Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool

Abstract
:1. Introduction
2. Materials and Methods
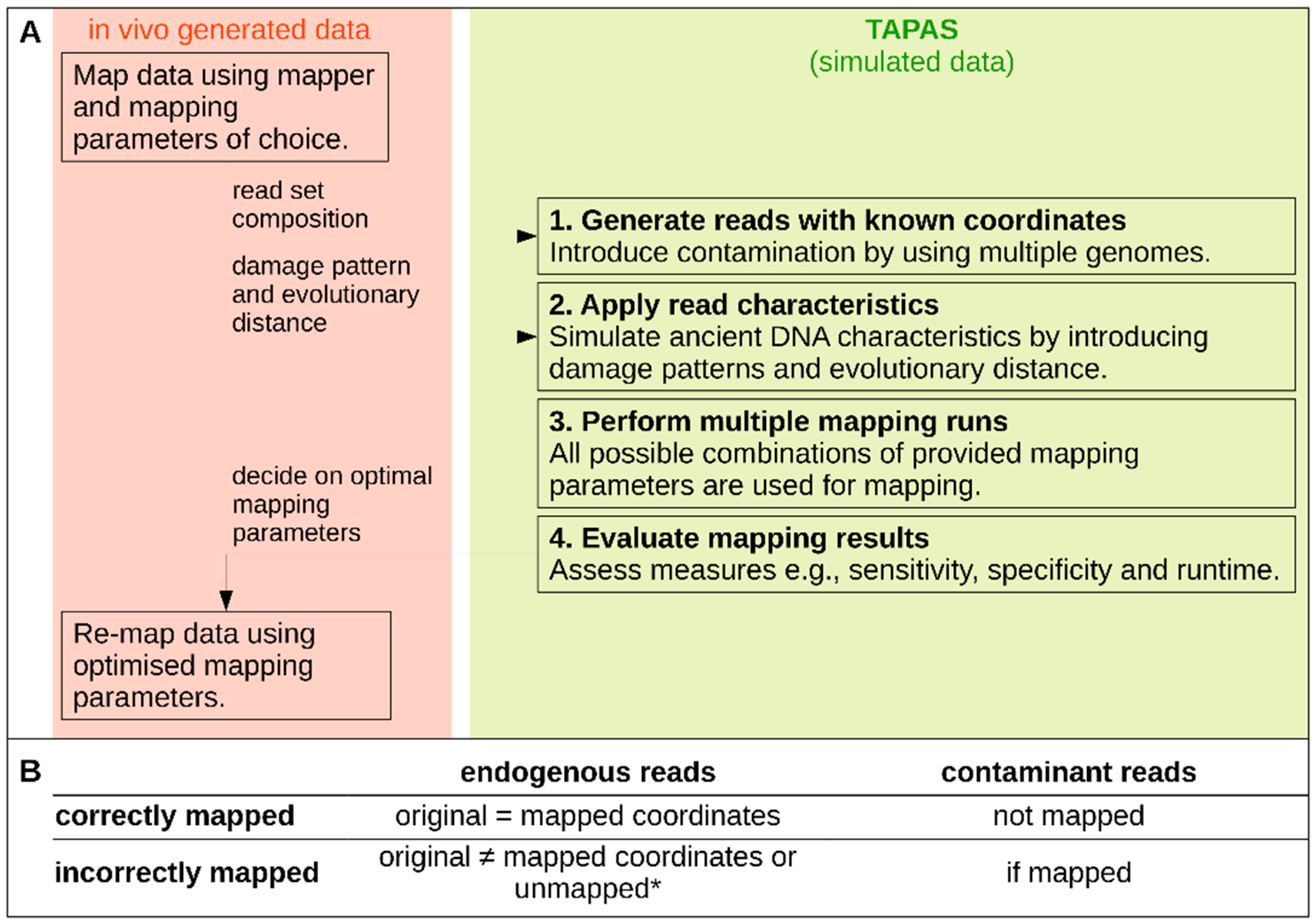
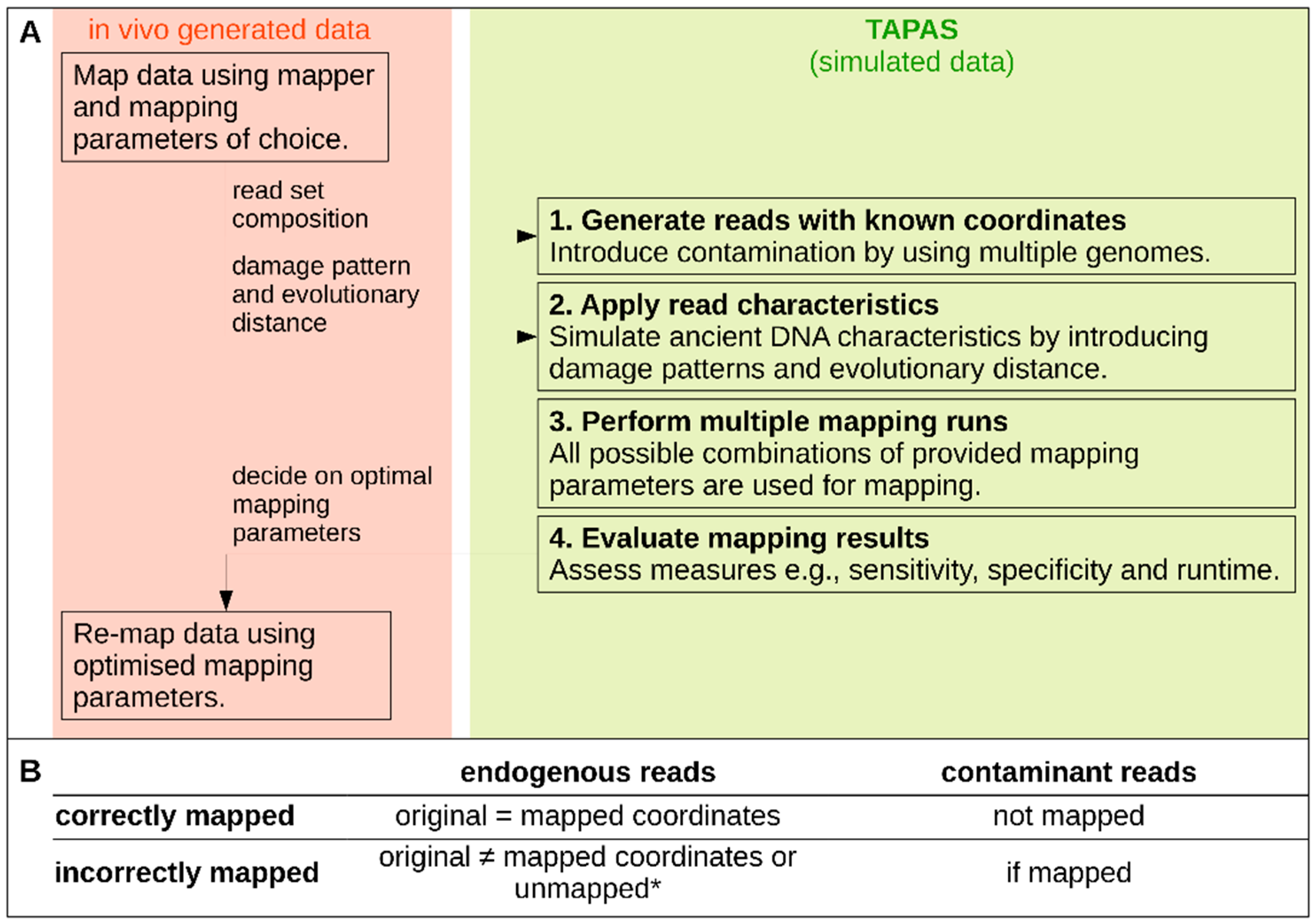
2.1. The TAPAS Tool
2.1.1. Data Simulation
2.1.2. Automated Read Mapping and Evaluation
2.2. In Vivo Generated Linsang Dataset
2.3. Published Bison Dataset
3. Results
3.1. In Vivo Linsang Data
3.2. TAPAS
3.2.1. Simulated Data Composition
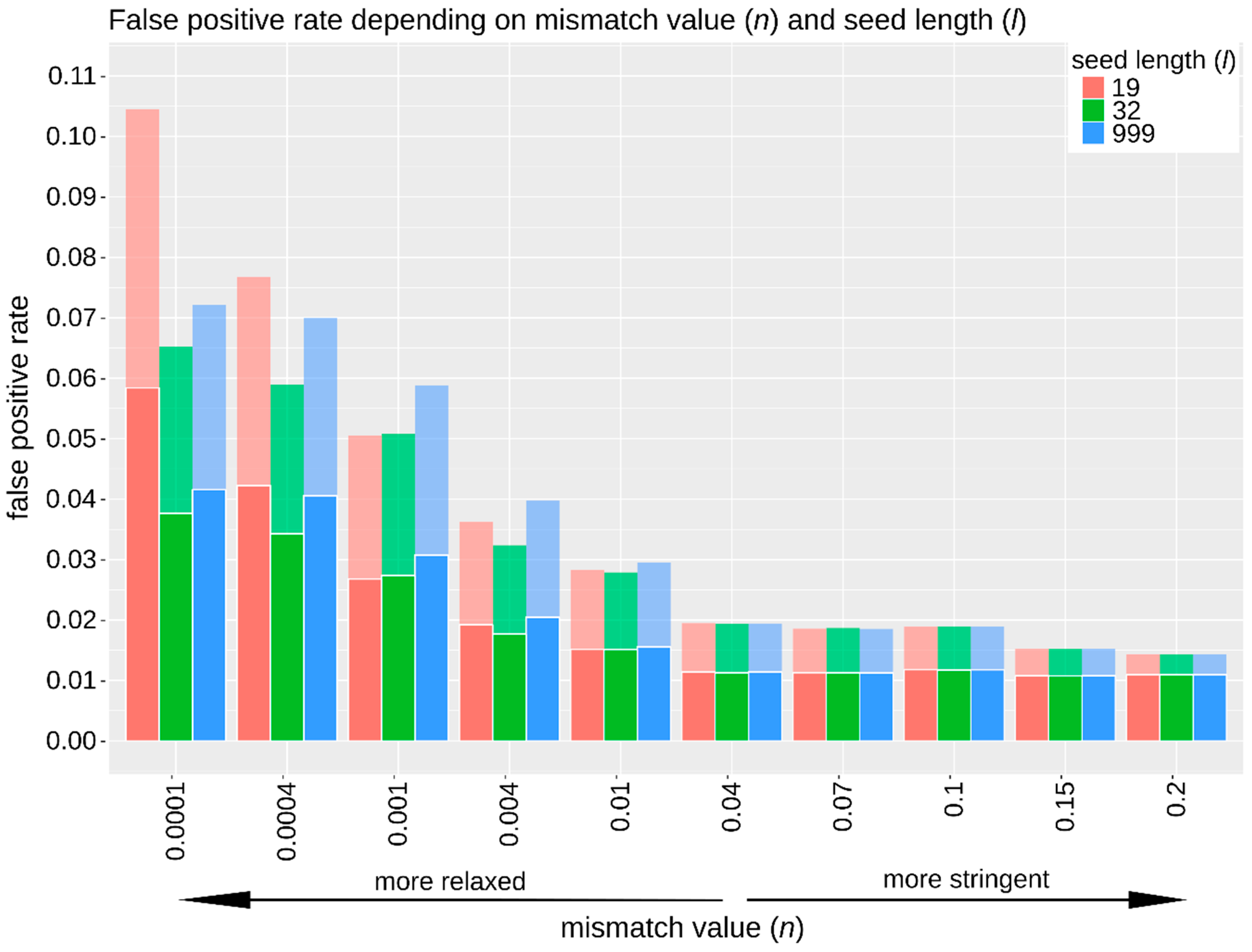
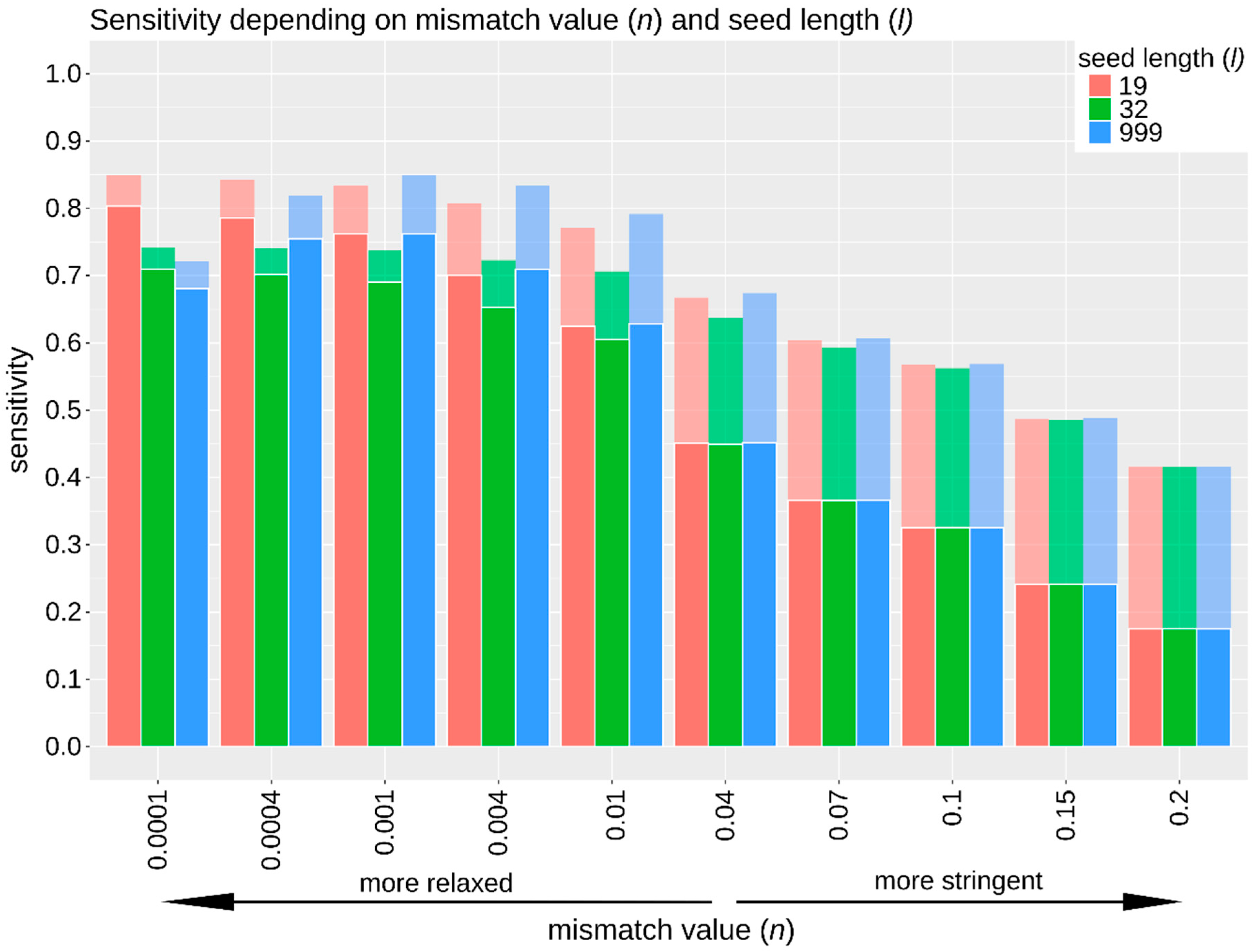
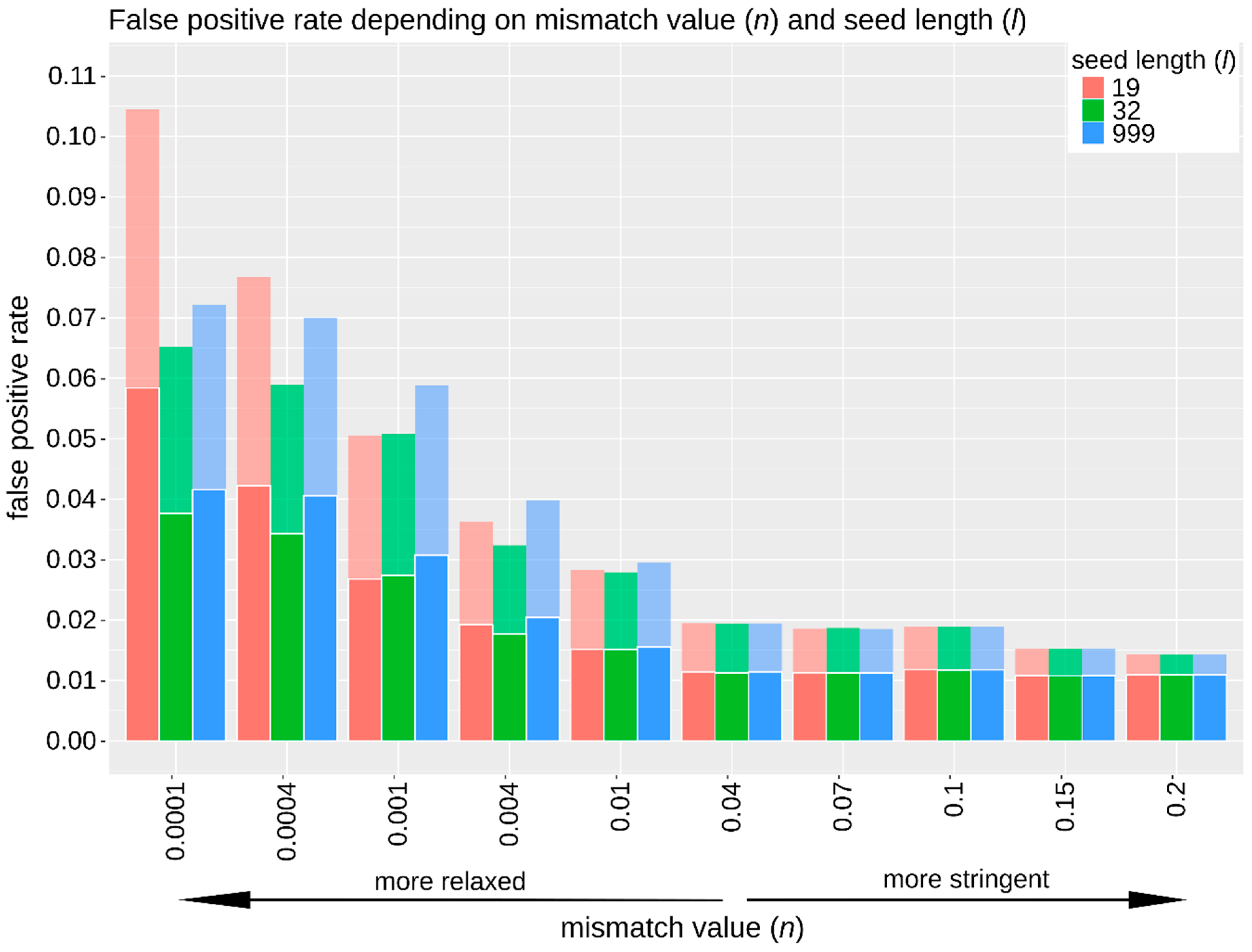
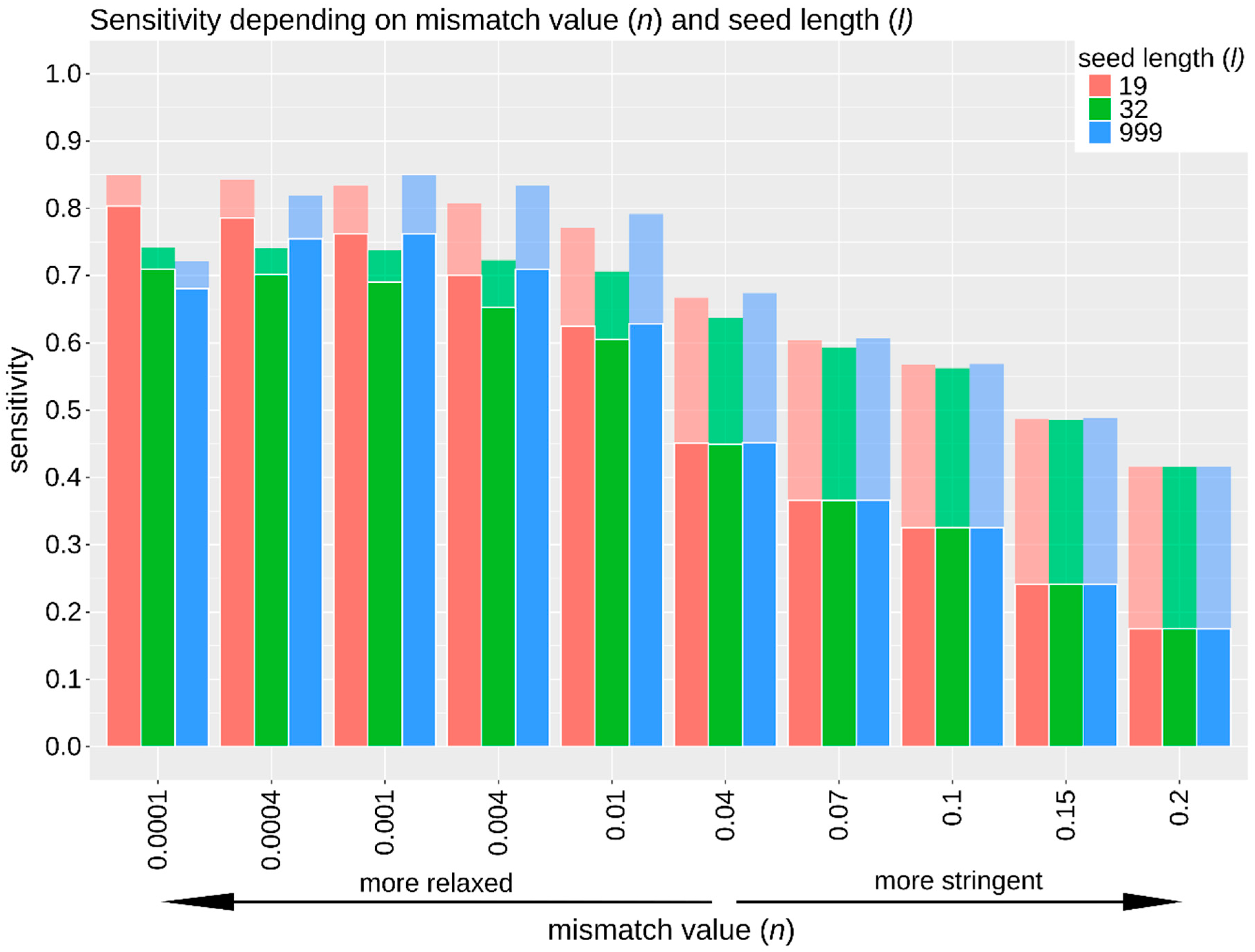
3.2.2. Sensitivity, Specificity, and False Positive Rate
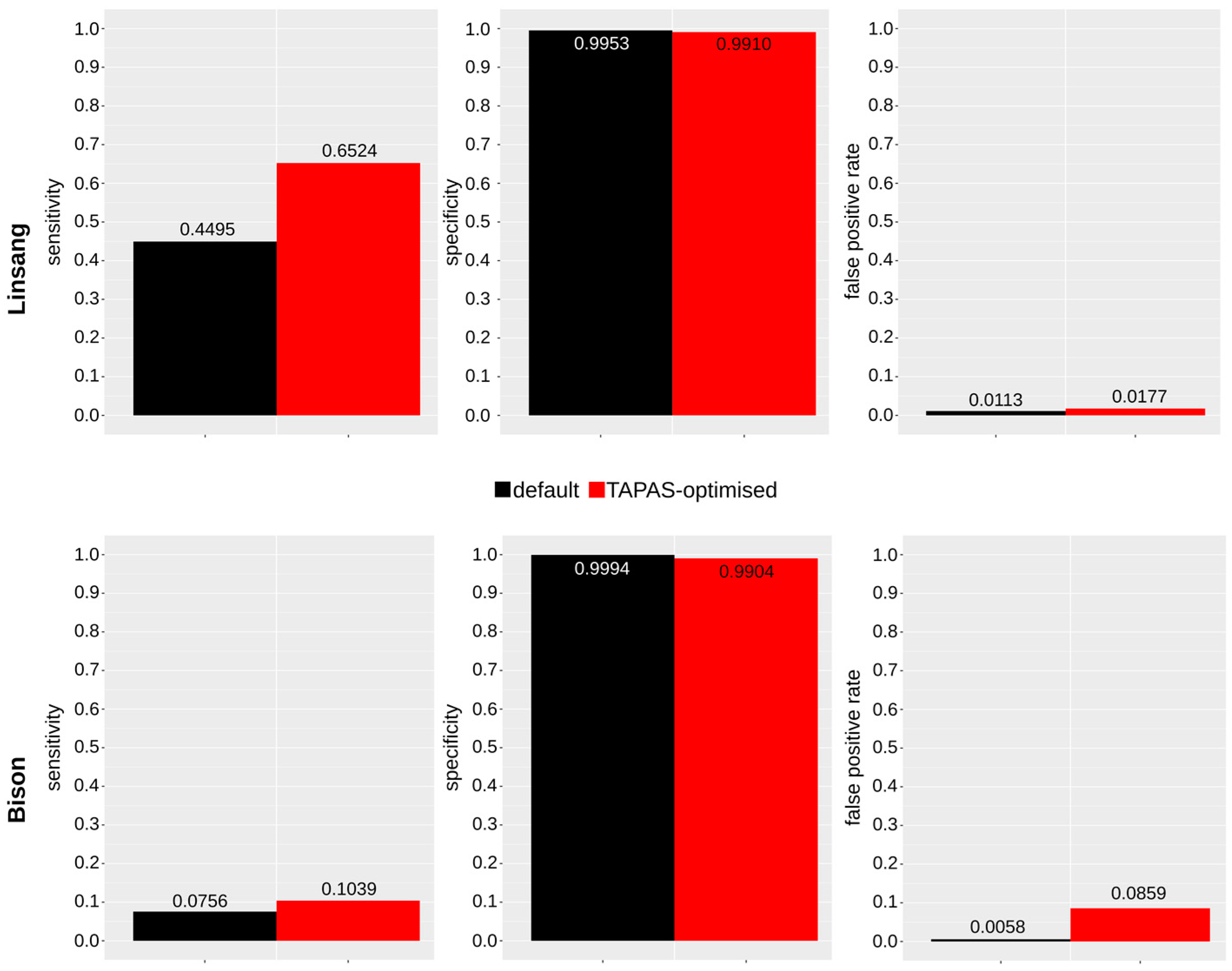
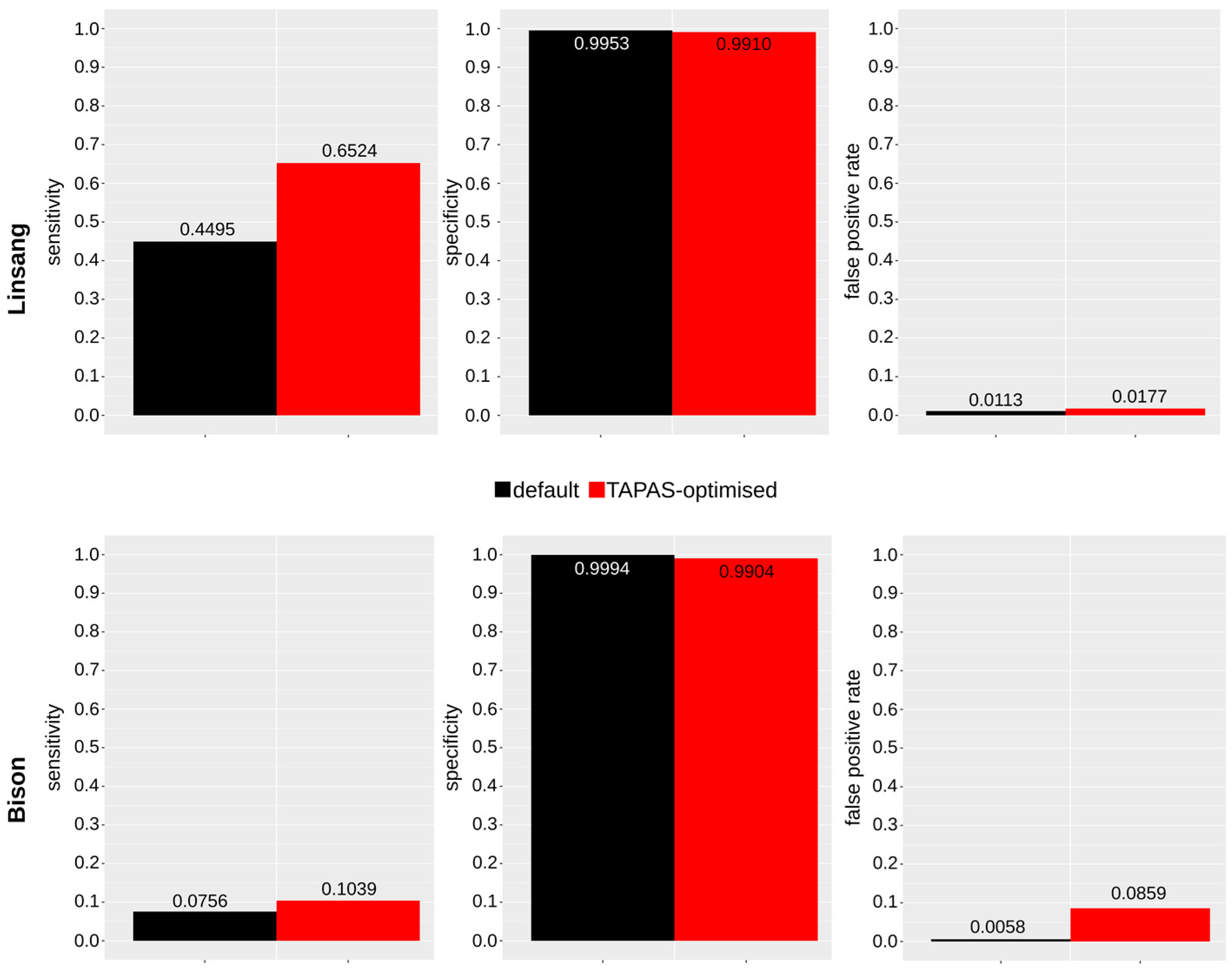
3.2.3. TAPAS-Optimized Parameters
3.3. Published Bison Data
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Data Availability
Conflicts of Interest
References
- Schubert, M.; Ginolhac, A.; Lindgreen, S.; Thompson, J.F.; AL-Rasheid, K.A.; Willerslev, E.; Krogh, A.; Orlando, L. Improving ancient DNA read mapping against modern reference genomes. BMC Genom. 2012, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Ermini, L.; Sarkissian, C.D.; Jónsson, H.; Ginolhac, A.; Schaefer, R.; Martin, M.D.; Fernández, R.; Kircher, M.; McCue, M.; et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014, 9, 1056–1082. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Malaspinas, A.-S.; Krause, J.; Briggs, A.W.; Johnson, P.L.F.; Uhler, C.; Meyer, M.; Good, J.M.; Maricic, T.; Stenzel, U.; et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 2008, 134, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, A.; Jäger, G.; Herbig, A.; Seitz, A.; Kniep, C.; Krause, J.; Nieselt, K. EAGER: Efficient ancient genome reconstruction. Genome Biol. 2016, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Key, F.M.; Posth, C.; Krause, J.; Herbig, A.; Bos, K.I. Mining Metagenomic Data Sets for Ancient DNA: Recommended Protocols for Authentication. Trends Genet. 2017, 33, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.A.; Rung, J.; Brazma, A.; Marioni, J.C. Tools for mapping high-throughput sequencing data. Bioinformatics 2012, 28, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.; Rescheneder, P.; Schatz, M.C.; von Haeseler, A.; Sedlazeck, F.J. Teaser: Individualized benchmarking and optimization of read mapping results for NGS data. Genome Biol. 2015, 16, 235. [Google Scholar] [CrossRef] [PubMed]
- Holtgrewe, M.; Emde, A.-K.; Weese, D.; Reinert, K. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinform. 2011, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Hatem, A.; Bozdağ, D.; Toland, A.E.; Çatalyürek, Ü.V. Benchmarking short sequence mapping tools. BMC Bioinform. 2013, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Renaud, G.; Hanghøj, K.; Willeslev, E.; Orlando, L. Gargammel: A sequence simulator for ancient DNA. Bioinformatics 2016, 33, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Westbury, M.; Baleka, S.; Barlow, A.; Hartmann, S.; Paijmans, J.L.A.; Kramarz, A.; Forasiepi, A.M.; Bond, M.; Gelfo, J.N.; Reguero, M.A.; et al. A mitogenomic timetree for Darwin’s enigmatic South American mammal Macrauchenia patachonica. Nat. Commun. 2017, 8, 15951. [Google Scholar] [CrossRef] [PubMed]
- Kehlmaier, C.; Barlow, A.; Hastings, A.K.; Vamberger, M.; Paijmans, J.L.A.; Steadman, D.W.; Albury, N.A.; Franz, R.; Hofreiter, M.; Fritz, U. Tropical ancient DNA reveals relationships of the extinct Bahamian giant tortoise Chelonoidis alburyorum. Proc. R Soc. B 2017, 284, 20162235. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; Stenzel, U.; Hofreiter, M.; Pääbo, S.; Kelso, J.; Green, R.E. Computational challenges in the analysis of ancient DNA. Genome Biol. 2010, 11, R47. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.A.; Shanks, O.; Hofreiter, M.; Kreuz, E.; Hodges, L.; Ream, W.; Wayne, R.K.; Fleischer, R.C. Animal DNA in PCR reagents plagues ancient DNA research. J. Archaeol. Sci. Rep. 2007, 34, 1361–1366. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.D.; Kim, S.K. Inconsistencies in Neanderthal genomic DNA sequences. PLoS Genet. 2007, 3, e175. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Briggs, A.W.; Krause, J.; Prüfer, K.; Burbano, H.A.; Siebauer, M.; Lachmann, M.; Pääbo, S. The Neandertal genome and ancient DNA authenticity. EMBO J. 2009, 28, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Highnam, G.; Wang, J.J.; Kusler, D.; Zook, J.; Vijayan, V.; Leibovich, N.; Mittelman, D. An analytical framework for optimizing variant discovery from personal genomes. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Wandeler, P.; Hoeck, P.E.A.; Keller, L.F. Back to the future: Museum specimens in population genetics. Trends Ecol. Evol. 2007, 22, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyakatura, K.; Bininda-Emonds, O.R. Updating the evolutionary history of Carnivora (Mammalia): A new species-level supertree complete with divergence time estimates. BMC Biol. 2012, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.; Clarke, A.C.; Horsburgh, K.A.; Matisoo-Smith, E.A. Setting the stage—Building and working in an ancient DNA laboratory. Ann. Anat. Anat. Anz. 2012, 194, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Paijmans, J.L.A.; Baleka, S.; Henneberger, K.; Taron, U.H.; Trinks, A.; Westbury, M.V.; Barlow, A. Sequencing single-stranded libraries on the Illumina NextSeq 500 platform. arXiv, 2017; arXiv:1711.11004. [Google Scholar]
- Rohland, N.; Siedel, H.; Hofreiter, M. Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens. BioTechniques 2004, 36, 814–816, 818–821. [Google Scholar] [PubMed]
- Gansauge, M.-T.; Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013, 8, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.P.; Hofreiter, M.; Smith, D.; Priest, J.R.; Rohland, N.; Rabeder, G.; Krause, J.; Detter, J.C.; Pääbo, S.; Rubin, E.M. Genomic sequencing of Pleistocene Cave Bears. Science 2005, 309, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Poinar, H.N.; Schwarz, C.; Qi, J.; Shapiro, B.; MacPhee, R.D.E.; Buigues, B.; Tikhonov, A.; Huson, D.H.; Tomsho, L.P.; Auch, A.; et al. Metagenomics to Paleogenomics: Large-Scale Sequencing of Mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M. Analysis of High-Throughput Ancient DNA Sequencing Data. Methods Mol. Biol. 2012, 840, 197–228. [Google Scholar] [PubMed]
- Fortes, G.G.; Grandal-d’Anglade, A.; Kolbe, B.; Fernandes, D.; Meleg, I.N.; García-Vázquez, A.; Pinto-Llona, A.C.; Constantin, S.; de Torres, T.J.; Ortiz, J.E.; et al. Ancient DNA reveals differences in behaviour and sociality between brown bears and extinct cave bears. Mol. Ecol. 2016, 25, 4907–4918. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Węcek, K.; Hartmann, S.; Paijmans, J.L.A.; Taron, U.; Xenikoudakis, G.; Cahill, J.A.; Heintzman, P.D.; Shapiro, B.; Baryshnikov, G.; Bunevich, A.N.; et al. Complex admixture preceded and followed the extinction of Wisent in the wild. Mol. Biol. Evol. 2017, 34, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Bibi, F. A multi-calibrated mitochondrial phylogeny of extant Bovidae (Artiodactyla, Ruminantia) and the importance of the fossil record to systematics. BMC Evol. Biol. 2013, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.P.; Palumbi, S.R. Body size, metabolic rate, generation time, and the molecular clock. Proc. Natl. Acad. Sci. USA 1993, 90, 4087–4091. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; vonHaeseler, A.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Paijmans, J.L.A.; Chang, F.; Wu, X.; Chen, G.; Lei, C.; Yang, X.; Wei, Z.; Bradley, D.G.; Orlando, L.; et al. Morphological and genetic evidence for early Holocene cattle management in northeastern China. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | l | Reads for Mapping | Duplicates (%) | Reads Mapped | Reads Mapped (%) | Mapped bp | Coverage | Median Read Length | Total CPU Time (h) | |
|---|---|---|---|---|---|---|---|---|---|---|
| fold increase | ||||||||||
| in vivo generated data | 0.04 * | 32 * | 6,178,108 | 1.92 | 580,773 | 9.40 | 26,240,720 | 0.011 | 42 | 9.23 |
| 0.004 | 32 * | 6,178,108 | 2.06 | 1,017,849 | 16.48 | 46,032,861 | 0.019 | 42 | 33.50 | |
| 1.1 | 1.8 | 1.8 | 1.8 | 1.8 | 1.0 | 3.6 | ||||
| simulated data | 0.04 * | 32 * | 1,000,000 | 0.78 | 117,379 | 11.74 | 5,802,202 | 0.002 | 44 | 0.10 |
| 0.004 | 32 * | 1,000,000 | 1.08 | 171,099 | 17.11 | 8,567,055 | 0.003 | 44 | 1.32 | |
| 1.4 | 1.5 | 1.5 | 1.5 | 1.5 | 1.0 | 13.5 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taron, U.H.; Lell, M.; Barlow, A.; Paijmans, J.L.A. Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool. Genes 2018, 9, 157. https://doi.org/10.3390/genes9030157
Taron UH, Lell M, Barlow A, Paijmans JLA. Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool. Genes. 2018; 9(3):157. https://doi.org/10.3390/genes9030157
Chicago/Turabian StyleTaron, Ulrike H., Moritz Lell, Axel Barlow, and Johanna L. A. Paijmans. 2018. "Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool" Genes 9, no. 3: 157. https://doi.org/10.3390/genes9030157