Phosphorylation-Dependent Inhibition of Akt1
by
Nileeka Balasuriya
1, 
McShane McKenna
1,
Xuguang Liu
1,
Shawn S. C. Li
1 and
Patrick O’Donoghue
1,2,* 1
Department of Biochemistry, Schulich School of Medicine and Dentistry, The University of Western Ontario, London, ON N6A 5C1, Canada
2
Department of Chemistry, Faculty of Science, The University of Western Ontario, London, ON N6A 5C1, Canada
*
Author to whom correspondence should be addressed.
Genes 2018, 9(9), 450; https://doi.org/10.3390/genes9090450
Submission received: 20 July 2018
/
Revised: 10 August 2018
/
Accepted: 10 August 2018
/
Published: 7 September 2018
(This article belongs to the Special Issue Synthetic DNA and RNA Programming)
Abstract
:Protein kinase B (Akt1) is a proto-oncogene that is overactive in most cancers. Akt1 activation requires phosphorylation at Thr308; phosphorylation at Ser473 further enhances catalytic activity. Akt1 activity is also regulated via interactions between the kinase domain and the N-terminal auto-inhibitory pleckstrin homology (PH) domain. As it was previously difficult to produce Akt1 in site-specific phosphorylated forms, the contribution of each activating phosphorylation site to auto-inhibition was unknown. Using a combination of genetic code expansion and in vivo enzymatic phosphorylation, we produced Akt1 variants containing programmed phosphorylation to probe the interplay between Akt1 phosphorylation status and the auto-inhibitory function of the PH domain. Deletion of the PH domain increased the enzyme activity for all three phosphorylated Akt1 variants. For the doubly phosphorylated enzyme, deletion of the PH domain relieved auto-inhibition by 295-fold. We next found that phosphorylation at Ser473 provided resistance to chemical inhibition by Akti-1/2 inhibitor VIII. The Akti-1/2 inhibitor was most effective against pAkt1T308 and showed four-fold decreased potency with Akt1 variants phosphorylated at Ser473. The data highlight the need to design more potent Akt1 inhibitors that are effective against the doubly phosphorylated and most pathogenic form of Akt1.
1. Introduction
Protein kinase B (Akt) is a human serine–threonine kinase and a member of the AGC family of protein kinases [1,2]. The pathway regulated by Akt is the most commonly activated signaling pathway in human cancers [3]. Given that more than 50% of human tumors contain hyperactivated Akt [2], effective inhibition of active Akt has the potential to treat several distinct cancers. There are three AKT genes in humans, encoding the isozymes Akt1, Akt2, and Akt3. The Akt1 isozyme has well-established roles in many human cancers. Overactive Akt1 is a hallmark of diverse human malignancies [3,4] and linked to reduced survival outcomes [5,6]. Indeed, Akt1 is as a leading drug target in cancer [7,8]. Over 300 clinical trials have been completed or are under way that involve targeting the Akt1 signaling pathway [9,10].
Akt1 is a key regulator of the phosphoinositide 3 kinase (PI3K)/Akt1 signaling cascade that controls cell growth and survival [1]. In human cells, the activation of Akt1 occurs in response to growth factor stimulation. Following activation by a receptor tyrosine kinase at the plasma membrane, PI3K phosphorylates its immediate downstream target, a lipid second messenger called phosphatidylinositol-4,5-bisphosphate (PIP2), converting PIP2 into phosphatidylinositol-3,4,5-triphosphate (PIP3) (Figure 1) [2]. Membrane-anchored PIP3 is a binding site for pleckstrin homology (PH) domain–containing proteins such as Akt1 and one of the upstream kinases that activates Akt1, phosphoinositide-dependent kinase 1 (PDK1) [11]. Co-localization of Akt1 with PDK1 leads to partial activation of Akt1 by PDK1-mediated phosphorylation of Thr308 in the kinase domain of Akt1. Mechanistic target of rapamycin complex 2 (mTORC2) is responsible for phosphorylating Akt1 at Ser473 in the C-terminal hydrophobic motif of Akt1 (Figure 1). The phosphorylation of Ser473 further increases the kinase activity of Akt1. Although the cellular role of Ser473 phosphorylation is not well defined, several studies point to the idea that pSer473 may impact Akt1 substrate selectivity (reviewed in [2]).
Leveraging our ability to produce Akt1 protein in specifically phosphorylated forms (Figure 2), we recently quantified the precise contribution of pThr308 and pSer473 to Akt1 activity in vitro and in mammalian cells [12]. In studies with purified full-length Akt1, we found that pAkt1T308 achieves 30% of the activity of the doubly phosphorylated kinase. We also observed in COS-7 cells that pSer473 is dispensable for Akt1 signaling [12]. In close agreement with our findings, studies in adipocytes found that half-maximal Akt1 cellular signaling activity is achieved when the kinase is 5–22% phosphorylated [13]. In studies of human tumors [12,13,14,15] and in clinical diagnostic settings [14,15], the phosphorylation status of Akt1 at Ser473 only is often used as a marker or proxy for Akt1 activity. Our work [12] and work by others [16] found that phosphorylation of Thr308 alone was sufficient for maximal Akt1 signal prorogation in cells. These results indicated that compared to Ser473, Thr308 phosphorylation status is a superior biomarker for Akt1 activity.
The N-terminal PH domain is auto-inhibitory to Akt1 activity. Due to previous roadblocks in preparing Akt1 with programmed phosphorylation(s), there are no reports that measure the contribution of a phosphate at each key regulatory site to the auto-inhibition of Akt1 by the PH domain. In the current model, Akt1 exists in an auto-inhibited (PH-in) and activated (PH-out) conformation. In its auto-inhibited conformation (PH-in), the PH domain binds between the N- and C-terminal lobes of the unphosphorylated Akt1 kinase domain. During growth factor–mediated activation of Akt1, the PH domain forms a new interaction with PIP3, causing it to move outward, away from the now more accessible kinase domain. This PH-out conformation is readily activated via phosphorylation at Thr308 by PDK1 and Ser473 by mTORC2. Disruption of the PH and kinase domain interaction was identified as a plausible cause of increased Akt1 phosphorylation and subsequent activity in cancer [18].
Here we quantified the ability of the PH domain to auto-inhibit Akt1 variants phosphorylated at either or both key regulatory sites Thr308 and Ser473. We produced novel recombinant Akt1 variants lacking the PH domain (ΔPH-Akt1) that also contained programmed phosphorylation at each site separately or with both sites phosphorylated. In the context of full-length phosphorylated Akt1 variants, we quantified the contribution of both regulatory phosphorylation sites to chemical inhibition with the PH domain–dependent allosteric inhibitor Akti-1/2.
2. Materials and Methods
2.1. Bacterial Strains and Plasmids
We designed a codon-optimized PH domain–deficient (ΔPH) human AKT1 gene (residues 109–480), which was synthesized by ATUM (Newark, CA, USA). The ΔPH-AKT1 gene was subcloned (NcoI/NotI) into an isopropyl β-D-1-thiogalactopyranoside (IPTG)–inducible T7lac promoter–driven pCDF-Duet1 vector with CloDF13-derived CDF replicon and streptomycin/spectinomycin resistance (pCDF-Duet1-ΔPHAkt1). The PDPK1 gene was purchased from the Harvard PlasmidID repository service (plasmid ID: HsCD00001584; Boston, MA, USA) and subcloned (KpnI/NdeI) into the second multicloning site (MCS) of pCDF-Duet-1. Full-length pAkt1 variants were produced from pCDF-Duet1 plasmids as described previously [12]. The genetic code expansion system for phosphoserine (pSer) is encoded on the pDS-pSer2 plasmid [12,19,20], which contains 5 copies of tRNASep [21], phosphoseryl-tRNA synthetase (SepRS9), and elongation factor Tu mutant (EFSep21) [22]. Incorporation of pSer also required site-directed mutagenesis of the Ser473 codon to TAG in the ΔPH-akt1 constructs. Successful cloning was verified by DNA sequencing at the London Regional Genomics Centre (London, ON, Canada) and Genewiz (Cambridge, MA, USA).
2.2. Protein and Phosphoprotein Production
Recombinant Akt1 protein variants were expressed in BL21(DE3) (ThermoFisher Scientific, Waltham, MA, USA) (Figure 2). The pDS-pSer2 plasmid [20] was used as before [12] to incorporate pSer in response to a UAG codon at position 473 in ΔPHAkt1 and full-length Akt1 variants. To produce both full-length and PH domain–deficient Akt1 variants containing pSer473 (Supplementary Table S1), the pCDFDuet-1 Akt1–bearing plasmid was co-transformed with pDS-pSer2 into Escherichia coli Bl21(DE3) and plated on Luria broth (LB) agar plates with 25 μg/mL kanamycin and 50 μg/mL streptomycin. To produce pAkt1T308 variants, a pCDF-Duet1 plasmid containing both the Akt1 variant (MSC 2) and PDK1 (MSC 1) was transformed into E. coli BL21(DE3) and plated on LB agar plates with 50 μg/mL streptomycin. To produce ppAkt1T308,S473 variants, a pCDF-Duet1 plasmid containing both the Akt1 variant, MSC 2, with a TAG codon at position 473 and PDK1 variant, MSC 1 [12] was co-transformed with pDS-pSer2 into E. coli Bl21(DE3) and plated on LB agar plates with 25 μg/mL kanamycin and 50 μg/mL streptomycin.
In all cases, a single colony was used to inoculate 70 mL of LB (with 50 μg/mL streptomycin and, if needed, 25 μg/mL kanamycin), which was grown, shaking, overnight at 37 °C. From this starter culture, a 10 mL inoculum was added to 1 L of LB with antibiotics (as above) and, for pSer473-containing variants only, O-phospho-l-serine (Sigma Aldrich, Oakville, ON, Canada) was added to a final concentration of 2.5 mM. The cultures were grown at 37 °C until OD600 = 0.6, at which point, for pSer473-containing variants only, 2.5 mM of additional pSer was added to the culture. Protein expression was induced by adding 300 µM of IPTG at OD600 = 0.8. Cultures were then incubated at 16 °C for 18 h. Cells were grown and pelleted at 5000× g and stored at −80 °C until further analysis. Akt1 protein variants were purified from the cell pellets using Ni-nitrilotriacetic acid affinity column chromatography (see Supplementary Material, affinity column chromatography description).
2.3. Parallel-Reaction Monitoring Mass Spectrometry of ppAkt1
The ppAkt1 protein produced as described above and the commercially available active Akt1 (Abcam, lot 1, Cambridge, MA, USA) were precipitated in ice-cold acetone/ethanol/acetic acid (50/50/0.1, vol/vol/vol). The protein precipitate was resuspended in 8 M urea, then reduced in 5 mM dithiothreitol (DTT) at 37 °C for 1 h and alkylated in 14 mM iodoacetamide (IAA) in darkness at room temperature for 1 h. Unreacted IAA was neutralized by adding 5 mM DTT. The final protein concentration was determined by Bradford assay. Glu-C digestion was performed at 37 °C overnight with a Glu-C–to–protein ratio of 1:20 (w/w). The digest was desalted in C18 column (Phenomenex, Torance, CA, USA) according to the manufacturer’s protocol and resuspended in mass spectrometry (MS)-grade water. A Q Exactive Hybrid Quadrupole Orbitrap MS (Thermo Fisher Scientific, Waltham, MA, USA) was used to analyze the peptides. Data were analyzed using Skyline software [23].
2.4. MALDI-TOF/TOF Mass Spectrometry Analysis
In-gel digestion was performed using MassPREP automated digester station (PerkinElmer, Downers Grove, IL, USA). Gel pieces were de-stained using 50 mM ammonium bicarbonate and 50% acetonitrile, which was followed by protein reduction using 10 mM DTT, alkylation using 55 mM IAA, and tryptic digestion in 50 mM ammonium bicarbonate, pH 8. Peptides were extracted using a solution of 1% formic acid and 2% acetonitrile and lyophilized. Prior to mass spectrometry analysis, dried peptide samples were re-dissolved in a 10% acetonitrile and 0.1% trifluoroacetic acid solution. MALDI matrix, a–cyano–4–hydroxycinnamic acid, was prepared as 5 mg/mL in 6 mM ammonium phosphate monobasic, 50% acetonitrile, 0.1% trifluoroacetic acid and mixed with the sample at a 1:1 ratio (v/v). Mass spectrometry data (Figure S1) were obtained using an AB Sciex 5800 MALDI TOF/TOF system (Framingham, MA, USA). Data acquisition and data processing were done using a TOF/TOF Series Explorer a nd Data Explorer (both from AB Sciex, Boston, MA, USA), respectively. The instrument was equipped with a 349 nm Nd:YLF OptiBeam On-Axis laser. The laser pulse rate was 400 Hz. Reflectron positive mode was used. Reflectron mode was externally calibrated at 50 ppm mass tolerance and internally at 10 ppm. Each mass spectrum was collected as a sum of 500 shots.
2.5. Akt1 Kinase Activity Assay
The activity of each Akt1 variant was characterized by performing kinase assays in the presence of 200 µM substrate peptide CKRPRAASFAE (SignalChem, Vancouver, BC, Canada) derived from the natural Akt1 substrate, glycogen synthase kinase (GSK-3β). Assays were performed in 3-(N-morpholino) propanesulfonic acid (MOPS, 25 mM, pH 7.0), β-glycerolphosphate (12.5 mM), MgCl2 (25 mM), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA, 5 mM, pH 8.0), ethylenediaminetetraacetic acid (EDTA) (2 mM), ATP (0.02 mM), and 0.4 µCi (0.033 μM) γ-[32P]-ATP in a 30 µL reaction volume. Reactions were incubated at 37 °C and time points were taken over 30 min time courses. As previously [12], reactions were initiated by the addition of 18 pmol of the indicated Akt1 variant to yield a final enzyme concentration of 600 nM and quenched by spotting on P81 paper [24]. For highly active Akt1 variants, the level of Akt1 was titrated to identify a linear range to accurately determine initial velocity (vo) (Figure S2). For this reason, Akt1 activity was compared based on the apparent catalytic rate (kapp) = vo/[Akt1]. Samples from each reaction (5 µL) were spotted on P81 paper at specified time points. Following washes with 1% phosphoric acid (3 × 10 min) and 95% ethanol (1 × 5 min), the P81 paper was air-dried. Incorporation of 32P into the substrate peptide was detected by exposing the P81 paper to a phosphor-imaging screen. The 32P-peptide products were imaged and quantitated using a Storm 860 Molecular Imager and ImageQuant TL software (GE Healthcare, Mississauga, ON, Canada).
2.6. Kinase Inhibition Assay
A concentration gradient (0.001, 0.05, 0.5, 1, 5, 10 µM) of Akti-1/2 inhibitor VIII (Sellekchem, Houston, TX, USA) [25] was selected to determine the concentration dependence of Akt1 inhibition. Kinase inhibition assays were performed exactly as described above (Section 2.5) and with the addition of dimethyl sulfoxide at 10% (vol/vol) (control) or the indicated concentration of Akti-1/2 inhibitor. For each condition, initial velocity was determined over a time course of 10 min with time points at 0, 2, 5, and 10 min (Figure S3). In the inhibition assays, following established protocols [18], the inhibitor was preincubated with the enzyme for 5 min at 37 °C before addition of ATP and Akt1 substrate peptide to start the reaction. The time courses were used to determine the initial velocity of each reaction. The fraction of enzyme inhibition was measured based on initial velocities of uninhibited and inhibited reactions. From these data, half maximal inhibitory concentration (IC50) values were calculated using Sigma Plot (Systat Software, Inc., San Jose, CA, USA).
3. Results
3.1. Production of Recombinant Akt1 Variants
We recently established a method to produce full-length Akt1 variants with programmed phosphorylations [12]. Here we applied this method to produced site-specifically phosphorylated Akt1 variants lacking the auto-inhibitory PH domain. The approach combines in vivo enzymatic phosphorylation with genetic code expansion to produce Akt1 variants containing either or both pThr308 and pSer473 (Figure 2).
Using genetic code expansion [12,20], we incorporated pSer in response to a UAG stop codon at position 473 in the relevant Akt1 constructs. Thr308 was site-specifically phosphorylated by co-expression of the upstream kinase PDK1 in E. coli. We [12] and others [26] have demonstrated that PDK1 is strictly specific in phosphorylating only the 308 site in Akt1. We used these methods in isolation or in combination to produce the three physiologically relevant Akt1 variants pAkt1S473, pAkt1T308, and ppAkt1T308,S473 (Figure 2). We observed that the ΔPH-Akt1 variants were all more soluble and were produced at 1.5- to 7-fold greater yield per liter of E. coli culture than the corresponding full-length Akt1 variants (Table 1). As previously reported [12], we confirmed phosphorylation of the Thr308 site (Figure 3C) and the Ser473 site (Figure S1) by mass spectrometry.
3.2. Recombinant Akt1 Produced in E. coli Versus Sf9 Cells
Earlier work established production of partially active and truncated Akt1 in E. coli, which was unsuccessful at producing a sufficient amount of full-length Akt1 to determine activity [27]. Although we recently overcame these difficulties and developed a robust protocol to produce full-length Akt1 from E. coli with programmed phosphorylation, studies in the intervening period relied on insect Sf9 cell culture to produce recombinant Akt1 [28]. The ability to generate ppAkt1 from insect cells, however, requires a complex and low-yield in vitro procedure to phosphorylate Akt with two additional purified upstream kinases in the presence of lipid vesicles [28]. Protein production in Sf9 cells failed to yeild Akt1 with site-specific or programmed phosphorylation. The resulting protein was a mixture of singly and doubly phosphorylated species [29].
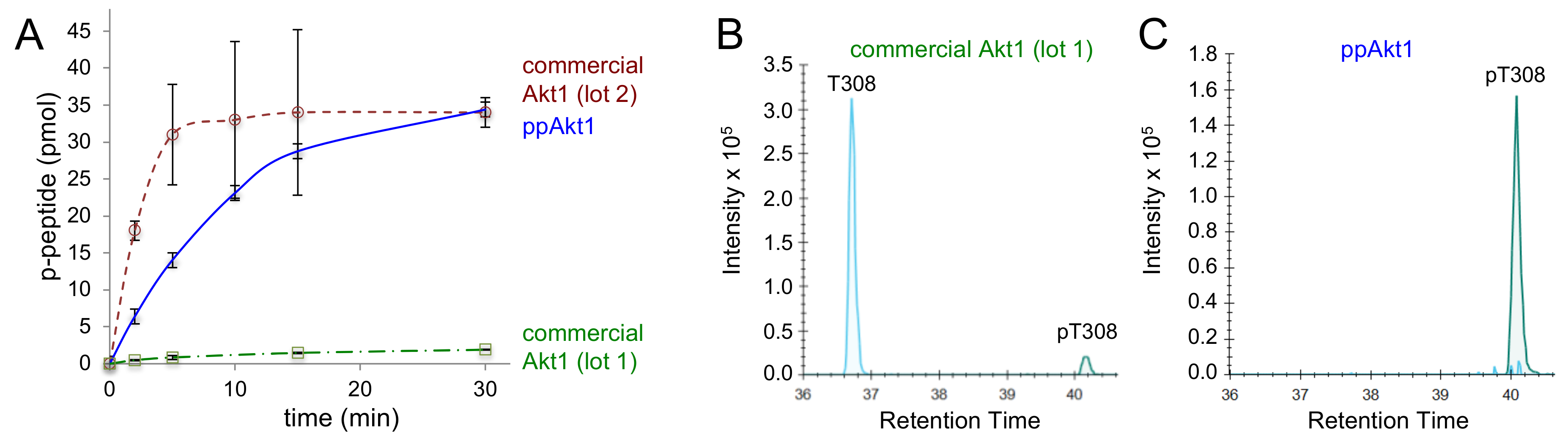
In order to benchmark the activity of the recombinant phosphorylated Akt1 protein we produced in E. coli, we compared the activity of full-length ppAkt1 to active Akt1 purchased from Abcam. The commercially available full-length Akt1 is made by a protocol similar to that established previously [28], in which Akt1 protein production in Sf9 cells is followed by in vitro phosphorylation of the purified Akt1 with purified PDK1. In testing an initial lot of active Akt1 (lot 1), we found that the commercially available Akt1 enzyme was catalytically deficient by eight-fold (Table 2) compared to the full-length ppAkt1 we produced (Figure 3A).
Parallel-reaction monitoring mass spectrometry (PRM-MS/MS) was used to determine the identity of the residue at position 308 and the level of phosphorylation in ppAkt1 we produced and in the purchased protein. Mass spectrometry revealed that the low activity of lot 1 enzyme was attributable to the low level of phosphorylation at position 308 (peak intensity 1.3 × 101) and a relatively high level of unphosphorylated Thr at position 308 (Figure 3B). In contrast, ppAkt1T308,S473 that we produced showed a high level (peak intensity 1.6 × 106) of Thr308 phosphorylation and unphosphorylated Thr308 was not detected (Figure 3C). We then purchased a second lot of Akt1 enzyme (lot 2), and this enzyme displayed significantly more but highly variable activity compared to lot 1 (Table 2). Presumably, the second lot was quantitatively phosphorylated during production. In contrast to the method we developed for pAkt1 production in E. coli (Figure 2), the protocol to generate active Akt1 relying on Sf9 cells and subsequent in vitro phosphorylation appears to lead to a variable level of phosphorylation and activity in the resulting Akt1 preparations.
3.3. Impact of PH Domain Deletion on Differentially Phosphorylated Akt1
In order to determine the impact of the PH domain on each phospho-form of Akt1, we next assayed the activity of specifically phosphorylated ΔPH-Akt1 variants using γ-[32P]-ATP and a substrate peptide for Akt1 (CKRPRAASFAE) that was derived from GSK-3β, a well-established Akt1 substrate [12,30]. In kinase assay conditions that we previously optimized [12] to measure a wide range of Akt1 activity, we determined the apparent catalytic rate of each ΔPH-Akt1 variant (unphosphorylated, pAkt1S473, pAkt1T308, ppAkt1T308,S473). We then compared the apparent catalytic rates to the measurements we made previously with full-length Akt1 and pAkt1 variants to ascertain the relative impact of each phosphorylation state on auto-inhibition by the PH domain.
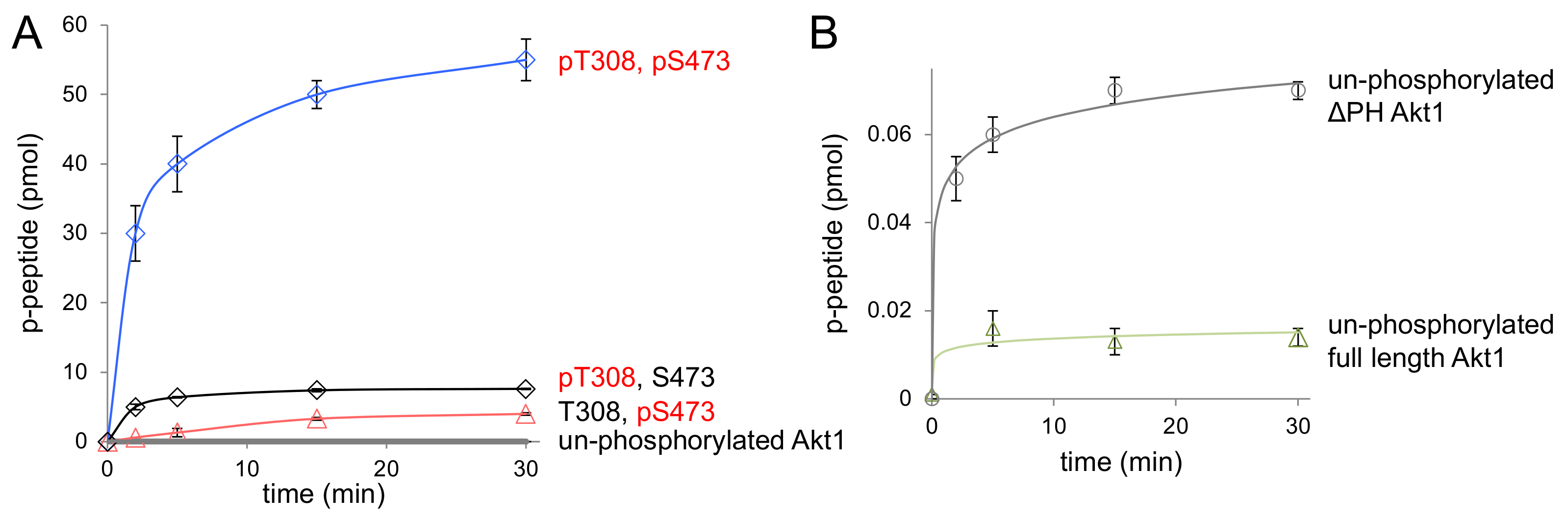
As anticipated, all Akt1 variants lacking the PH domain were significantly more active than their full-length counterparts (Table 2). Doubly phosphorylated ΔPH-Akt1 (ppAktT308,S473) showed the highest activity among all three variants (Figure 4). The unphosphorylated ΔPH-Akt1 showed a basal level of activity that was significantly higher (2.7 ± 0.3-fold) than the background activity we recorded for full-length unphosphorylated Akt1 (Table 3). Although both unphosphorylated Akt1s would not have sufficient activity to induce Akt1-dependent signaling in cells [2,12], it is interesting to note that the auto-inhibitory effect of the PH domain is indeed measurable in the increased minimal activity of unphosphorylated ΔPH-Akt1 (Figure 4B).
Compared to unphosphorylated ΔPH-Akt1, the most active doubly phosphorylated Akt1 variant (ΔPH-ppAktT308,S473) showed a 150,000-fold increase in apparent catalytic rate (Table 2, Figure 4). Since rapid enzyme kinetics were observed with 18 pmol of enzyme with both of these variants containing Thr308 phosphorylation, the enzyme concentrations were subsequently reduced by 10-fold and 100-fold to obtain a highly accurate initial velocity with which to determine the apparent rate (kapp = vo/[enzyme]) (Figure S2).
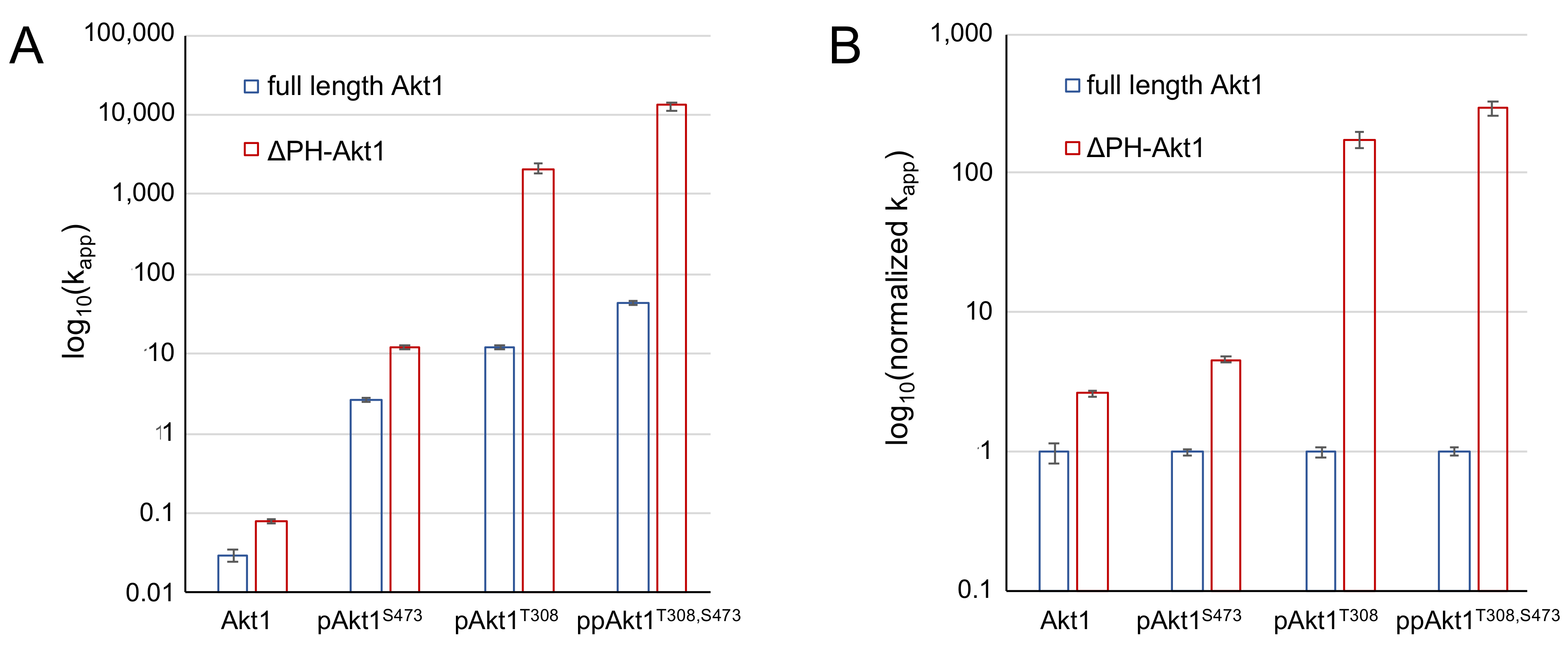
Interestingly, dual phosphorylation of ΔPH-Akt1 led to a 100-fold greater increase in activity than that observed upon dual phosphorylation of the full-length enzyme, which was 1500-fold more active than the unphosphorylated full-length enzyme. The data indicate that the PH domain significantly dampens the catalytic activity endowed by phosphorylation at Thr308 and Ser473 (Figure 5). The ΔPH-Akt1 variant with a single phosphorylation at the Thr308 site was robustly active, 2500-fold above the unphosphorylated ΔPH-Akt1, yet with 60-fold reduced activity compared to the doubly phosphorylated enzyme ΔPH-ppAktT308/S473 (Table 2). Phosphorylation at the C-terminal site Ser473 activated the enzyme 140-fold above the unphosphorylated control, but to a lesser extent (18-fold) than ΔPH-pAkt1T308. We previously observed a quantitatively similar pattern of activity with the full-length Akt1 variants (Table 2, [12]).
We found that the PH domain exerts an auto-inhibitory effect, the strength of which depends on the phosphorylation status of the Akt1 enzyme. We compared apparent catalytic rates, kapp, (Figure 5A) and normalized relative catalytic rates (Figure 5B) between the full-length [12] and PH domain–deficient Akt1 variants. In the context of a single phosphorylation at Ser473, the PH domain is associated with an approximately five-fold reduction in activity (Table 3, Figure 5). In the singly phosphorylated pAkt1T308 enzyme, the auto-inhibition was far stronger (175-fold). The two phosphorylations together led to a super-additive inhibitory effect (295-fold) in the doubly phosphorylated enzyme (Figure 5).
3.4. Chemical Inhibition of Phosphorylated Akt1 Variants
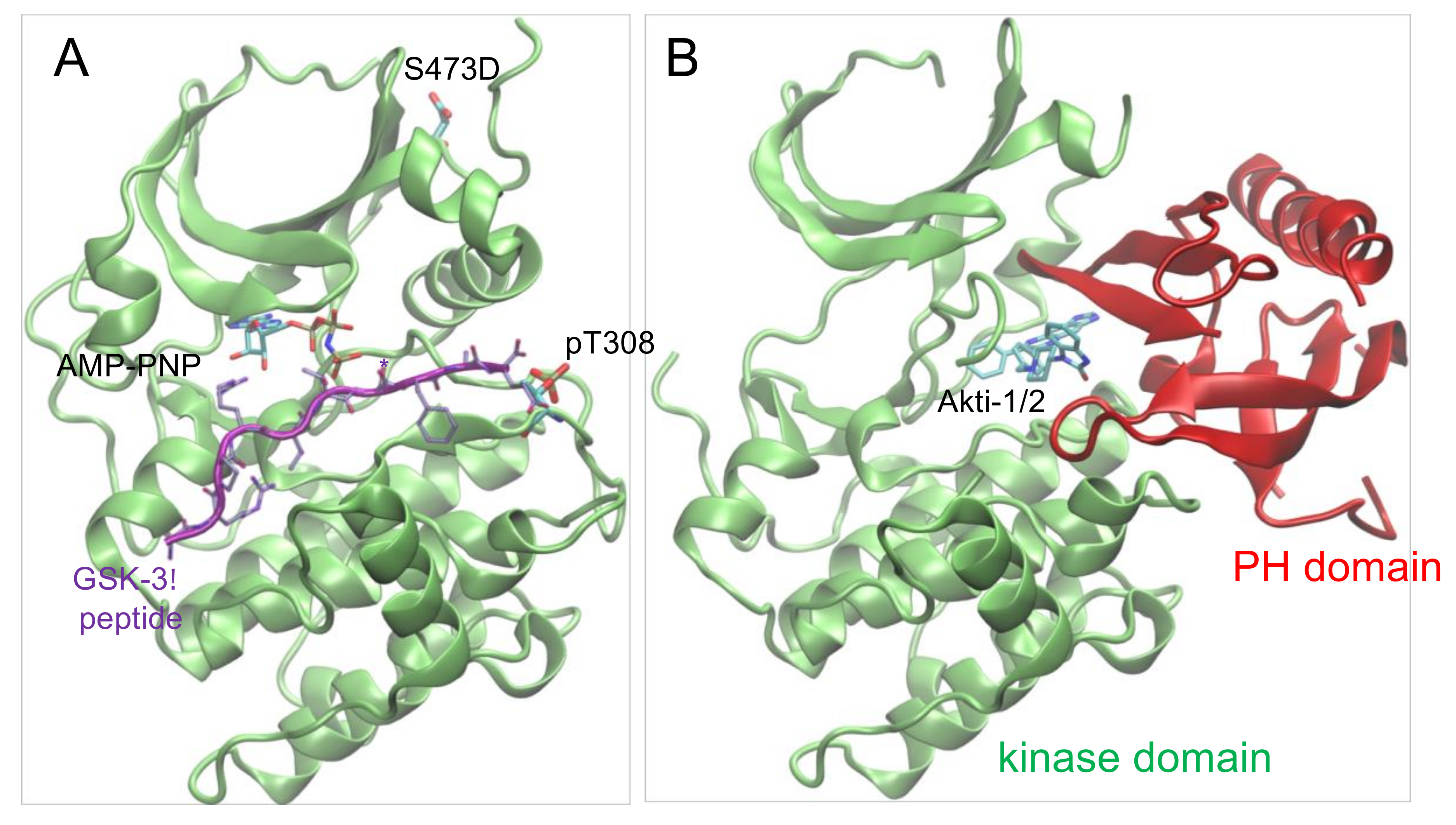
Given the differential impact of Akt1 phosphorylation on auto-inhibition, we next identified phosphorylation dependence in the interaction between Akt1 and a clinically relevant drug scaffold, Akti-1/2 inhibitor VIII. Several classes of chemical inhibitors were developed to repress aberrant Akt1 activity in cancer cells [10]. Early Akt1 inhibitors focused on ATP competitive compounds [31], yet these molecules suffered from significant cross-reactivity with other AGC family kinases [32]. For this reason, subsequent efforts focused on allosteric Akt1 inhibitors. Akti-1/2 is an allosteric inhibitor that binds Akt1 in a cleft between the kinase and PH domains, locking the enzyme in a noncatalytic conformation (Figure 6). Biochemical data with different Akt1 preparations has provided estimates of IC50 of Akt1 for the Akti-1/2 inhibitor ranging from 58 nM to 2.7 μM [24,33,34] (Table 4). We suspected that this range of values resulted from preparations of Akt1 with various levels of phosphorylation, with less active Akt1 preparations leading to an underestimate of IC50. In addition, previous studies were unable to isolate the contribution of each regulatory phosphorylation site to inhibition by Akti-1/2.
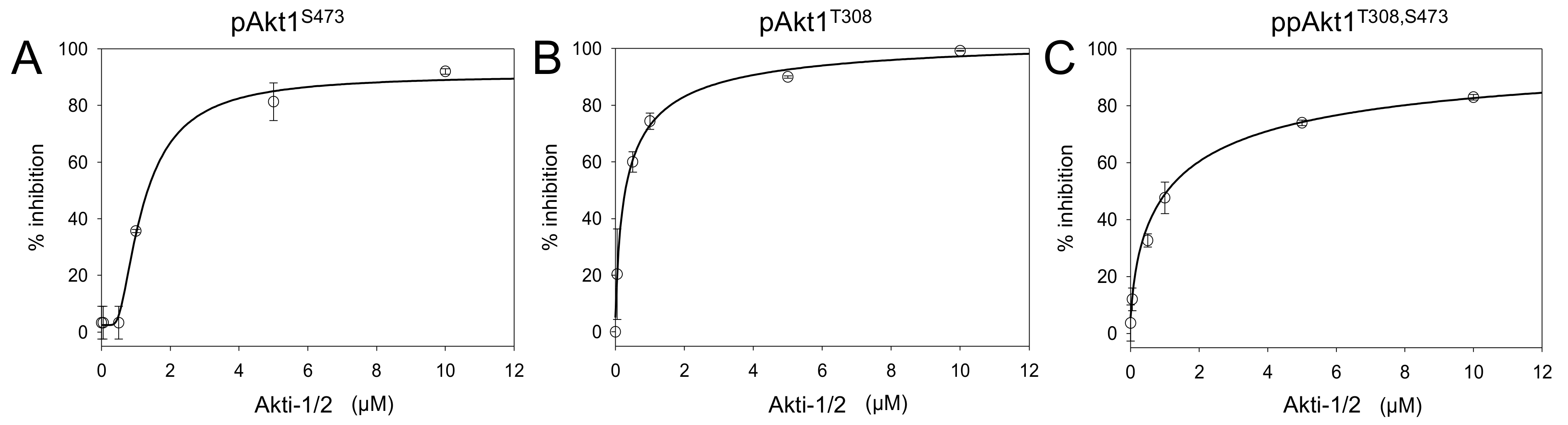
We conducted Akt1 inhibition assays using Akti-1/2 concentrations varying from 0.01 to 10 μM. All three full-length phosphorylated Akt1 variants showed concentration-responsive enzyme inhibition (Figure 7, Figures S3–S6). Akti-1/2 was most potent (IC50 = 300 nM) when only the Thr308 site was phosphorylated (Table 4). Interestingly, at the lower concentrations of Akti-1/2 (0.001, 0.05, 0.5 μM), the compound was not effective at inhibiting the activity of pAkt1S473. Akt1 phosphorylated at the C-terminal Ser473 site was overall more resistant to inhibitor VIII inhibition, resulting in an IC50 value of 1.3 μM. In a surprising finding, the doubly phosphorylated enzyme had an indistinguishable IC50 from the pAkt1S473 enzyme. Together the data indicate that Ser473 phosphorylation provides a four-fold increase in resistance to the Akti-1/2 inhibitor even in the presence of phosphorylation at Thr308. This is the first report demonstrating phosphorylation dependence in the interaction between Akt1 and an inhibitor.
4. Discussion
Akt1 is a prime target for therapeutic intervention due to its involvement in regulating a multitude of cellular pathways [37,38]. Aberrant Akt function has been linked to cancers and a variety of human diseases related to metabolic regulation, immune function, and neurological development [2]. The broad range of cellular processes governed by Akt, therefore, presents an opportunity for single-target therapeutic intervention in a variety of conditions. To capitalize on this opportunity, compounds that selectively attenuate Akt1 activity rather than global kinase inactivation are required. This need is reflected in clinical trials showing that nonspecific Akt inhibition in cancer therapy, due to inadvertent off-targeting of closely related kinases or multiple Akt isozymes, can result in detrimental side effects including liver damage and metabolic disorders [39,40,41,42]. Accordingly, the development of small molecule inhibitors for Akt reflects this sentiment of engineering greater target specificity [10]. From the earliest ATP-competitive inhibitors that unintentionally inhibited related AGC kinases, to more recent allosteric inhibitors that preferentially target Akt or even a subset of Akt isozymes, the continued pursuit for Akt1 inhibitor specificity requires an expanded understanding of the molecular basis of Akt1 activation and inhibition.
Previous work was unable to uncover the role of each regulatory site (Thr308 and Ser473) in the inhibition kinetics of Akt1. Given this critical knowledge gap, we investigated the role of Akt1 phosphorylation status on the auto-inhibitory effect of the PH domain and on allosteric inhibition by Akti-1/2.
4.1. Phosphorylation-Dependent Auto-Inhibition of Akt1
The N-terminal PH domain is shared among all three Akt isozymes and its direct function is to mediate protein–lipid interactions. It is well documented that PH domain binding to a lipid second messenger PIP3 in cell membranes releases the inhibitory effect of the PH domain [2]. Here, we produced differentially phosphorylated Akt1 variants with and without the PH domain in E. coli using genetic code expansion to incorporate pSer473, and the upstream Thr308 kinase PDK1 was co-expressed in E. coli, leading to quantitative phosphorylation of the recombinant Akt1 protein. Compared to unphosphorylated Akt1, all phosphorylated variants showed increased enzyme activity.
Previously, live cell imaging studies revealed that, once phosphorylated, Akt1 favors a PH-out conformation that increases Akt1 activity in the downstream phosphorylation of substrate proteins in both the cytoplasm and the nucleus. In addition, binding of Akt1 to PIP3 in the plasma membrane further releases the auto-inhibitory effect of the PH domain [43,44]. Although phosphorylation of the kinase domain at Thr308 is known to reduce the PH domain’s affinity for the kinase domain [42], the contribution of Thr308 and Ser473 phosphorylation to auto-inhibition by the PH domain was unknown.
We quantitated the release of PH domain–mediated inhibition corresponding to each phosphorylation site and both sites in combination. Our data revealed that the magnitude of the auto-inhibitory effect of the PH domain was sensitively dependent on the phosphorylation status of Akt1. In comparison to the activity of full-length Akt1 phospho-variants, deletion of the PH domain significantly increased activity. In comparison to singly phosphorylated Akt1 variants, the auto-inhibitory effect of the PH domain was strongest (295-fold) in the most active and doubly phosphorylated Akt1 variant. Interestingly, the doubly phosphorylated Akt1 displayed increased activity upon PH domain deletion that was greater than the sum of the effects observed for the singly phosphorylated Akt1 variants. The data suggest a novel finding that the PH domain dampens the activity of Akt1 variants in a phosphorylation-dependent manner.
The combination of phosphorylation and PH domain release regulates the kinase activity of Akt1 over a 400,000-fold range. The data indicate the degree to which the unphosphorylated PH-in conformation of Akt1 is suppressed in comparison to the PH-out doubly phosphorylated version of the enzyme that exists at the plasma membrane in living cells. Recent in vitro biochemical work suggested that association with PIP3-containing lipid vesicles stimulated the activity of a full-length Akt1 protein by seven-fold [44]. The authors suggested that this may be an underestimate due to low levels of phosphorylation (10% pThr308, 1% pSer473) in their Akt1 prepared from Sf9 cells. Our data suggest that release of the PH domain in the context of doubly phosphorylated Akt1 would lead to a maximal activity increase of 300-fold; a PH domain–dependent reduction lower than this value would indicate that there may be significant interaction between the Akt1 kinase and PH domains even in the PIP3-bound state at the plasma membrane.
Indeed, a recent debate in the literature [45] is under way to explain how Akt1 activity can be maintained far from the membrane. Recent findings suggest that Akt1 is dephosphorylated on a time scale of ~10 min following dissociation from PIP3 on the plasma membrane [46]. Multiple nuclear substrates of Akt1 are, however, known to be phosphorylated much more rapidly, on a time scale of seconds [47]. Although the authors suggested that all or perhaps most of the active Akt1 is membrane-associated [46], this finding disputes several independent studies identifying many cytosolic and nuclear targets of Akt1-dependent phosphorylation. Multiple studies clearly identified both phospho-Akt1 (e.g., [47,48,49]) and directly measured Akt1 activity in the nucleus [17]. Using a fluorescent reporter that quantifies Akt1 activity in live cells in real time, Kunkel and Newton observed Akt1 activity distributed in both the cytoplasm and nucleus on a time scale of seconds to minutes following growth factor stimulation. Their data further show similar levels of Akt1 activity in the nucleus and cytoplasm, with a delay in the peak activity in the nucleus of ~2 min. Additional experiments confirmed that this lag is related to the time it takes for active Akt1 to translocate from the plasma membrane to the nucleus [17]. In light of our data and these findings, membrane-associated Akt1 is likely significantly more active than doubly phosphorylated Akt1 found in the nucleus and cytosol. Phosphorylated Akt1, nevertheless, does persist away from the membrane, retaining significant activity in comparison to unphosphorylated Akt1.
4.2. Phosphorylation-Dependent Chemical Inhibition of Akt1
Posttranslational modifications, of which phosphorylation is the most common, are ubiquitous mechanisms that cells use to tune the activity of individual proteins and increase the functional diversity of the proteome. Phosphorylation can have a significant impact on the activity of proteins (kinases are a prime example), and the phosphorylation status of a protein target can accordingly impact the ability of small molecules to inhibit enzyme activity. In the experiments presented here, we determined precisely how the phosphorylation status of Akt1 influences the repression of kinase activity by chemical inhibition. We investigated the Akt-specific allosteric inhibitor Akti-1/2. Based on the results of our assays, the phosphorylation status of Akt1 can indeed influence the degree to which Akti-1/2 is able to act on Akt1. Akti-1/2 was most effective at inhibiting pAkt1T308, whereas phosphorylation at position Ser473 in either pAktS473 or ppAktT308/S473 reduced the relative effectiveness of the inhibitor by four-fold.
Ser473 phosphorylation appears to act as a built-in mechanism of drug resistance for Akt1. It is well established that hyperphosphorylation of Akt1 at both regulatory sites is linked to poor prognosis and therapeutic resistance [3,4,5,6,50]. The accepted model for Akti-1/2 activity suggests that the compound locks Akt1 into the auto-inhibited or PH-in conformation and prevents Akt1 from binding PIP3 in the plasma membrane, inhibiting phosphorylation at Thr308 and Ser473 [51]. Our data also suggest that, although limited by Ser473 phosphorylation, Akti-1/2 has significant activity in inhibiting the pre-phosphorylated Akt1 variants that the inhibitor initially encounters in the cell. Studies in cell culture and mouse models show that the short-term impact of Akti-1/2 treatment is that pThr308 and pSer473 levels are depleted within hours. A related allosteric Akt inhibitor, and the most clinically promising [52] compound, MK-2206 significantly reduces Ser473 phosphorylation after 10 h in mouse models with patient-derived xenografts of endometrial tumors [53].
Surprisingly, breast cancer cells (BT474) treated with Akti-1/2 (1 μM) showed a rebound effect in Akt1 phosphorylation status following Akti-1/2 treatment. These cells have amplified HER2 genes and constitutively activate the PI3K/Akt signaling cascade. In short-term experiments (minutes to hours), Akt1 phosphorylation reduced in response to inhibitor treatment, but at longer times (2–3 days) Akt1 phosphorylation status returned to stimulated levels in these cancer cells. In this longer time frame, phosphorylation of the downstream Akt1 target S6K was not restored, but PRAS40 phosphorylation was partially restored concomitant with increased Akt1 phospho-status. The study provided evidence that Akti-1/2 ultimately induces the expression and activation of receptor tyrosine kinases EGFR, HER3, and HER4, which in turn reactivate Akt1 [54]. Interestingly, in mouse models a similar rebound in Ser473 phosphorylation following treatment with MK-2206 was observed on time courses of 5 to 20 days [55]. In these cases, Ser473 phosphorylation may provide the tumor cell with a means to reduce the effectiveness of Akt1 inhibitors. The data suggest a need to develop an inhibitor that is more effective against the most active and doubly phosphorylated form of Akt1.
4.3. Synthetic Biology Approach to Generate Active Akt1
In our current study and previous work [12,19,20,21], we demonstrated that genetic code expansion with pSer in E. coli provides a simple route to produce designer phosphoproteins. Here, we produced differentially phosphorylated Akt1 variants with and without the PH domain in E. coli using genetic code expansion and enzymatic phosphorylation in E. coli. We found that the variants lacking the PH domain were produced at significantly higher yield compared to the full-length Akt1 protein. The ability to produce recombinant Akt1 protein with programmed phosphorylation(s) was essential for our investigation into the role of each phospho-site in Akt1 inhibition.
In agreement with our previous work [12], we found that the system we developed to produce Akt1 with either or both regulatory phosphorylation sites (Figure 2) leads to a consistently active Akt1 variant with indistinguishable batch-to-batch variability. This is in contrast to the commercially available Akt1 produced using the Sf9 cell system followed by in vitro PDK1 phosphorylation of Thr308 [28]. We observed vast batch-to-batch variability of enzyme activity in the commercially available Akt1, which warrants cautious attention in its experimental use. Previous work also established that Akt1 produced in Sf9 cells leads to a mixture of singly and doubly phosphorylated Akt1 variants [29], at times leading to low stoichiometry of phosphorylation [44] (Figure 3B). Our approach consistently results in site-specifically phosphorylated and active Akt1 variants that provide a reliable and consistent source of protein for biochemical studies and applications in inhibitor screening.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/9/9/450/s1, Supplementary Materials and Methods, Table S1: Plasmids used in this study, Figure S1: Mass spectra confirming genetically encoded phosphoserine in ΔPH-ppAkt1, Figure S2: Initial velocity measurements for highly active ΔPH-Akt1 variants, Figure S3: Inhibition of full-length Akt1 variants incubated with Akti-1/2 inhibitor VIII, Figure S4: Autoradiographs of Akt1 inhibitor assays with ppAkt1T308, S473, Figure S5: Autoradiographs of Akt1 inhibitor assays with pAkt1T308, Figure S6: Autoradiographs of Akt1 inhibitor assays with pAkt1S473.
Author Contributions
Conceptualization, P.O., S.S.C.L.; methodology, N.B., X.L.; validation, N.B., M.M., X.L.; formal analysis, N.B.; investigation, N.B., M.M., X.L., P.O.; resources, P.O., S.S.C.L.; data curation, N.B., M.M.; writing—original draft preparation, N.B., M.M., P.O.; writing—review and editing, N.B., M.M., P.O.; visualization, N.B., M.M., X.L., P.O.; supervision, P.O., S.S.C.L.; project administration, P.O.; funding acquisition, P.O., S.S.C.L.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN 04282-2014 to P.O.), a Canadian Cancer Society Research Institute innovation grant (704324 to P.O. and S.S.C.L.), the Canada Foundation for Innovation (229917 to P.O.), the Ontario Research Fund (229917 to P.O.), Canada Research Chairs (950-229917 to P.O.), and the Canadian Breast Cancer Foundation (to S.S.C.L.).
Acknowledgments
We are grateful to Ilka Heinemann and Murray Junop for critical discussions and suggestions. We also thank Kristina Jurcic and Chaochao Chen at the MALDI Mass Spectrometry Facility at the University of Western Ontario for assistance with mass spectrometry analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, E.; Brattain, M.G.; Chowdhury, S. Cell survival and metastasis regulation by Akt signaling in colorectal cancer. Cell. Signal 2013, 25, 1711–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, A.; Yoon, S.S.; Harrison, S.J.; Morris, S.R.; Smith, D.A.; Brigandi, R.A.; Gauvin, J.; Kumar, R.; Opalinska, J.B.; Chen, C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014, 124, 2190–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelli, M.; Massimino, M.; Morra, I.; Garre, M.L.; Gardiman, M.P.; Buttarelli, F.R.; Arcella, A.; Giangaspero, F. Expression of pERK and pAKT in pediatric high grade astrocytomas: Correlation with YKL40 and prognostic significance. Neuropathology 2012, 32, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shirai, K.; Oka, K.; Mobaraki, A.; Yoshida, Y.; Noda, S.E.; Okamoto, M.; Suzuki, Y.; Itoh, J.; Itoh, H.; et al. Higher pAkt expression predicts a significant worse prognosis in glioblastomas. J. Radiat. Res. 2010, 51, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Blachly, J.S.; Baiocchi, R.A. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br. J. Haematol. 2014, 167, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin. Lymphoma Myeloma Leuk 2014, 14, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Jung-Testas, I.; Hu, Z.Y.; Baulieu, E.E.; Robel, P. Neurosteroids: Biosynthesis of pregnenolone and progesterone in primary cultures of rat glial cells. Endocrinology 1989, 125, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasuriya, N.; Kunkel, M.T.; Liu, X.; Biggar, K.K.; Li, S.S.; Newton, A.C.; O’Donoghue, P. Genetic code expansion and live cell imaging reveal that Thr308 phosphorylation is irreplaceable and sufficient for Akt1 activity. J. Biol. Chem. 2018, 293, 10744–10756. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.X.; Ng, Y.; Meoli, C.C.; Kumar, A.; Khoo, P.S.; Fazakerley, D.J.; Junutula, J.R.; Vali, S.; James, D.E.; Stockli, J. Amplification and demultiplexing in insulin-regulated Akt protein kinase pathway in adipocytes. J. Biol. Chem. 2012, 287, 6128–6138. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.; Levinger, I.; Mousa, A.; Howlett, K.; de Courten, B. Plasma 25-hydroxyvitamin D is related to protein signaling involved in glucose homeostasis in a tissue-specific manner. Nutrients 2016, 8, 631. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wu, Y.; Liu, M.; Qin, Y.; Wang, H.; Wang, L.; Li, S.; Zhu, H.; He, Z.; Luo, J.; et al. SEMA3B improves the survival of patients with esophageal squamous cell carcinoma by upregulating p53 and p21. Oncol. Rep. 2016, 36, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.R.; Vogt, P.K. Phosphorylation of AKT: A mutational analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, M.T.; Ni, Q.; Tsien, R.Y.; Zhang, J.; Newton, A.C. Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem. 2005, 280, 5581–5587. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.; Janakiraman, V.; Wu, W.I.; Foo, C.K.; Kljavin, N.M.; Chaudhuri, S.; Stawiski, E.; Lee, B.; Lin, J.; Li, H.; et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 19368–19373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.; Wang, S.M.; Bi, Y.; Treidlinger, M.; Barber, K.R.; Shaw, G.S.; O’Donoghue, P. Ubiquitin phosphorylated at Ser57 hyper-activates parkin. Biochim. Biophys. Acta 2017, 1861, 3038–3046. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Aguirre, J.D.; Spratt, D.E.; Bi, Y.; Jeffery, M.; Shaw, G.S.; O’Donoghue, P. Generation of phospho-ubiquitin variants by orthogonal translation reveals codon skipping. FEBS Lett. 2016, 590, 1530–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerni, H.R.; Shifman, M.A.; Rogulina, S.; O’Donoghue, P.; Rinehart, J. Revealing the amino acid composition of proteins within an expanded genetic code. Nucleic Acids Res. 2015, 43, e8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Oh, S.; Yang, A.; Kim, J.; Soll, D.; Lee, D.; Park, H.S. A facile strategy for selective incorporation of phosphoserine into histones. Angew. Chem. Int. Ed. Engl. 2013, 52, 5771–5775. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Turowec, J.P.; Duncan, J.S.; French, A.C.; Gyenis, L.; St Denis, N.A.; Vilk, G.; Litchfield, D.W. Protein kinase CK2 is a constitutively active enzyme that promotes cell survival: Strategies to identify CK2 substrates and manipulate its activity in mammalian cells. Method Enzymol. 2010, 484, 471–493. [Google Scholar]
- Lindsley, C.W.; Zhao, Z.; Leister, W.H.; Robinson, R.G.; Barnett, S.F.; Defeo-Jones, D.; Jones, R.E.; Hartman, G.D.; Huff, J.R.; Huber, H.E.; et al. Allosteric Akt (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Klein, S.; Geiger, T.; Linchevski, I.; Lebendiker, M.; Itkin, A.; Assayag, K.; Levitzki, A. Expression and purification of active PKB kinase from Escherichia coli. Protein Expr. Purif. 2005, 41, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, S.; Yamane, H.; Wahl, R.; Ali, A.; Lofgren, J.A.; Kendall, R.L. Kinetic mechanism of AKT/PKB enzyme family. J. Biol. Chem. 2006, 281, 13949–13956. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Batt, D.; Rose, P.; Schacher, B.; Roberts, T.M.; Ferrari, S. Homogeneous purification of human recombinant GST-Akt/PKB from Sf9 cells. Protein Expr. Purif. 1999, 17, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Caudwell, F.B.; Andjelkovic, M.; Hemmings, B.A.; Cohen, P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996, 399, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Reuveni, H.; Livnah, N.; Geiger, T.; Klein, S.; Ohne, O.; Cohen, I.; Benhar, M.; Gellerman, G.; Levitzki, A. Toward a PKB inhibitor: Modification of a selective PKA inhibitor by rational design. Biochemistry 2002, 41, 10304–10314. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Walton, M.I.; Grimshaw, K.M.; Te Poele, R.H.; Eve, P.D.; Valenti, M.R.; de Haven Brandon, A.K.; Martins, V.; Zetterlund, A.; Heaton, S.P.; et al. AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin. Cancer Res. 2012, 18, 3912–3923. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Goransson, O.; Kular, G.S.; Leslie, N.R.; Gray, A.; Alessi, D.R.; Sakamoto, K.; Hundal, H.S. Use of Akt inhibitor and a drug-resistant mutant validates a critical role for protein kinase B/Akt in the insulin-dependent regulation of glucose and system A amino acid uptake. J. Biol. Chem. 2008, 283, 27653–27667. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.F.; Defeo-Jones, D.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Jones, R.E.; Kahana, J.A.; Kral, A.M.; Leander, K.; Lee, L.L.; et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 2005, 385, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef] [PubMed]
- Massihnia, D.; Avan, A.; Funel, N.; Maftouh, M.; van Krieken, A.; Granchi, C.; Raktoe, R.; Boggi, U.; Aicher, B.; Minutolo, F.; et al. Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slipicevic, A.; Holm, R.; Nguyen, M.T.; Bohler, P.J.; Davidson, B.; Florenes, V.A. Expression of activated Akt and PTEN in malignant melanomas: Relationship with clinical outcome. Am. J. Clin. Pathol. 2005, 124, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Chen, X.; Hay, N. Akt as a target for cancer therapy: More is not always better (lessons from studies in mice). Br. J. Cancer 2017, 117, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wan, M.; Leavens, K.F.; Chu, Q.; Monks, B.R.; Fernandez, S.; Ahima, R.S.; Ueki, K.; Kahn, C.R.; Birnbaum, M.J. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med. 2012, 18, 388–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouthamel, M.C.; Kahana, J.A.; Korenchuk, S.; Zhang, S.Y.; Sundaresan, G.; Eberwein, D.J.; Brown, K.K.; Kumar, R. Mechanism and management of AKT inhibitor-induced hyperglycemia. Clin. Cancer Res. 2009, 15, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Alcor, D.; Laguerre, M.; Park, J.; Vojnovic, B.; Hemmings, B.A.; Downward, J.; Parker, P.J.; Larijani, B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007, 5, e95. [Google Scholar] [CrossRef] [PubMed]
- Lucic, I.; Rathinaswamy, M.K.; Truebestein, L.; Hamelin, D.J.; Burke, J.E.; Leonard, T.A. Conformational sampling of membranes by Akt controls its activation and inactivation. Proc. Natl. Acad. Sci. USA 2018, 115, E3940–E3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.K. How to explain the AKT phosphorylation of downstream targets in the wake of recent findings. Proc. Natl. Acad. Sci. USA 2018, 115, E6099–E6100. [Google Scholar] [CrossRef] [PubMed]
- Ebner, M.; Lucic, I.; Leonard, T.A.; Yudushkin, I. PI(3,4,5)P3 engagement restricts akt activity to cellular membranes. Mol. Cell 2017, 65, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Azimifar, S.B.; Mann, M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015, 33, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Hu, C.; Li, J.; Xue, P.; He, X.; Ge, C.; Qin, W.; Yao, G.; Gu, J. Real-time imaging nuclear translocation of Akt1 in HCC cells. Biochem. Biophys. Res. Commun. 2007, 356, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Hixon, M.L.; Boekelheide, K. Expression and localization of total Akt1 and phosphorylated Akt1 in the rat seminiferous epithelium. J. Androl. 2003, 24, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Freudlsperger, C.; Horn, D.; Weissfuss, S.; Weichert, W.; Weber, K.J.; Saure, D.; Sharma, S.; Dyckhoff, G.; Grabe, N.; Plinkert, P.; et al. Phosphorylation of AKT(Ser473) serves as an independent prognostic marker for radiosensitivity in advanced head and neck squamous cell carcinoma. Int. J. Cancer 2015, 136, 2775–2785. [Google Scholar] [CrossRef] [PubMed]
- Okuzumi, T.; Fiedler, D.; Zhang, C.; Gray, D.C.; Aizenstein, B.; Hoffman, R.; Shokat, K.M. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 2009, 5, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisinski, K.B.; Tevaarwerk, A.J.; Burkard, M.E.; Rampurwala, M.; Eickhoff, J.; Bell, M.C.; Kolesar, J.M.; Flynn, C.; Liu, G. Phase I study of an AKT inhibitor (MK-2206) combined with lapatinib in adult solid tumors followed by dose expansion in advanced HER2+ breast cancer. Clin. Cancer Res. 2016, 22, 2659–2667. [Google Scholar] [CrossRef] [PubMed]
- Winder, A.; Unno, K.; Yu, Y.; Lurain, J.; Kim, J.J. The allosteric AKT inhibitor, MK2206, decreases tumor growth and invasion in patient derived xenografts of endometrial cancer. Cancer Biol. Ther. 2017, 18, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Piao, H.L.; Zhuang, L.; Sarbassov dos, D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
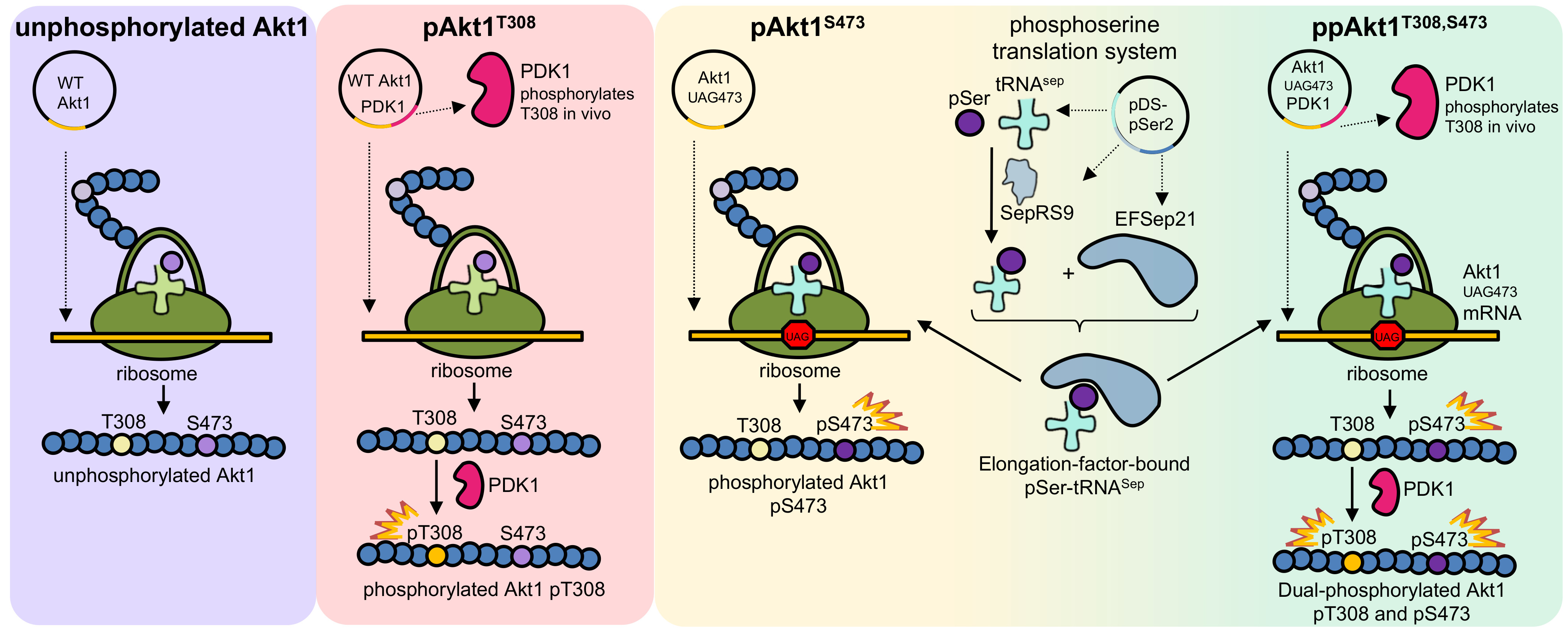
Figure 1.
Simplified schematic of protein kinase B (Akt1) activation via phosphorylation of sites Thr308 and Ser473. The transition from Akt1’s inactive state (PH-in) to its fully active state (ppAkt1T308/S473) requires the release of pleckstrin homology (PH) domain–mediated auto-inhibition. This release occurs when Akt1’s PH domain interacts with PIP3 (PH-out). In the PH-out conformation, Akt1 is more susceptible to phosphorylation at Thr308 and Ser473 by phosphoinositide dependent kinase 1 (PDK1) and mechanistic target of rapamycin complex 2 (mTORC2), respectively. Upon release from PIP3, Akt1 distributes rapidly in the cytosol and translocates to the nucleus to phosphorylate >100 cellular proteins [2,17].
Figure 1.
Simplified schematic of protein kinase B (Akt1) activation via phosphorylation of sites Thr308 and Ser473. The transition from Akt1’s inactive state (PH-in) to its fully active state (ppAkt1T308/S473) requires the release of pleckstrin homology (PH) domain–mediated auto-inhibition. This release occurs when Akt1’s PH domain interacts with PIP3 (PH-out). In the PH-out conformation, Akt1 is more susceptible to phosphorylation at Thr308 and Ser473 by phosphoinositide dependent kinase 1 (PDK1) and mechanistic target of rapamycin complex 2 (mTORC2), respectively. Upon release from PIP3, Akt1 distributes rapidly in the cytosol and translocates to the nucleus to phosphorylate >100 cellular proteins [2,17].

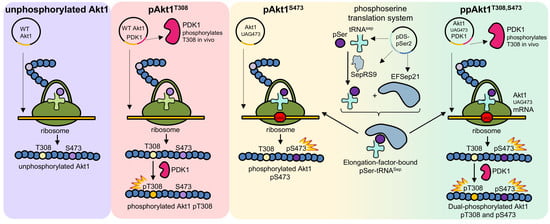
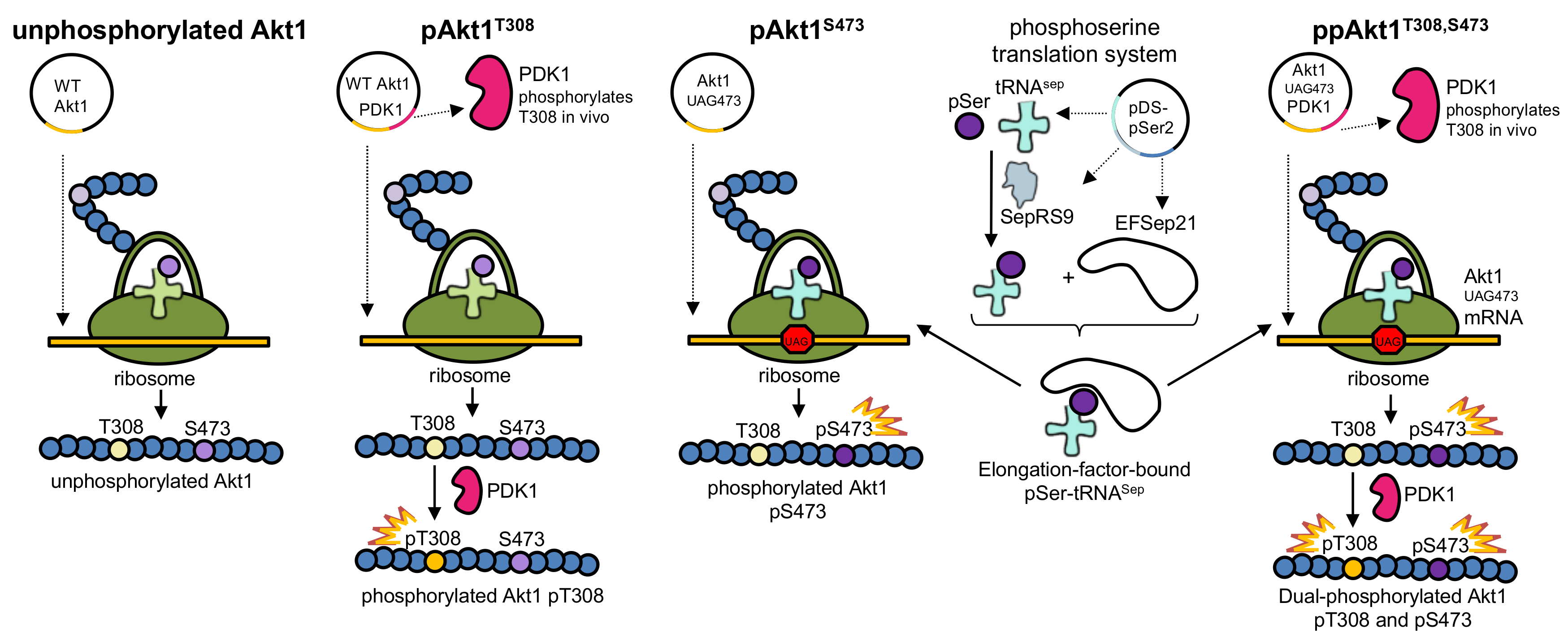
Figure 2.
Production of Akt1 variants with programmed phosphorylation. To produce pAkt1T308, PDK1 (Akt1’s natural upstream kinase) was co-expressed along with Akt1. To produce pAkt1S473, the phosphoserine orthogonal translation system was used to genetically incorporate phosphoserine at position 473 in response to an amber (UAG) codon. The ppAkt1T308,S473 variant was produced by combining both methods. WT: wild type.
Figure 2.
Production of Akt1 variants with programmed phosphorylation. To produce pAkt1T308, PDK1 (Akt1’s natural upstream kinase) was co-expressed along with Akt1. To produce pAkt1S473, the phosphoserine orthogonal translation system was used to genetically incorporate phosphoserine at position 473 in response to an amber (UAG) codon. The ppAkt1T308,S473 variant was produced by combining both methods. WT: wild type.

Figure 3.
Activity of full-length ppAkt1 and commercially available active Akt1. (A) Kinase activity assays over a 30 min time course show substantially reduced activity of commercial Akt1 (lot 1, green squares) compared to full-length ppAkt1 (blue diamonds) produced in E. coli. Commercial Akt1 lot 2 (red circles) showed highly variable but similar activity to full-length ppAkt1. (B) Tryptic peptides from commercial Akt1 and ppAkt1 were analyzed by parallel-reaction monitoring mass spectrometry (PRM-MS). The purchased active Akt1 (lot 1) showed a low-intensity peak for phosphorylation at Thr308 (green peak at a retention time of ~40 min) and high-intensity peak for non-phosphorylated Thr308 (cyan peak, retention time of ~37 min). (C) PRM-MS analysis of ppAkt1T308,S473 showed a high-intensity peak for phosphorylation at Thr308 (green peak at a retention time of 40 min) and the non-phosphorylated Thr308 was undetectable.
Figure 3.
Activity of full-length ppAkt1 and commercially available active Akt1. (A) Kinase activity assays over a 30 min time course show substantially reduced activity of commercial Akt1 (lot 1, green squares) compared to full-length ppAkt1 (blue diamonds) produced in E. coli. Commercial Akt1 lot 2 (red circles) showed highly variable but similar activity to full-length ppAkt1. (B) Tryptic peptides from commercial Akt1 and ppAkt1 were analyzed by parallel-reaction monitoring mass spectrometry (PRM-MS). The purchased active Akt1 (lot 1) showed a low-intensity peak for phosphorylation at Thr308 (green peak at a retention time of ~40 min) and high-intensity peak for non-phosphorylated Thr308 (cyan peak, retention time of ~37 min). (C) PRM-MS analysis of ppAkt1T308,S473 showed a high-intensity peak for phosphorylation at Thr308 (green peak at a retention time of 40 min) and the non-phosphorylated Thr308 was undetectable.

Figure 4.
Enzyme activity of ΔPH-Akt1 variants. (A) The activity of differentially phosphorylated ΔPH Akt1 variants with the GSK-3β substrate peptide was measured over a 30 min time course. Akt1 phosphorylated at 308 and 473 (ΔPH-ppAktS473,T308, blue diamonds) showed maximal activity compared to unphosphorylated ΔPH-Akt1 (gray circles) and singly phosphorylated Akt1 variants ΔPH-pAkt1T308 (black diamonds) and ΔPH-pAkt1S473 (pink triangles). (B) The basal activity of unphosphorylated ΔPH-Akt1 (gray circles) was compared to full-length unphosphorylated Akt1 (green triangles). All reported values represent the mean of triplicate experiments, with error bars indicating one standard deviation.
Figure 4.
Enzyme activity of ΔPH-Akt1 variants. (A) The activity of differentially phosphorylated ΔPH Akt1 variants with the GSK-3β substrate peptide was measured over a 30 min time course. Akt1 phosphorylated at 308 and 473 (ΔPH-ppAktS473,T308, blue diamonds) showed maximal activity compared to unphosphorylated ΔPH-Akt1 (gray circles) and singly phosphorylated Akt1 variants ΔPH-pAkt1T308 (black diamonds) and ΔPH-pAkt1S473 (pink triangles). (B) The basal activity of unphosphorylated ΔPH-Akt1 (gray circles) was compared to full-length unphosphorylated Akt1 (green triangles). All reported values represent the mean of triplicate experiments, with error bars indicating one standard deviation.

Figure 5.
Impact of PH domain on the activity of differentially phosphorylated Akt1. (A) Apparent catalytic rates (kapp) and (B) normalized kapp values of full-length Akt1 variants (blue) and ΔPH-Akt1 variants (red) are shown. Error bars represent one standard deviation of triplicate measurements.
Figure 5.
Impact of PH domain on the activity of differentially phosphorylated Akt1. (A) Apparent catalytic rates (kapp) and (B) normalized kapp values of full-length Akt1 variants (blue) and ΔPH-Akt1 variants (red) are shown. Error bars represent one standard deviation of triplicate measurements.

Figure 6.
Structure of Akt1 in active and inhibitor bound forms. (A) Structure of ΔPH-pAkt1Thr308 Ser473Asp (PDB 1O6K [35]) is shown in complex with ATP analog (ANP-PNP) and substrate peptide (purple). (B) Structure of the full-length Akt1 (unphosphorylated) is shown in complex with the Akti-1/2 inhibitor VIII (PDB 1O96 [36]) binding in the cleft between the kinase domain (green) and the N-terminal PH domain (red).
Figure 6.
Structure of Akt1 in active and inhibitor bound forms. (A) Structure of ΔPH-pAkt1Thr308 Ser473Asp (PDB 1O6K [35]) is shown in complex with ATP analog (ANP-PNP) and substrate peptide (purple). (B) Structure of the full-length Akt1 (unphosphorylated) is shown in complex with the Akti-1/2 inhibitor VIII (PDB 1O96 [36]) binding in the cleft between the kinase domain (green) and the N-terminal PH domain (red).

Figure 7.
Chemical inhibition of full-length phosphorylated Akt1 variants. Inhibition of (A) pAkt1S473, (B) pAkt1T308, and (C) ppAkt1T308,S473 with varying concentrations (0.001, 0.05, 0.5, 1, 5, 10 μM) of Akti-1/2 inhibitor VIII. The resulting IC50 values are in Table 4.
Figure 7.
Chemical inhibition of full-length phosphorylated Akt1 variants. Inhibition of (A) pAkt1S473, (B) pAkt1T308, and (C) ppAkt1T308,S473 with varying concentrations (0.001, 0.05, 0.5, 1, 5, 10 μM) of Akti-1/2 inhibitor VIII. The resulting IC50 values are in Table 4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Protein yields for Akt1 variants.
| Protein Yields (μg/L Escherichia coli Culture) | ||
|---|---|---|
| Akt1 Variant | Full Length | ΔPH Akt1 |
| Akt1 (unphosphorylated) | 46 | 330 |
| pAkt1S473 | 100 | 150 |
| pAkt1T308 | 37 | 235 |
| ppAkt1T308,S473 | 45 | 224 |
Table 2.
Activity of Akt1 variants.
| Akt1 Variant | Akt1 Amount (pmol) | Initial Velocity vo (fmol/min) | Apparent Catalytic Rate kapp (fmol/min/pmol Akt1) | Activation (Fold Increase) | Reference |
|---|---|---|---|---|---|
| Akt1 (unphosphorylated) | 18 | 0.6 ± 0.2 | 0.03 ± 0.01 | 1.0 ± 0.3 | [12] |
| pAkt1S473 | 18 | 46 ± 5 | 2.6 ± 0.3 | 85 ± 9 | [12] |
| pAkt1T308 | 1.8 | 22 ± 4 | 12 ± 2 | 400 ± 70 | [12] |
| ppAkt1T308,S473 | 1.8 | 79 ± 11 | 44 ± 6 | 1500 ± 200 | [12] |
| ΔPHAkt1 (unphosphorylated) | 18 | 1.4 ± 0.1 | 0.079 ± 0.006 | 2.7 ± 0.2 | this study |
| ΔPH pAkt1S473 | 18 | 210 ± 20 | 12 ± 1 | 390 ± 40 | this study |
| ΔPH pAkt1T308 | 0.18 | 370 ± 100 | 2100 ± 600 | 6900 ± 1800 | this study |
| ΔPH ppAkt1T308,S473 | 0.18 | 2300 ± 600 | (13 ± 3) × 103 | (4 ± 1) × 105 | this study |
| Commercial Akt1 (Abcam) | |||||
| Lot 1 | 18 | 100 ± 20 | 6 ± 1 | 200 ± 30 | this study |
| Lot 2 | 18 | 2200 ± 700 | 120 ± 40 | 4000 ± 1000 | this study |
Table 3.
Relative activity of full-length Akt1 versus variants lacking the PH domain (ΔPH Akt1) *.
| Akt1 Variant | Full-Length Akt1 | ΔPH-Akt1 |
|---|---|---|
| Akt1 (unphosphorylated) | 1.0 ± 0.3 | 2.7 ± 0.2 |
| pAkt1S473 | 1.0 ± 0.1 | 4.6 ± 0.4 |
| pAkt1T308 | 1.0 ± 0.2 | 175 ± 50 |
| ppAkt1T308,S473 | 1.0 ± 0.1 | 295 ± 68 |
* The fold increase in kapp for each ΔPH-Akt1 enzyme variant was calculated by normalizing to the corresponding full-length Akt1 variant kapp.
Table 4.
Akti-1/2 inhibitor half maximal inhibitory concentration (IC50) for pAkt1 variants.
| Akt1 Variant | Inhibitor Type | IC50 (μM) | Expression System | Reference |
|---|---|---|---|---|
| ppAkt1T308,S473 | Akti-1/2 inhibitor VIII | 1.2 ± 0.3 | E. coli | this study |
| pAkt1T308 | Akti-1/2 inhibitor VIII | 0.3 ± 0.1 | E. coli | this study |
| pAkt1S473 | Akti-1/2 inhibitor VIII | 1.3 ± 0.1 | E. coli | this study |
| active | Akti-1/2 inhibitor VIII | 0.058 | Drosophila S2 cells | [25] |
| active | Akti-1/2 inhibitor VIII | 0.1 | HEK 293 cells | [33] |
| active | Akti-1/2 inhibitor | 2.7 | Drosophila S2 cells | [34] |
| active | Akti-1 inhibitor | 4.6 | Drosophila S2 cells | [34] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Balasuriya, N.; McKenna, M.; Liu, X.; Li, S.S.C.; O’Donoghue, P. Phosphorylation-Dependent Inhibition of Akt1. Genes 2018, 9, 450. https://doi.org/10.3390/genes9090450
AMA Style
Balasuriya N, McKenna M, Liu X, Li SSC, O’Donoghue P. Phosphorylation-Dependent Inhibition of Akt1. Genes. 2018; 9(9):450. https://doi.org/10.3390/genes9090450
Chicago/Turabian StyleBalasuriya, Nileeka, McShane McKenna, Xuguang Liu, Shawn S. C. Li, and Patrick O’Donoghue. 2018. "Phosphorylation-Dependent Inhibition of Akt1" Genes 9, no. 9: 450. https://doi.org/10.3390/genes9090450
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.