
Evaluating Summer-Time Ozone Enhancement Events in the Southeast United States


Abstract
:1. Introduction
2. Methods
2.1. TOLNet Ozone Lidar Measurements
2.2. Ground-Based and Airborne in Situ Measurements
2.3. GEOS-Chem Model
2.4. Model-Predicted Sources of Ozone
3. Results and Discussion
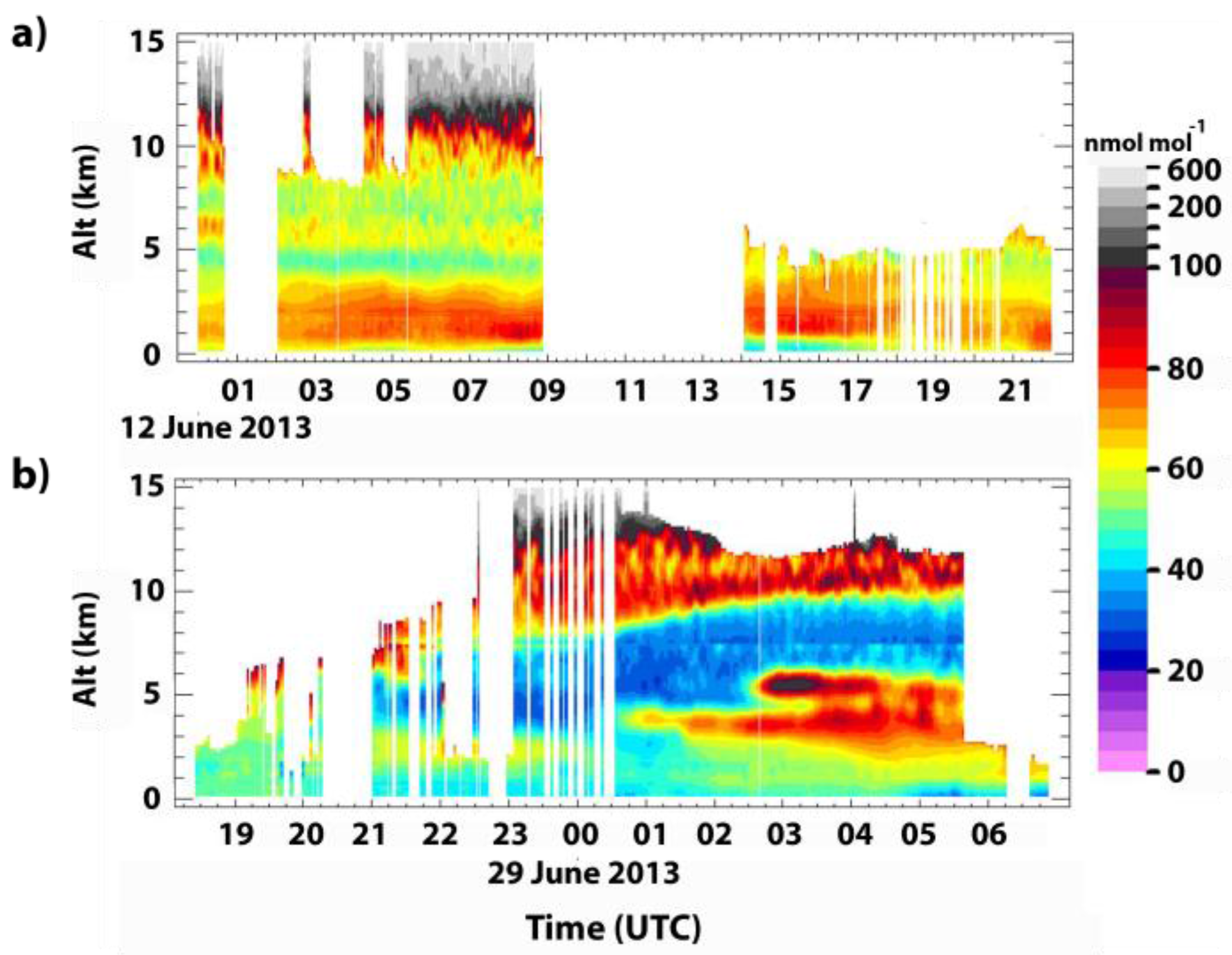
3.1. TOLNet Observations of Enhanced O3 during June 2013
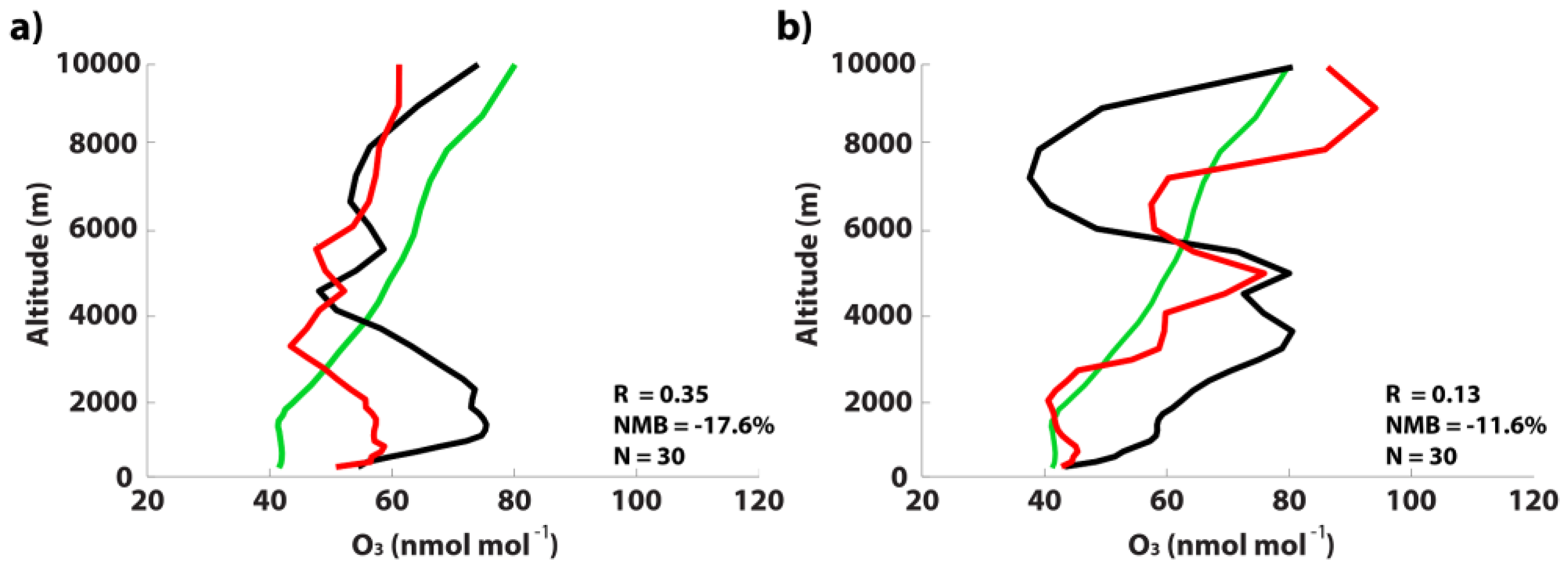
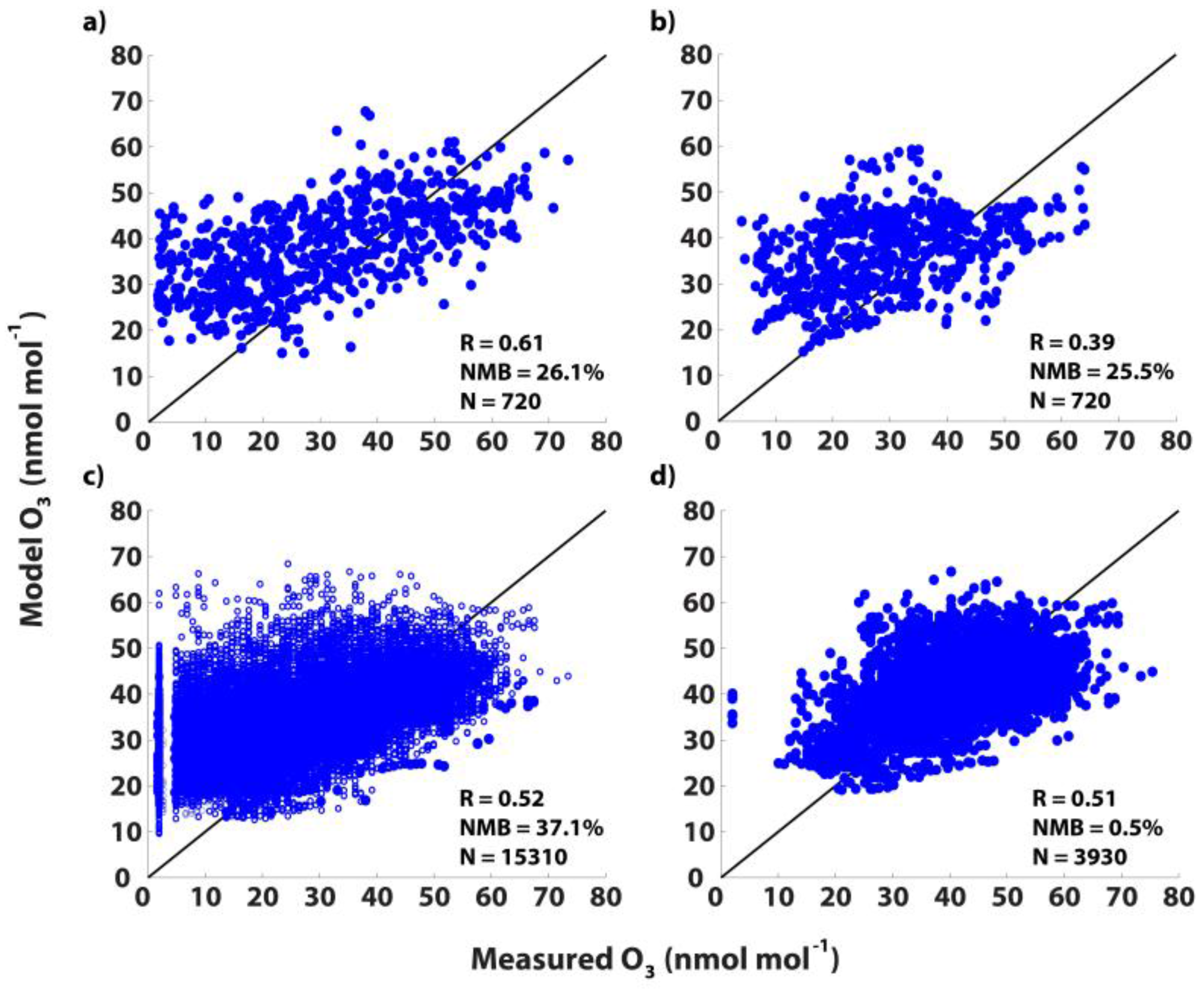
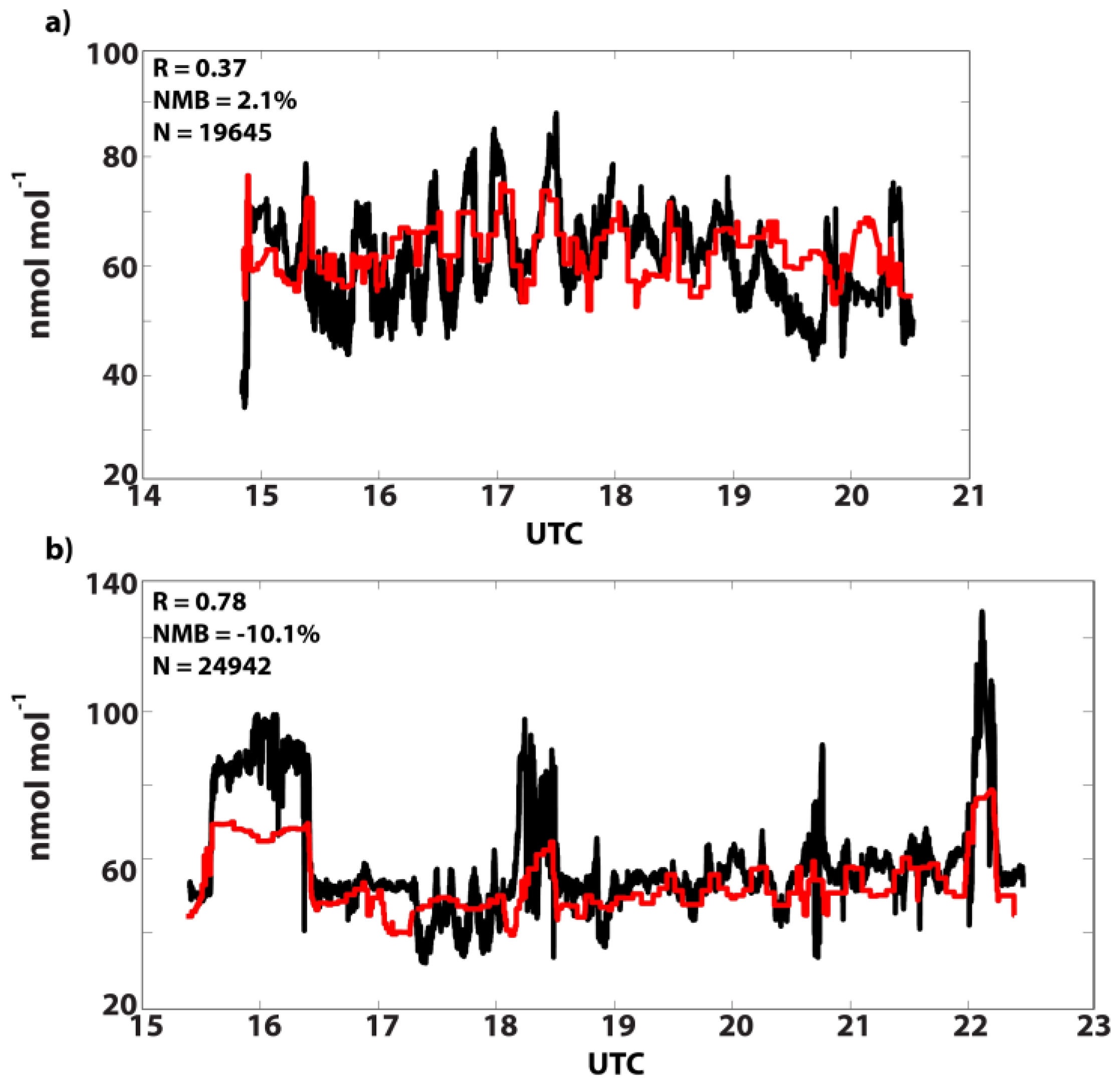
3.2. GEOS-Chem Evaluation
3.3. Sources of O3 Measured by TOLNet
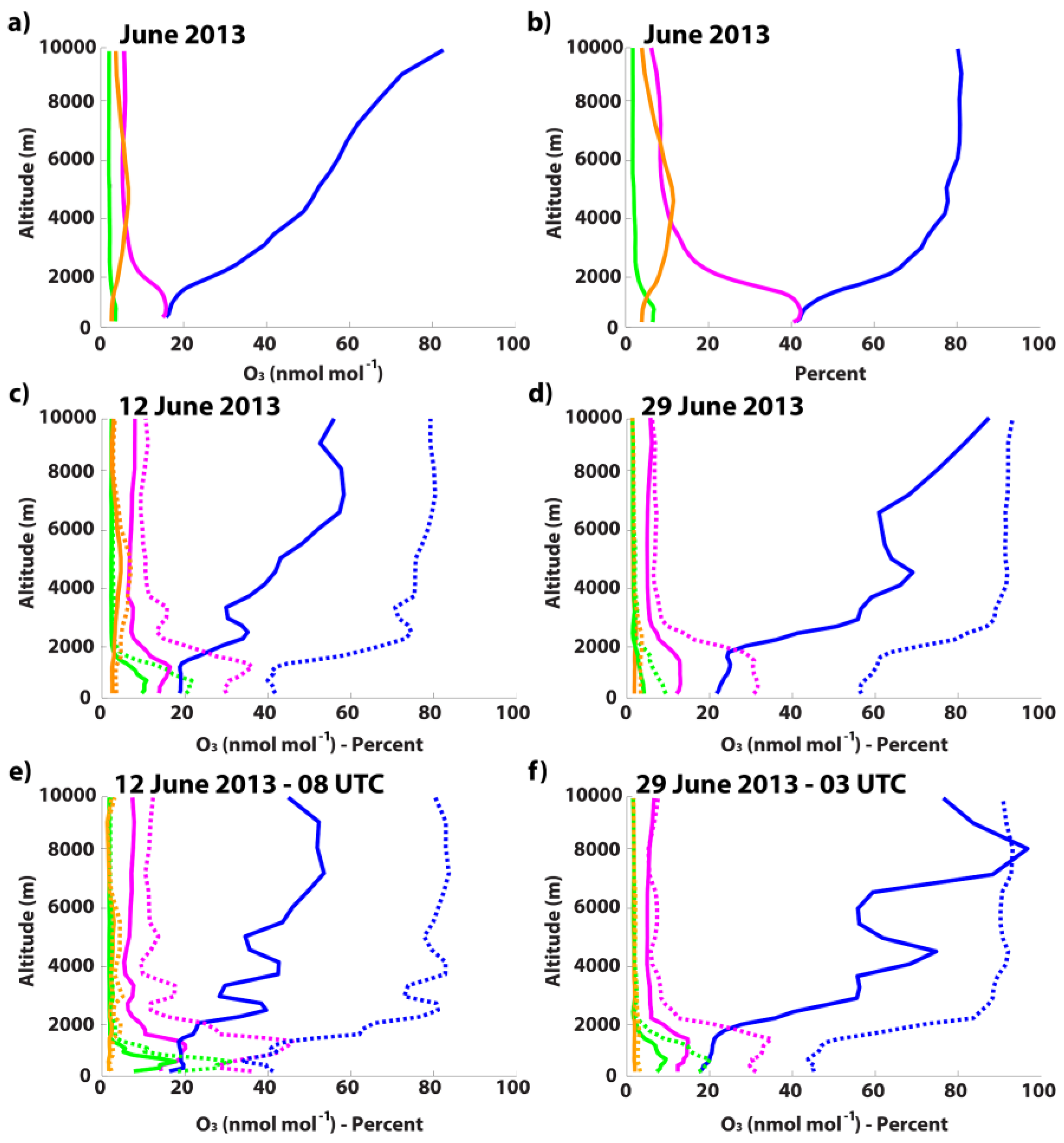
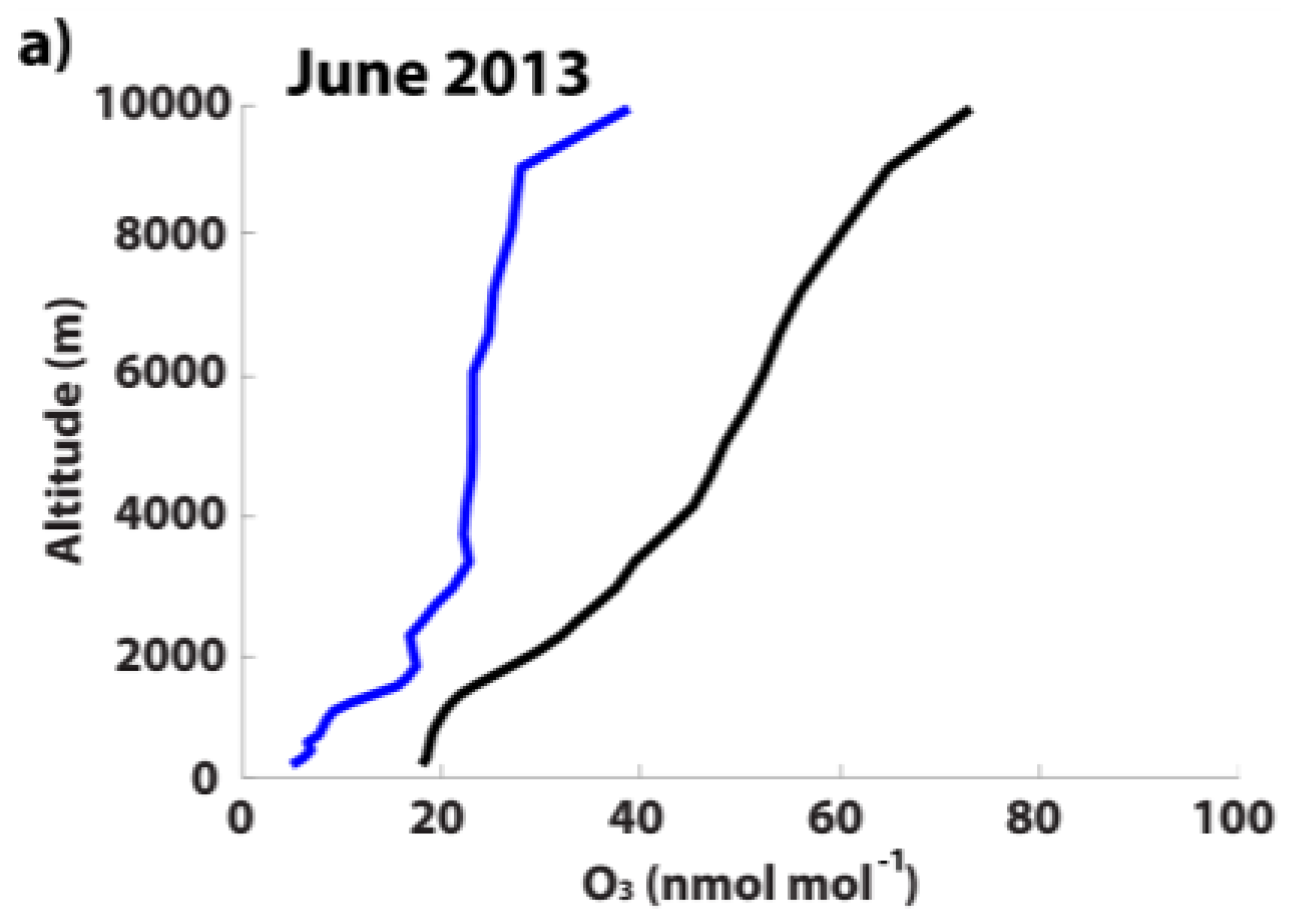
3.3.1. Monthly-Averaged Source Attribution during June 2013
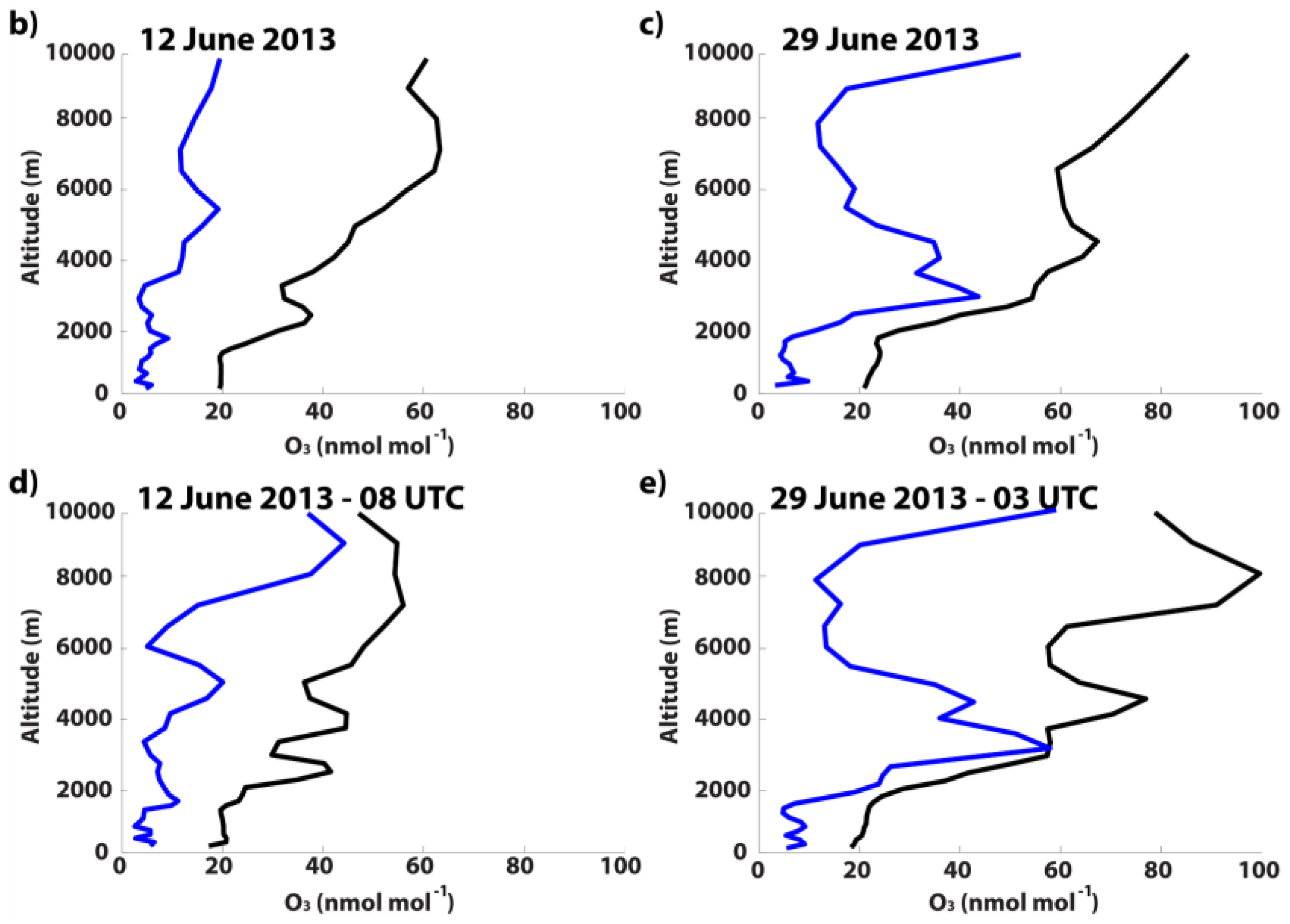
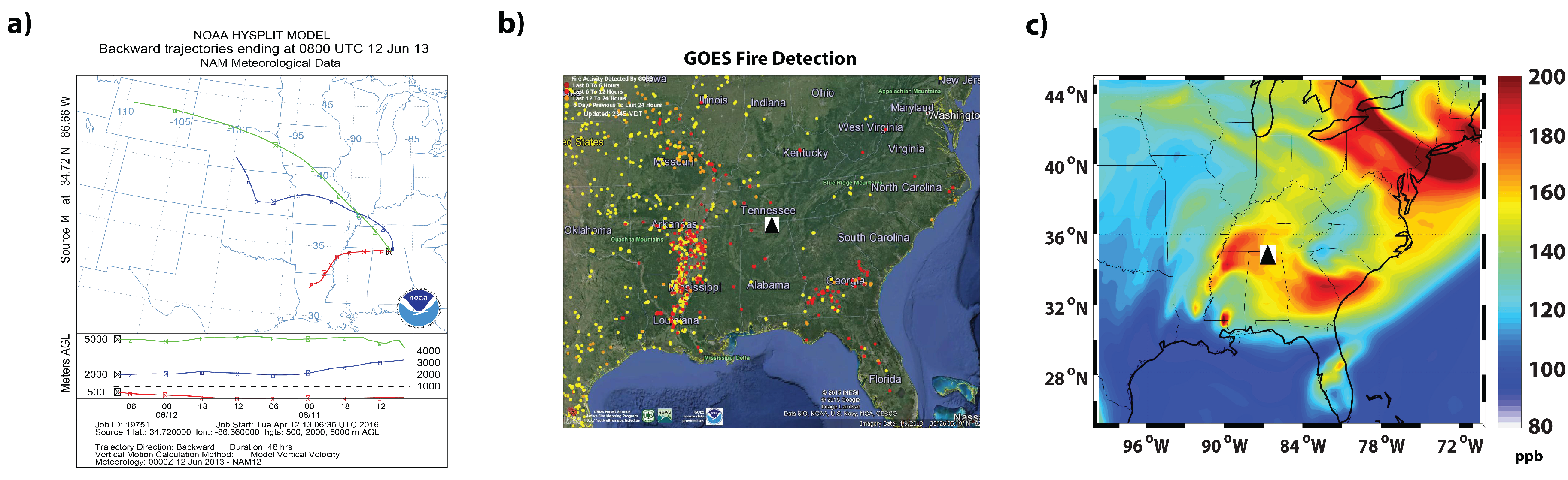
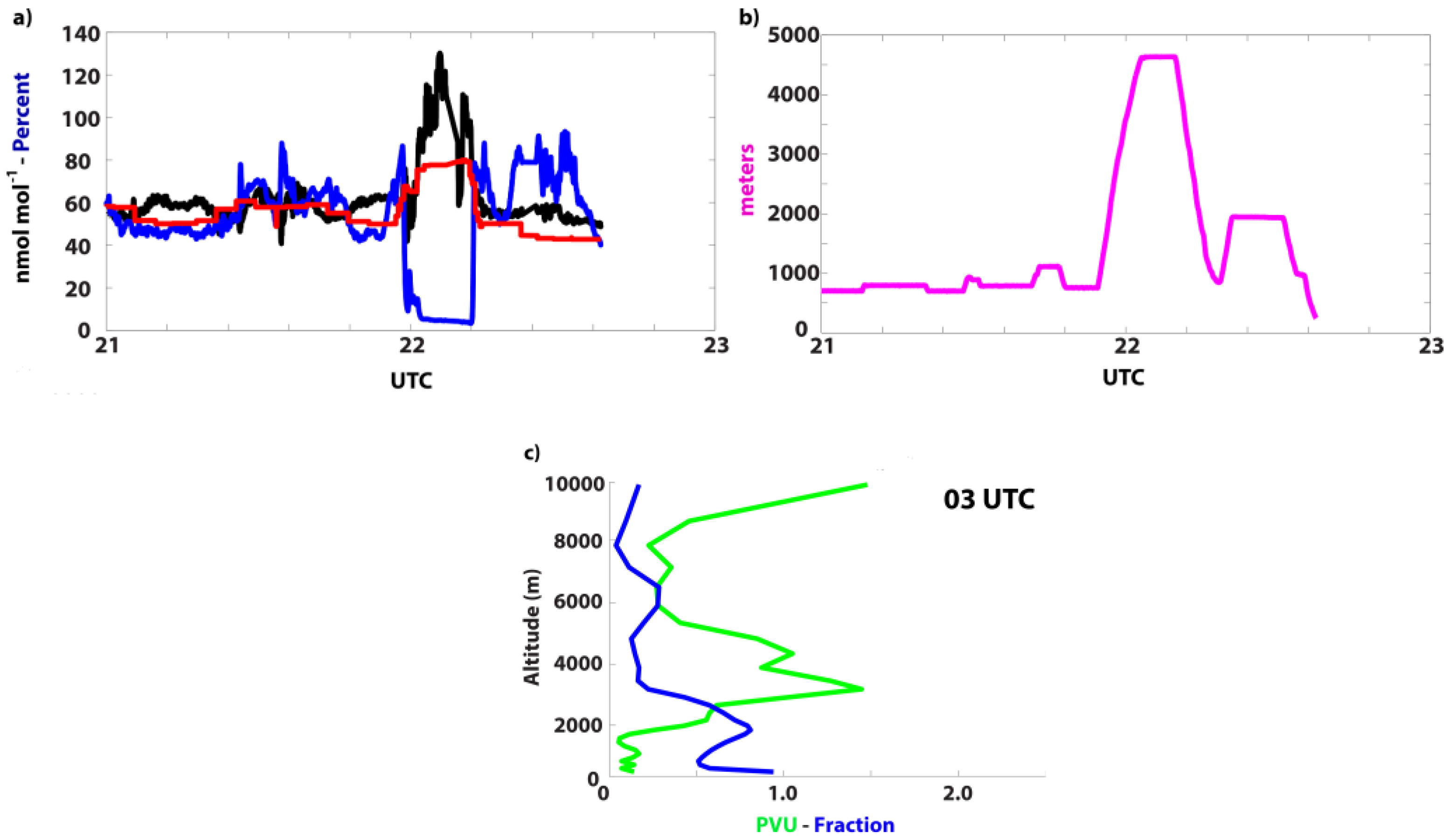
3.3.2. 12 June 2013
3.3.3. 29 June 2013
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- U.S. Environmental Protection Agency. Air Quality Criteria for Ozone and Related Photochemical Oxidants (2006 Final); EPA/600/R-05/004aF-cF; U.S. Environmental Protection Agency: Washington, DC, USA, 2006.
- Worden, H.M.; Bowman, K.W.; Worden, J.R.; Eldering, A.; Beer, R. Satellite measurements of the clear-sky greenhouse effect from tropospheric ozone. Nat. Geosci. 2008, 1, 305–308. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. National Ambient Air Quality Standards for Ozone—Final Rule; Federal Register 80; U.S. Environmental Protection Agency: Washington, DC, USA, 2015.
- Fiore, A.M.; Jacob, D.J.; Liu, H.; Yantosca, R.M.; Fairlie, T.D.; Li, Q. Variability in surface ozone background over the United States: Implications for air quality policy. J. Geophys. Res. Atmos. 2003. [Google Scholar] [CrossRef]
- Vingarzan, R. A review of surface ozone background levels and trends. Atmos. Environ. 2004, 38, 3431–3442. [Google Scholar] [CrossRef]
- Lefohn, A.S.; Wernli, H.; Shadwick, D.; Limbach, S.; Oltmans, S.J.; Shapiro, M. The importance of stratospheric-tropospheric transport in affecting surface ozone concentrations in the western and northern tier of the United States. Atmos. Environ 2011, 45, 4845–4857. [Google Scholar] [CrossRef]
- Lin, M.; Fiore, A.M.; Cooper, O.R.; Horowitz, L.W.; Langford, A.O.; Levy, H.; Johnson, B.J.; Naik, V.; Oltmans, S.J.; Senff, C.J. Springtime high surface ozone events over the western United States: Quantifying the role of stratospheric intrusions. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef]
- Yates, E.L.; Iraci, L.T.; Roby, M.C.; Pierce, R.B.; Johnson, M.S.; Reddy, P.J.; Tadić, J.M.; Loewenstein, M.; Gore, W. Airborne observations and modeling of springtime stratosphere-to-troposphere transport over California. Atmos. Chem. Phys. 2013, 13, 12481–12494. [Google Scholar] [CrossRef]
- Zhang, L.; Jacob, D.J.; Yue, X.; Downey, N.V.; Wood, D.A.; Blewitt, D. Sources contributing to background surface ozone in the US Intermountain West. Atmos. Chem. Phys. 2014, 14, 5295–5309. [Google Scholar] [CrossRef]
- Lin, M.; Fiore, A.M.; Horowitz, L.W.; Langford, A.O.; Oltmans, S.J.; Tarasick, D.; Reider, H.E. Climate variability modulates western US ozone air quality in spring via deep stratospheric intrusions. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hidy, G.M.; Blanchard, C.L.; Baumann, K.; Edgerton, E.; Tanenbaum, S.; Shaw, S.; Knipping, E.; Tombach, I.; Jansen, J.; Walters, J. Chemical climatology of the southeastern United States. Atmos. Chem. Phys. 2014, 14, 11893–11914. [Google Scholar] [CrossRef]
- Pfister, G.G.; Emmons, L.K.; Hess, P.G.; Lamarque, J.-F.; Thompson, A.M.; Yorks, J.E. Analysis of the Summer 2004 ozone budget over the United States using Intercontinental Transport Experiment Ozonesonde Network Study (IONS) observations and Model of Ozone and Related Tracers (MOZART-4) simulations. J. Geophys. Res. 2008, 113. [Google Scholar] [CrossRef]
- Hudman, R.C.; Murray, L.T.; Jacob, D.J.; Turquety, S.; Wu, S.; Millet, D.B.; Avery, M.; Goldstein, A.H.; Holloway, J. North American influence on tropospheric ozone and the effects of recent emission reductions: Constraints from ICARTT observations. J. Geophys. Res. 2009, 114. [Google Scholar] [CrossRef]
- TOLNet-Tropospheric Ozone Lidar Network. Available online: http://www-air.larc.nasa.gov/missions/TOLNet/ (accessed on 15 July 2015).
- Kuang, S.; Burris, J.F.; Newchurch, M.J.; Johnson, S.; Long, S. Differential absorption Lidar to measure subhourly variation of tropospheric ozone profiles. IEEE Trans. Geosci. Remote Sens. 2011, 49, 557–571. [Google Scholar] [CrossRef]
- Kuang, S.; Newchurch, M.J.; Burris, J.; Liu, X. Ground-based Lidar for atmospheric boundary layer ozone measurements. Appl. Opt. 2013, 52, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Newchurch, M.J.; Burris, J.; Wang, L.; Buckley, P.; Johnson, S.; Knupp, K.; Huang, G.; Phillips, D. Nocturnal ozone enhancement in the lower troposphere observed by Lidar. Atmos. Environ. 2011, 45, 6078–6084. [Google Scholar] [CrossRef]
- Kuang, S.; Newchurch, M.J.; Burris, J.; Wang, L.; Knupp, K.; Huang, G. Stratosphere-to-troposphere transport revealed by ground-based Lidar and ozonesonde at a midlatitude site. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef]
- Wang, L.; Follette-Cook, M.; Newchurch, M.; Pickering, K.; Pour-Biazar, A.; Kuang, S.; Koshak, W.; Peterson, H. Evaluation of lightning-induced tropospheric ozone enhancements observed by ozone Lidar and simulated by WRF/Chem. Atmos. Environ. 2015, 115, 185–191. [Google Scholar] [CrossRef]
- SouthEastern Aerosol Research and Characterization (SEARCH) Network. Available online: http://www.atmospheric-research.com/studies/SEARCH/ (accessed on 20 September 2015).
- Environmental Protection Agency—AirData. Available online: https://www3.epa.gov/airquality/airdata/ (accessed on 20 September 2015).
- Earth System Research Laboratory—Chemical Sciences Division—SENEX Data. Available online: http://www.esrl.noaa.gov/csd/groups/csd7/measurements/2013senex/ (accessed on 13 January 2016).
- Bey, I.; Jacob, D.J.; Yantosca, R.M.; Logan, J.A.; Field, B.; Fiore, A.M.; Li, Q.; Liu, H.; Mickley, L.J.; Schultz, M. Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation. J. Geophys. Res. 2001, 106, 23073–23095. [Google Scholar] [CrossRef]
- Lin, S.J.; Rood, R.B. Multidimensional flux form semi-Lagrangian transport schemes. Mon. Weather Rev. 1996, 124, 2046–2070. [Google Scholar] [CrossRef]
- Lin, J.-T.; McElroy, M. Impacts of boundary layer mixing on pollutant vertical profiles in the lower troposphere: Implications to satellite remote sensing. Atmos. Environ. 2010, 44, 1726–1739. [Google Scholar] [CrossRef]
- Liu, H.; Jacob, D.J.; Bey, I.; Yantosca, R.M. Constraints from 210Pb and 7Be on wet deposition and transport in a global three dimensional chemical tracer model driven by assimilated meteorological fields. J. Geophys. Res. 2001, 106, 12109–12128. [Google Scholar] [CrossRef]
- Amos, H.M.; Jacob, D.J.; Holmes, C.D.; Fisher, J.A.; Wang, Q.; Yantosca, R.M.; Corbitt, E.S.; Galarneau, E.; Rutter, A.P.; Gustin, M.S.; et al. Gas-Particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition. Atmos. Chem. Phys. 2012, 12, 591–603. [Google Scholar] [CrossRef]
- Wang, Y.H.; Jacob, D.J.; Logan, J.A. Global simulation of tropospheric O3-NOx-hydrocarbon chemistry: 1. Model formulation. J. Geophys. Res. 1998, 103, 10713–10725. [Google Scholar] [CrossRef]
- Travis, K.R.; Jacob, D.J.; Fisher, J.A.; Kim, P.S.; Marais, E.A.; Zhu, L.; Yu, K.; Miller, C.C.; Yantosca, R.M.; Sulprizio, M.P.; et al. NOx emissions, isoprene oxidation pathways, vertical mixing, and implications for surface ozone in the Southeast United States. Atmos. Chem. Phys. Discuss. 2016. [Google Scholar] [CrossRef]
- Environmental Protection Agency—Air Pollutant Emissions Trends Data. Available online: https://www.epa.gov/air-emissions-inventories/air-pollutant-emissions-trends-data (accessed on 5 August 2015).
- Fujita, E.M.; Campbell, D.E.; Zielinska, B.; Chow, J.C.; Lindhjem, C.E.; DenBleyker, A.; Bishop, G.A.; Schuchmann, B.G.; Stedman, D.H.; Lawson, D.R. Comparison of the MOVES2010a, MOBILE6.2, and EMFAC2007 mobile source emission models with on-road traffic tunnel and remote sensing measurements. J. Air Waste Manag. Assoc. 2012, 62, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Brioude, J.; Angevine, W.M.; Ahmadov, R.; Kim, S.W.; Evan, S.; McKeen, S.A.; Hsie, E.Y.; Frost, G.J.; Neuman, J.A.; Pollack, I.B.; et al. Top-down estimate of surface flux in the Los Angeles Basin using a mesoscale inverse modeling technique: assessing anthropogenic emissions of CO, NOx and CO2 and their impacts. Atmos. Chem. Phys. 2013, 13, 3661–3677. [Google Scholar] [CrossRef]
- Anderson, D.C.; Loughner, C.P.; Diskin, G.; Weinheimer, A.; Canty, T.P.; Salawitch, R.J.; Worden, H.M.; Fried, A.; Mikoviny, T.; Wisthaler, A.; et al. Measured and modeled CO and NOy in DISCOVER-AQ: An evaluation of emissions and chemistry over the eastern US. Atmos. Environ. 2014, 96, 78–87. [Google Scholar] [CrossRef]
- Canty, T.P.; Hembeck, L.; Vinciguerra, T.P.; Anderson, D.C.; Goldberg, D.L.; Carpenter, S.F.; Allen, D.J.; Loughner, C.P.; Salawitch, R.J.; et al. Ozone and NOx chemistry in the eastern US: Evaluation of CMAQ/CB05 with satellite (OMI) data. Atmos. Chem. Phys. 2015, 15, 10965–10982. [Google Scholar] [CrossRef]
- Darmenov, A.S.; da Silva, A. The Quick Fire Emissions Dataset (QFED): Documentation of Versions 2.1, 2.2 and 2.4. Available online: https://gmao.gsfc.nasa.gov/pubs/docs/Darmenov796.pdf (accessed on 19 August 2016).
- Murray, L.T.; Jacob, D.J.; Logan, J.A.; Hudman, R.C.; Koshak, W.J. Optimized regional and interannual variability of lightning in a global chemical transport model constrained by LIS/OTD satellite data. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef]
- Hudman, R.C.; Jacob, D.J.; Turquety, S.; Leibensperger, E.M.; Murray, L.T.; Wu, S.; Gilliland, A.B.; Avery, M.; Bertram, T.H.; Brune, W.; et al. Surface and lightning sources of nitrogen oxides over the United States: Magnitudes, chemical evolution, and outflow. J. Geophys. Res. 2007, 112. [Google Scholar] [CrossRef]
- McLinden, C.A.; Olsen, S.C.; Hannegan, B.; Wild, O.; Prather, M.J.; Sundet, J. Stratospheric ozone in 3-D models: A simple chemistry and the cross-tropopause flux. J. Geophys. Res. 2000, 105, 14653–14665. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Prather, M.J.; Wild, O. Diagnosing the stratosphere-to-troposphere flux of ozone in a chemistry transport model. J. Geophys. Res. 2005, 110. [Google Scholar] [CrossRef] [Green Version]
- Ott, L.E.; Duncan, B.N.; Thompson, A.M.; Diskin, G.; Fasnacht, Z.; Langford, A.O.; Lin, M.; Molod, A.M.; Nielsen, J.E.; Pusede, S.E.; et al. Frequency and impact of summertime stratospheric intrusions over Maryland during DISCOVER-AQ (2011): New evidence from NASA’s GEOS-5 simulations. J. Geophys. Res. Atmos. 2016, 121. [Google Scholar] [CrossRef]
- Cohan, D.S.; Napelenok, S.L. Air quality response modeling for decision support. Atmosphere 2011, 2, 407–425. [Google Scholar] [CrossRef]
- Kwok, R.H.F.; Baker, K.R.; Napelenok, S.L.; Tonnesen, G.S. Photochemical grid model implementation and application of VOC, NOx, and O3 source apportionment. Geosci. Model Dev. 2015, 8, 99–114. [Google Scholar] [CrossRef]
- Stohl, A.; Spichtinger-Rakowsky, N.; Bonasoni, P.; Feldmann, H.; Memmesheimer, M.; Scheel, H.E.; Trickl, T.; Hübener, S.; Ringer, W.; Mandl, M. The influence of stratospheric intrusions on Alpine ozone concentrations. Atmos. Environ. 2000, 34, 1323–1354. [Google Scholar] [CrossRef]
- Rao, T.N.; Kirkwood, S.; Arvelius, J.; Von der Gathen, P.; Kivi, R. Climatology of UTLS ozone and the ratio of ozone and potential vorticity over northern Europe. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Sullivan, J.T.; McGee, T.J.; Thompson, A.M.; Pierce, R.B.; Sumnicht, G.K.; Twigg, L.W.; Eloranta, E.; Hoff, R.M. Characterizing the lifetime and occurrence of stratospheric-tropospheric exchange events in the rocky mountain region using high-resolution ozone measurements. J. Geophys. Res. Atmos. 2015, 120, 12410–12424. [Google Scholar] [CrossRef]
- Beekmann, M.; Ancellet, G.; Mégie, G. Climatology of tropospheric ozone in southern Europe and its relation to potential vorticity. J. Geophys. Res. 1994, 99, 12841–12853. [Google Scholar] [CrossRef]
- Hoskins, B.J.; McIntyre, M.E.; Robertson, A.W. On the use and significance of isentropic potential vorticity maps. Q. J. R. Meteorol. Soc. 1985, 111, 877–946. [Google Scholar] [CrossRef]
- Adamson, D.S.; Belcher, S.E.; Hoskins, B.J.; Plant, R.S. Boundary-layer friction in the midlatitude cyclone. Q. J. R. Meteorol. Soc. 2006, 132, 101–124. [Google Scholar] [CrossRef]
- Jaffe, D.A.; Wigder, N.L. Ozone production from wildfires: A critical review. Atmos. Environ. 2012, 51, 1–10. [Google Scholar] [CrossRef]
- Earth System Research Laboratory—Chemical Sciences Division—FLEXPART Backtrajectories. Available online: http://esrl.noaa.gov/csd/groups/csd4/forecasts/backward/ (accessed on 18 November 2015).
- Kim, P.S.; Jacob, D.J.; Fisher, J.A.; Travis, K.; Yu, K.; Zhu, L.; Yantosca, R.M.; Sulprizio, M.P.; Jimenez, J.L.; Campuzano-Jost, P.; et al. Sources, seasonality, and trends of southeast US aerosol: An integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model. Atmos. Chem. Phys. 2015, 15, 10411–10433. [Google Scholar] [CrossRef]
- Earth System Research Laboratory—Chemical Sciences Division—FLEXPART Forecasts for SENEX 2013. Available online: http://esrl.noaa.gov/csd/groups/csd4/forecasts/senex/ (accessed on 18 November 2015).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SENEX Flight—12 June | ||||
| CO | NO2 | NO | ISOP | |
| Measure (Mean) * | 153.5 | 0.40 | 0.06 | 1.00 |
| Model (Mean) * | 120.8 | 0.39 | 0.10 | 1.61 |
| NMB (%) | −21.33 | −3.11 | 55.93 | 60.50 |
| Correlation (R) | 0.89 | 0.67 | 0.42 | 0.63 |
| SENEX Flight—29 June | ||||
| CO | NO2 | NO | ISOP | |
| Measure (Mean) * | 123.14 | 0.24 | 0.05 | 0.65 |
| Model (Mean) * | 94.02 | 0.20 | 0.06 | 1.20 |
| NMB (%) | −23.65 | −18.67 | 23.94 | 84.82 |
| Correlation (R) | 0.76 | 0.31 | 0.19 | 0.59 |
| Source (0–3 km) | June 2013 | 12 June 2013 | 12 June 2013 08 UTC |
| Anthro | 14.3 (35.6) | 12.8 (30.1) | 14.5 (33.9) |
| Wildfires | 1.5 (3.8) | 5.5 (13.0) | 5.6 (13.1) |
| Lightning | 1.8 (4.6) | 1.0 (2.3) | 0.7 (1.6) |
| LR + Strat * | 22.4 (55.9) | 23.3 (54.6) | 22.1 (51.5) |
| Source (0–1 km) | June 2013 | 12 June 2013 | 12 June 2013 08 UTC |
| Anthro | 16.8 (43.8) | 14.5 (33.1) | 16.8 (35.7) |
| Wildfires | 2.2 (5.9) | 9.1 (20.8) | 10.9 (23.1) |
| Lightning | 1.1 (2.9) | 0.8 (1.8) | 0.5 (1.0) |
| LR + Strat * | 18.1 (47.4) | 19.4 (44.4) | 18.9 (40.2) |
| Source (3–6 km) | June 2013 | 29 June 2013 | 29 June 2013 03 UTC |
| Anthro | 6.4 (11.6) | 4.4 (6.5) | 4.4 (6.4) |
| Wildfires | 0.4 (0.7) | 0.8 (1.2) | 0.7 (1.0) |
| Lightning | 5.1 (9.2) | 0.8 (1.2) | 0.7 (1.0) |
| LR + Strat * | 43.3 (78.5) | 60.9 (91.1) | 63.5 (91.6) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, M.S.; Kuang, S.; Wang, L.; Newchurch, M.J. Evaluating Summer-Time Ozone Enhancement Events in the Southeast United States. Atmosphere 2016, 7, 108. https://doi.org/10.3390/atmos7080108
Johnson MS, Kuang S, Wang L, Newchurch MJ. Evaluating Summer-Time Ozone Enhancement Events in the Southeast United States. Atmosphere. 2016; 7(8):108. https://doi.org/10.3390/atmos7080108
Chicago/Turabian StyleJohnson, Matthew S., Shi Kuang, Lihua Wang, and M. J. Newchurch. 2016. "Evaluating Summer-Time Ozone Enhancement Events in the Southeast United States" Atmosphere 7, no. 8: 108. https://doi.org/10.3390/atmos7080108