Natural Killer (NK)- and T-Cell Engaging Antibody-Derived Therapeutics

Abstract
:1. Introduction
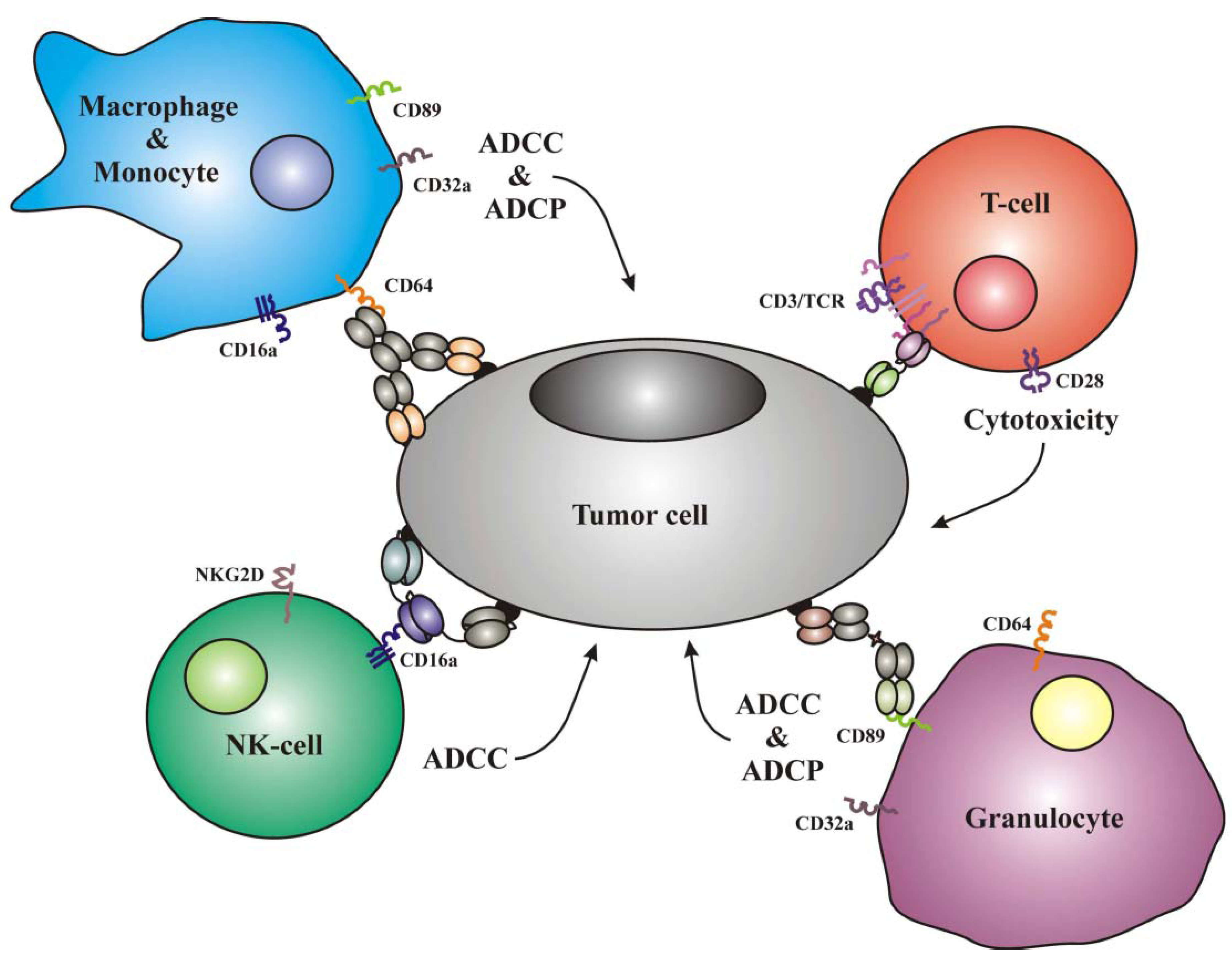
1.1. Frequently Used Trigger Molecules and Classes of Effector Cells

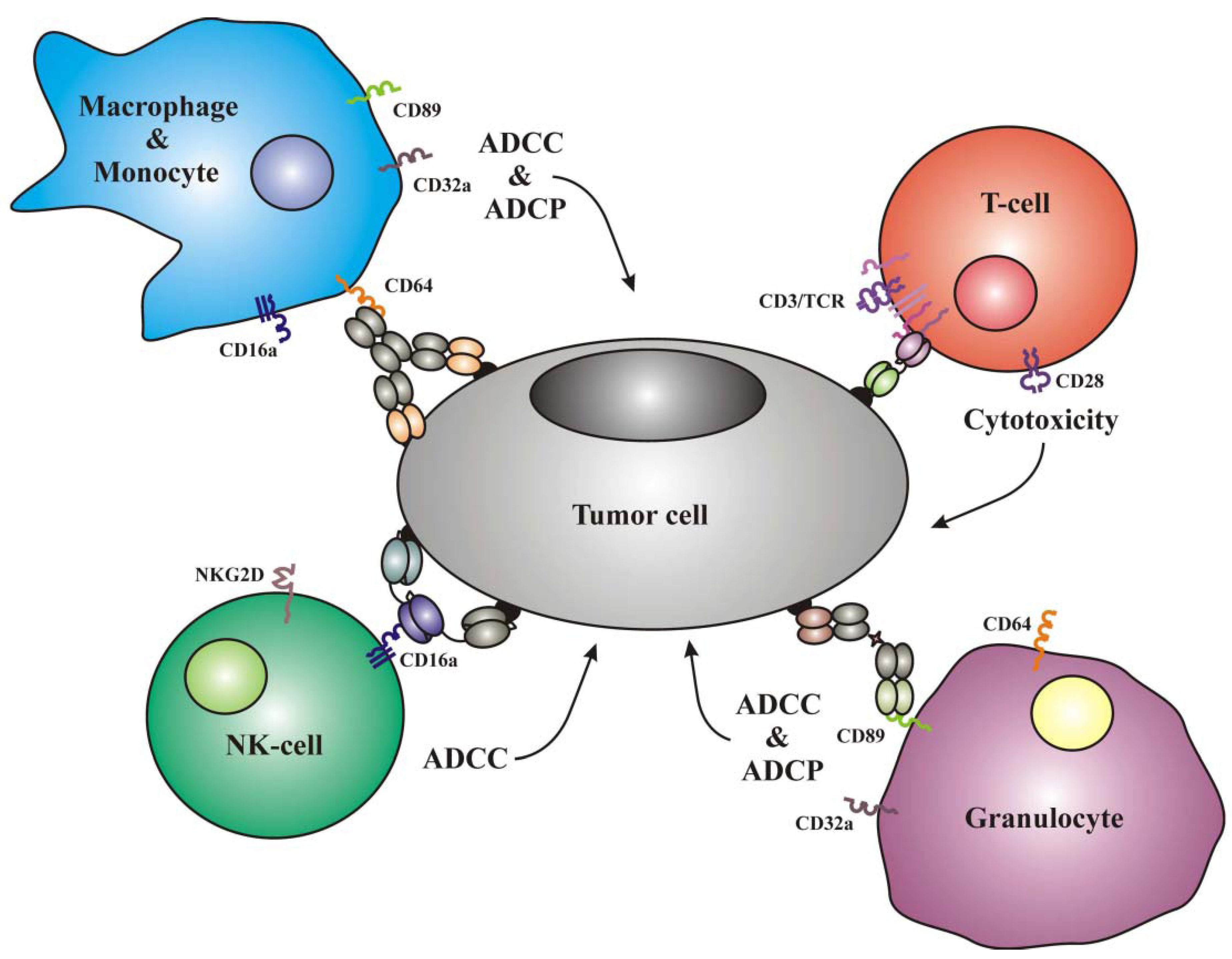{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effector cells | Trigger molecules |
|---|---|
| T-cells | CD3, TCRα,β, CTLA4, PD1, CD28 |
| NK-cells | CD16a, NKG2D, NKp30, NKp40, LFA1 |
| Monocytes | CD89, CD64, CD32a, CD16a |
| Macrophages | CD89, CD64, CD32a, CD16a |
| Neutrophilic Granulocytes | CD89, CD64, CD32a |
1.2. Examples of ab-Derived Agents Recruiting Effector Cells Other than NK- and T-Cells
| Specificity | Format | Disease | Development status | Company | Trial |
|---|---|---|---|---|---|
| CD19, CD3 | BsscFv | NHL, B-ALL | Phase I/II | Amgen | NCT01207388, NCT00274742 |
| EpCAM, CD3 | BsscFv | Various tumors | Phase I | Amgen | NCT00635596. |
| CEA, CD3 | BsscFv | Gastrointestinal cancers | Phase I | Amgen | NCT01284231. |
| EpCAM, CD3 | Hybrid IgG | Ovarian and gastric cancer, malignant ascites | Phase II/market approval | Trion | NCT00464893 |
| HER2/neu, CD3 | Hybrid IgG | Breast cancer | Phase II | Trion | NCT00522457 |
| CD20, CD3 | Hybrid IgG | B-cell lymphoma | Phase I/II | Trion | NCT01138579 |
| CD30, CD16a | TandAb | Hodgkin lymphoma | Phase I/IIa | Affimed | NCT01221571 |
2. Redirecting NK- and T-Cells for Cancer Therapy
2.1. T-Cells as Effectors for Cancer Therapy
2.1.1. Optimization of T-Cell Activity
2.1.2. Recruiting T-Cells via CD3
| Cell type | Leukocytes per µl blood |
|---|---|
| T-cells | 300–900 |
| NK-cells | 120–350 |
| Monocytes | 200–600 |
| Macrophages | - |
| Granulocytes | 1800–7000 |
2.2. NK-Cells for Cancer Therapy
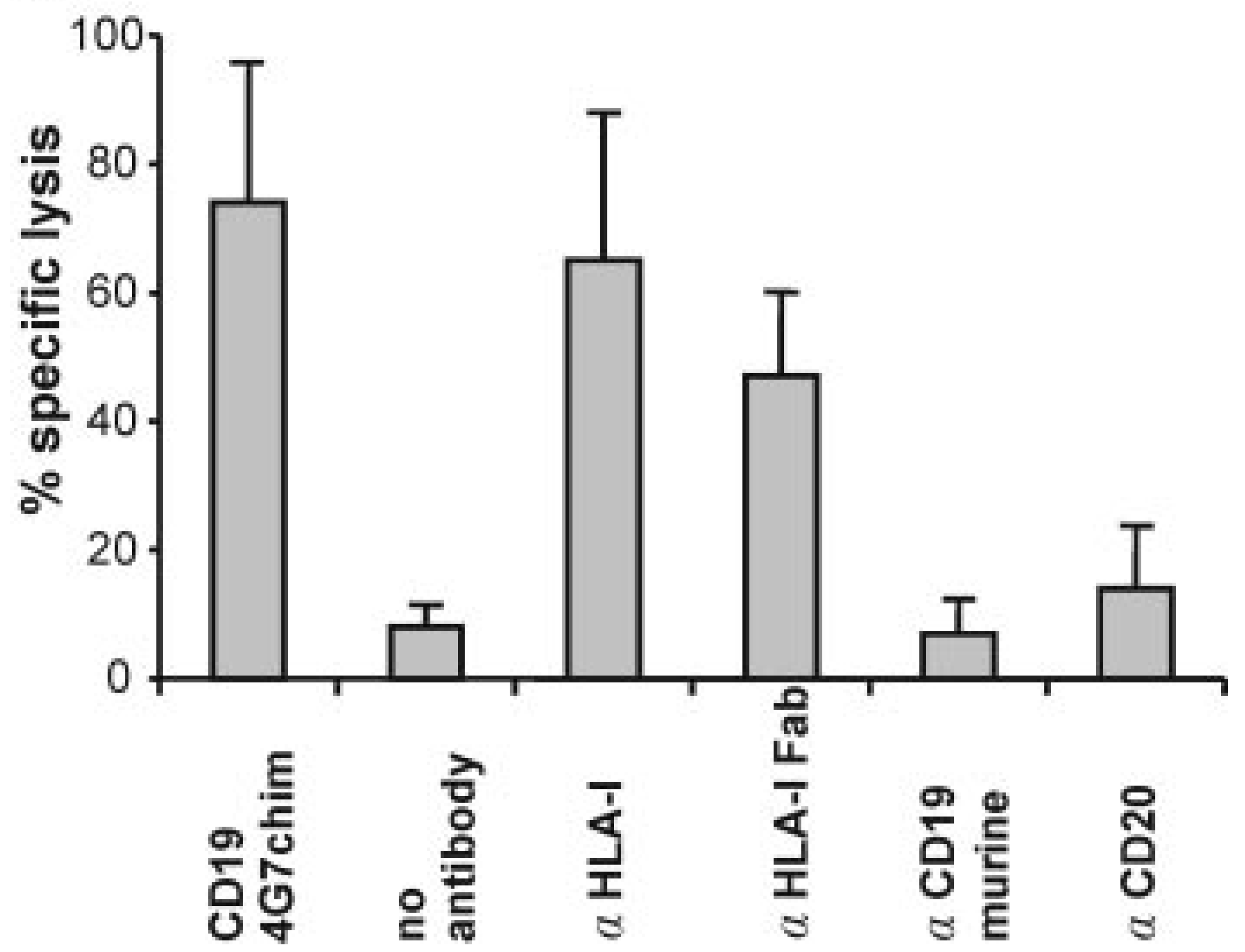
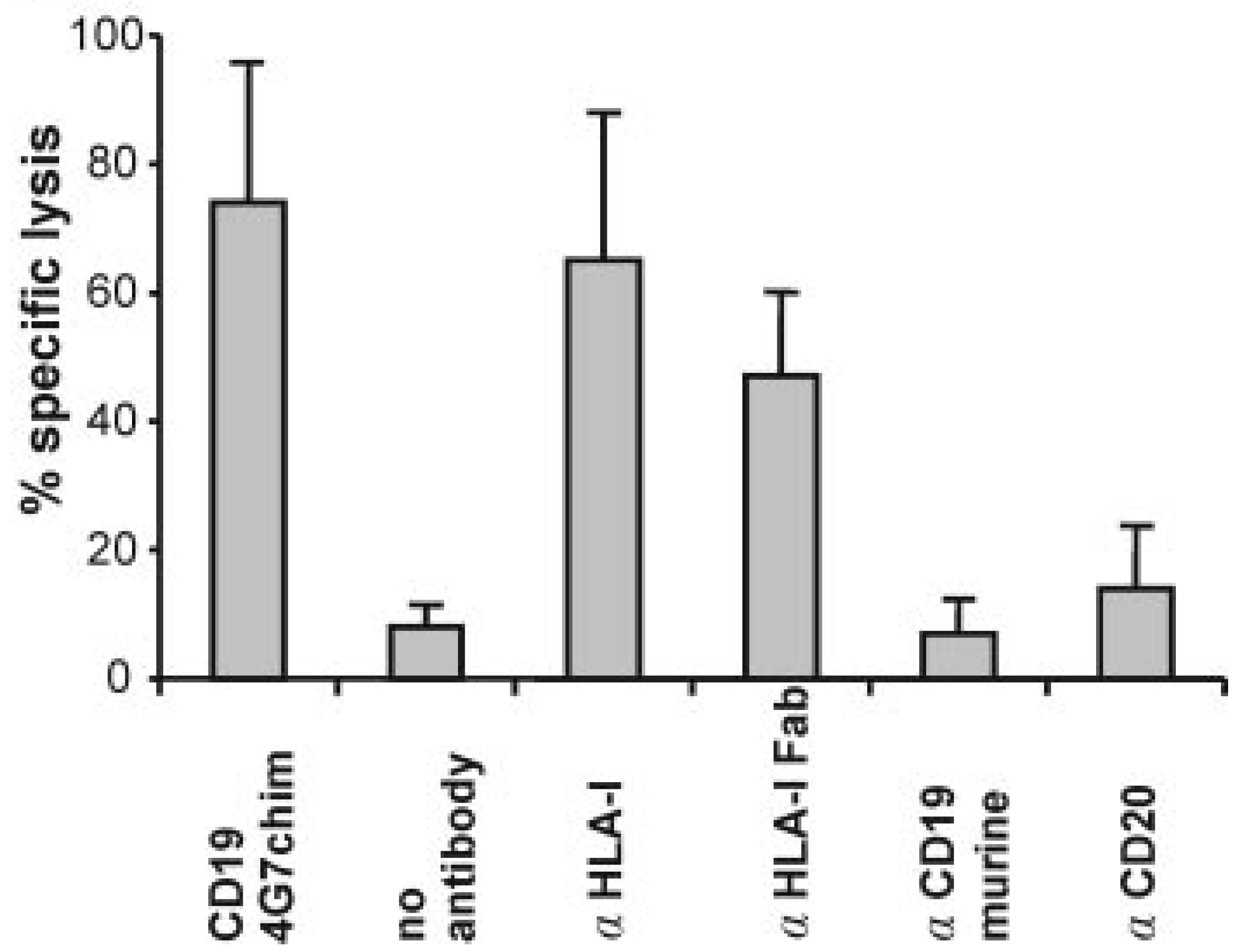
2.2.1. Recruitment of NK-Cells by Anti-tumor Abs
2.2.2. NK-Cell Recruiting ab-Derivates
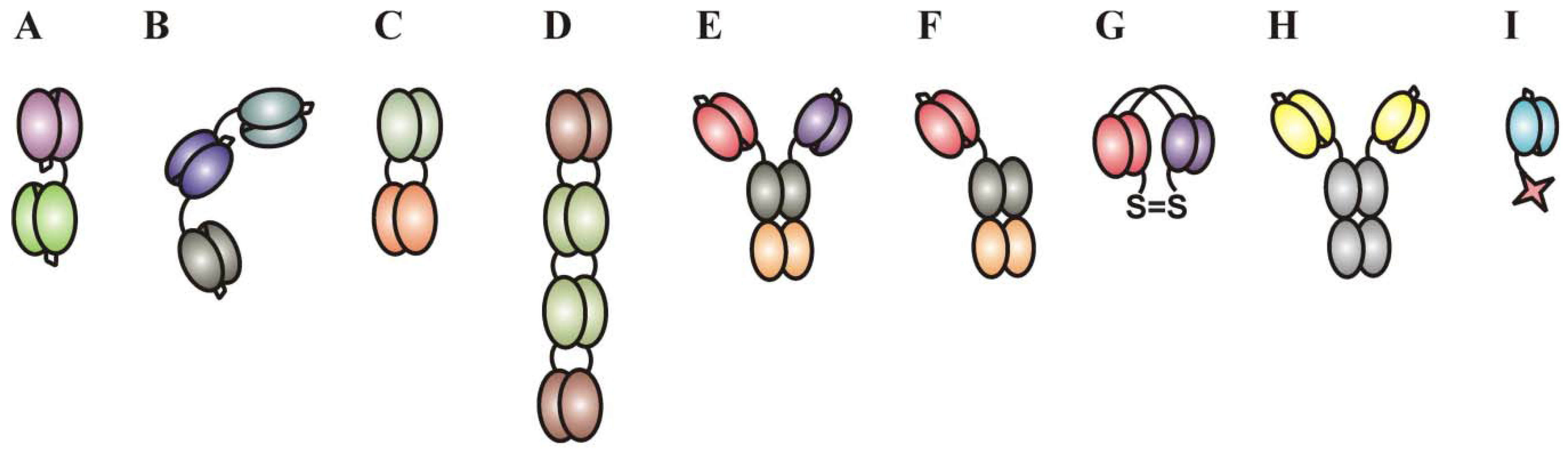
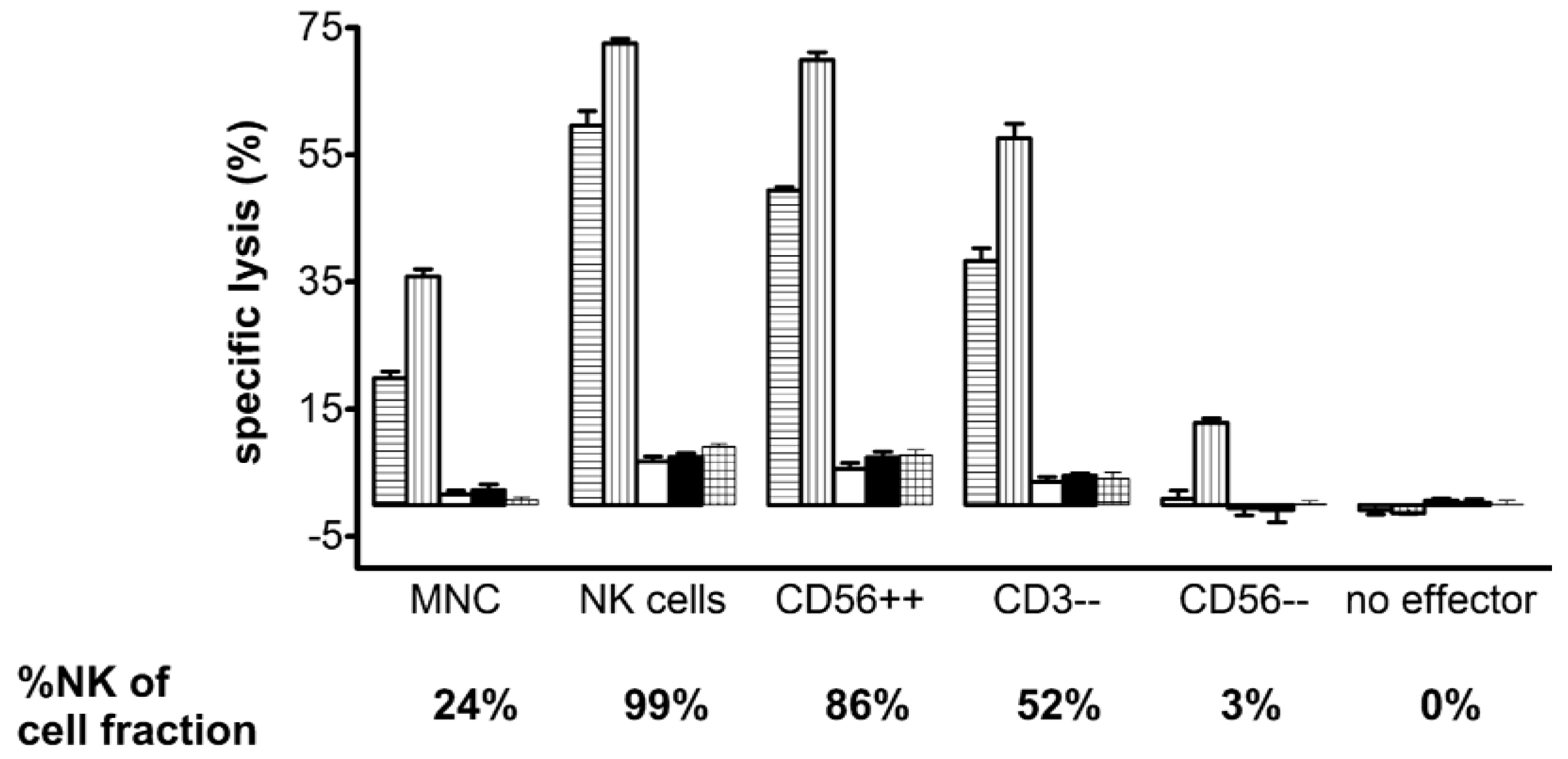
2.2.3. Immunoligands for the Recruitment of NK-Cells
3. Conclusions
Acknowledgments
References and Notes
- Rahlff, J.; Trusch, M.; Haag, F.; Bacher, U.; Horst, A.; Schluter, H.; Binder, M. Antigen-specificity of oligoclonal abnormal protein bands in multiple myeloma after allogeneic stem cell transplantation. Cancer Immunol. Immunother. 2012. [Google Scholar]
- Sahin, U.; Tureci, O.; Schmitt, H.; Cochlovius, B.; Johannes, T.; Schmits, R.; Stenner, F.; Luo, G.; Schobert, I.; Pfreundschuh, M. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc. Natl. Acad. Sci. USA 1995, 92, 11810–11813. [Google Scholar]
- Scanlan, M.J.; Chen, Y.T.; Williamson, B.; Gure, A.O.; Stockert, E.; Gordan, J.D.; Tureci, O.; Sahin, U.; Pfreundschuh, M.; Old, L.J. Characterization of human colon cancer antigens recognized by autologous antibodies. Int. J. Cancer 1998, 76, 652–658. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2011, 14, 529–537. [Google Scholar]
- Carter, P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 2001, 1, 118–129. [Google Scholar] [CrossRef]
- Dalken, B.; Giesubel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar]
- Stasi, R. Gemtuzumab ozogamicin: An anti-CD33 immunoconjugate for the treatment of acute myeloid leukaemia. Expert Opin. Biol. Ther. 2008, 8, 527–540. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Reinhardt, D.; Zimmerman, M.; Hasle, H.; Stary, J.; Stark, B.; Dworzak, M.; Creutzig, U.; Kaspers, G.J. Salvage treatment for children with refractory first or second relapse of acute myeloid leukaemia with gemtuzumab ozogamicin: Results of a phase II study. Br. J. Haematol. 2010, 148, 768–776. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef]
- Redner, R.L. Why doesn't imatinib cure chronic myeloid leukemia? Oncologist 2010, 15, 182–186. [Google Scholar] [CrossRef]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar]
- Ferrara, C.; Grau, S.; Jager, C.; Sondermann, P.; Brunker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar]
- Vidal, L.; Gafter-Gvili, A.; Salles, G.; Dreyling, M.H.; Ghielmini, M.; Hsu Schmitz, S.F.; Pettengell, R.; Witzens-Harig, M.; Shpilberg, O. Rituximab maintenance for the treatment of patients with follicular lymphoma: An updated systematic review and meta-analysis of randomized trials. J. Natl. Cancer Inst. 2011, 103, 1799–1806. [Google Scholar]
- Preithner, S.; Elm, S.; Lippold, S.; Locher, M.; Wolf, A.; da Silva, A.J.; Baeuerle, P.A.; Prang, N.S. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol. Immunol. 2006, 43, 1183–1193. [Google Scholar] [CrossRef]
- Lopez-Guillermo, A.; Mercadal, S. The clinical use of antibodies in haematological malignancies. Ann. Oncol. 2007, 18 Suppl. 9, ix51–ix57. [Google Scholar] [CrossRef]
- Indik, Z.K.; Park, J.G.; Hunter, S.; Schreiber, A.D. The molecular dissection of Fc gamma receptor mediated phagocytosis. Blood 1995, 86, 4389–4399. [Google Scholar]
- Boruchov, A.M.; Heller, G.; Veri, M.C.; Bonvini, E.; Ravetch, J.V.; Young, J.W. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J. Clin. Invest. 2005, 115, 2914–2923. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007, 19, 239–245. [Google Scholar] [CrossRef]
- Selvaraj, P.; Fifadara, N.; Nagarajan, S.; Cimino, A.; Wang, G. Functional regulation of human neutrophil Fc gamma receptors. Immunol. Res. 2004, 29, 219–230. [Google Scholar] [CrossRef]
- Xiang, Z.; Cutler, A.J.; Brownlie, R.J.; Fairfax, K.; Lawlor, K.E.; Severinson, E.; Walker, E.U.; Manz, R.A.; Tarlinton, D.M.; Smith, K.G. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat. Immunol. 2007, 8, 419–429. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef]
- Fanger, M.W.; Segal, D.M.; Romet-Lemonne, J.L. Bispecific antibodies and targeted cellular cytotoxicity. Immunol. Today 1991, 12, 51–54. [Google Scholar] [CrossRef]
- Keler, T.; Graziano, R.F.; Mandal, A.; Wallace, P.K.; Fisher, J.; Guyre, P.M.; Fanger, M.W.; Deo, Y.M. Bispecific antibody-dependent cellular cytotoxicity of HER2/neu-overexpressing tumor cells by Fc gamma receptor type I-expressing effector cells. Cancer Res. 1997, 57, 4008–4014. [Google Scholar]
- Muller, D.; Kontermann, R.E. Recombinant bispecific antibodies for cellular cancer immunotherapy. Curr. Opin. Mol. Ther. 2007, 9, 319–326. [Google Scholar]
- Ochiai, K.; Wang, B.; Rieger, A.; Kilgus, O.; Maurer, D.; Fodinger, D.; Kinet, J.P.; Stingl, G.; Tomioka, H. A review on Fc epsilon RI on human epidermal Langerhans cells. Int. Arch. Allergy Immunol. 1994, 104 Suppl. 1, 63–64. [Google Scholar] [CrossRef]
- Prussin, C.; Metcalfe, D.D. 5. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 2006, 117 Suppl. 2, Mini-Primer. S450–S456. [Google Scholar] [CrossRef]
- von Bubnoff, D.; Novak, N.; Kraft, S.; Bieber, T. The central role of FcepsilonRI in allergy. Clin. Exp. Dermatol. 2003, 28, 184–187. [Google Scholar] [CrossRef]
- Kikutani, H.; Yokota, A.; Uchibayashi, N.; Yukawa, K.; Tanaka, T.; Sugiyama, K.; Barsumian, E.L.; Suemura, M.; Kishimoto, T. Structure and function of Fc epsilon receptor II (Fc epsilon RII/CD23): A point of contact between the effector phase of allergy and B cell differentiation. Ciba Found. Symp. 1989, 147, 23–31, discussion 31-35. [Google Scholar]
- Hamre, R.; Farstad, I.N.; Brandtzaeg, P.; Morton, H.C. Expression and modulation of the human immunoglobulin A Fc receptor (CD89) and the FcR gamma chain on myeloid cells in blood and tissue. Scand. J. Immunol. 2003, 57, 506–516. [Google Scholar] [CrossRef]
- Stockmeyer, B.; Dechant, M.; van Egmond, M.; Tutt, A.L.; Sundarapandiyan, K.; Graziano, R.F.; Repp, R.; Kalden, J.R.; Gramatzki, M.; Glennie, M.J.; et al. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J. Immunol. 2000, 165, 5954–5961. [Google Scholar]
- Stockmeyer, B.; Valerius, T.; Repp, R.; Heijnen, I.A.; Buhring, H.J.; Deo, Y.M.; Kalden, J.R.; Gramatzki, M.; van de Winkel, J.G. Preclinical studies with Fc(gamma)R bispecific antibodies and granulocyte colony-stimulating factor-primed neutrophils as effector cells against HER-2/neu overexpressing breast cancer. Cancer Res. 1997, 57, 696–701. [Google Scholar]
- Honeychurch, J.; Tutt, A.L.; Valerius, T.; Heijnen, I.A.; Van De Winkel, J.G.; Glennie, M.J. Therapeutic efficacy of FcgammaRI/CD64-directed bispecific antibodies in B-cell lymphoma. Blood 2000, 96, 3544–3552. [Google Scholar]
- Ranft, K.; Thepen, T.; Fischer, R.; Barth, S.; Stocker, M. Recombinant bispecific single chain antibody fragments induce Fc gamma-receptor-mediated elimination of CD30+ lymphoma cells. Cancer Lett. 2009, 282, 187–194. [Google Scholar] [CrossRef]
- Sundarapandiyan, K.; Keler, T.; Behnke, D.; Engert, A.; Barth, S.; Matthey, B.; Deo, Y.M.; Graziano, R.F. Bispecific antibody-mediated destruction of Hodgkin's lymphoma cells. J. Immunol. Methods 2001, 248, 113–123. [Google Scholar] [CrossRef]
- Ely, P.; Wallace, P.K.; Givan, A.L.; Graziano, R.F.; Guyre, P.M.; Fanger, M.W. Bispecific-armed, interferon gamma-primed macrophage-mediated phagocytosis of malignant non-Hodgkin's lymphoma. Blood 1996, 87, 3813–3821. [Google Scholar]
- Borchmann, P.; Schnell, R.; Fuss, I.; Manzke, O.; Davis, T.; Lewis, L.D.; Behnke, D.; Wickenhauser, C.; Schiller, P.; Diehl, V.; et al. Phase 1 trial of the novel bispecific molecule H22xKi-4 in patients with refractory Hodgkin lymphoma. Blood 2002, 100, 3101–3107. [Google Scholar] [CrossRef]
- Fury, M.G.; Lipton, A.; Smith, K.M.; Winston, C.B.; Pfister, D.G. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol. Immunother. 2008, 57, 155–163. [Google Scholar]
- Posey, J.A.; Raspet, R.; Verma, U.; Deo, Y.M.; Keller, T.; Marshall, J.L.; Hodgson, J.; Mazumder, A.; Hawkins, M.J. A pilot trial of GM-CSF and MDX-H210 in patients with erbB-2-positive advanced malignancies. J. Immunother. 1999, 22, 371–379. [Google Scholar] [CrossRef]
- James, N.D.; Atherton, P.J.; Jones, J.; Howie, A.J.; Tchekmedyian, S.; Curnow, R.T. A phase II study of the bispecific antibody MDX-H210 (anti-HER2 x CD64) with GM-CSF in HER2+ advanced prostate cancer. Br. J. Cancer 2001, 85, 152–156. [Google Scholar] [CrossRef]
- Guettinger, Y.; Barbin, K.; Peipp, M.; Bruenke, J.; Dechant, M.; Horner, H.; Thierschmidt, D.; Valerius, T.; Repp, R.; Fey, G.H.; Stockmeyer, B. A recombinant bispecific single-chain fragment variable specific for HLA class II and Fc alpha RI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. J. Immunol. 2010, 184, 1210–1217. [Google Scholar]
- Stadick, H.; Stockmeyer, B.; Kuhn, R.; Schrott, K.M.; Kalden, J.R.; Glennie, M.J.; van de Winkel, J.G.; Gramatzki, M.; Valerius, T.; Elsasser, D. Epidermal growth factor receptor and g250: Useful target antigens for antibody mediated cellular cytotoxicity against renal cell carcinoma? J. Urol. 2002, 167, 707–712. [Google Scholar]
- Valerius, T.; Stockmeyer, B.; van Spriel, A.B.; Graziano, R.F.; van den Herik-Oudijk, I.E.; Repp, R.; Deo, Y.M.; Lund, J.; Kalden, J.R.; Gramatzki, M.; et al. FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood 1997, 90, 4485–4492. [Google Scholar]
- van Egmond, M.; Hanneke van Vuuren, A.J.; van de Winkel, J.G. The human Fc receptor for IgA (Fc alpha RI, CD89) on transgenic peritoneal macrophages triggers phagocytosis and tumor cell lysis. Immunol. Lett. 1999, 68, 83–87. [Google Scholar] [CrossRef]
- van Egmond, M.; van Vuuren, A.J.; Morton, H.C.; van Spriel, A.B.; Shen, L.; Hofhuis, F.M.; Saito, T.; Mayadas, T.N.; Verbeek, J.S.; van de Winkel, J.G. Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood 1999, 93, 4387–4394. [Google Scholar]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Maniecki, M.B.; Etzerodt, A.; Ulhoi, B.P.; Steiniche, T.; Borre, M.; Dyrskjot, L.; Orntoft, T.F.; Moestrup, S.K.; Moller, H.J. Tumor-promoting macrophages induce the expression of the macrophage-specific receptor CD163 in malignant cells. Int. J. Cancer 2012. [Google Scholar]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63–ra94. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar]
- Kellner, C.; Bruenke, J.; Stieglmaier, J.; Schwemmlein, M.; Schwenkert, M.; Singer, H.; Mentz, K.; Peipp, M.; Lang, P.; Oduncu, F.; et al. A novel CD19-directed recombinant bispecific antibody derivative with enhanced immune effector functions for human leukemic cells. J. Immunother. 2008, 31, 871–884. [Google Scholar] [CrossRef]
- Kugler, M.; Stein, C.; Kellner, C.; Mentz, K.; Saul, D.; Schwenkert, M.; Schubert, I.; Singer, H.; Oduncu, F.; Stockmeyer, B.; et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br. J. Haematol. 2010, 150, 574–586. [Google Scholar] [CrossRef]
- Stevenson, F.K.; Stevenson, G.T. Follicular lymphoma and the immune system: From pathogenesis to antibody therapy. Blood 2012, 119, 3659–3667. [Google Scholar] [CrossRef]
- Linsley, P.S.; Brady, W.; Grosmaire, L.; Aruffo, A.; Damle, N.K.; Ledbetter, J.A. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 1991, 173, 721–730. [Google Scholar] [CrossRef]
- Rudd, C.E.; Schneider, H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat. Rev. Immunol. 2003, 3, 544–556. [Google Scholar] [CrossRef]
- van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef]
- Hodi, F.S.; O'Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar]
- Khong, H.T.; Wang, Q.J.; Rosenberg, S.A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: Tumor escape by antigen loss and loss of MHC expression. J. Immunother. 2004, 27, 184–190. [Google Scholar] [CrossRef]
- Cabrera, T.; Lara, E.; Romero, J.M.; Maleno, I.; Real, L.M.; Ruiz-Cabello, F.; Valero, P.; Camacho, F.M.; Garrido, F. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol. Immunother. 2007, 56, 709–717. [Google Scholar] [CrossRef]
- Lanier, L.L. Follow the leader: NK cell receptors for classical and nonclassical MHC class I. Cell 1998, 92, 705–707. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Tosti, G.; di Pietro, A.; Passoni, C.; Pari, C.; Tedeschi, I.; Cataldo, F.; Martinoli, C.; Testori, A. Newly identified tumor antigens as promising cancer vaccine targets for malignant melanoma treatment. Curr. Top. Med. Chem. 2012, 12, 11–31. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar]
- Madrenas, J.; Chau, L.A.; Teft, W.A.; Wu, P.W.; Jussif, J.; Kasaian, M.; Carreno, B.M.; Ling, V. Conversion of CTLA-4 from inhibitor to activator of T cells with a bispecific tandem single-chain Fv ligand. J. Immunol. 2004, 172, 5948–5956. [Google Scholar]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One 2011, 6, e19499. [Google Scholar]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar]
- Fisher, T.S.; Kamperschroer, C.; Oliphant, T.; Love, V.A.; Lira, P.D.; Doyonnas, R.; Bergqvist, S.; Baxi, S.M.; Rohner, A.; Shen, A.C.; et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol. Immunother. 2012. [Google Scholar]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellstrom, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar]
- Attarwala, H. TGN1412: From discovery to disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Otz, T.; Grosse-Hovest, L.; Hofmann, M.; Rammensee, H.G.; Jung, G. A bispecific single-chain antibody that mediates target cell-restricted, supra-agonistic CD28 stimulation and killing of lymphoma cells. Leukemia 2009, 23, 71–77. [Google Scholar] [CrossRef]
- Bruenke, J.; Barbin, K.; Kunert, S.; Lang, P.; Pfeiffer, M.; Stieglmaier, K.; Niethammer, D.; Stockmeyer, B.; Peipp, M.; Repp, R.; et al. Effective lysis of lymphoma cells with a stabilised bispecific single-chain Fv antibody against CD19 and FcgammaRIII (CD16). Br. J. Haematol. 2005, 130, 218–228. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; Chiang, S.C.; Darmanin, S.; Fauriat, C.; Schlums, H.; Theorell, J.; Wood, S.M. Molecular mechanisms of natural killer cell activation. J. Innate Immun. 2011, 3, 216–226. [Google Scholar] [CrossRef]
- de Bruyn, M.; Bremer, E.; Helfrich, W. Antibody-based fusion proteins to target death receptors in cancer. Cancer Lett. 2011. [Google Scholar]
- de Bruyn, M.; Wei, Y.; Wiersma, V.R.; Samplonius, D.F.; Klip, H.G.; van der Zee, A.G.; Yang, B.; Helfrich, W.; Bremer, E. Cell surface delivery of TRAIL strongly augments the tumoricidal activity of T cells. Clin. Cancer Res. 2011, 17, 5626–5637. [Google Scholar] [CrossRef]
- Majeti, R.; Becker, M.W.; Tian, Q.; Lee, T.L.; Yan, X.; Liu, R.; Chiang, J.H.; Hood, L.; Clarke, M.F.; Weissman, I.L. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3396–3401. [Google Scholar]
- ten Cate, B.; de Bruyn, M.; Wei, Y.; Bremer, E.; Helfrich, W. Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr. Drug Targets 2011, 11, 95–110. [Google Scholar]
- Kontermann, R.E. Bispecific Antibodies: Developments and current perspectives. In Bispecific Antibodies, 1st ed.; Kontermann, R.E., Ed.; Springer-Verlag: Berlin, Germany, 2011; p. 373. [Google Scholar]
- Haas, C.; Krinner, E.; Brischwein, K.; Hoffmann, P.; Lutterbuse, R.; Schlereth, B.; Kufer, P.; Baeuerle, P.A. Mode of cytotoxic action of T cell-engaging BiTE antibody MT110. Immunobiology 2009, 214, 441–453. [Google Scholar] [CrossRef]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocyte. Blood 2000, 95, 2098–2103. [Google Scholar]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef]
- Topp, M.S.; Kufer, P.; Gokbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar]
- Handgretinger, R.; Zugmaier, G.; Henze, G.; Kreyenberg, H.; Lang, P.; von Stackelberg, A. Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leukemia 2011, 25, 181–184. [Google Scholar] [CrossRef]
- Pasquetto, M.V.; Vecchia, L.; Covini, D.; Digilio, R.; Scotti, C. Targeted drug delivery using immunoconjugates: principles and applications. J. Immunother. 2011, 34, 611–628. [Google Scholar] [CrossRef]
- Hopp, J.; Hornig, N.; Zettlitz, K.A.; Schwarz, A.; Fuss, N.; Muller, D.; Kontermann, R.E. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng. Des. Sel. 2010, 23, 827–834. [Google Scholar] [CrossRef]
- Kellner, C.; Peipp, M.; Valerius, T. Effector cell recruitment by bispecific antibodies. In Bispecific Antibodies, 1st ed.; Kontermann, R.E., Ed.; Springer-Verlag: Berlin, Germany, 2011; p. 373. [Google Scholar]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef]
- Kischel, R. Characterization in primates of MCSP- and CD33-specific human BiTE antibodies for treatment of Melanoma and AML. Proc. Am. Assoc. Cancer Res. 2008, 99, 2404. [Google Scholar]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Lutterbuese, P.; Schlereth, B.; Schaller, E.; Mangold, S.; Rau, D.; Meier, P.; Kiener, P.A.; et al. Potent control of tumor growth by CEA/CD3-bispecific single-chain antibody constructs that are not competitively inhibited by soluble CEA. J. Immunother. 2009, 32, 341–352. [Google Scholar] [CrossRef]
- Osada, T.; Hsu, D.; Hammond, S.; Hobeika, A.; Devi, G.; Clay, T.M.; Lyerly, H.K.; Morse, M.A. Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T-cell killing mediated by CEA/CD3-bispecific T-cell-engaging BiTE antibody. Br. J. Cancer 2010, 102, 124–133. [Google Scholar] [CrossRef]
- Torisu-Itakura, H.; Schoellhammer, H.F.; Sim, M.S.; Irie, R.F.; Hausmann, S.; Raum, T.; Baeuerle, P.A.; Morton, D.L. Redirected lysis of human melanoma cells by a MCSP/CD3-bispecific BiTE antibody that engages patient-derived T cells. J. Immunother. 2011, 34, 597–605. [Google Scholar] [CrossRef]
- Jager, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Strohlein, M.; Theissen, B.; Heiss, M.M.; Lindhofer, H. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res. 2012, 72, 24–32. [Google Scholar] [CrossRef]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef]
- Kiewe, P.; Hasmuller, S.; Kahlert, S.; Heinrigs, M.; Rack, B.; Marme, A.; Korfel, A.; Jager, M.; Lindhofer, H.; Sommer, H.; et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 3085–3091. [Google Scholar]
- Schroeder, P.; Lindemann, C.; Dettmar, K.; Brieger, J.; Gosepath, J.; Pogorzelski, B.; Seimetz, D.; Atz, J. Trifunctional antibodies induce efficient antitumour activity with immune cells from head and neck squamous cell carcinoma patients after radio-chemotherapy treatment. Clin. Transl. Oncol. 2011, 13, 889–898. [Google Scholar] [CrossRef]
- Zhang, T.; Sentman, C.L. Cancer immunotherapy using a bispecific NK receptor fusion protein that engages both T cells and tumor cells. Cancer Res. 2011, 71, 2066–2076. [Google Scholar] [CrossRef]
- Barber, A.; Rynda, A.; Sentman, C.L. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J. Immunol. 2009, 183, 6939–6947. [Google Scholar] [CrossRef]
- Thakur, A.; Sorenson, C.; Norkina, O.; Schalk, D.; Ratanatharathorn, V.; Lum, L.G. Activated T cells from umbilical cord blood armed with anti-CD3 x anti-CD20 bispecific antibody mediate specific cytotoxicity against CD20+ targets with minimal allogeneic reactivity: A strategy for providing antitumor effects after cord blood transplants. Transfusion 2012, 52, 63–75. [Google Scholar] [CrossRef]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Morgado, S.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Arcos, M.J.; Bengochea, M.L.; Duran, E.; Solana, R.; Tarazona, R. Human NK cells in acute myeloid leukaemia patients: analysis of NK cell-activating receptors and their ligands. Cancer Immunol Immunother 2011, 60, 1195–1205. [Google Scholar] [CrossRef]
- Alderson, K.L.; Sondel, P.M. Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J. Biomed. Biotechnol. 2011, 379123. [Google Scholar]
- Lundqvist, A.; Abrams, S.I.; Schrump, D.S.; Alvarez, G.; Suffredini, D.; Berg, M.; Childs, R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: A novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006, 66, 7317–7325. [Google Scholar]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Leibson, P.J. Signal transduction during natural killer cell activation: Inside the mind of a killer. Immunity 1997, 6, 655–661. [Google Scholar] [CrossRef]
- Terunuma, H.; Deng, X.; Dewan, Z.; Fujimoto, S.; Yamamoto, N. Potential role of NK cells in the induction of immune responses: implications for NK cell-based immunotherapy for cancers and viral infections. Int. Rev. Immunol. 2008, 27, 93–110. [Google Scholar] [CrossRef]
- Jaiswal, S.; Chao, M.P.; Majeti, R.; Weissman, I.L. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010, 31, 212–219. [Google Scholar]
- Lang, P.; Barbin, K.; Feuchtinger, T.; Greil, J.; Peipp, M.; Zunino, S.J.; Pfeiffer, M.; Handgretinger, R.; Niethammer, D.; Fey, G.H. Chimeric CD19 antibody mediates cytotoxic activity against leukemic blasts with effector cells from pediatric patients who received T-cell-depleted allografts. Blood 2004, 103, 3982–3985. [Google Scholar]
- Fuchs, A.; Colonna, M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin. Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Malmberg, K.J. Prospects for the use of NK cells in immunotherapy of human cancer. Nat. Rev. Immunol. 2007, 7, 329–339. [Google Scholar] [CrossRef]
- Lang, P.; Pfeiffer, M.; Teltschik, H.M.; Schlegel, P.; Feuchtinger, T.; Ebinger, M.; Klingebiel, T.; Bader, P.; Schlegel, P.G.; Beck, J.; et al. Natural killer cell activity influences outcome after T cell depleted stem cell transplantation from matched unrelated and haploidentical donors. Best Pract. Res. Clin. Haematol. 2011, 24, 403–411. [Google Scholar] [CrossRef]
- Locatelli, F.; Vinti, L.; Palumbo, G.; Rossi, F.; Bertaina, A.; Mastronuzzi, A.; Bernardo, M.E.; Rutella, S.; Dellabona, P.; Giorgiani, G.; et al. Strategies to optimize the outcome of children given T-cell depleted HLA-haploidentical hematopoietic stem cell transplantation. Best Pract. Res. Clin. Haematol. 2011, 24, 339–349. [Google Scholar] [CrossRef]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar]
- Ruggeri, L.; Mancusi, A.; Capanni, M.; Urbani, E.; Carotti, A.; Aloisi, T.; Stern, M.; Pende, D.; Perruccio, K.; Burchielli, E.; et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: Challenging its predictive value. Blood 2007, 110, 433–440. [Google Scholar] [CrossRef]
- Velardi, A.; Ruggeri, L.; Mancusi, A.; Aversa, F.; Christiansen, F.T. Natural killer cell allorecognition of missing self in allogeneic hematopoietic transplantation: a tool for immunotherapy of leukemia. Curr. Opin. Immunol. 2009, 21, 525–530. [Google Scholar] [CrossRef]
- Kitamoto, K.; Machida, Y.; Uchida, J.; Izumi, Y.; Shiota, M.; Nakao, T.; Iwao, H.; Yukimura, T.; Nakatani, T.; Miura, K. Effects of liposome clodronate on renal leukocyte populations and renal fibrosis in murine obstructive nephropathy. J. Pharmacol. Sci. 2009, 111, 285–292. [Google Scholar] [CrossRef]
- Giorgini, A.; Brown, H.J.; Lock, H.R.; Nimmerjahn, F.; Ravetch, J.V.; Verbeek, J.S.; Sacks, S.H.; Robson, M.G. Fc gamma RIII and Fc gamma RIV are indispensable for acute glomerular inflammation induced by switch variant monoclonal antibodies. J. Immunol. 2008, 181, 8745–8752. [Google Scholar]
- Hamerman, J.A.; Ogasawara, K.; Lanier, L.L. Cutting edge: Toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J. Immunol. 2004, 172, 2001–2005. [Google Scholar]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8 T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frebourg, T.; Michel, P.; et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar]
- Elsasser, D.; Stadick, H.; Stark, S.; Van de Winkel, J.G.; Gramatzki, M.; Schrott, K.M.; Valerius, T.; Schafhauser, W. Preclinical studies combining bispecific antibodies with cytokine-stimulated effector cells for immunotherapy of renal cell carcinoma. Anticancer Res. 1999, 19, 1525–1528. [Google Scholar]
- Weiner, L.M.; Holmes, M.; Adams, G.P.; LaCreta, F.; Watts, P.; Garcia de Palazzo, I. A human tumor xenograft model of therapy with a bispecific monoclonal antibody targeting c-erbB-2 and CD16. Cancer Res. 1993, 53, 94–100. [Google Scholar]
- Weiner, L.M.; Holmes, M.; Richeson, A.; Godwin, A.; Adams, G.P.; Hsieh-Ma, S.T.; Ring, D.B.; Alpaugh, R.K. Binding and cytotoxicity characteristics of the bispecific murine monoclonal antibody 2B1. J. Immunol. 1993, 151, 2877–2886. [Google Scholar]
- Lu, H.; Shi, M.; Wang, M.; Xie, Z.; Hu, M.; Yu, M.; Shen, B.; Ma, Y.; Guo, N. In vitro and in vivo antitumor effect of a trivalent bispecific antibody targeting ErbB2 and CD16. Cancer Biol. Ther. 2008, 7, 1744–1750. [Google Scholar] [CrossRef]
- Bruenke, J.; Fischer, B.; Barbin, K.; Schreiter, K.; Wachter, Y.; Mahr, K.; Titgemeyer, F.; Niederweis, M.; Peipp, M.; Zunino, S.J.; et al. A recombinant bispecific single-chain Fv antibody against HLA class II and FcgammaRIII (CD16) triggers effective lysis of lymphoma cells. Br. J. Haematol. 2004, 125, 167–179. [Google Scholar] [CrossRef]
- Singer, H.; Kellner, C.; Lanig, H.; Aigner, M.; Stockmeyer, B.; Oduncu, F.; Schwemmlein, M.; Stein, C.; Mentz, K.; Mackensen, A.; Fey, G.H. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J. Immunother. 2010, 33, 599–608. [Google Scholar] [CrossRef]
- Schubert, I.; Kellner, C.; Stein, C.; Kugler, M.; Schwenkert, M.; Saul, D.; Mentz, K.; Singer, H.; Stockmeyer, B.; Hillen, W.; et al. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. MAbs 2011, 3, 21–30. [Google Scholar] [CrossRef]
- Schubert, I.; Kellner, C.; Stein, C.; Kugler, M.; Schwenkert, M.; Saul, D.; Stockmeyer, B.; Berens, C.; Oduncu, F.S.; Mackensen, A.; et al. A recombinant triplebody with specificity for CD19 and HLA-DR mediates preferential binding to antigen double-positive cells by dual-targeting. MAbs 2012, 4, 45–56. [Google Scholar] [CrossRef]
- Schubert, I.; Stein, C.; Fey, G.H. Dual-Targeting for the Elimination of Cancer Cells with Increased Selectivity. Antibodies 2012, 1, 2–18. [Google Scholar] [CrossRef]
- Stein, C.; Schubert, I.; Fey, G.H. Trivalent and trispecific antibody derivatives for cancer therapy. In Bispecific Antibodies, 1st ed.; Kontermann, R.E., Ed.; Springer-Verlag: Berlin, Germany, 2011; p. 373. [Google Scholar]
- Kellner, C.; Bruenke, J.; Horner, H.; Schubert, J.; Schwenkert, M.; Mentz, K.; Barbin, K.; Stein, C.; Peipp, M.; Stockmeyer, B.; et al. Heterodimeric bispecific antibody-derivatives against CD19 and CD16 induce effective antibody-dependent cellular cytotoxicity against B-lymphoid tumor cells. Cancer Lett. 2011, 303, 128–139. [Google Scholar] [CrossRef]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef]
- Arndt, M.A.; Krauss, J.; Kipriyanov, S.M.; Pfreundschuh, M.; Little, M. A bispecific diabody that mediates natural killer cell cytotoxicity against xenotransplantated human Hodgkin's tumors. Blood 1999, 94, 2562–2568. [Google Scholar]
- Kipriyanov, S.M.; Cochlovius, B.; Schafer, H.J.; Moldenhauer, G.; Bahre, A.; Le Gall, F.; Knackmuss, S.; Little, M. Synergistic antitumor effect of bispecific CD19 x CD3 and CD19 x CD16 diabodies in a preclinical model of non-Hodgkin's lymphoma. J. Immunol. 2002, 169, 137–44. [Google Scholar]
- Schlenzka, J.; Moehler, T.M.; Kipriyanov, S.M.; Kornacker, M.; Benner, A.; Bahre, A.; Stassar, M.J.; Schafer, H.J.; Little, M.; Goldschmidt, H.; et al. Combined effect of recombinant CD19 x CD16 diabody and thalidomide in a preclinical model of human B cell lymphoma. Anticancer Drugs 2004, 15, 915–919. [Google Scholar] [CrossRef]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar]
- Germain, C.; Larbouret, C.; Cesson, V.; Donda, A.; Held, W.; Mach, J.P.; Pelegrin, A.; Robert, B. MHC class I-related chain A conjugated to antitumor antibodies can sensitize tumor cells to specific lysis by natural killer cells. Clin. Cancer Res. 2005, 11, 7516–7522. [Google Scholar]
- Dhodapkar, M.V.; Sanderson, R.D. Syndecan-1 (CD 138) in myeloma and lymphoid malignancies: A multifunctional regulator of cell behavior within the tumor microenvironment. Leuk. Lymphoma 1999, 34, 35–43. [Google Scholar]
- Dhodapkar, K.M.; Krasovsky, J.; Williamson, B.; Dhodapkar, M.V. Antitumor monoclonal antibodies enhance cross-presentation ofcCellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J. Exp. Med. 2002, 195, 125–133. [Google Scholar] [CrossRef]
- von Strandmann, E.P.; Hansen, H.P.; Reiners, K.S.; Schnell, R.; Borchmann, P.; Merkert, S.; Simhadri, V.R.; Draube, A.; Reiser, M.; Purr, I.; et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 2006, 107, 1955–1962. [Google Scholar]
- Cho, H.M.; Rosenblatt, J.D.; Tolba, K.; Shin, S.J.; Shin, D.S.; Calfa, C.; Zhang, Y.; Shin, S.U. Delivery of NKG2D ligand using an anti-HER2 antibody-NKG2D ligand fusion protein results in an enhanced innate and adaptive antitumor response. Cancer Res. 2010, 70, 10121–10130. [Google Scholar]
- Jachimowicz, R.D.; Fracasso, G.; Yazaki, P.J.; Power, B.E.; Borchmann, P.; Engert, A.; Hansen, H.P.; Reiners, K.S.; Marie, M.; von Strandmann, E.P.; et al. Induction of in vitro and in vivo NK cell cytotoxicity using high-avidity immunoligands targeting prostate-specific membrane antigen in prostate carcinoma. Mol. Cancer Ther. 2011, 10, 1036–1045. [Google Scholar] [CrossRef]
- Kellner, C.; Hallack, D.; Glorius, P.; Staudinger, M.; Mohseni Nodehi, S.; de Weers, M.; van de Winkel, J.G.; Parren, P.W.; Stauch, M.; Valerius, T.; et al. Fusion proteins between ligands for NKG2D and CD20-directed single-chain variable fragments sensitize lymphoma cells for natural killer cell-mediated lysis and enhance antibody-dependent cellular cytotoxicity. Leukemia 2011, 26, 830–834. [Google Scholar]
- Barkholt, L.; Alici, E.; Conrad, R.; Sutlu, T.; Gilljam, M.; Stellan, B.; Christensson, B.; Guven, H.; Bjorkstrom, N.K.; Soderdahl, G.; et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: A phase I clinical study. Immunotherapy 2009, 1, 753–764. [Google Scholar] [CrossRef]
- Klingemann, H. G.; Martinson, J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy 2004, 6, 15–22. [Google Scholar] [CrossRef]
- Koehl, U.; Esser, R.; Zimmermann, S.; Tonn, T.; Kotchetkov, R.; Bartling, T.; Sorensen, J.; Gruttner, H.P.; Bader, P.; Seifried, E.; et al. Ex vivo expansion of highly purified NK cells for immunotherapy after haploidentical stem cell transplantation in children. Klin. Padiatr. 2005, 217, 345–350. [Google Scholar] [CrossRef]
- Schubert, I. Department Biology, University of Erlangen-Nuremberg. 2012, unpublished work.
- Alici, E.; Sutlu, T.; Bjorkstrand, B.; Gilljam, M.; Stellan, B.; Nahi, H.; Quezada, H.C.; Gahrton, G.; Ljunggren, H.G.; Dilber, M.S. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood 2008, 111, 3155–3162. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stein, C.; Schubert, I.; Fey, G.H. Natural Killer (NK)- and T-Cell Engaging Antibody-Derived Therapeutics. Antibodies 2012, 1, 88-123. https://doi.org/10.3390/antib1010088
Stein C, Schubert I, Fey GH. Natural Killer (NK)- and T-Cell Engaging Antibody-Derived Therapeutics. Antibodies. 2012; 1(1):88-123. https://doi.org/10.3390/antib1010088
Chicago/Turabian StyleStein, Christoph, Ingo Schubert, and Georg H. Fey. 2012. "Natural Killer (NK)- and T-Cell Engaging Antibody-Derived Therapeutics" Antibodies 1, no. 1: 88-123. https://doi.org/10.3390/antib1010088