The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis


1
University of Texas Health Science Center at Houston School of Dentistry, Houston, TX 77054, USA
2
Department of Diagnostic and Biomedical Sciences, University of Texas Health Science Center at Houston School of Dentistry, Houston, TX 77054, USA
3
Center for Craniofacial Research, University of Texas Health Science Center at Houston School of Dentistry, Houston, TX 77054, USA
4
Pediatric Research Center, University of Texas Health Science Center at Houston McGovern Medical School, Houston, TX 77030, USA
*
Author to whom correspondence should be addressed.
Genes 2018, 9(5), 255; https://doi.org/10.3390/genes9050255
Submission received: 3 April 2018
/
Revised: 1 May 2018
/
Accepted: 9 May 2018
/
Published: 16 May 2018
(This article belongs to the Special Issue DNA Variations in Evolution and Human Diseases)
Abstract
:Despite much progress in understanding the genetics of syndromic tooth agenesis (TA), the causes of the most common, isolated TA remain elusive. Recent studies have identified novel genes and variants contributing to the etiology of TA, and revealed new pathways in which tooth development genes belong. Further, the use of new research approaches including next-generation sequencing has provided increased evidence supporting an oligogenic inheritance model for TA, and may explain the phenotypic variability of the condition. In this review, we present current knowledge about the genetic mechanisms underlying syndromic and isolated TA in humans, and highlight the value of incorporating next-generation sequencing approaches to identify causative and/or modifier genes that contribute to the etiology of TA.
1. Introduction
Tooth development requires a sequential and reciprocal series of signaling interactions between the oral epithelium and the neural crest-derived mesenchyme, which are under strict genetic control by a number of signaling molecules and their downstream signaling pathways. During these stages, the continuous interplay of inductive signals between epithelia and mesenchyme in a precisely organized manner results in the formation of distinct and highly specialized structures, such as incisor, canine, premolar and molar teeth [1]. Disturbances during any step of tooth development may affect growth, differentiation and pattern formation [2].
Tooth agenesis (TA) is the congenital absence of one or more permanent teeth, and results from disturbances during early stages of tooth development. TA is recognized as the most common abnormality of dental development and may appear as a characteristic feature of ≈150 syndromes, although it is more frequent as an isolated trait that may appear sporadically or segregate in families [3,4]. Autosomal dominant, autosomal recessive, or X-linked patterns of inheritance have been described for TA, with considerable variation in penetrance and expressivity. Based on the number of missing teeth other than third molars, TA is referred to as hypodontia (≤5 teeth missing) or oligodontia (≥6 teeth missing). Anodontia refers to the absence of all permanent teeth and is associated with syndromic forms of TA [5].
There are large differences in the prevalence of TA, whether it refers to hypodontia or oligodontia, and also among genders and different racial populations. In general, population studies have revealed that the prevalence of isolated hypodontia ranges from 3–10%, while oligodontia is less common with a reported prevalence of 0.1–0.5%, excluding third molars. Upon inclusion of third molars, the prevalence rises to 25% [4,6]. The most commonly missing teeth are permanent mandibular second premolars, maxillary lateral incisors, and maxillary second premolars [3]. Females are more affected than males in a 3:2 ratio [5].
TA may be occasionally caused by exogenous factors, such as infections, trauma, chemotherapy, or radiotherapy, however, the majority of cases are due to genetic factors [7]. Studies of odontogenesis in mice have elegantly shown that tooth development is under strict genetic control, which determines the positions, numbers and shapes of different teeth [8]. Over many years, studies using transgenic animals provided functional data showing that disruption of genes in the bone morphogenetic protein (BMP), fibroblast growth factor (FGF), sonic hedgehog (SHH), and wingless-related integration site (WNT) signaling pathways resulted in severe abnormalities of tooth development ranging from defects in tooth patterning to complete arrest of tooth development [1,4].
In humans, mutations in several genes have been reported as causal for TA. However, known mutations explain a restricted number of cases and much remains to be learned about the underlying molecular mechanisms in TA [4]. Epigenetic regulation affecting a network of interconnected signaling pathways that included tooth development was also suggested to play a role in the etiology of TA [9]. Recently, new technologies such as next-generation sequencing have proven to be valuable tools in the identification of novel TA genes and variants, and begun to elucidate the genetic defects responsible for this condition [10,11].
The purpose of this review is to present an overview of genes and pathways identified as having a role in human TA, and discuss recent findings from next-generation sequencing studies. We also present new perspectives on the potential molecular mechanisms underlying human TA that may bring new possibilities for future prevention and treatment.
2. Genetic Basis of Tooth Agenesis
2.1. Syndromes Associated with Tooth Agenesis
More than 150 syndromes include TA as a phenotype. These include mostly oral-facial cleft syndromes and ectodermal dysplasia syndromes [12,13]. Teeth share developmental mechanisms with other ectodermal organs in terms of timing and the cellular processes involved, therefore genes involved in odontogenesis may play a role in other developmental processes [12,14]. A summary of the syndromic forms of TA and their etiologic genes is presented in Table 1.
2.1.1. Ectodermal Dysplasia Syndromes
Ectodermal dysplasias (ED) comprise a group of disorders characterized by a combination of findings involving defects in the skin, hair, nails, teeth, exocrine and sebaceous glands. Distinct types of ED have been reported, caused by mutations in different genes. X-linked recessive hypohidrotic (HED) is the most common ED and caused by mutations in the EDA (ectodysplasin A) gene. Currently, more than 200 mutations in EDA have been found in individuals with HED. The EDA protein is a type-II trimeric transmembrane protein belonging to the tumor necrosis factor (TNF) ligand superfamily which functions as a signaling molecule during epithelial morphogenesis [98]. Upon stimulation, EDA binds to its receptor, EDAR (ectodysplasin A receptor), which in turn binds to the adaptor EDARADD (EDAR-associated death domain) for activation of downstream target proteins. Mutations in EDAR and EDARADD cause autosomal dominant and recessive forms of HED [4,7]. In addition to variable involvement of teeth in distinct forms of ED, minor ectodermal anomalies are often observed.
Witkop syndrome is a rare autosomal dominant ED involving the teeth and nails, caused by mutations in the MSX1 (muscle segment homeobox, homolog 1) gene. Although a few reported cases have sparse or fine hair, most affected individuals have normal hair and sweat glands [99]. MSX1 is a homeobox gene that belongs to a family of transcription factors that are expressed in overlapping patterns at multiple sites of tissue interactions during vertebrate development [2]. In mice, MSX1 is essential for initiation of the tooth germ during the bud stage and then promotes the odontogenic potential of the dental mesenchyme [2]. Mice lacking MSX1 protein function present cleft palate, deficient mandibular and maxillary alveolar bones, and arrest of tooth development [100]. Further, MSX1 is a direct downstream target of WNT/ß-catenin signaling during craniofacial development and regulates the expression of additional genes within this pathway, including BMP2 (bone morphogenetic protein 2) and BMP4 (bone morphogenetic protein 4) [4]. Over 20 MSX1 mutations have been reported in association with both syndromic and nonsyndromic TA phenotypes in humans.
Odontoonychodermal dysplasia (OODD) and Schopf–Schulz–Passarge syndrome (SSPS) are autosomal recessive disorders caused by homozygous or compound heterozygous missense mutations in WNT10A (wingless-type MMTV integration site family, member 10A). Affected individuals have dry hair, severe TA, smooth tongue, palmoplantar keratosis, and dystrophic nails [93,94]. WNT10A is thought to function through the canonical Wnt-ß catenin signaling pathway and activates target genes in the nucleus through binding to LEF1 transcription factor [101]. During tooth development, continuous Wnt-β catenin signaling in the dental epithelium and mesenchyme is required for tooth formation and morphogenesis [102]. During mouse tooth development, WNT10A is first detected in the enamel knot and then shifting to the secondary enamel knot to the underlying mesenchyme and developing odontoblasts [103,104]. In WNT10A null mice, however, supernumerary teeth and altered molar crown morphology are observed, in contrast to the TA phenotype in humans [105]. Numerous reports have shown the involvement of WNT10A mutations in OODD and SSPS, and in a wide spectrum of autosomal recessive ectodermal dysplasias [93,94,106,107] as well as in isolated TA cases [11,108,109,110].
A homozygous missense mutation (c.626T > C, p.Phe209Ser) in KREMEN1 (kringle containing transmembrane protein 1), a negative regulator of the Wnt pathway, was identified in four consanguineous Palestinian families with ED. This variant in its heterozygous state was also identified in 6 out of 39 unaffected control individuals [64]. Recently, two additional variants in KREMEN1 (c.146C > G and c.773_778del) were identified as pathogenic in two Turkish families with suspected ED [11]. KREMEN1 encodes a kringle domain-containing transmembrane protein that binds to DKK1, creating a DKK1-Kremen-LRP6 ligand-receptor complex critical for Wnt signaling [111]. While in this complex, Kremen triggers the internalization of LRP6 inhibiting Wnt signaling. In the absence of DKK1, however, Kremen can increase Wnt signaling through LRP6 binding. Targeted disruption of the Wnt regulator Kremen in mice induces limb defects and high bone density but no other obvious phenotypes [111].
2.1.2. Oral-Facial Cleft Syndromes
Van der Woude syndrome (VWS) is an autosomal dominant disorder and one of the most common clefting syndromes. Affected individuals present with cleft lip with or without cleft palate (CL/P) and a range of associated features including lower lip pits and TA, which is present in ≈70% of the cases. VWS is caused by mutations in the IRF6 (interferon regulatory factor 6) gene that encodes a transcription factor highly expressed during craniofacial development and a regulator of keratinocyte proliferation and differentiation [57]. Mice deficient for IRF6 have abnormal skin, limb and craniofacial development, resultant from a primary defect in keratinocyte differentiation and proliferation. Furthermore, mice homozygous for the IRF6 null allele have a cleft palate which seems to be caused by a defect in elevation, either as a primary defect or secondary to crowding of the craniofacial structures owing to the constrictive action of the skin or oral adhesions [112]. IRF6 mutations are recognized as primary genetic causes of isolated and syndromic CL/P [58,113,114].
Cleft lip/palate-ectodermal dysplasia syndrome is a rare, autosomal recessive disorder caused by homozygous loss-of-function mutations of the PVRL1 (poliovirus receptor-like 1) gene encoding nectin-1 [79]. Nectin-1 is a cell–cell adhesion molecule that is important for the initial step in the formation of adherens junctions and tight junctions; it is expressed in keratinocytes, neurons, and the developing face and palate. Clinical manifestations comprise a unique facial appearance with cleft lip/palate, ectodermal dysplasia, cutaneous syndactyly of the fingers and/or toes, hypodontia and in some cases, mental retardation [80].
2.1.3. Axenfeld–Rieger Syndrome
Axenfeld–Rieger syndrome (MIM #180500) is an autosomal dominant disorder characterized by abnormal development of the anterior segment of the eye, and results in blindness from glaucoma in approximately 50% of affected individuals [115]. Systemic anomalies are associated and include failure of involution of the umbilicus, hypospadias and dental anomalies ranging from microdontia, TA, and tooth malformations [116]. The cause of Axenfeld–Rieger syndrome has been attributed to mutations in the PITX2 (paired like homeodomain 2) gene, which encodes the earliest transcription marker of tooth development and is expressed in the oral epithelium and dental placode [117]. PITX2 null mice have tooth development arrested at E12.5 [118]. Additionally, PITX2 has been shown to play a critical role in establishing left–right asymmetry in vertebrates [119].
2.1.4. Familial Adenomatous Polyposis Syndrome
Familial adenomatous polyposis (FAP) is an autosomal dominant condition characterized by the development of multiple adenomatous polyps in the colon and rectum with high risk of subsequent malignant transformation. In addition, extracolonic changes occur in many affected subjects. These include epidermoid cysts, desmoid tumours, congenital hypertrophy of retinal pigment epithelium (CHRPE), osseous changes in the jaws and skeleton, and dental anomalies. FAP results from germline mutations in the APC (adenomatous polyposis coli) gene on chromosome 5q21 [120]. Approximately 17% of individuals with APC gene mutations have dental anomalies, particularly supernumerary teeth and compound odontomas, although cases of TA as well as impacted teeth have also been reported. Importantly, in addition to the established association between certain dental anomalies and FAP, an association between TA and genetic predisposition to colon cancer was suggested [22,121,122].
2.1.5. Oligodontia–Colorectal Cancer Syndrome
A germline nonsense mutation in AXIN2 (c.1996C > T, p.Arg656*) was identified as the likely cause of autosomal dominant oligodontia (severe TA) and colorectal cancer segregating in a large, four-generation Finnish family. Eleven members of the family lacked at least eight permanent teeth, two of whom developed only three permanent teeth. Colorectal cancer or precancerous lesions of variable types were found in eight of the patients with oligodontia [22]. In this same study, a second frameshift mutation leading to a heterozygous 1-bp insertion (c.1994_1995insG, p.706*) in exon 7 of the AXIN2 gene was identified in an unrelated individual with oligodontia, suggesting that this gene may contribute to both tooth development and tumor development later in life [22]. A recent study showed that variations in colorectal cancer-related genes (ATF1 and DUSP10) were significantly associated with TA (albeit in isolated cases); further, Atf1 and Dusp10 expression was detected in the mouse developing teeth from early bud stages to the formation of the complete tooth, suggesting a potential role for these genes and their encoded proteins in toothdevelopment [123]. Taken together, these findings continue to support a potential overlap in the molecular etiology of TA and colorectal cancer, although no cause-effect relationship can yet be established and more research is warranted in this area.
2.2. Isolated (Nonsyndromic) Tooth Agenesis
Syndromic forms of TA and genes implicated during normal tooth development in animal models have provided important clues to identify candidate genes for isolated TA in humans. To date, numerous genes have been proposed as etiologic for isolated TA (Table 2).
2.2.1. MSX1
Mutations in MSX1 were the first to be described in individuals with isolated TA [152]. Since then, over 20 mutations in MSX1 have been reported in association with isolated TA, most of which are nonsense or missense mutations located in the homeobox domain, and which suggest that haploinsufficiency of MSX1 underlies TA phenotypes [153] (Figure 1). Mutations in the homeobox domain disrupt DNA-binding and preferentially cause isolated TA, meanwhile variants in the natively unfolded N-terminal part of the protein generally cause oral-facial clefts. These observations suggest that the effect of MSX1 mutations are directly related to the affected protein domain. MSX1-associated TA typically includes missing maxillary and mandibular second premolars and maxillary first premolars.
2.2.2. PAX9
PAX9 belongs to the paired box (PAX) family of transcription factors that are essential for normal development in several multicellular organisms. In addition to MSX1, PAX9 has also long been implicated in isolated TA phenotypes and is one of the most widely studied genes in odontogenesis [4]. PAX9 is expressed in the presumptive dental mesenchyme to activate signals and initiate tooth development. Absence of PAX9 in mice results in arrest of tooth development at the bud stage [154,155].
To date, more than 30 variations in PAX9 have been described in association with TA, most of which are insertions/deletions or missense mutations located in exon 2 of the gene and affecting the paired domain of the PAX9 protein [156] (Figure 1). The presence of PAX9 variation has primarily been associated with agenesis of permanent second molars, followed by second premolars; a few reports of agenesis of anterior teeth also exist [7,156]. In general, the severity of the TA phenotype is associated with the type of mutation and its impact on PAX9 function. Individuals with nonsense/frameshift mutations present with a more severe phenotype when compared to those with missense mutations. In TA, known PAX9 mutations are heterozygous and show autosomal dominant inheritance, indicating that haploinsufficiency is likely contributing to the phenotype. Smaller tooth crown dimensions throughout the dentition have also been reported in TA patients with PAX9 mutations [157].
2.2.3. AXIN2
Rare and common variants in AXIN2 have been found in association with isolated TA, presenting a mixed pattern of affected teeth [98,124,125]. Agenesis of molars, lower incisors and upper lateral incisors have been described in TA individuals, and the absence of at least one incisor is frequently reported [4,98]. Five AXIN2 mutations have been widely reported in the literature, including four missense (c.956 + 16A > G; p.Pro50Ser, c.2051C > T; p.Ala684Val, c.2062C > T; p.Leu688Leu, and c.2272G > A; p.Ala758Thr), and one frameshift (c.1994insG; p.Asn666GlyfsX41). The presence of this frameshift mutation was associated with more missing teeth than missense mutations in all affected individuals [98,158].
2.2.4. WNT10A
WNT10A has been the focus of many genetic studies of TA. Over 50 heterozygous, homozygous as well as compound heterozygous variants in WNT10A have been identified in 15.8% of TA patients with 1 to 3 missing teeth, and in ≈52% of patients with >4 missing teeth [109]. Recent genotype–phenotype correlations have provided insights for the role of WNT10A in TA. Overall, WNT10A compound heterozygous mutations have been found in association with severe TA and a larger number of missing teeth in comparison to individuals with a single variant [108,109,110,148]. While there are no preferential patterns of missing teeth in individuals with WNT10A variants, the absence of maxillary and mandibular molars, as well as mandibular incisors is often reported [149]. Of note, heterozygous WNT10A variants have also been identified in unaffected individuals in TA families, as well as in unrelated control individuals with no TA or family history of TA [108,109]. It has been estimated that approximately 41% of individuals showing a single heterozygous variant in WNT10A will not have TA [148].
Figure 1 shows WNT10A variants identified in TA patients. A few WNT10A variants have been suggested to be common ‘hotspots’ for mutations in specific populations. For example, the c.637G > A (p.Gly213Ser) variant has been found more frequently in Asian populations [108,149], meanwhile the c.682T > A (p.Phe228Ile) variant has been widely reported in homozygous or heterozygous forms in Caucasian individuals with TA, but also in normal controls at a frequency of 2.3% [109]. The Phe228Ile variant is the most commonly found variant, and often described in combination with additional variants in WNT10A or in other genes [159,160]. These findings support an oligogenic inheritance model for TA as discussed later in this review.
2.2.5. LRP6
LRP6 (LDL receptor related protein 6) is a co-receptor in the Wnt/β-catenin pathway and has been recently reported to contribute to isolated TA in different studies [11,136,137]. Six variants, including a nonsense variant (c.1779dupT, p.Glu594*), two insertion (c.2224_2225dupTT, p.Leu742Phefs*7 and c.1144_1145dupAG, p.Ala383Glyfs*8) and a splice-site (c.3607 + 3_6del, p.?) variant resulting in a truncated mRNA product, as well as a missense variant (c.56C > T, p.Ala19Val) were found in individuals with sporadic TA and/or segregating with TA in families [11,136,137]. In mice, LRP6 expression was noted in the tooth follicle and inner enamel epithelium [137], while homozygous deletion of LRP6 led to severe skeletal abnormalities and lethality [161].
2.2.6. Other Genes Recently Implicated in TA
Mutations in GREM2, which encodes GREMLIN2, were identified in 7 patients with TA (hypodontia) and additional malformations, including taurodontism, sparse and slow-growing hair and dry and itchy skin [132]. GREMLIN2 is known to regulate BMPs in embryonic development. Specifically, BMP4 has an important role in tooth development and its knockdown resulted in the arrest of tooth development in mice [8]. Interestingly, GREM2 knockout mice have small and malformed teeth but do not have tooth development arrested. However, these findings suggest a potential role for GREM2 during tooth development [162]. Three missense mutations in GREM2 (p.Ala13Val, p.Glu136Asp and p.Gln76Glu) were identified as pathogenic in individuals with isolated TA and have not been reported in association with other structural malformations. GREM2 mutations exhibit variable expressivity even within the same families [132].
EDA, EDAR and EDARADD have also been suggested to contribute to isolated TA [98,129,163]. In a genotype–phenotype correlation study, all EDA mutations in individuals with isolated TA were missense mutations and most likely to be located in the TNF domain [163].
Another WNT pathway gene, WNT10B (wingless-type MMTV integration site family, member 10B), has also been implicated in isolated TA, albeit mostly in families from China and Thailand. Three heterozygous missense mutations (c.632G > A, p.Arg211Gln; c.569C > G, p.Pro190Arg; and c.851T > G, p.Phe284Cys) and one nonsense mutation (c.786G > A, p.Trp262*) in WNT10B were identified in Chinese individuals with TA, especially those missing the upper lateral incisors [151]. More recently, two additional heterozygous missense mutations (c.475G > C, p.Ala159Pro and c.1052G > A, p.Arg351His) were identified in five Thai families, and associated with isolated TA and other dental anomalies including microdontia and taurodontism [150].
A homozygous missense variant c.1312C > T (p.Arg438Cys) in ANTXR1 (anthrax toxin receptor 1) was identified in association with TA (oligodontia) in a Turkish family [10]. Homozygous and biallelic variants in ANTXR1 have been associated with Growth retardation, Alopecia, Pseudoanodontia, and Optic atrophy (GAPO) syndrome, characterized by delayed growth, alopecia, failure of tooth eruption, and optic atrophy segregating as an AR trait [18,19,164]. Targeted disruption of Antxr1 in mice resulted in viable mice without major structural defects, although dental overgrowth, incisor misalignment, and dental dysplasia were observed, due to an accumulation of extracellular matrix in various tissues [165]. Antxr1 expression was detected in the epithelium of developing tongue, maxillary and mandibular processes, as well as in the dental epithelium and mesenchyme at early stages of tooth development. At later stages, Antxr1 expression was noted in the epithelium of the enamel organ and in the dental papilla, and then shifted to the polarized layer of ameloblasts and differentiating odontoblasts. ANTXR1 is a tumor-specific endothelial marker implicated in colorectal cancer, and upregulated in tumor angiogenesis [166,167,168]. Previously, Lammi et al. [22] showed that variants in the tumor suppressor gene AXIN2 segregated in a family with severe TA (oligodontia) and colorectal cancer, and suggested that TA and colorectal cancer may have a common genetic etiology. Numerous studies have since reported on variations in cancer-related genes in association with TA [123,169,170]. These findings highlight the complex nature of TA and emphasize the need to consider modifier genes and/or gene-gene interactions in studies of this condition.
A recent genome-wide association study (GWAS) that included over 1900 TA cases and 330,000 controls of European ancestry identified 4 novel risk variants that associate with TA, and 5 that associate with a combined phenotype of TA plus oral-facial clefts [171]. Dental anomalies are frequent findings in children with oral-facial clefts, and cleft subphenotypes have been proposed based on the pattern of the associated dental anomalies [172]. Of the 9 variants found, 5 were located in or close to Wnt pathway genes that have been implicated in tooth development and/or development of other ectodermal structures (EDA, EDAR, FOXI3, FORXP1 and LEF1), and 4 were located in or close to genes that have not been implicated in TA or tooth development (ASCL5/CACNA1S, ARHGAP15, NOL11 and FAM49A). In addition, two known variants in WNT10A (p.Phe228Ile and p.Cys107*) were also found to be significantly associated with TA in this GWAS [171].
3. Monogenic vs. Oligogenic Inheritance Models
In recent years, oligogenic inheritance and multi-locus variation models have been proposed for a number of Mendelian diseases, further establishing the concept of mutational load in human genetic disease [173]. For TA, evidence for oligogenic inheritance is emerging, supported by the findings of recent whole exome sequencing studies and/or direct sequencing studies with more than one candidate gene [10,11].
Digenic mutations in MSX1 and PAX9 had been reported as associated with a more severe TA phenotype (15–17 missing teeth) [142], and the interactions between these genes had begun to be elucidated. Studies have shown that PAX9 interacts with MSX1 to synergistically activate the expression of downstream tooth development genes, i.e., BMP4, which is essential for proper tooth morphogenesis [2]. The presence of digenic mutations in these genes might abolish their interactions and thus lead to more severe TA phenotypes [142].
More recently, biallelic or heterozygous genotypes of WNT10A were found in TA patients who also presented homozygous or heterozygous genotypes of EDA, EDAR or EDARADD, suggesting the combined phenotypic effects of alleles in distinct genes as contributing to TA [174]. Additionally, compound heterozygous mutations in WNT10A (IVS2 + 1G > A and c.637G > A) were identified segregating together with a missense heterozygous variant in GREM2 (c.38C > T) in a patient with TA of maxillary permanent canines [132].
Additional heterozygous splicing mutations in DKK1 (dickkopf WNT signaling pathway inhibitor 1; c.548-4G > T) and in COL17A1 (collagen type XVII alpha 1 chain; c.3277 + 3G > C), and a heterozygous missense variant in LAMA3 (laminin subunit alpha 3; c.2798G > T) were identified segregating with TA in one consanguineous Turkish family [11]. Pathogenic mutations in these genes had not yet been identified in individuals with TA, although they can be considered biologically plausible candidate genes due to their biological roles and/or disease-associated phenotypes. DKK1 encodes a high-affinity dickkopf homolog 1 transmembrane receptor that cooperates with LRP6 to block Wnt signaling during development and other cellular processes [175]. In mice, DKK1 is expressed in the dental mesenchyme, odontoblasts and osteoblasts, and its ectopic expression in the oral epithelia of transgenic mouse embryos resulted in blocked epithelial and mesenchymal signaling leading to arrest of tooth development at the early bud stage [176]. A common single-nucleotide polymorphism in DKK1 (rs11001553) was previously associated with isolated TA in a Chinese Han population [126].
Mutations in LAMA3 cause junctional epidermolysis bullosa (OMIM #226650), an autosomal recessive skin disorder characterized by the presence of multiple blisters and erosions, dystrophic nails, enamel hypoplasia and hypodontia [177]. Further, targeted disruption of LAMA3 in mice resulted in defects of ameloblast differentiation [178]. Variations in COL17A1 have also been described in epidermolysis bullosa patients with enamel defects [179].
A homozygous variant (c.-387delC > G) in the 5’ UTR of the PITX2 gene, described above as etiologic for Axenfeld–Rieger syndrome, and a homozygous missense variant in BMP4 (c.T455C, p.Val152Ala) were identified segregating with isolated TA in two siblings from an Italian family [180].
The finding of likely pathogenic alleles in more than one locus suggests the potential for oligogenic inheritance and multilocus variation models in isolated TA, likely contributing to the variable phenotypes. With advances in genome-wide sequencing studies of well-characterized TA individuals and families, and careful genotype-phenotype correlations, new TA genes acting individually or interactively with other genes are likely to be identified.
4. Genetic Pathways as the Focus of Future Studies
Over the years, studies using transgenic animals demonstrated that defects in genes belonging to BMP, FGF and WNT signaling pathways resulted in severe abnormalities of tooth development ranging from defects in tooth patterning to complete arrest of tooth development [1,4,8]. Meanwhile, mutations in genes belonging to the FGF family have not yet been described in association with TA, whereas a single variant in BMP4 (see above) was found in one TA family.
The current available evidence supports a significant role for WNT pathway genes in isolated TA, mostly supported by the higher frequency of pathogenic mutations in AXIN2, WNT10A, WNT10B and LRP6 in TA individuals [11,22,150,171]. WNT signaling molecules are essential for patterning, proliferation and differentiation of multiple cell types during embryonic development. Secretion of WNTs, particularly WNT4, WNT6 and WNT10, from the dental epithelium has been reported as critical for tooth development, as the absence of WNT signaling leads to a dysfunctional enamel knot and subsequently in arrest of tooth development [103,181].
EDA, EDAR and EDARADD, with roles in both syndromic and isolated TA, belong to the NF-kB signaling pathway. Additional genes in the NF-kB pathway include NEMO (inhibitor of nuclear factor kappa B kinase subunit gamma), an important pathway modulator, and TRAF6 (TNF receptor associated factor 6), although little is known about the exact roles of these genes in tooth development and variations in these two genes were reported in individuals with syndromic TA [4].
Based on the aforementioned observations of multilocus variation as a potential explanation for some TA cases, with candidate genes belonging to the same or different signaling pathways, it is presumable to hypothesize that isolated TA may be the result of variation in more than one gene, acting individually or in combination with other genes and contributing to the variable expressivity of the condition [182]. Determining the full spectrum of putative defective genes in TA, the pathways in which they belong (Figure 2), their functions and interactive partners, will allow for improving our understanding of the underlying mechanisms in TA and may be the basis of future prevention and tooth replacement strategies.
5. Conclusions
Isolated TA is a heterogeneous condition with variable expressivity. While variations in numerous genes have been attributed as causal for TA, the etiology of TA in many individuals is still unsolved and may reflect mutations in genes yet unknown to tooth development, or the presence of multilocus variation. Moreover, environmental and epigenetic factors may also be considered likely contributors to TA phenotypes and should be explored in future studies. Next-generation sequencing studies of well-characterized individuals and families present the unique ability to identify all of the TA-predisposing variants throughout the genome while revealing important genetic and network interactions that may be critical for tooth development. Further genetic and functional studies focusing on newly identified genes and pathways have the potential to elucidate the genetic landscape of isolated TA and provide insights into preventive and treatment strategies. Targeted therapeutics for TA-relevant genes and/or pathways may represent future tooth replacement therapies.
Author Contributions
M.A.W. contributed with literature search, provided analysis and tables, critically revised the paper; A.L. conceived and wrote the paper; critically revised the paper.
Acknowledgments
This study was supported by NIH/NIDCR R03-DE024596 to A.L.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bei, M. Molecular genetics of tooth development. Curr. Opin. Genet. Dev. 2009, 19, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, I.; Vaahtokari, A.; Vainio, S.; Jowett, A. Molecular mechanisms of cell and tissue interactions during early tooth development. Anat. Rec. 1996, 245, 151–161. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Cohen, M.M., Jr.; Levin, S.L. Syndromes of the Head and Neck, 3rd ed.; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Yin, W.; Bian, Z. The gene network underlying hypodontia. J. Dent. Res. 2015, 94, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Polder, B.J.; Van’t Hof, M.A.; Van der Linden, F.P.; Kuijpers-Jagtman, A.M. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Commun. Dent. Oral Epidemiol. 2004, 32, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Carter, K.; Worthington, S. Morphologic and demographic predictors of third molar agenesis: A systematic review and meta-analysis. J. Dent. Res. 2015, 94, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Attaie, A.B. genetic basis of nonsyndromic and syndromic tooth agenesis. J. Pediatr. Genet. 2016, 5, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Jia, S.; Jiang, R. Molecular patterning of the mammalian dentition. Semin. Cell Dev. Biol. 2014, 25, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, K.; Shen, Y.; Xu, Y.; Xie, J.; Huang, R.; Zhang, Y.; Xu, C.; Zhang, X.; Wang, R.; et al. DNA methylation is critical for tooth agenesis: Implications for sporadic non-syndromic anodontia and hypodontia. Sci. Rep. 2016, 6, 19162. [Google Scholar] [CrossRef] [PubMed]
- Dinckan, N.; Du, R.; Akdemir, Z.C.; Bayram, Y.; Jhangiani, S.N.; Doddapaneni, H.; Hu, J.; Muzny, D.M.; Guven, Y.; Aktoren, O.; et al. A biallelic ANTXR1 variant expands the anthrax toxin receptor associated phenotype to tooth agenesis. Am. J. Med. Genet. A 2018, 176, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Dinckan, N.; Du, R.; Petty, L.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Paine, I.; Baugh, E.H.; Erdem, A.P.; Kayserili, H.; Doddapaneni, H.; et al. Whole-exome sequencing identifies novel variants for tooth agenesis. J. Dent. Res. 2018, 97, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P. Genetic basis of tooth agenesis. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312, 320–342. [Google Scholar] [CrossRef] [PubMed]
- Howe, B.J.; Cooper, M.E.; Vieira, A.R.; Weinberg, S.M.; Resick, J.M.; Nidey, N.L.; Wehby, G.L.; Marazita, M.L.; Moreno Uribe, L.M. Spectrum of dental phenotypes in nonsyndromic orofacial clefting. J. Dent. Res. 2015, 94, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R. Oral clefts and syndromic forms of tooth agenesis as models for genetics of isolated tooth agenesis. J. Dent. Res. 2003, 82, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Nuytinick, L.; Hausser, I.; van Esse, A.J.; Thiry, M.; Herens, C.; Adès, L.C.; Malfait, F.; Paepe, A.D.; Franck, P.; et al. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (type VIIC) and common polymorphisms in the ADAMTS2 gene. J. Invest. Dermatol. 2004, 123, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Sieron, A.L.; Li, S.W.; Schwarze, U.; Petty, E.; Wertelecki, W.; Wilcox, W.; Krakow, D.; Cohn, D.H.; Reardon, W.; et al. Human Ehlers-Danlos syndrome type VIIC and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am. J. Hum. Genet. 1999, 65, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; Pehlivan, D.; Karaca, E.; Gambin, T.; Jhangiani, S.N.; Erdin, S.; Elcioglu, N.H. Whole exome sequencing identifies three novel mutations in ANTXR1 in families with GAPO syndrome. Am. J. Med. Genet. A 2014, 164, 2328–2334. [Google Scholar] [CrossRef] [PubMed]
- Stranecky, V.; Hoischen, A.; Hartmannová, H.; Zaki, M.S.; Chaudhary, A.; Zudaie, E.; Nosková, L.; Baresová, V.; Pristoupilová, A.; Hodanová, K.; et al. Mutations in ANTXR1 cause GAPO syndrome. Am. J. Hum. Genet. 2013, 92, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Salas-Alanis, J.C.; Scott, C.A.; Fajardo-Ramírez, O.R.; Duran, C.; Moreno-Treviño, M.G.; Kelsell, D.P. New ANTXR1 gene mutation for GAPO syndrome: A case report. Mol. Syndromol. 2016, 7, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Marvin, M.L.; Mazzoni, S.M.; Herron, C.M.; Edwards, S.; Gruber, S.B.; Petty, E.M. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am. J. Med. Genet. A 2011, 155, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Lammi, L.; Arte, S.; Somer, M.; Järvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, B.; Andersson, K.; Lindahl, K.; Kindmark, A.; Grigelioniene, G.; Zachariadis, V.; Dahllöf, G.; Aström, E. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis. 2017, 23, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Stachtou, J.; Emmanouil, D.; Arveiler, B.; Griffiths, D.; Lacombe, D. Oro-dental features as useful diagnostic tool in Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 2007, 143, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Zonana, J.; Ferguson, B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Gong, Y.; Wu, H.; Zhang, X.; Yan, M.; Wang, X.; Qu, H.; Feng, H.; Song, S. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur. J. Med. Genet. 2008, 51, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Lexner, M.O.; Bardow, A.; Juncker, I.; Jensen, L.G.; Almer, L.; Kreiborg, S.; Hertz, J.M. X-linked hypohidrotic ectodermal dysplasia. Genetic and dental findings in 67 Danish patients from 19 families. Clin. Genet. 2008, 74, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Jin, B.; Guo, S.Z.; Qing, W.; Feng, G.Y.; Brooks, D.G.; Liu, L.; Xu, J.; Li, T.; Yan, Y. A novel missense mutation of the EDA gene in a Mongolian family with congenital hypodontia. J. Hum. Genet. 2006, 51, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Henningsen, E.; Svendsen, M.T.; Lidballe, D.L.; Jensen, P.K. A novel mutation in the EDAR gene causes severe autosomal recessive hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. A 2014, 164, 2059–2061. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Muhammad, D.; Ahmad, W. Novel mutations in the EDAR gene in two Pakistani consanguineous families with autosomal recessive hypohidrotic ectodermal dysplasia. Br. J. Dermatol. 2005, 153, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.K.; Wasif, N.; Javaid, H.; Ahmad, W. Two novel mutations in the gene EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia. Orthod. Craniofac. Res. 2011, 14, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Sato, N.; Miyashita, A.; Hashimoto, T.; Ito, M.; Kuwano, R. A rare case of hypohidrotic ectodermal dysplasia caused by compound heterozygous mutations in the EDAR gene. J. Invest. Dermatol. 2004, 123, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhao, Q.; Li, S.; Lu, H.; Lu, J.; Ma, L.; Zhao, W.; Yu, D. Novel EDA or EDAR Mutations identified in patients with X-linked hypohidrotic ectodermal dysplasia or non-syndromic tooth agenesis. Genes 2017, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Bal, E.; Baala, L.; Cluzeau, C.; El Kerch, F.; Ouldim, K.; Hadj-Rabia, S.; Bodemer, C.; Munnich, A.; Courtois, G.; Sefiani, A. Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum. Mutat. 2007, 28, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Söder, S.; Smahi, A.; Schneider, H. A novel missense mutation in the gene EDARADD associated with an unusual phenotype of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. A 2016, 170, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.L.; Ide, S.E.; Strom, T.M.; Lorenz, B.; Wilson, D.; Woods, K.; King, L.; Francomano, C.; Freisinger, P.; Spranger, S.; et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000, 24, 283–286. [Google Scholar] [CrossRef] [PubMed]
- D’Asdia, M.C.; Torrent, I.; Consoli, F.; Ferese, R.; Magliozzi, M.; Bernardini, L.; Guida, V.; Diglio, M.C.; Marino, B.; Dallapiccola, B.; et al. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet. 2013, 56, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Song, G.; Fan, M.; Shi, L.; Jabs, E.W.; Huang, S.; Guo, R.; Bian, Z. A novel heterozygous deletion in the EVC2 gene causes Weyers acrofacial dysostosis. Hum Genet 2006, 119, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Han, D.; Zhang, J.; Zhao, H.; Feng, H. Two novel heterozygous mutations of EVC2 cause a mild phenotype of Ellis-van Creveld syndrome in a Chinese family. Am. J. Med. Genet. A 2011, 155, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Entesarian, M.; Matsson, H.; Klar, J.; Bergendal, B.; Olson, L.; Arakaki, R.; Hayashi, Y.; Ohuchi, H.; Falahat, B.; Bolstad, A.I.; et al. Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat Genet 2005, 37, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Milunsky, J.M.; Zhao, G.; Maher, T.A.; Colby, R.; Everman, D.B. LADD syndrome is caused by FGF10 mutations. Clin. Genet. 2006, 69, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Rohmann, E.; Brunner, H.G.; Kayserili, H.; Uyguner, O.; Nürnberg, G.; Lew, E.D.; Dobbie, A.; Eswarakumar, V.P.; Uzumcu, A.; Ulubil-Emeroglu, A.; et al. Mutations in different components of FGF signaling in LADD syndrome. Nat. Genet. 2006, 38, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Bailleul-Forestier, I.; Gros, C.; Zenaty, D.; Bennaceur, S.; Leger, J.; de Roux, N. Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. Int. J. Paediatr. Dent. 2010, 20, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Letra, A.; de Almeida, A.L.; Kaizer, R.; Esper, L.A.; Sgarbosa, S.; Granjeiro, J.M. Intraoral features of Apert’s syndrome. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2007, 103, e38–e41. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, D.; Bartzela, T.; Bronkhorst, E.; Mohlin, B.; Hagberg, C. Dental agenesis patterns of permanent teeth in Apert syndrome. Eur. J. Oral Sci. 2011, 119, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, O.A.; Chiu, E.S.; McCarthy, J.G.; Mohammadi, M. Understanding the molecular basis of Apert syndrome. Plast. Reconstr. Surg. 2005, 115, 264–270. [Google Scholar] [PubMed]
- Meyers, G.A.; Orlow, S.J.; Munro, I.R.; Przylepa, K.A.; Jabs, E.W. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat. Genet. 1995, 11, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Reardon, W.; Wilkes, D.; Rutland, P.; Pulleyn, L.J.; Malcolm, S.; Dean, J.C.; Evans, R.D.; Jones, B.M.; Hayward, R.; Hall, C.M. Craniosynostosis associated with FGFR3 pro250arg mutation results in a range of clinical presentations including unisutural sporadic craniosynostosis. J. Med. Genet. 1997, 34, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.; Rutland, P.; Pulleyn, L.J.; Reardon, W.; Moss, C.; Ellis, J.P.; Winter, R.M.; Malcolm, S. A recurrent mutation, ala391glu, in the transmembrane region of FGFR3 causes Crouzon syndrome and acanthosis nigricans. J. Med. Genet. 1996, 33, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.C.; Ling, J.Y.; King, N.M.; Chow, S.K. Oral and craniofacial morphology of a patient with Larsen syndrome. J. Craniofac. Genet. Dev. Biol. 1986, 6, 357–362. [Google Scholar] [PubMed]
- Berenstein-Aizman, G.; Hazan-Molina, H.; Drori, D.; Aizenbud, D. Axenfeld-Rieger syndrome: Dentofacial manifestation and oral rehabilitation considerations. Pediatr. Dent. 2011, 33, 440–444. [Google Scholar] [PubMed]
- O’Dwyer, E.M.; Jones, D.C. Dental anomalies in Axenfeld-Rieger syndrome. Int. J. Paediatr. Dent. 2005, 15, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, K.W.; Hansen, L.; Eiberg, H.; Leicht, P.; Opitz, J.N.; Tommerup, N. Novel Connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. Am. J. Med. Genet. A 2004, 127, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Paznekas, W.A.; Karczeski, B.; Vermeer, S.; Lowry, R.B.; Delatycki, M.; Laurence, F.; Koivisto, P.A.; Van Maldergem, L.; Boyadjiev, S.A.; Bodurtha, J.N. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 2009, 30, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Petrof, G.; Nanda, A.; Howden, J.; Takeichi, T.; McMillan, J.R.; Aristodemou, S.; Ozoemena, L.; Liu, L.; South, A.P.; Purreyron, C.; et al. Mutations in GRHL2 result in an autosomal-recessive ectodermal Dysplasia syndrome. Am. J. Hum. Genet. 2014, 95, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Schutte, B.C.; Richardsson, R.J.; Bjork, B.C.; Knight, A.S.; Watanabe, Y.; Howard, E.; de Lima, R.L.; Daack-Hirsch, S.; Sander, A.; et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002, 32, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Zucchero, T.M.; Cooper, M.E.; Maher, B.S.; Daak-Hirsch, S.; Nepomuceno, B.; Ribeiro, L.; Caprau, D.; Christensen, K.; Suzuki, Y.; Machida, J.; et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N. Engl. J. Med. 2004, 351, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.C.; Lacbawan, F.; Francomano, C.A.; Ho, V. Severe hypodontia and oral xanthomas in Alagille syndrome. Am. J. Med. Genet. 2000, 93, 250–252. [Google Scholar] [CrossRef]
- Lederer, D.; Shears, D.; Benoit, V.; Verellen-Dumoulin, C.; Maystadt, I. A three generation X-linked family with Kabuki syndrome phenotype and a frameshift mutation in KDM6A. Am. J. Med. Genet. A 2014, 164, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaik, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Lerone, M.; Priolo, M.; Naselli, A.; Vignolo, A.; Romeo, G.; Silengo, M.C. Ectodermal abnormalities in Kabuki syndrome. Am. J. Med. Genet. 1997, 73, 263–266. [Google Scholar] [CrossRef]
- Matsune, K.; Matsune, K.; Shimizu, T.; Tohma, T.; Asada, Y.; Ohashi, H.; Maeda, T. Craniofacial and dental characteristics of Kabuki syndrome. Am. J. Med. Genet. 2001, 98, 185–190. [Google Scholar] [CrossRef]
- Issa, Y.A.; Kamal, L.; Rayyan, A.A.; Dweik, D.; Pierce, S.; Lee, M.K.; King, M.C.; Walsh, T.; Kanaan, M. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur. J. Hum. Genet. 2016, 24, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl syndrome-now and in the future. Front Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Jumlongras, D.; Bei, M.; Stimson, J.M.; Wen-Fang, W.; DePalma, S.R.; Seidman, C.E.; Felbor, U.; Maas, R.; Seidman, J.G. A nonsense mutation in MSX1 causes Witkop syndrome. Am. J. Hum. Genet. 2001, 69, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Aradhya, S.; Woffendin, H.; Jakins, T.; Bardaro, T.; Esposito, T.; Smahi, A.; Shaw, C.; Levy, M.; Munnich, A.; D’Urso, M. A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum. Mol. Genet. 2001, 10, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.L.; Dupuis-Girod, S.; Dittrich, A.M.; Bustamante, J.; Santos, O.F.; Schulze, I.; Bertrand, Y.; Couly, G.; Bodemer, C.; Bossuy, X. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics 2005, 115, e615–e619. [Google Scholar] [CrossRef] [PubMed]
- Hirai, N.; Matsune, K.; Ohashi, H. Craniofacial and oral features of Sotos syndrome: Differences in patients with submicroscopic deletion and mutation of NSD1 gene. Am. J. Med. Genet. A 2011, 155, 2933–2939. [Google Scholar] [CrossRef] [PubMed]
- Kotilainen, J.; Pohjola, P.; Pirinen, S.; Arte, S.; Niminen, P. Premolar hypodontia is a common feature in Sotos syndrome with a mutation in the NSD1 gene. Am. J. Med. Genet. A 2009, 149, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Shotelersuk, V.; Tifft, C.J.; Vacha, S.; Peters, K.F.; Biesecker, L.G. Discordance of oral-facial-digital syndrome type 1 in monozygotic twin girls. Am. J. Med. Genet. 1999, 86, 269–273. [Google Scholar] [CrossRef]
- Larralde de Luna, M.; Raspa, M.L.; Ibargoyen, J. Oral-facial-digital type 1 syndrome of Papillon-Leage and Psaume. Pediatr. Dermatol. 1992, 9, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.G.; Hamel, B.C.; Bokhoven, H.V. P63 gene mutations and human developmental syndromes. Am. J. Med. Genet. 2002, 112, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Rodini, E.O.; Freitas, J.A.; Richieri-Costa, A. Rapp-Hodgkin syndrome: Report of a Brazilian family. Am. J. Med. Genet. 1990, 36, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Sripathomsawat, W.; Tanpaiboon, P.; Heering, J.; Dötsch, V.; Hennekam, R.C.; Kantaputra, P. Phenotypic analysis of Arg227 mutations of TP63 with emphasis on dental phenotype and micturition difficulties in EEC syndrome. Am. J. Med. Genet. A 2011, 155, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.P.; Moroi, S.E.; Downs, C.A.; Wiltse, S.; Othman, M.I.; Semina, E.V.; Richards, J.E. A novel mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome. Ophthalmic Genet. 2004, 25, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.F.; Pressman, C.; Dyer, R.; Johnson, R.L.; Martin, J.F. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature 1999, 401, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Venugopalan, S.R.; Cao, H.; Pinho, F.O.; Paine, M.L.; Snead, M.L.; Semina, E.V.; Amendt, B.A. A model for the molecular underpinnings of tooth defects in Axenfeld-Rieger syndrome. Hum. Mol. Genet. 2014, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Bustos, T.; Simosa, V.; Pinto-Cisternas, J.; Abramovits, W.; Jolay, L.; Rodriguez, L.; Fernandez, L.; Ramela, M. Autosomal recessive ectodermal dysplasia: I. An undescribed dysplasia/malformation syndrome. Am. J. Med. Genet. 1991, 41, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Sozen, M.A.; Suzuki, K.; Tolarova, M.M.; Bustos, T.; Fernandez Iglesias, J.E.; Spritz, R.A. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat. Genet. 2001, 29, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Starr, D.G.; McClure, J.P.; Connor, J.M. Non-dermatological complications and genetic aspects of the Rothmund-Thomson syndrome. Clin. Genet. 1985, 27, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.G. Coffin-Lowry syndrome: A 20-year follow-up and review of long-term outcomes. Am. J. Med. Genet. 2002, 111, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Ming, J.E.; Du, Y.; Hall, R.K.; Aldred, M.; Bankier, A.; Muenke, M. SHH mutation is associated with solitary median maxillary central incisor: A study of 13 patients and review of the literature. Am. J. Med. Genet. 2001, 102, 1–10. [Google Scholar] [CrossRef]
- Petryk, A.; Graf, D.; Marcucio, R. Holoprosencephaly: Signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Le, T.; Watjkins, W.S.; Dixon, M.E.; Kramer, B.E.; Roeder, A.D.; Carey, J.C.; Root, S.; Schinzel, A.; Van Maldergem, L. The spectrum of mutations in TBX3: Genotype/Phenotype relationship in ulnar-mammary syndrome. Am. J. Hum. Genet. 1999, 64, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Lowry, R.B.; Morgan, K.; Holmes, T.M.; Metcalf, P.J.; Stauffer, G.F. Mandibulofacial dysostosis in Hutterite sibs: A possible recessive trait. Am. J. Med. Genet. 1985, 22, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mernagh, J.; Bourgeois, J. Novel craniofacial and extracraniofacial findings in a case of Treacher Collins syndrome with a pathogenic mutation and a missense variant in the TCOF1 gene. Clin. Dysmorphol. 2009, 18, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Radhakrishnan, J.; Farhi, A.; Carew, K.S.; Waarnes, C.A.; Nelson-Williams, C.; Day, R.W.; Pober, B.; State, M.W.; Lifton, R.P. Syndromic patent ductus arteriosus: Evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder. Proc. Natl. Acad. Sci. USA 2005, 102, 2975–2979. [Google Scholar] [CrossRef] [PubMed]
- Korenberg, J.R. Toward a molecular understanding of Down syndrome. Prog. Clin. Biol. Res. 1993, 384, 87–115. [Google Scholar] [PubMed]
- Palaska, P.K.; Antonarakis, G.S. Prevalence and patterns of permanent tooth agenesis in individuals with Down syndrome: A meta-analysis. Eur. J. Oral Sci. 2016, 124, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Sarig, O.; Samuelov, L.; Bertolini, M.; Ziv, L.; Weissglas-Volkov, D.; Eskin-Schwartz, M.; Adase, C.A.; Malchin, N.; Bochner, R.; et al. Mutations in TSPEAR, encoding a regulator of notch signaling, affect tooth and hair follicle morphogenesis. PLoS Genet. 2016, 12, e1006369. [Google Scholar] [CrossRef] [PubMed]
- Almashraki, N.; Abdulnabee, M.Z.; Sukalo, M.; Alrajoudi, A.; Sharafadeen, I.; Zenker, M. Johanson-Blizzard syndrome. World J. Gastroenterol. 2011, 17, 4247–4250. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Megarbane, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Megarbane, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Klar, J.; Wajid, M.; Aslam, M.; Tariq, M.; Schuster, J.; Baig, S.M.; Dahl, N. WNT10A missense mutation associated with a complete odonto-onycho-dermal dysplasia syndrome. Eur. J. Hum. Genet. 2009, 17, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.E.; Pieris, S.; Soni, V. Schopf-Schulz-Passarge syndrome. Br. J. Dermatol. 1992, 127, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Schopf, E.; Schulz, H.J.; Passarge, E. Syndrome of cystic eyelids, palmo-plantar keratosis, hypodontia and hypotrichosis as a possible autosomal recessive trait. Birth Defects Orig. Artic. Ser. 1971, 7, 219–221. [Google Scholar] [PubMed]
- Bergendal, B.; Klar, J.; Stecksén-Blicks, C.; Norderyd, J.; Dahl, N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am. J. Med. Genet. A 2011, 155, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.D.; Witkop, C.J. Autosomal dominant hypodontia with nail dysgenesis. Report of twenty-nine cases in six families. Oral Surg. Oral Med. Oral Pathol. 1975, 39, 409–423. [Google Scholar] [CrossRef]
- Satokata, I.; Maas, R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994, 6, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, E.; Salazar-Ciudad, I.; Birchmeier, W.; Taketo, M.M.; Jernvall, J.; Thesleff, I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18627–18632. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Millar, S.E. Wnt/beta-catenin signaling in oral tissue development and disease. J. Dent. Res. 2010, 89, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Zheng, L.; Shitaku, Y.; Saito, M.; Tsubakimoto, T.; Takada, K.; Takano-Yamamoto, T.; Thesleff, I. Wnt10a regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differentiation 2007, 75, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lany, Y.; Baek, J.A.; Gao, Y.; Jiang, R. Wnt/beta-catenin signaling plays an essential role in activation of odontogenic mesenchyme during early tooth development. Dev. Biol. 2009, 334, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, S.K.; Choi, M.; Reid, B.M.; Hu, Y.; Lee, Y.L.; Herzog, C.R.; Kim-Berman, H.; Lee, M.; Benke, P.J.; et al. Taurodontism, variations in tooth number, and misshapened crowns in Wnt10a null mice and human kindreds. Mol. Genet. Genomic Med. 2015, 3, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaisancie, J.; Bailleul-Forestier, I.; Gaston, V.; Vaysse, F.; Lacombe, D.; Holder-Espinasse, M.; Abramowicz, M.; Coubes, C.; Plessis, G.; Faivre, L.; et al. Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am. J. Med. Genet. A 2013, 161, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhao, R.; He, H.; Zhang, J.; Feng, H.; Lin, L. WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum. Genet. 2014, 133, 117–124. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.J.; Créton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; van Amstel, H.K.P. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.; Sripathomsawat, W. WNT10A and isolated hypodontia. Am. J. Med. Genet. A 2011, 155, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, K.; Saito, H.; Clément-Lacroix, P.; Maltry, N.; Niedermeyer, J.; Lee, W.K.; Baron, R.; Rawadi, G.; Westphal, H.; Niehrs, C. Targeted disruption of the Wnt regulator Kremen induces limb defects and high bone density. Mol. Cell Biol. 2008, 28, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, C.R.; Kinoshita, A.; Kondo, S.; Yang, B.; Sajan, S.; Trout, K.J.; Malik, M.I.; Dunnwald, M.; Goudy, S.L.; Lovett, M.; et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 2006, 38, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Mancuso, J.L.; Schutte, B.C.; Cooper, M.E.; Durda, K.M.; L’heureux, J.; Zucchero, T.M.; Marazita, M.L.; Murray, J.C. Search for genetic modifiers of IRF6 and genotype-phenotype correlations in Van der Woude and popliteal pterygium syndromes. Am. J. Med. Genet. A 2013, 161, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.U.; Mangold, E.; Herms, S.; Nowak, S.; Reutter, H.; Paul, A.; Becker, J.; Herberz, R.; AlChawa, T.; Nasser, E.; Böhmer, A.C. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 2012, 44, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Fitch, N.; Kaback, M. The Axenfeld syndrome and the Rieger syndrome. J. Med. Genet. 1978, 15, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, I.A.; Chudley, A.E. Autosomal dominant iridogoniodysgenesis with associated somatic anomalies: Four-generation family with Rieger’s syndrome. Br. J. Ophthalmol. 1983, 67, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Reiter, R.; Leysens, N.J.; Alward, W.L.M.; Small, K.W.; Datson, N.A.; Siegel-Bartelt, J.; Bierke-Nelson, D.; Bitoun, P.; Zabel, B.U.; et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996, 14, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Selever, J.; Lu, M.F.; Martin, J.F. Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development 2003, 130, 6375–6385. [Google Scholar] [CrossRef] [PubMed]
- Hjalt, T.A.; Semina, E.V.; Amendt, B.A.; Murray, J.C. The Pitx2 protein in mouse development. Dev. Dyn. 2000, 218, 195–200. [Google Scholar] [CrossRef]
- Gardner, E.J. Follow-up study of a family group exhibiting dominant inheritance for a syndrome including intestinal polyps, osteomas, fibromas and epidermal cysts. Am. J. Hum. Genet. 1962, 14, 376–390. [Google Scholar] [PubMed]
- Letra, A.; Bjork, B.; Cooper, M.E.; Szabo-Rogers, H.; Deleyiannis, F.W.B.; Field, L.L.; Czeizel, A.E.; Ma, L.; Garlet, G.P.; Poletta, F.A.; et al. Association of AXIN2 with non-syndromic oral clefts in multiple populations. J. Dent. Res. 2012, 91, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Win, A.K.; Gallinger, S.; Daftary, D.; Thibodeau, S.N.; Silva, R.; Letra, A. Colorectal cancer and self-reported tooth agenesis. Hered. Cancer Clin. Pract. 2014, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Biguetti, C.; Romero-Bustillos, M.; Maheshwari, K.; Dinckan, N.; Cavalla, F.; Vieira, A.R. Colorectal cancer-associated genes are associated with tooth agenesis and may have a role in tooth development. Sci. Rep. 2018, 8, 2979. [Google Scholar] [CrossRef] [PubMed]
- Callahan, N.; Modesto, A.; Meira, R.; Seymen, F.; Patir, A.; Vieira, A.R. Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch. Oral Biol. 2009, 54, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Biedziak, B.; Jagodzinski, P.P. Axis inhibition protein 2 (AXIN2) polymorphisms may be a risk factor for selective tooth agenesis. J. Hum. Genet. 2006, 51, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Zhang, J.; Wong, S.; Han, D.; Zhao, H.S.; Feng, H.L. Association between rs11001553 of DKK1 and non-syndromic tooth agenesis in the Chinese Han population. Genet. Mol. Res. 2014, 13, 7133–7139. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Tardivel, A.; Willen, L.; Kapadia, H.; Seaman, R.; Frazier-Bowers, S.; Schneider, P.; D’Souza, R.N. Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur. J. Hum. Genet. 2010, 18, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, Y.; Liu, Y.; Liu, H.; Zhao, H.; Zhang, G.; Snead, M.L.; Han, D.; Feng, H. Functional study of ectodysplasin-a mutations causing non-syndromic tooth agenesis. PLoS ONE 2016, 11, e0154884. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Han, D.; Qu, H.; Gong, Y.; Wu, H.; Zhang, X.; Zhong, N.; Feng, H. EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 2009, 88, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Bansal, R.; Das, P. Whole genome sequencing reveals novel non-synonymous mutation in ectodysplasin A (EDA) associated with non-syndromic X-linked dominant congenital tooth agenesis. PLoS ONE 2014, 9, e106811. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Modesto, A.; Meira, R.; Barbosa, A.R.; Lidral, A.C.; Murray, J.C. Interferon regulatory factor 6 (IRF6) and fibroblast growth factor receptor 1 (FGFR1) contribute to human tooth agenesis. Am. J. Med. Genet. A 2007, 143, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Kaewgahya, M.; Hatsadaloi, A.; Vogel, P.; Kawasaki, K.; Ohazama, A.; Ketudat Cairns, J.R. GREMLIN 2 mutations and dental anomalies. J. Dent. Res. 2015, 94, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Seymen, F.; Patir, A.; Menezes, R. Evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and isolate tooth agenesis, in a Turkish population. Arch. Oral Biol. 2008, 53, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Lidral, A.C.; Reising, B.C. The role of MSX1 in human tooth agenesis. J. Dent. Res. 2002, 81, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Vastardis, H.; Karimbux, N.; Guthua, S.W.; Seidman, J.G.; Seidman, C.E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat. Genet. 1996, 13, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Massink, M.P.; Créton, M.A.; Spanevello, F.; Fennis, W.M.; Cune, M.S.; Savelberg, S.M.; Nijman, I.J.; Maurice, M.M.; van den Boogaard, M.J.H.; van Haaften, G. Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am. J. Hum. Genet. 2015, 97, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Ockeloen, C.W.; Khandelwal, K.D.; Dreesen, K.; Ludwig, K.U.; Sullivan, R.; Van Rooij, I.A.; Thonissen, M.; Swinnen, S.; Phan, M.; Conte, F.; et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet. Med. 2016, 18, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Dugan, S.L.; Temme, R.T.; Olson, R.A.; Mikhailov, A.; Law, R.; Mahmood, H.; Noor, A.; Vincent, J.B. New recessive truncating mutation in LTBP3 in a family with oligodontia, short stature, and mitral valve prolapse. Am. J. Med. Genet. A 2015, 167, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Windpassinger, C.; Vitcu, I.; Orlic, M.; Rafiq, M.A.; Khalid, M.; Malik, M.N.; Ayub, M.; Alman, B.; Vincent, J.B. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am. J. Hum. Genet. 2009, 84, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Bansal, R.; Das, P. A novel G to A transition at initiation codon and exon-intron boundary of PAX9 identified in association with familial isolated oligodontia. Gene 2017, 635, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Meira, R.; Modesto, A.; Murray, J.C. MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J. Dent. Res. 2004, 83, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Han, D.; Zhang, H.; Liu, Y.; Zhang, X.; Miao, M.Z.; Wang, Y.; Zhao, N.; Zeng, L.; Bai, B.; et al. Nine novel PAX9 mutations and a distinct tooth agenesis genotype-phenotype. J. Dent. Res. 2018, 97, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Bowers, S.A.; Guo, D.C.; Cavender, A.; Xue, L.; Evans, B.; King, T.; Milewicz, D.; D’Souza, R.N. A novel mutation in human PAX9 causes molar oligodontia. J. Dent. Res. 2002, 81, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, S.N.; Yasue, A.; Masuda, K.; Watanabe, K.; Horiuchi, S.; Imopto, I.; Tanaka, E. Novel PAX9 mutations cause non-syndromic tooth agenesis. J. Dent. Res. 2014, 93, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Stockton, D.W.; Das, P.; Goldenberg, M.; D’Souza, R.N.; Patel, P.I. Mutation of PAX9 is associated with oligodontia. Nat. Genet. 2000, 24, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Alfawaz, S.; Fong, F.; Plagnol, V.; Wong, F.S.; Fearne, J.; Kelsell, D.P. Recessive oligodontia linked to a homozygous loss-of-function mutation in the SMOC2 gene. Arch. Oral Biol. 2013, 58, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Jamet, X.; Etard, C.; Lauget, V.; Muller, J.; Geoffroy, V.; Straus, J.P.; Pelletier, V.; Marion, V.; Poch, O.; et al. Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in SMOC2, causing major dental developmental defects. Am. J. Hum. Genet. 2011, 89, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for (1/4) of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. A 2014, 164, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhao, M.; Tandon, B.; Maili, L.; Liu, X.; Zhang, A.; Baugh, E.H.; Tran, T.; Silva, R.M.; Hecht, J.T.; et al. Role of WNT10A in failure of tooth development in humans and zebrafish. Mol. Genet. Genomic. Med. 2017, 5, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Hutsadaloi, A.; Kaewgahya, M.; Intachai, W.; German, R.; Koparal, M.; Leethanakul, C.; Tolun, A.; Ketudat Cairns, J.R. WNT10B mutations associated with isolated dental anomalies. Clin. Genet. 2018, 93, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Yang, W.; Han, D.; Wang, X.; Guo, S.; Li, J.; Li, F.; Zhang, X.; Wong, S.W.; Bai, B.; et al. Mutations in WNT10B are identified in individuals with oligodontia. Am. J. Hum. Genet. 2016, 99, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Vastardis, H. The genetics of human tooth agenesis: New discoveries for understanding dental anomalies. Am. J. Orthod. Dentofacial Orthop. 2000, 117, 650–656. [Google Scholar] [CrossRef]
- Liang, J.; Von den Hoff, J.; Lange, J.; Ren, Y.; Bian, Z.; Carels, C.E. MSX1 mutations and associated disease phenotypes: Genotype-phenotype relations. Eur. J. Hum. Genet. 2016, 24, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Neubüser, A.; Kratochwil, K.; Balling, R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998, 12, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Neubuser, A.; Balling, R. Pax genes and organogenesis: Pax9 meets tooth development. Eur. J. Oral. Sci. 1998, 106, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Bonczek, O.; Balcar, V.J.; Sery, O. PAX9 gene mutations and tooth agenesis: A review. Clin. Genet. 2017, 92, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Brook, A.H.; Elcock, C.; Aggarwal, M.; Lath, D.L.; Russell, J.M.; Patel, P.I.; Smith, R.N. Tooth dimensions in hypodontia with a known PAX9 mutation. Arch. Oral Biol. 2009, 54, S57–S62. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jian, F.; Chen, J.; Wang, H.; Lin, Y.; Yang, Z.; Pan, X.; Lai, W. Sequence analysis of PAX9, MSX1 and AXIN2 genes in a Chinese oligodontia family. Arch. Oral Biol. 2011, 56, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.P.; Ockeloen, C.W.; Ten Kate, S.; Koolen, D.A.; Van Amstel, J.K.P.; Kuijpers-Jagtman, A.M.; Van Heumen, C.C.; Kleefstra, T.; Carels, C.E. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur. J. Hum. Genet. 2014, 22, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Han, D.; Feng, H.; Qu, H.; Song, S.; Bai, B.; Zhang, Z. Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS ONE 2013, 8, e80393. [Google Scholar] [CrossRef] [PubMed]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [PubMed]
- Vogel, P.; Liu, J.; Platt, K.A.; Read, R.W.; Thiel, M.; Vance, R.B.; Brommage, R. Malformation of incisor teeth in Grem2−/− mice. Vet. Pathol. 2015, 52, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, D.; Song, S.; Wang, Y.; Zhao, H.; Pan, S.; Bai, B.; Feng, H. Correlation between the phenotypes and genotypes of X-linked hypohidrotic ectodermal dysplasia and non-syndromic hypodontia caused by ectodysplasin-A mutations. Eur. J. Med. Genet. 2011, 54, e377–e382. [Google Scholar] [CrossRef] [PubMed]
- Tipton, R.E.; Gorlin, R.J. Growth retardation, alopecia, pseudo-anodontia, and optic atrophy—The GAPO syndrome: Report of a patient and review of the literature. Am. J. Med. Genet. 1984, 19, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Seaman, S.; Chaudhary, A.; Yang, M.Y.; Hilton, M.B.; Logsdon, D.; Croix, B.S. Host-derived tumor endothelial marker 8 promotes the growth of melanoma. Cancer Res. 2009, 69, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Carson-Walter, E.B.; Watkins, D.N.; Nanda, A.; Vogelstein, B.; Kinzler, K.W.; Croix, B.S. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001, 61, 6649–6655. [Google Scholar] [PubMed]
- Fernando, S.; Fletcher, B.S. Targeting tumor endothelial marker 8 in the tumor vasculature of colorectal carcinomas in mice. Cancer Res. 2009, 69, 5126–5132. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xie, G.; Geng, P.; Zheng, C.; Li, J.; Pan, F.; Liang, H. Anti-tumor angiogenesis effect of genetic fusion vaccine encoding murine β-defensin 2 and tumor endothelial marker-8 in a CT-26 murine colorectal carcinoma model. Int. J. Clin. Exp. Med. 2015, 8, 4744–4752. [Google Scholar] [PubMed]
- Yue, H.; Liang, J.; Yang, K.; Hua, B.; Bian, Z. Functional analysis of a novel missense mutation in AXIN2 associated with non-syndromic tooth agenesis. Eur. J. Oral Sci. 2016, 124, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hosomichi, K.; Yano, K.; Kim, Y.I.; Nakaoka, H.; Kimura, R.; Shirota, T. Comprehensive genetic exploration of selective tooth agenesis of mandibular incisors by exome sequencing. Hum. Genome Var. 2017, 4, 17005. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, L.; Magnusson, T.E.; Thordarson, A.; Jonsson, T.; Geller, F.; Feenstra, B.; Rivadeneira, F. Rare and common variants conferring risk of tooth agenesis. J. Dent. Res. 2018, 97, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Letra, A.; Menezes, R.; Granjeiro, J.M.; Vieira, A.R. Defining subphenotypes for oral clefts based on dental development. J. Dent. Res. 2007, 86, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Xia, F. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Arte, S.; Parmanen, S.; Pirinen, S.; Alaluusua, S.; Nieminen, P. Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PLoS ONE 2013, 8, e73705. [Google Scholar] [CrossRef] [PubMed]
- Fedi, P.; Bafico, A.; Soria, A.N.; Burgess, W.H.; Miki, T.; Bottaro, D.P.; Aaronson, S.A. Isolation and biochemical characterization of the human Dkk-1 homologue, a novel inhibitor of mammalian Wnt signaling. J. Biol. Chem. 1999, 274, 19465–19472. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, X.; Xu, X.; Mayo, J.; Bringas, P. Jr.; Jiang, R.; Wang, S.; Chai, Y. SMAD4-mediated WNT signaling controls the fate of cranial neural crest cells during tooth morphogenesis. Development 2011, 138, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A.; Kivirikko, S.; Ciatti, S.; Moss, C.; Dunnill, M.G.S.; Eady, R.A.; Uitto, J. A homozygous nonsense mutation in the α3 chain gene of laminin 5 (LAMA3) in Herlitz junctional epidermolysis bullosa: Prenatal exclusion in a fetus at risk. Genomics 1995, 29, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Lee, K.; Miyashita, Y.; Carter, W.G. Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J. Cell Biol. 1999, 145, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Tasanen, K.; Eble, J.A.; Aumailley, M.; Schumann, H.; Baetge, J.; Tu, H.; Bruckner-Tuderman, L. Collagen XVII is destabilized by a glycine substitution mutation in the cell adhesion domain Col15. J. Biol. Chem. 2000, 275, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Salvi, A.; Giacopuzzi, E.; Bardellini, E.; Amadori, F.; Ferrari, L.; De Petro, G.; Majorana, A. Mutation analysis by direct and whole exome sequencing in familial and sporadic tooth agenesis. Int. J. Mol. Med. 2016, 38, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, P.; Liu, Y.; Zhang, X.; Fu, J.; Yu, H.M.I.; Zhang, Z. Intra-epithelial requirement of canonical Wnt signaling for tooth morphogenesis. J. Biol. Chem. 2013, 288, 12080–12089. [Google Scholar] [CrossRef] [PubMed]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: Genetic and clinical perspectives. J. Oral Pathol. Med. 2009, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Phenolyzer. Available online: http://phenolyzer.wglab.org (accessed on 3 May 2018).
Figure 1.
Location of predicted missense, frameshift, and nonsense mutations in MSX1, PAX9, and WNT10A genes. Green boxes represent exons, horizontal lines between exons represent introns. HD, corresponds to homeodomain in MSX1. PD, corresponds to paired domain in PAX9. UTR: Untranslated region.
Figure 1.
Location of predicted missense, frameshift, and nonsense mutations in MSX1, PAX9, and WNT10A genes. Green boxes represent exons, horizontal lines between exons represent introns. HD, corresponds to homeodomain in MSX1. PD, corresponds to paired domain in PAX9. UTR: Untranslated region.

Figure 2.
Tooth agenesis gene network as predicted by Phenolyzer [183]. The network shown includes the top 50 prioritized genes, and their predicted relations with seed genes. Larger dark blue nodes indicate seed genes, medium royal blue nodes indicate interacting genes. Green font indicates predicted genes. Blue lines indicate protein–protein interactions.
Figure 2.
Tooth agenesis gene network as predicted by Phenolyzer [183]. The network shown includes the top 50 prioritized genes, and their predicted relations with seed genes. Larger dark blue nodes indicate seed genes, medium royal blue nodes indicate interacting genes. Green font indicates predicted genes. Blue lines indicate protein–protein interactions.


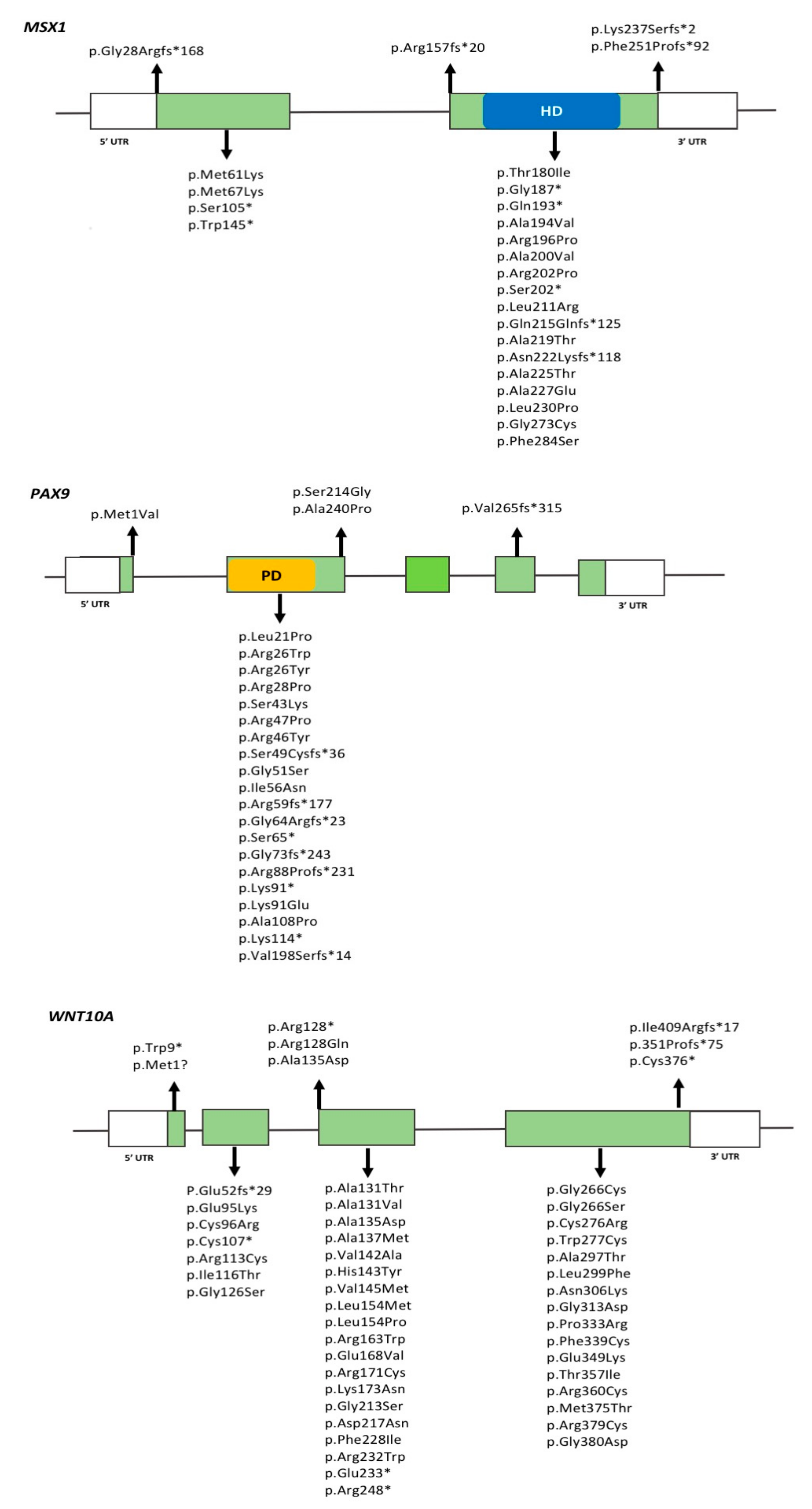{kind=link}
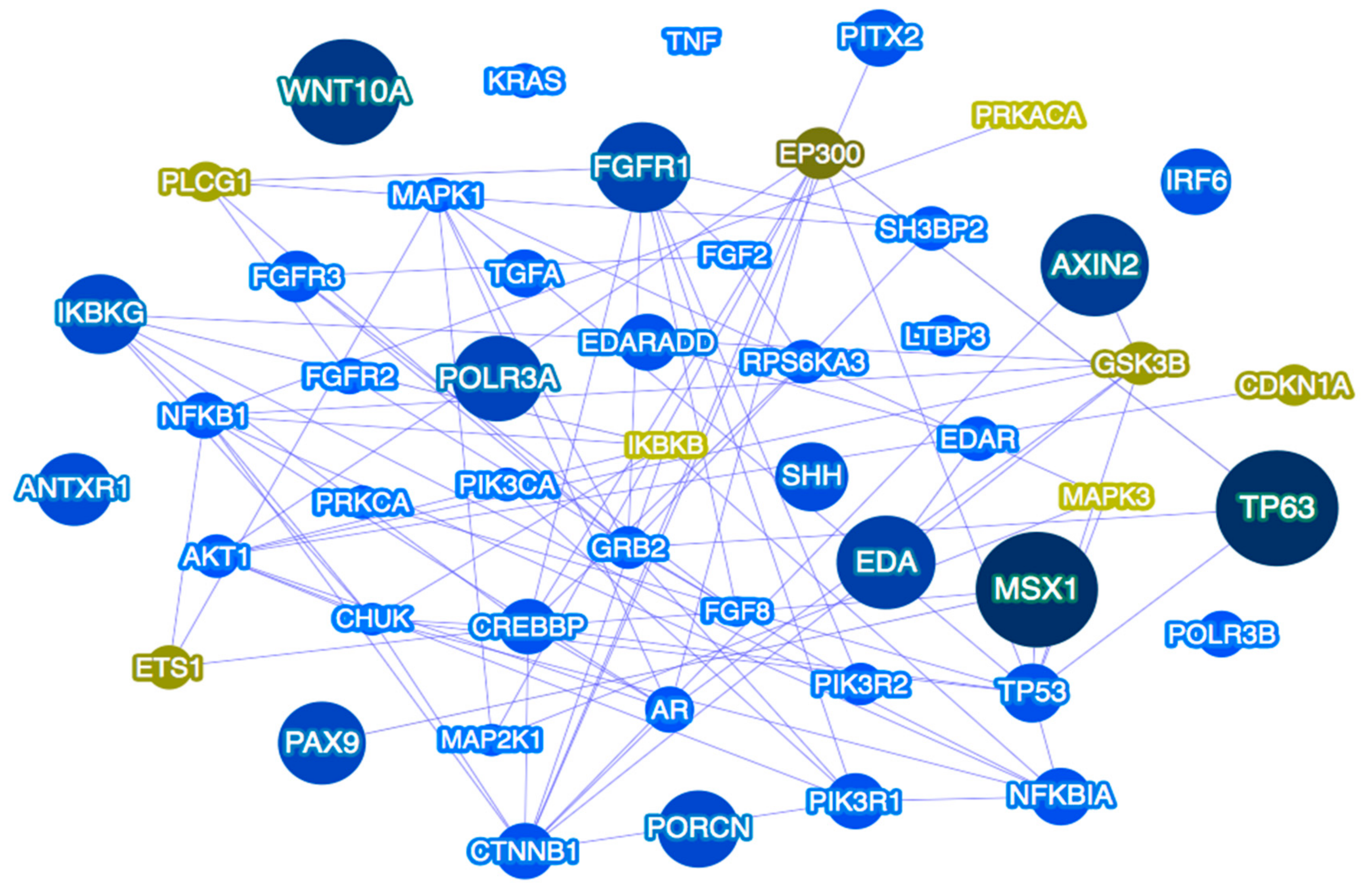{kind=link}
Table 1.
Genes involved in syndromic tooth agenesis and associated phenotypes.
| Gene/Locus | OMIM | Chromosome | Syndrome | Inheritance | Dental/Oral Phenotypes | Animal Model | Animal Model Phenotype | Reference |
|---|---|---|---|---|---|---|---|---|
| ADAMTS2 | 604539 | 5q35.3 | Ehlers–Danlos syndrome | AR | Hypodontia, microdontia, tooth discoloration | Yes Bovine | Dermatosparatic phenotype resembling EDS type VII C | [15,16,17] |
| ANTXR1 | 606410 | 2p13.3 | Growth retardation, alopecia, pseudoanodontia, and optic atrophy (GAPO) syndrome | AR | Hypodontia, delayed eruption | Yes | Growth delay, bone loss, shortened skulls with frontal bossing, and midfacial hypoplasia | [18,19,20] |
| AXIN2 | 604025 | 17q24.1 | Oligodontia-colorectal cancer syndrome | AD | Oligodontia | Yes | Abnormal cranium morphology | [21,22] |
| COL1A1/2 | 120150 | 17q21.33 | Osteogenesis imperfecta type 1 | AD | Hypodontia, oligodontia | Yes | Lethal, bone fractures | [23] |
| CREBBP | 600140 | 16p13.3 | Rubinstein–Taybi syndrome | AD | Hypodontia, retrognathia, micrognathia, arched/narrow palate, talon cusps, dental crowding, screwdriver incisors, cross bite, and enamel hypoplasia | Yes | Skeletal malformations | [24] |
| EDA | 300451 | Xq13.1 | Ectodermal dysplasia, hypohidrotic | XLR | Anodontia, hypodontia, misshapen teeth, microdontia | Yes Canine | Incomplete set of conically shaped teeth | [25,26,27,28] |
| EDAR | 604095 | 2q13 | Ectodermal dysplasia, hypohidrotic/hair/tooth type | AR | Anodontia, hypodontia, oligodontia | Yes Mouse | Decreased molar number, small incisor, small molars, abnormal enamel knot morphology | [29,30,31,32,33] |
| EDARADD | 606603 | 1q42-q43 | Ectodermal dysplasia, hypohidrotic/hair/tooth type | AD | Anodontia, hypodontia, taurodontism, microdontia | Yes Mouse | Abnormal tooth morphology, decreased molar number, small molars, abnormal enamel morphology | [34,35,36] |
| EVC | 604831 | 4p16.2 | Ellis–van Creveld syndrome and Weyers acrofacial dysostosis | AR/AD | Natal teeth, enamel abnormalities, hypodontia, microdontia | Yes Mouse | Enamel defects, abnormal tooth morphology | [37,38,39] |
| EVC2 | 607261 | 4p16.2 | Ellis–van Creveld syndrome and Weyers acrofacial dysostosis | AR/AD | Natal teeth, enamel abnormalities, hypodontia, oligodontia, microdontia | Yes Mouse | Microdontia, small upper incisors, small cranium | [37,38,39,40] |
| FGF10 | 602115 | 5p12 | Lacrimoauriculodentodigital syndrome | AD | Hypodontia (maxillary incisors), microdontia, delayed eruption, enamel dysplasia | Yes Mouse | Abnormal tooth morphology, short incisors, small molars, abnormal palatal development, abnormal tongue morphology | [41,42,43] |
| FGFR1 | 136350 | 8p11.23 | Kallmann syndrome | XLR | Hypodontia, cleft lip/palate | Yes Mouse | Abnormal cranium morphology, facial asymmetry, long incisors | [44] |
| FGFR2 | 176943 | 10q26.13 | Lacrimoauriculodentodigital syndrome | AD | Hypodontia (maxillary incisors), microdontia peg laterals, delayed eruption, enamel dysplasia | Yes Mouse | Arrest of tooth development, long incisors, decreased molar number, micrognathia | [43] |
| Apert syndrome | AD | Hypodontia (maxillary canines), enamel opacities, ectopic eruptions, gingival hyperplasia | Yes Mouse | Arrest of tooth development, long incisors, decreased molar number, micrognathia | [45,46,47] | |||
| FGFR3 | 134934 | 4p16.3 | Crouzon syndrome with acanthosis nigricans | AD | Hypodontia, malocclusion, cementomas, delayed eruption, midface hypoplasia | Yes Mouse | Tooth misalignment, long incisors, malocclusion, prognathia, maxillary retrognathia | [48,49,50] |
| FLNB | 603381 | 3p14.3 | Larsen syndrome | AD | Hypodontia, delayed dental development, class 3 occlusion, morphological anomalies | Yes Mouse | Abnormal cranium morphology | [51] |
| FOXC1 | 601090 | 6p25.3 | Axenfeld–Rieger syndrome type 3 | AD | Hypodontia, microdontia, taurodontism | Yes, Mouse | Short mandible | [52,53] |
| GJA1 | 121014 | 6q22.31 | Oculodentodigital dysplasia | AD, AR | Microdontia, enamel hypoplasia, hypodontia, delayed eruption | Yes Mouse | Abnormal tooth morphology, microdontia, small mandible and maxilla, reduced enamel thickness | [54,55] |
| GRHL2 | 608576 | 8q22.3 | Ectodermal dysplasia/short stature syndrome | AR | Delayed eruption, hypodontia, enamel hypoplasia | Yes, Mouse | Abnormal cranium morphology, facial and midline clefts | [56] |
| IRF6 | 607199 | 1q32.2 | van der Woude syndrome | AD | Hypodontia, cleft lip/palate | Yes Mouse | Abnormal tooth morphology, abnormal palatal development, small mandible | [57,58] |
| JAG1 | 601920 | 20p12.2 | Alagille Syndrome | AD | Hypodontia, enamel hypoplasia and opacities, hypomineralization | Yes mouse | Short maxilla, malocclusion, abnormal palate morphology | [59] |
| KDM6A | 300128 | Xp11.3 | Kabuki syndrome 2 | XLD | High-arched palate, malocclusion, microdontia, a small dental arch, hypodontia, severe maxillary recession, conical teeth | Yes Mouse | Cranioschisis | [60,61] |
| KMT2D | 602113 | 12q13.12 | Kabuki syndrome 1 | AD | High-arched palate, malocclusion, microdontia, a small dental arch, hypodontia, severe maxillary retrognathia, conical teeth | Yes Mouse | Short maxilla, flattened snout | [62,63] |
| KREMEN1 | 609898 | 22q12.1 | Ectodermal dysplasia, hair/tooth type | AR | Oligodontia, hypodontia, alveolar ridge deficiency, increased palatal depth | Yes | No craniofacial phenotype | [64] |
| MKKS | 604896 | 20p12.2 | Bardet-Biedl syndrome | AR | Dental crowding, high-arched palate, hypodontia, malocclusion, enamel hypoplasia, retrognathia | Yes Mouse | Abnormal olfactory epithelium | [65] |
| MSX1 | 142983 | 4p16.1 | Witkop syndrome | AD | Hypodontia, oligodontia | Yes Mouse | Arrest of tooth development, nail bed and nail plates defective, cleft palate | [66] |
| NEMO | 300248 | Xq28 | Incontinentia pigmenti | XLD | Hypodontia, anodontia, microdontia | Yes Mouse | No craniofacial phenotype | [67,68] |
| NSD1 | 606681 | 5q35.3 | Sotos syndrome I | AD | Hypodontia, enamel defects, malocclusion | Yes Mouse | No craniofacial phenotypes | [69,70] |
| OFD1 | 300170 | Xp22.2 | Orofaciodigital syndrome I | XLD | Hypodontia, missing lateral incisors, canine malposition, micrognathia | Yes | Primary cilia formed then disappeared, renal cysts | [71,72] |
| P63 | 603273 | 3q28 | Orofacial cleft 8, Rapp-Hodgkin, and Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 | AD | Hypodontia, enamel hypoplasia, extensive dental caries, hypodontia of the mandibular canines, generalized microdontia, prominent marginal ridges of permanent maxillary incisors, round-shaped permanent molars, and barrel-shaped permanent maxillary central incisors | Yes Mouse | Arrest of tooth development, small mandible and maxilla, abnormal craniofacial development, cleft palate | [73,74,75] |
| PITX2 | 601542 | 4q25 | Axenfeld–Rieger syndrome, type 1 | AD | Hypodontia, microdontia, enamel hypoplasia | Yes Mouse | Abnormal maxilla and mandible morphology, arrested tooth development | [53,76,77,78] |
| PVRL1 | 600644 | 11q23.3 | Cleft lip/palate-ectodermal dysplasia | AR | Hypodontia, cleft lip and palate, abnormal dental morphology, microdontia | Yes Mouse | Abnormal tooth morphology | [79,80] |
| RECQL4 | 603780 | 8q24.3 | Rothmund–Thomson syndrome | AR | Hypodontia, microdontia, hypoplastic teeth | Yes Mouse | Delayed tooth eruption, cleft palate | [81] |
| RSK2 | 300075 | Xp22.12 | Coffin–Lowry syndrome | XLD | High narrow palate, midline lingual furrow, hypodontia, and microdontia | Yes Mouse | Abnormal tooth morphology, supernumerary teeth | [82] |
| SHH | 600725 | 7q36.3 | Holoprosencephaly | AD | Cleft lip and palate, single central incisor, micrognathia | Yes | Abnormal tooth morphology, microdontia | [83,84] |
| TBX3 | 601621 | 12q24.21 | Ulnar-mammary syndrome | AD | Hypodontia, ectopic and hypoplastic canines | Yes | Secondary palate clefting | [85] |
| TCOF1 | 606847 | 5q32-q33 | Treacher Collins syndrome | AD | Hypodontia, micrognathia, malocclusion, spaced teeth | Yes | Short mandible and maxilla | [86,87] |
| TFAP2B | 601601 | 6p12.3 | Char syndrome | AD | Oligodontia, hypodontia, thick lips, retention of primary teeth | Yes | No craniofacial phenotype | [88] |
| Trisomy 21 | 190685 | 21q22.13 | Down syndrome | IC | Hypodontia, delayed eruption, barrel-shaped permanent maxillary central incisors | Yes | General hypoplasia and developmental delay, hydronephrosis, heart and neurologic defects | [89,90] |
| TSPEAR | 612920 | 21q22.3 | Ectodermal dysplasia | AR | Hypodontia, microdontia | Yes | No craniofacial phenotypes | [91] |
| UBR1 | 605981 | 15q15.2 | Johanson–Blizzard syndrome | AR | Oligodontia | Yes | No craniofacial phenotypes | [92] |
| WNT10A | 606268 | 2q35 | Odontoonychodermal dysplasia | AR | Oligodontia, hypodontia, microdontia | Yes | Arrested tooth development of molars, supernumerary molars, abnormal tooth morphology | [93,94,95] |
| Schopf–Schulz–Passarge syndrome | AR | Oligodontia, hypodontia, microdontia | Yes | Arrested tooth development of molars, supernumerary molars, abnormal tooth morphology | [94,96,97] |
OMIM, Online Mendelian Inheritance in Man (https://www.ncbi.nlm.nih.gov/omim/) AR, autosomal recessive; AD, autosomal dominant, XLD, x-linked dominant, XLR, x-linked recessive; IC, isolated cases.
Table 2.
Genes involved in isolated tooth agenesis.
| Gene | OMIM | Chromosome | Dental Phenotypes | Reference(s) |
|---|---|---|---|---|
| AXIN2 | 604025 | 17q24.1 | Oligodontia, hypodontia | [22,98,124,125] |
| ANTXR1 | 606410 | 2p13.3 | Oligodontia, hypodontia | [10] |
| COL17A1 | 113811 | 10q25.1 | Hypodontia | [11] |
| DKK1 | 605189 | 10q21.1 | Hypodontia | [11,126] |
| EDA | 300451 | Xq13.1 | Oligodontia, hypodontia | [33,127,128,129,130] |
| EDAR | 604095 | 2q13 | Oligodontia, hypodontia | [33] |
| EDARADD | 606603 | 1q42-q43 | Oligodontia, hypodontia | [33] |
| FGFR1 | 136350 | 8p11.23 | Hypodontia | [131] |
| GREM2 | 608832 | 1q43 | Hypodontia, microdontia, taurodontia | [132] |
| IRF6 | 607199 | 1q32.2 | Hypodontia, lip pits | [131,133] |
| MSX1 | 142983 | 4p16.2 | Oligodontia, hypodontia | [98,134,135] |
| LAMA3 | 600805 | 18q11.2 | Hypodontia | [11] |
| LRP6 | 603507 | 12p13.2 | Oligodontia | [11,136,137] |
| LTBP3 | 602090 | 11q13.1 | Oligodontia, hypodontia | [138,139] |
| PAX9 | 167416 | 14q13.3 | Oligodontia, hypodontia, microdontia | [98,140,141,142,143,144,145] |
| SMOC2 | 607223 | 6q27 | Oligodontia, microdontia, abnormal morphology | [146,147] |
| WNT10A | 606268 | 2q35 | Oligodontia, hypodontia | [11,108,109,110,148,149] |
| WNT10B | 601906 | 12q13.12 | Oligodontia, microdontia | [150,151] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Williams, M.A.; Letra, A. The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes 2018, 9, 255. https://doi.org/10.3390/genes9050255
AMA Style
Williams MA, Letra A. The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes. 2018; 9(5):255. https://doi.org/10.3390/genes9050255
Chicago/Turabian StyleWilliams, Meredith A., and Ariadne Letra. 2018. "The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis" Genes 9, no. 5: 255. https://doi.org/10.3390/genes9050255
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.