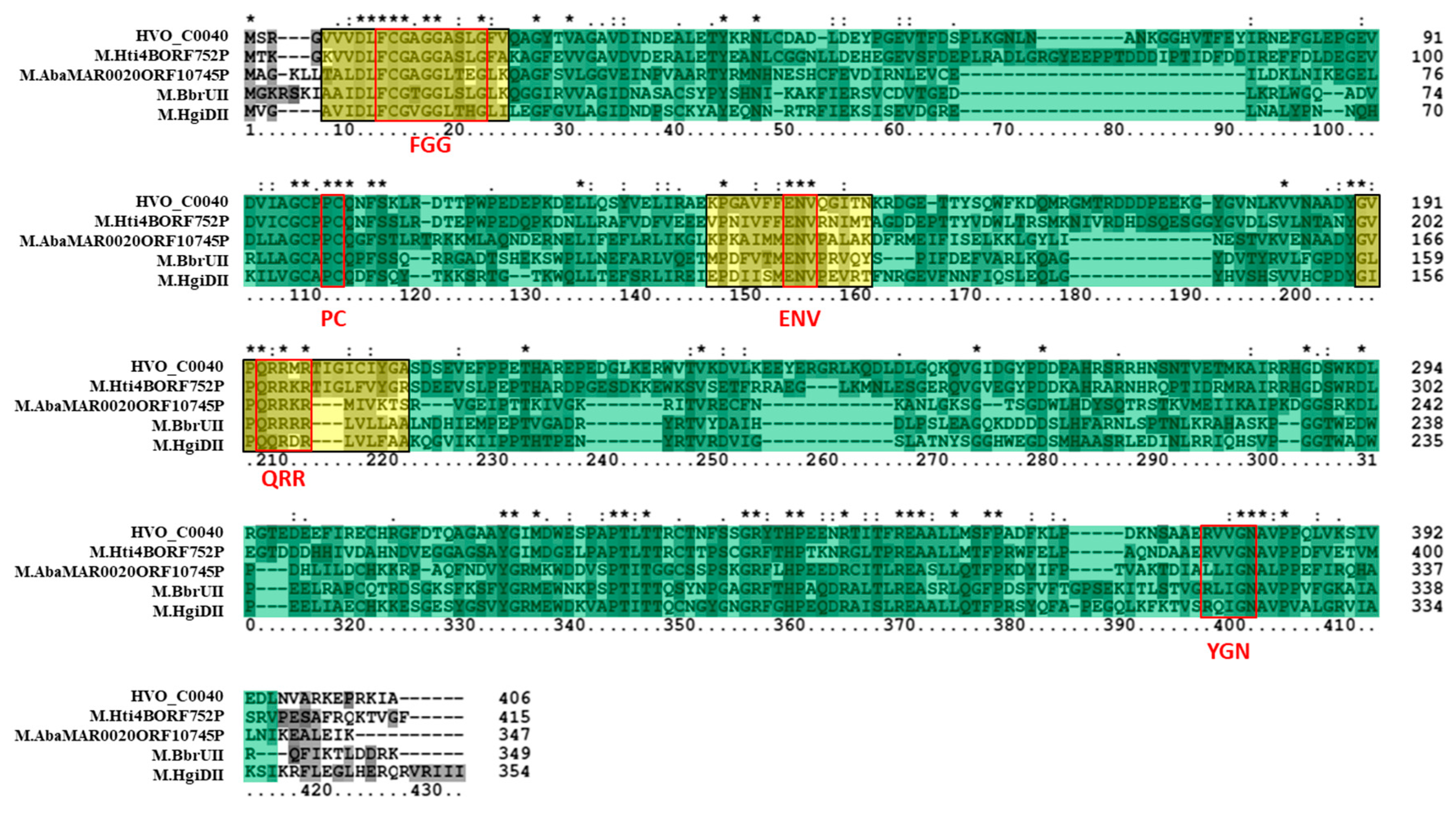
3.3. Bioinformatics Analysis Supports Annotation of HVO_C0040 as a 5mC MTase and HVO_A0079 as a 6mA MTase
3.4. Deletion of HVO_0794, HVO_A0006, and HVO_A0237 Eliminates 4mC Methylation and Does Not Effect 6mA Methylation
3.5. Deletion of the rmeRMS Operon Abolishes 6mA Methylation
3.6. Multi-RM Deletion Eliminates Detection of All DNA Methylation
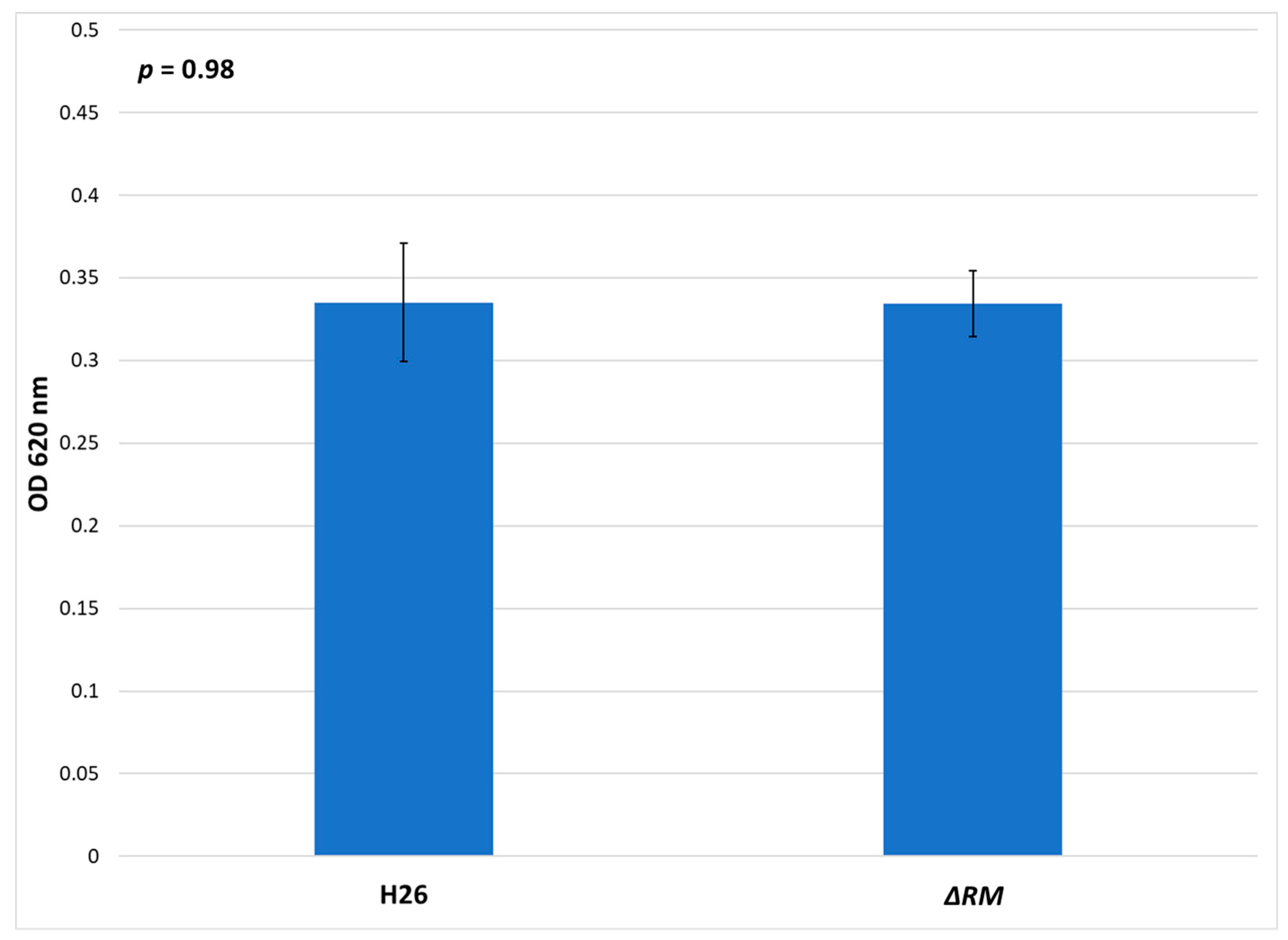
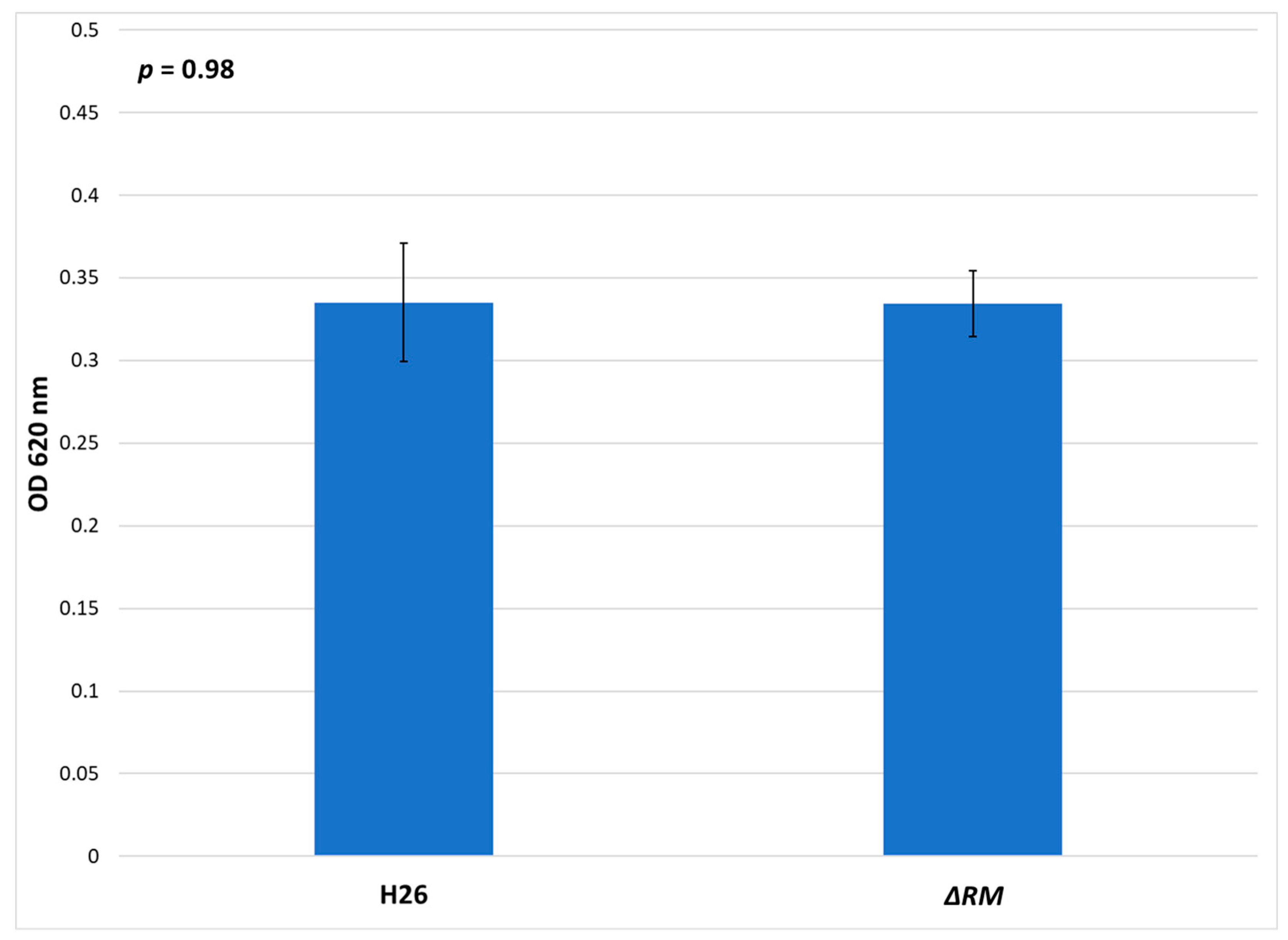
3.7. No Defect in Growth Occurs in the Multi-RM Deletion Compared to the Parental Strain
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bickle, T.A.; Krüger, D.H. Biology of DNA restriction. Microbiol. Rev. 1993, 57, 434–450. [Google Scholar] [PubMed]
- Tock, M.R.; Dryden, D.T. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005, 8, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001, 29, 3742–3756. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Handa, N.; Watanabe-Matsui, M.; Takahashi, N.; Kobayashi, I. Maintenance forced by a restriction-modification system can be modulated by a region in its modification enzyme not essential for methyltransferase activity. J. Bacteriol. 2008, 190, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Korlach, J.; Turner, S.W. Going beyond five bases in DNA sequencing. Curr. Opin. Struct Biol. 2012, 22, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Bheemanaik, S.; Reddy, Y.V.; Rao, D.N. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem. J. 2006, 399, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Malone, T.; Blumenthal, R.M.; Cheng, X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Bujnicki, J.M. Sequence permutations in the molecular evolution of DNA methyltransferases. BMC Evol. Biol. 2002, 2, 3. [Google Scholar] [CrossRef]
- Bujnicki, J.M.; Radlinska, M. Molecular evolution of DNA-(cytosine-N4) methyltransferases: Evidence for their polyphyletic origin. Nucleic Acids Res. 1999, 27, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2010, 38, D234–D236. [Google Scholar] [CrossRef] [PubMed]
- Posfai, J.; Bhagwat, A.S.; Posfai, G.; Roberts, R.J. Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res. 1989, 17, 2421–2435. [Google Scholar] [CrossRef] [PubMed]
- Militello, K.T.; Simon, R.D.; Qureshi, M.; Maines, R.; VanHorne, M.L.; Hennick, S.M.; Jayakar, S.K.; Pounder, S. Conservation of Dcm-mediated cytosine DNA methylation in Escherichia coli. FEMS Microbiol. Lett. 2012, 328, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Belfort, M.; Bestor, T.; Bhagwat, A.S.; Bickle, T.A.; Bitinaite, J.; Blumenthal, R.M.; Degtyarev, S.; Dryden, D.T.; Dybvig, K.; et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003, 31, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Ershova, A.S.; Rusinov, I.S.; Spirin, S.A.; Karyagina, A.S.; Alexeevski, A.V. Role of restriction-modification systems in prokaryotic evolution and ecology. Biochemistry 2015, 80, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014, 42, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Tang, Q.; Zhang, J.Z.; Tian, L.F.; Gao, P.; Yan, X.X. Structural basis underlying complex assembly and conformational transition of the type I R-M system. Proc. Natl. Acad. Sci. USA 2017, 114, 11151–11156. [Google Scholar] [CrossRef] [PubMed]
- Pingoud, A.; Wilson, G.G.; Wende, W. Type II restriction endonucleases—A historical perspective and more. Nucleic Acids Res. 2014, 42, 7489–7527. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.D.; Bhatia, T.K.; Lovasco, L.; Davis, T.B. MmeI: A minimal type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 2008, 36, 6558–6570. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.N.; Dryden, D.T.; Bheemanaik, S. Type III restriction-modification enzymes: A historical perspective. Nucleic Acids Res. 2014, 42, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Raleigh, E.A. The other face of restriction: Modification-dependent enzymes. Nucleic Acids Res. 2014, 42, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Curtis, P.D. DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 2016, 40, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Romero, M.A.; Busby, S.J.; Dyer, N.P.; Ott, S.; Millard, A.D.; Grainger, D.C. Dynamic distribution of SeqA protein across the chromosome of Escherichia coli K-12. MBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, H.; Han, J.S.; Hwang, D.S. Interaction of SeqA and Dam methylase on the hemimethylated origin of Escherichia coli chromosomal DNA replication. J. Biol. Chem. 1999, 274, 11463–11468. [Google Scholar] [CrossRef] [PubMed]
- Waldminghaus, T.; Skarstad, K. The Escherichia coli SeqA protein. Plasmid 2009, 61, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Welsh, K.M.; Lu, A.L.; Clark, S.; Modrich, P. Isolation and characterization of the Escherichia coli mutH gene product. J. Biol. Chem. 1987, 262, 15624–15629. [Google Scholar] [PubMed]
- Au, K.G.; Welsh, K.; Modrich, P. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 1992, 267, 12142–12148. [Google Scholar] [PubMed]
- Putnam, C.D. Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair 2016, 38, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zweiger, G.; Marczynski, G.; Shapiro, L. A Caulobacter DNA methyltransferase that functions only in the predivisional cell. J. Mol. Biol. 1994, 235, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Domian, I.J.; Reisenauer, A.; Shapiro, L. Feedback control of a master bacterial cell-cycle regulator. Proc. Natl. Acad. Sci. USA 1999, 96, 6648–6653. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Naito, Y.; Handa, N.; Kobayashi, I. A DNA methyltransferase can protect the genome from postdisturbance attack by a restriction-modification gene complex. J. Bacteriol. 2002, 184, 6100–6108. [Google Scholar] [CrossRef] [PubMed]
- Seshasayee, A.S.; Singh, P.; Krishna, S. Context-dependent conservation of DNA methyltransferases in bacteria. Nucleic Acids Res. 2012, 40, 7066–7073. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The epigenomic landscape of prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed]
- Nolling, J.; de Vos, W.M. Identification of the CTAG-recognizing restriction-modification systems MthZI and MthFI from Methanobacterium thermoformicicum and characterization of the plasmid-encoded mthZIM gene. Nucleic Acids Res. 1992, 20, 5047–5052. [Google Scholar] [CrossRef] [PubMed]
- Grogan, D.W. Cytosine methylation by the suai restriction-modification system: Implications for genetic fidelity in a hyperthermophilic archaeon. J. Bacteriol. 2003, 185, 4657–4661. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Watanabe, M.; Kuroita, T.; Uchiyama, I.; Bujnicki, J.M.; Kawakami, B.; Tanokura, M.; Kobayashi, I. Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5′-GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res. 2005, 33, e112. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Yuzawa, H.; Handa, N.; Kobayashi, I. Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Appl. Environ. Microbiol. 2006, 72, 5367–5375. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.; Jackson, L.; Chimileski, S.; Papke, R.T. Genome-wide DNA methylation analysis of Haloferax volcanii H26 and identification of DNA methyltransferase related PD-(D/E)XK nuclease family protein HVO_A0006. Front. Microbiol. 2015, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Allers, T.; Mevarech, M. Archaeal genetics—The third way. Nat. Rev. Genet. 2005, 6, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Blaby, I.K.; Phillips, G.; Blaby-Haas, C.E.; Gulig, K.S.; El Yacoubi, B.; de Crecy-Lagard, V. Towards a systems approach in the genetic analysis of archaea: Accelerating mutant construction and phenotypic analysis in Haloferax volcanii. Archaea 2010, 2010, 426239. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Murray, I.A.; Morgan, R.D.; Kislyuk, A.O.; Spittle, K.E.; Boitano, M.; Fomenkov, A.; Roberts, R.J.; Korlach, J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012, 40, e29. [Google Scholar] [CrossRef] [PubMed]
- Allers, T.; Ngo, H.P.; Mevarech, M.; Lloyd, R.G. Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl. Environ. Microbiol. 2004, 70, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M. The Halohandbook: Protocols for Haloarchaeal Genetics Ver. 7.1. Available online: http://www.haloarchaea.com/resources/halohandbook/index.html (accessed on 6 September 2013).
- Mullakhanbhai, M.F.; Larsen, H. Halobacterium volcanii spec. Nov., a dead sea halobacterium with a moderate salt requirement. Arch. Microbiol. 1975, 104, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Bitan-Banin, G.; Ortenberg, R.; Mevarech, M. Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J. Bacteriol. 2003, 185, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Allers, T.; Barak, S.; Liddell, S.; Wardell, K.; Mevarech, M. Improved strains and plasmid vectors for conditional overexpression of his-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 2010, 76, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Detecting DNA Base Modifications: SMRT Analysis of Microbial Methylomes. Available online: https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Methylome-Analysis-Technical-Note (accessed on 16 June 2015).
- Hartman, A.L.; Norais, C.; Badger, J.H.; Delmas, S.; Haldenby, S.; Madupu, R.; Robinson, J.; Khouri, H.; Ren, Q.; Lowe, T.M.; et al. The complete genome sequence of Haloferax volcanii DS2, a model archaeon. PLoS ONE 2010, 5, e9605. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. Seaview version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.; Malla, S.; Blythe, M.J.; Nieduszynski, C.A.; Allers, T. Accelerated growth in the absence of DNA replication origins. Nature 2013, 503, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Haberman, A.; Heywood, J.; Meselson, M. DNA modification methylase activity of Escherichia coli restriction endonucleases K and P. Proc. Natl. Acad. Sci. USA 1972, 69, 3138–3141. [Google Scholar] [CrossRef] [PubMed]
- Kennaway, C.K.; Obarska-Kosinska, A.; White, J.H.; Tuszynska, I.; Cooper, L.P.; Bujnicki, J.M.; Trinick, J.; Dryden, D.T. The structure of M.EcoKI type I DNA methyltransferase with a DNA mimic antirestriction protein. Nucleic Acids Res. 2009, 37, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Clark, T.A.; Morgan, R.D.; Boitano, M.; Anton, B.P.; Luong, K.; Fomenkov, A.; Turner, S.W.; Korlach, J.; Roberts, R.J. The methylomes of six bacteria. Nucleic Acids Res. 2012, 40, 11450–11462. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, J.E.; Daniel, A.S.; Murray, N.E. Mutations that confer de novo activity upon a maintenance methyltransferase. J. Mol. Biol. 1991, 221, 431–440. [Google Scholar] [CrossRef]
- O’Neill, M.; Powell, L.M.; Murray, N.E. Target recognition by EcoKI: The recognition domain is robust and restriction-deficiency commonly results from the proteolytic control of enzyme activity. J. Mol. Biol. 2001, 307, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tang, Q.; An, X.; Yan, X.; Liang, D. Structure of HsdS subunit from Thermoanaerobacter. tengcongensis sheds lights on mechanism of dynamic opening and closing of type I methyltransferase. PLoS ONE 2011, 6, e17346. [Google Scholar] [CrossRef] [PubMed]
- Janscak, P.; Bickle, T.A. The DNA recognition subunit of the type IB restriction-modification enzyme EcoAI tolerates circular permutions of its polypeptide chain. J. Mol. Biol. 1998, 284, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Dusterhoft, A.; Kroger, M. Cloning, sequence and characterization of m5C-methyltransferase-encoding gene, hgiDIIM (GTCGAC), from Herpetosiphon. giganteus strain Hpa2. Gene 1991, 106, 87–92. [Google Scholar] [CrossRef]
- O'Connell Motherway, M.; O'Driscoll, J.; Fitzgerald, G.F.; Van Sinderen, D. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microb. Biotechnol. 2009, 2, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Janulaitis, A.; Vaisvila, R.; Timinskas, A.; Klimasauskas, S.; Butkus, V. Cloning and sequence analysis of the genes coding for Eco57I type IV restriction-modification enzymes. Nucleic Acids Res. 1992, 20, 6051–6056. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Lu, X.; Luong, K.; Dai, Q.; Boitano, M.; Turner, S.W.; He, C.; Korlach, J. Enhanced 5-methylcytosine detection in single-molecule, real-time sequencing via Tet1 oxidation. BMC Biol. 2013, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Westphal, L.L.; Sauvey, P.; Champion, M.M.; Ehrenreich, I.M.; Finkel, S.E. Genomewide Dam methylation in Escherichia coli during long-term stationary phase. mSystems 2016, 1, e00130-16. [Google Scholar] [CrossRef] [PubMed]
- Charlebois, R.L.; Lam, W.L.; Cline, S.W.; Doolittle, W.F. Characterization of pHV2 from Halobacterium volcanii and its use in demonstrating transformation of an archaebacterium. Proc. Natl. Acad. Sci. USA 1987, 84, 8530–8534. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. Bioessays. 2011, 33, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.K.; Dyall-Smith, M.; Marchfelder, A. The adaptive immune system of Haloferax volcanii. Life 2015, 5, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Stachler, A.E.; Turgeman-Grott, I.; Shtifman-Segal, E.; Allers, T.; Marchfelder, A.; Gophna, U. High tolerance to self-targeting of the genome by the endogenous CRISPR-Cas system in an archaeon. Nucleic Acids Res. 2017, 45, 5208–5216. [Google Scholar] [CrossRef] [PubMed]
- García-Heredia, I.; Martín-Cuadrado, A.B.; Mojica, F.J.; Santos, F.; Mira, A.; Antón, J.; Rodríguez-Valera, F. Reconstructing viral genomes from the environment using fosmid clones: The case of haloviruses. PLoS ONE 2012, 7, e33802. [Google Scholar] [CrossRef] [PubMed]
- Luk, A.W.; Williams, T.J.; Erdmann, S.; Papke, R.T.; Cavicchioli, R. Viruses of haloarchaea. Life (Basel) 2014, 4, 681–715. [Google Scholar] [CrossRef] [PubMed]
- Naor, A.; Lapierre, P.; Mevarech, M.; Papke, R.T.; Gophna, U. Low species barriers in halophilic archaea and the formation of recombinant hybrids. Curr. Biol. 2012, 22, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Roer, L.; Aarestrup, F.M.; Hasman, H. The EcoKI type I restriction-modification system in Escherichia coli affects but is not an absolute barrier for conjugation. J. Bacteriol. 2015, 197, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.A.; Zhang, X.S.; Levine, S.M.; Gill, S.R.; Falush, D.; Blaser, M.J. Natural transformation of Helicobacter pylori involves the integration of short DNA fragments interrupted by gaps of variable size. PLoS Pathog. 2009, 5, e1000337. [Google Scholar] [CrossRef] [PubMed]
- Erwin, A.L.; Sandstedt, S.A.; Bonthuis, P.J.; Geelhood, J.L.; Nelson, K.L.; Unrath, W.C.; Diggle, M.A.; Theodore, M.J.; Pleatman, C.R.; Mothershed, E.A.; et al. Analysis of genetic relatedness of Haemophilus influenzae isolates by multilocus sequence typing. J. Bacteriol. 2008, 190, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Budroni, S.; Siena, E.; Dunning Hotopp, J.C.; Seib, K.L.; Serruto, D.; Nofroni, C.; Comanducci, M.; Riley, D.R.; Daugherty, S.C.; Angiuoli, S.V.; et al. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc. Natl. Acad. Sci. USA 2011, 108, 4494–4499. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.D.; Dwinell, E.A.; Bhatia, T.K.; Lang, E.M.; Luyten, Y.A. The MmeI family: Type II restriction-modification enzymes that employ single-strand modification for host protection. Nucleic Acids Res. 2009, 37, 5208–5221. [Google Scholar] [CrossRef] [PubMed]
- Babski, J.; Haas, K.A.; Nather-Schindler, D.; Pfeiffer, F.; Forstner, K.U.; Hammelmann, M.; Hilker, R.; Becker, A.; Sharma, C.M.; Marchfelder, A.; et al. Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-seq (dRNA-seq). BMC Genomics 2016, 17, 629. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Abe, K.; Kobayashi, I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010, 38, 2428–2443. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Plasmid Name | Description | Source |
|---|---|---|
| E. coli HST08 | Cloning strain of E. coli | Clontech, Cat. # 636763 |
| H. volcanii DS2 | Wild-type strain | Mullakhanbhai and Larsen [43] |
| H. volcanii H26 | ∆pyrE2; uracil auxotroph derived from DS2 | Bitan-Banin et al. [44] |
| H. volcanii H1206 | ∆pyrE2/∆mrr; derived from H26 | Allers et al. [45] |
| H. volcanii ∆rmeRMS | rmeRMS deletion strain; derived from H1206 | This study |
| H. volcanii ∆HVO_0794 ∆HVO_A0006 ∆HVO_A0237 | Deletion strain of HVO_0794, HVO_A0006, and HVO_A0237; Derived from H1206 | This study |
| H. volcanii ∆RM | Deletion strain of HVO_0794, rmeRMS, HVO_A0006, HVO_A0074, HVO_A0079, and HVO_A0237; derived from H1206 | This study |
| pTA131 | Vector used for gene deletion. Contains lacZ cloning site for blue-white screening, ampR ampicillin resistance gene for selectivity in E. coli, and pyrE2 for screening in H. volcanii. | Allers, Ngo, Mevarech and Lloyd [41] |
| p∆HVO_A0006 | Derivative of pTA131 with flanking regions of HVO_A0006 inserted into lacZ cloning site for gene deletion | Ouellette, Jackson, Chimileski and Papke [37] |
| p∆HVO_0794 | Derivative of pTA131 with flanking regions of HVO_0794 inserted into lacZ cloning site for gene deletion | This study |
| p∆rmeRMS | Derivative of pTA131 with flanking regions of rmeRMS operon inserted into lacZ cloning site for gene deletion | This study |
| p∆HVO_A0074 | Derivative of pTA131 with flanking regions of HVO_A0074 inserted into lacZ cloning site for gene deletion | This study |
| p∆HVO_A0079 | Derivative of pTA131 with flanking regions of HVO_A0079 inserted into lacZ cloning site for gene deletion | This study |
| p∆HVO_A0237 | Derivative of pTA131 with flanking regions of HVO_A0237 inserted into lacZ cloning site for gene deletion | This study |
| Gene Locus Tag | Gene Symbol | Putative RM Classification | Gene Size (bp) | Location in the Genome | Notes |
|---|---|---|---|---|---|
| HVO_0682 | mrr | Type IV | 1005 | Chromosome | Type IV restriction endonuclease |
| HVO_0794 | zim | Type II | 1095 | Chromosome | Putative 4mC CTAG methyltransferase |
| HVO_2269-2271 | rmeRMS | Type I | 2223, 1395, 1233 | Chromosome | Operon which contains a putative Type I RM system with 6mA methyltransferase |
| HVO_C0040 | - | Type II | 1221 | pHV1 | Putative 5mC GTCGAC methyltransferase |
| HVO_A0006 | - | Type IIG | 660 | pHV4 | Putative restriction endonuclease fragment of HVO_A0237 [37] |
| HVO_A0074 | - | Type IV | 3315 | pHV4 | Putative Type IV restriction endonuclease |
| HVO_A0079 | - | Type IIG | 3267 | pHV4 | Putative 6mA Type IIG RM protein |
| HVO_A0237 | - | Type IIG | 2199 | pHV4 | Putative 6mA methyltransferase and target recognition protein |
| Primer Name | Primer Sequence | Primer Description |
|---|---|---|
| HVO_A0006 FR1F | 5’- CGG GCC CCC CCT CGA GTC AAG CAG TAC CTC AAC ACG GAA CA -3’ | Used to amplify the flanking regions of HVO_A0006 for insertion into pTA131 linearized with XhoI and XbaI (Primer designs from Ouellette et al. [37]) |
| HVO_A0006 FR1R | 5’- ATT CGA TAT CAA GCT GTC CTC AAG GAC GGC CTG CA -3’ | |
| HVO_A0006 FR2F | 5’- GAC GCG TTG ATA TCC CGA AGA ATC CAG TTG CTG TCT GTT G -3’ | |
| HVO_A0006 FR2R | 5’- GGA TAT CAA CGC GTC GGC ATT ATG CAA TTC -3’ | |
| HVO_0794 FR1F | 5’- GCT TGA TAT CGA ATT CCC CGC GAG AAA GAC GAG AAG -3’ | Used to amplify the flanking regions of HVO_0794 for insertion into pTA131 linearized with EcoRI and BamHI |
| HVO_0794 FR1R | 5’- GCC TGG TAG AAT TCC CGT ACG GAC GTA TTT CCC CCG A -3’ | |
| HVO_0794 FR2F | 5’- GGA ATT CTA CCA GGC CGA CGA CGA CCG ACT GAG GTC -3’ | |
| HVO_0794 FR2R | 5’- TAG AAC TAG TGG ATC CGA ACG GCA GCA CCC GCG A -3’ | |
| rmeRMS FR1F | 5’- CGG GCC CCC CCT CGA GTC GGT GTT TCG CAG GTC ATT C -3’ | Used to amplify the flanking regions of the rmeRMS operon for insertion into pTA131 linearized with XhoI and ClaI |
| rmeRMS FR1R | 5’- GGG CGC CAT CCA GGC TAC TCA CTA TAT TTC ACT CGG GGT A -3’ | |
| rmeRMS FR2F | 5’- GCC TGG ATG GCG CCC CTC ACC TAT TCA CAA AGA GAG GAA -3’ | |
| rmeRMS FR2R | 5’- ATA TCA AGC TTA TCG ATT GCC GGG TTT CCT GTT ATT TT CT -3’ | |
| HVO_A0074 FR1F | 5’ GCT TGA TAT CGA ATT CTG CTC GTC GTG GTA CTT GTC -3’ | Used to amplify the flanking regions of HVO_A0074 for insertion into pTA131 linearized with EcoRI and XbaI |
| HVO_A0074 FR1R | 5’- CGG TAC CGA CAT GTT ATC TCA ATG CAG CGC TTC TC -3’ | |
| HVO_A0074 FR2F | 5’- AAC ATG TCG GTA CCG TTG AGG ACT GGG AGC GTA TC -3’ | |
| HVO_A0074 FR2R | 5’- TGG CGG CCG CTC TAG TTG AAG GTC TGT GTC GCA TC -3’ | |
| HVO_A0079 FR1F | 5’- GCG AAT TGG GTA CCG GCC CCG ACC TGC CTT GG -3’ | Used to amplify the flanking regions of HVO_A0079 for insertion into pTA131 linearized with ApaI and EcoRV |
| HVO_A0079 FR1R | 5’- GCC TGG TAG AAT TCC CCG TGT TCG GTT AAG CGG A -3’ | |
| HVO_A0079 FR2F | 5’- GGA ATT CTA CCA GGC AAT GGG ATC TGA CGA AGG AGG -3’ | |
| HVO_A0079 FR2R | 5’- CTG CAG GAA TTC GAT CAT AAA GGT CTT CTC AGC GGT T -3’ | |
| HVO_A0237 FR1F | 5’- CGG GCC CCC CCT CGA GGT TCG CGC TCT TGC TCA GGT -3’ | Used to amplify the flanking regions of HVO_A0237 for insertion into pTA131 linearized with XhoI and XbaI |
| HVO_A0237 FR1R | 5’- GGG ATC CAA AGC TTG AGG CGT TGC TGA CAT TAT ATC GAA G -3’ | |
| HVO_A0237 FR2F | 5’- CAA GCT TTG GAT CCC GCC TTT CTG CTG GCG AGT TTC C -3’ | |
| HVO_A0237 FR2R | 5’- TGG CGG CCG CTC TAG AAT ATC GCG CAG CTC TAT CGG G -3’ | |
| M13(-21) F | 5’- GTA AAA CGA CGG CCA GT -3’ | Used for amplifying the multiple cloning site of pTA131 for screening |
| M13 R | 5’- AGG AAA CAG CTA TGA CCA T -3’ |
| Accession Number, Organism and Chromosome Number | Total CTAG | Total GATC | CTAG/kb | GATC/kb | GATC/CTAG | Match to E. coli Dam $ |
|---|---|---|---|---|---|---|
| NC 013967.1 Haloferax volcanii DS2 | 671 | 1851 | 0.24 | 0.65 | 2.8 | |
| NZ CP007551.1 Haloferax mediterranei ATCC 33500 | 1130 | 1472 | 0.38 | 0.50 | 1.3 | |
| NZ CP011947.1 Haloferax gibbonsii strain ARA6 | 556 | 1510 | 0.19 | 0.51 | 2.7 | |
| NC 017941.2 Haloferax mediterranei ATCC 33500 | 1142 | 1500 | 0.39 | 0.51 | 1.3 | |
| NC 023013.1 Haloarcula hispanica N601 chr.1 | 1497 | 7523 | 0.50 | 2.50 | 5.0 | |
| NC 023010.2 Haloarcula hispanica N601 chr.2 | 340 | 1675 | 0.94 | 4.61 | 4.9 | |
| NZ CP010529.1 Haloarcula sp. CBA1115 | 1849 | 9333 | 0.54 | 2.73 | 5.0 | |
| NC 006396.1 Haloarcula marismortui ATCC 43049 chr.I | 1816 | 6564 | 0.58 | 2.10 | 3.6 | |
| NC 006397.1 Haloarcula marismortui ATCC 43049 chr.II | 274 | 1011 | 0.95 | 3.51 | 3.7 | |
| NC 015948.1 Haloarcula hispanica ATCC 33960 chr.I | 1493 | 7462 | 0.50 | 2.49 | 5.0 | |
| NC 015943.1 Haloarcula hispanica ATCC 33960 chr.II | 479 | 2210 | 0.98 | 4.52 | 4.6 | |
| NZ LN831302.1 Halobacterium hubeiense strain JI20-1 | 795 | 1820 | 0.32 | 0.72 | 2.3 | |
| NZ CP007060.1 Halobacterium sp. DL1 | 1168 | 4634 | 0.41 | 1.63 | 4.0 | |
| NC 002607.1 Halobacterium sp. NRC-1 | 551 | 11047 | 0.27 | 5.48 | 20.0 | |
| NC 010364.1 Halobacterium salinarum R1 | 537 | 10991 | 0.27 | 5.49 | 20.5 | |
| NC 012029.1 Halorubrum lacusprofundi ATCC 49239 chr.1 | 756 | 25016 | 0.28 | 9.15 | 33.1 | + |
| NC 012028.1 Halorubrum lacusprofundi ATCC 49239 chr.2 | 389 | 3306 | 0.74 | 6.29 | 8.5 | + |
| NC 007426.1 Natronomonas pharaonis DSM 2160 | 1016 | 1839 | 0.39 | 0.71 | 1.8 | |
| NC 008212.1 Haloquadratum walsbyi DSM 16790 | 2290 | 14449 | 0.73 | 4.61 | 6.3 | |
| NC 017459.1 Haloquadratum walsbyi C23 | 2281 | 14681 | 0.72 | 4.66 | 6.4 | |
| NC 014729.1 Halogeometricum borinquense DSM 11551 | 1085 | 8407 | 0.38 | 2.98 | 7.7 | |
| CP024845.1 Halophilic archaeon True-ADL | 1786 | 23542 | 0.54 | 7.07 | 13.2 | |
| NC 021921.1 Halorhabdus tiamatea SARL4B | 892 | 27010 | 0.32 | 9.59 | 30.3 | + |
| NC 013158.1 Halorhabdus utahensis DSM 12940 | 964 | 32101 | 0.31 | 10.30 | 33.3 | |
| NC 013202.1 Halomicrobium mukohataei DSM 12286 | 918 | 27978 | 0.30 | 8.99 | 30.5 | |
| NC 013743.1 Haloterrigena turkmenica DSM 5511 | 1347 | 37472 | 0.35 | 9.64 | 27.8 | |
| NC 013922.1 Natrialba magadii ATCC 43099 | 1592 | 25139 | 0.42 | 6.70 | 15.8 | |
| NC 014297.1 Halalkalicoccus jeotgali B3 | 1106 | 29489 | 0.39 | 10.50 | 26.7 | |
| NC 015666.1 Halopiger xanaduensis SH-6 | 1090 | 33560 | 0.30 | 9.15 | 30.8 | |
| NC 018224.1 Natrinema sp. J7-2 | 1393 | 33801 | 0.38 | 9.14 | 24.3 | |
| NC 019792.1 Natronobacterium gregoryi SP2 | 2330 | 31628 | 0.62 | 8.35 | 13.6 | |
| NC 019962.1 Natrinema pellirubrum DSM 15624 | 1384 | 36667 | 0.37 | 9.67 | 26.5 | |
| NC 019964.1 Halovivax ruber XH-70 | 1256 | 30664 | 0.39 | 9.51 | 24.4 | |
| NC 019974.1 Natronococcus occultus SP4 | 1534 | 42563 | 0.38 | 10.61 | 27.7 | |
| NC 020388.1 Natronomonas moolapensis 8.8.11 | 1088 | 23003 | 0.37 | 7.90 | 21.1 | |
| NC 021313.1 Salinarchaeum sp. Harcht-Bsk1 | 948 | 33056 | 0.29 | 10.15 | 34.9 | |
| NZ AP017558.1 Halopenitus persicus DNA CBA1233 | 695 | 31995 | 0.23 | 10.78 | 46.0 | + |
| NZ AP017569.1 Halorubrum trapanicum DNA CBA1232 | 426 | 19948 | 0.15 | 7.03 | 46.8 | + |
| NZ CP007055.1 Halostagnicola larsenii XH-48 | 1094 | 24861 | 0.39 | 8.91 | 22.7 | |
| NZ CP008874.1 Halanaeroarchaeum sulfurireducens HSR2 | 596 | 15696 | 0.29 | 7.53 | 26.3 | |
| NZ CP011564.1 Halanaeroarchaeum sulfurireducens M27-SA2 | 637 | 16067 | 0.30 | 7.55 | 25.2 | |
| NZ CP016070.1 Halodesulfurarchaeum formicicum HTSR1 | 639 | 18453 | 0.32 | 9.36 | 28.9 | |
| NZ CP016804.1 Halodesulfurarchaeum formicicum HSR6 | 696 | 19038 | 0.33 | 9.13 | 27.4 | |
| NZ CP019067.1 Halorientalis sp. IM1011 | 1046 | 25987 | 0.31 | 7.68 | 24.8 | + |
| NZ CP019285.1 Halobiforma lacisalsi AJ5 | 1235 | 40138 | 0.30 | 9.64 | 32.5 | |
| NZ CP019327.1 Haloterrigena daqingensis JX313 | 1124 | 25935 | 0.33 | 7.63 | 23.1 | |
| NZ CP019893.1 Natrialbaceae archaeon JW/NM-HA 15 | 1177 | 35113 | 0.30 | 8.93 | 29.8 | + |
| Mean value per chromosome | 0.42 | 6.34 | 18.4 | |||
| Standard Deviation | 0.19 | 3.35 | 12.7 | |||
| NC 000913.3 Escherichia coli str K-12 substr MG1655 | 885 | 19124 | 0.19 | 4.12 | 21.6 | + |
| ∆HVO_0794 ∆HVO_A0006 ∆HVO_A0237 | ΔrmeRMS | ΔRM | ||||
|---|---|---|---|---|---|---|
| Motif | GCAm6BNNNNNNVTGC | Cm4TAG | GCAm6BNNNNNNVTGC | Cm4TAG | GCAm6BNNNNNNVTGC | Cm4TAG |
| Methylated position | 3 | 1 | 3 | 1 | 3 | 1 |
| Methylation type | 6mA | 4mC | 6mA | 4mC | 6mA | 4mC |
| Number of methylated motifs | 410 | 0 | 0 | 1199 | 0 | 0 |
| Number of motifs in genome | 410 | 1342 | 410 | 1342 | 410 | 1342 |
| Percent of methylated motifs | 100 | 0 | 0 | 89 | 0 | 0 |
| Mean modification QV score | 213.0 | - | - | 104.1 | - | - |
| Mean motif coverage | 130.4 | - | - | 113.0 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouellette, M.; Gogarten, J.P.; Lajoie, J.; Makkay, A.M.; Papke, R.T. Characterizing the DNA Methyltransferases of Haloferax volcanii via Bioinformatics, Gene Deletion, and SMRT Sequencing. Genes 2018, 9, 129. https://doi.org/10.3390/genes9030129
Ouellette M, Gogarten JP, Lajoie J, Makkay AM, Papke RT. Characterizing the DNA Methyltransferases of Haloferax volcanii via Bioinformatics, Gene Deletion, and SMRT Sequencing. Genes. 2018; 9(3):129. https://doi.org/10.3390/genes9030129
Chicago/Turabian StyleOuellette, Matthew, J. Peter Gogarten, Jessica Lajoie, Andrea M. Makkay, and R. Thane Papke. 2018. "Characterizing the DNA Methyltransferases of Haloferax volcanii via Bioinformatics, Gene Deletion, and SMRT Sequencing" Genes 9, no. 3: 129. https://doi.org/10.3390/genes9030129