Current and Potential Uses of Immunocytokines as Cancer Immunotherapy

1
The Departments of Pediatrics, Human Oncology and Genetics and the UW Carbone Cancer Center, University of Wisconsin, Madison, WI 53792, USA
2
Provenance Biopharmaceuticals Corp, Burlington, MA 01805, USA
*
Author to whom correspondence should be addressed.
Antibodies 2012, 1(2), 149-171; https://doi.org/10.3390/antib1020149
Submission received: 13 June 2012
/
Revised: 26 June 2012
/
Accepted: 26 June 2012
/
Published: 4 July 2012
(This article belongs to the Special Issue Modes of Antibody Action for Cancer Therapy)
Abstract
:Immunocytokines (ICs) are a class of molecules created by linking tumor-reactive monoclonal antibodies to cytokines that are able to activate immune cells. Tumor selective localization is provided by the ability of the mAb component to bind to molecules found on the tumor cell surface or molecules found selectively in the tumor microenvronment. In this way the cytokine component of the immunocytokine is selectively localized to sites of tumor and can activate immune cells with appropriate receptors for the cytokine. Immunocytokines have been made and tested by us, and others, using a variety of tumor-reactive mAbs linked to distinct cytokines. To date, the majority of clinical progress has been made with ICs that have linked human interleukin-2 (IL2) to a select number of tumor reactive mAbs that had already been in prior clinical testing as non-modified mAbs. Here we briefly review the background for the creation of ICs, summarize current clinical progress, emphasize mechanisms of action for ICs that are distinct from those of their constituent components, and present some directions for future development and testing.
1. Cancer Immunotherapy: Broad Application
Despite over 50 years of compelling preclinical evidence for effective cancer immunotherapy, as of 2009 there were only 4 widely used, approved, effective clinical immunotherapies being used broadly to treat cancer. They included: (A) allogeneic bone marrow transplantation for leukemia, largely successful via the “graft vs. leukemia” effect [1]; (B) tumor reactive monoclonal antibodies (mAbs), effective in part via antibody dependent cell-mediated cytotoxicity (ADCC) [2]; (C) intravesical BCG for superficial bladder cancer, likely active via toll-like receptors on leukocytes [3]; and (D) administration of IL2 for melanoma and renal cell cancer [4].
Over the past 2 years Phase III trials of 3 distinct forms of immunotherapy demonstrated clinical benefit. Each was published in the New England Journal of Medicine. Two are now FDA approved and the 3rd is under FDA review. They are: (1) Vaccination of patients (pts) with advanced prostate cancer, with a preparation of PAP-loaded autologous dendritic cells [5]; (2) Augmenting endogenous anti-melanoma immunity via anti-CTLA-4 mAb [6,7]; and (3) Enhancing ADCC in children with neuroblastoma (NBL) by combining an anti-GD2 mAb with NK activation (via IL2) and neutrophil/monocyte activation (via GM-CSF) [8].
The use of ICs for cancer treatment is, at least in part, based on augmenting ADCC by the more effective localization of cytokines able to activate ADCC.
2. ADCC and FcRs
Preclinical studies have shown that tumor-reactive mAbs can mediate in vitro tumor destruction via ADCC [9,10]. While there is a family of FcγRs on leukocytes [11], the most important for ADCC are the FcγR2a (CD32) FcRs expressed primarily on neutrophils, monocytes and macrophages, and the FcγRγ3a (CD16) FcRs expressed primarily on NK cells. The affinity of these human FcRs is highest for human IgG1 immunoglobulins [12]. When a sufficient concentration of a tumor-reactive mAb encounters a tumor cell (e.g., Rituximab and a CD20+ tumor cell), the mAb binds to the cell, presenting a lattice of surface bound mAb molecules with exposed Fccomponents. When an effector expressing FcRs (such as an NK cell) encounters this mAb-coated tumor cell, the FcRs simultaneously engage multiple Fc moieties via multipoint binding. The multipoint binding enables the effector cell to transiently adhere to the mAb-coated tumor cell (akin to the multipoint adhesion facilitated by “a Velcro effect”), and then to activate the effector cell via the signaling pathways induced by the FcRs [13]. This mAb-FcR mediated activation of the tumor-bound effector cell can result in cytokine and chemokine release by the effector cell, antigen ingestion/presentation, and “downstream” immune recruitment and activation of other antitumor effector cells [14]. In addition, this mAb-FcR mediated activation of the tumor-bound effector cell can result in activation of ADCC, which may involve granule induced cell death, death-signal induced apoptosis, or biochemical induced toxicity, depending upon the effector cell type and state of activation [15,16]. For simplicity, we will refer to these pathways (direct ADCC and FcR induced “downstream” effects) as “in vivo ADCC”. In mice, antitumor effects induced by mAbs that mediate ADCC are abrogated when using mAbs without functional Fc components [9], or in mice lacking functional FcRs [17].
The importance of FcR affinity in the in vivo efficacy of mAbs has been demonstrated in several clinical studies. The human CD16 FcγR3a molecule has 2 primary alleles; a lower affinity allele that bears a phenylalanine (F) at amino acid (a.a.) 158, and a higher affinity allele that bears a valine (V) at a.a. 158. Analogously, the human CD32 FcγR2a molecule also has 2 primary alleles; a lower affinity allele that bears an arginine (R) at a.a. 131, and a higher affinity allele that bears a histidine (H) at a.a. 131. Landmark studies demonstrated that lymphoma patients with high affinity FcR alleles were more likely to benefit from Rituximab than patients without high affinity FcRs [18,19]. Furthermore, the presence or absence of complement inactivators [20] (such as CD59) on lymphomas does not seem to influence Rituximab efficacy [21]. The CD16 FcγR3a allelic polymorphism affects rituximab-mediated ADCC of autologous EBV transformed B lymphocytes in vitro [22]. As expected, this cytotoxicity is attenuated by inhibitory killer-immunoglobulin-like receptors (KIR) found primarily on NK cells, but to a variable degree in different individuals [22]. Together, these observations indicate that the in vivo efficacy of Rituximab is likely dependent on in vivo ADCC rather than complement activation [23] and that the degree of cytotoxicity may also be regulated by KIR.
Herceptin and Erbitux are FDA-approved anti-tumor-receptor (HER2 and EGFR) mAbs that inhibit ligand-induced activation of these receptors from stimulating tumor cell growth [24]. In addition, both these mAbs mediate ADCC in vitro. Most, but not all, analyses of FcRs in clinical studies for these mAbs (and for Rituxan) have demonstrated greater benefit in individuals with high affinity alleles for FcγR2a and FcγR3a [25,26]. These results suggest that at least some of the antitumor benefits of Herceptin and Erbitux come from in vivo ADCC, although increased signal inhibition and apoptosis resulting from the cross-linking of cell-bound antibody by FcR-expressing cells cannot be ruled out [27].
While not all analyses of Rituximab, Herceptin and Erbitux have shown identical results, most have demonstrated benefit for individuals with high affinity FcR alleles [28]. In addition, there is still controversy regarding the “gene dose” effect for these alleles. Namely the high affinity genotypes (VV for FcγR3a and HH for FcγR2a) show better antitumor effects than the lowest affinity genotypes (FF and RR respectively). However it is not clear whether the heterozygotes (i.e., VF and HR) show effects that are similar to the low affinity homozygotes (FF and RR), or potentially function somewhere in between the homozygotes (i.e., VF functioning between VV and FF, and HR functioning between HH and RR) [26].
In addition, FcR affinity influences the antitumor efficacy of antitumor antibodies induced by vaccination regimens. Having high affinity FcγR2a and FcγR3a genotypes is associated with beneficial antitumor effects for immunization to idiotypic antigens on B cell tumors [29] and to antigens on colon cancer [30].
3. Augmenting ADCC with Effector Activation, Ch 14.18 + Cytokines
IL2 is a potent activator of NK cells [31,32], which have FcRs and mediate ADCC [13]. In vitro treatment of NK cells with IL2 augments NK ADCC [33]. We demonstrated that in vivo administration of IL2 to patients augmented the ability of their circulating NK cells to mediate ADCC in vitro [34]. We proposed that activating NK cells in vivo with IL2 would thus enhance in vivo ADCC from concomitant mAb treatment [35,36]. Our studies utilized mAbs that recognize the GD2 disialoganglioside on melanoma and NBL [37]. Our preclinical data suggested efficacy would be most apparent when this approach was used to treat individuals with smaller amounts of cancer (non-bulky disease) [38,39]. We performed a series of pilot and phase I/II trials of this approach for patients with neuroblastoma (NBL) or melanoma using the 14.G2a murine antibody and its derivative, the ch14.18 chimeric mAb [40,41,42]. We worked with the Children’s Oncology Group (COG) to test this approach clinically in the minimal disease setting in a pilot COG Phase I trial for children with high-risk NBL that were in remission after autologous HSCT (ASCT) but likely to relapse [43]. To augment ADCC we incorporated IL2 to activate NK cells and GM-CSF to activate neutrophils/macrophages [44,45,46]. The regimen was tolerated acceptably, and clinical results appeared better than historical controls [43]. We then moved this same regimen into a large Children’s Oncology Group (COG) phase III trial [8]. With only 61% of anticipated accrual, our biostatisticians stopped and “unblinded” the study, since the immunotherapy treatment was statistically superior to the control treatment for both event-free survival (66% vs. 46% p = 0.01), and for overall survival (86% vs. 75% p = 0.02). A separate German study of this same ch14.18 mAb, in a similar dose and regimen, but without the use of IL2 and GM-CSF initially reported no immunotherapeutic advantage for the ch14.18 mAb [47]. This suggests, although does not prove, that adding the IL2 and GM-CSF (to augment ADCC) to the ch14.18 treatment was responsible for the clinical benefit. Furthermore, these data from this COG study [8] suggest that other ADCC-inducing mAbs (i.e., Rituximab, Herceptin and Erbitux) might be considered for trials in which high risk patients likely to relapse receive these mAbs in combination with agents known to activate ADCC (like IL2 + GM-CSF).
4. Immunocytokines: Linking IL2 to Anti-GD2 mAb; Preclinical Development
Despite the efficacy of anti-GD2 mAb + cytokines in our recent NBL trial [8], only 66% are NBL-free at 2 years. We aim to further enhance the clinical potency of ADCC, in order to obtain even better clinical results. Preclinical data with the hu14.18-IL2 IC indicate that this should be possible. ADCC depends, in part, upon the number and function of FcRs on the effector cells [25,26,27,28,29,30]. When NK cells are stimulated with IL2 in vivo, they mediate augmented ADCC [34]. However, we have shown that up to 50% of the activated NK cells circulating in cancer patients following in vivo treatment with IL2 do not have FcRs, in contrast to most resting NK cells [48]. These FcR- activated NK cells are more lytic to tumor cells in direct assays not dependent on mAb and FcRs. We also found that NK cells activated in vivo by IL2 show augmented expression of the IL2Rβ [49] and enhanced in vitro responses to IL2 [50]. Furthermore, IL2R‑bearing T cells that may not be able to specifically recognize tumors (with their TCRs) should still be responsive to IL2. It may be beneficial to treat these IL2R+ effector cells with a molecule that bridges them to tumor cells and then activates them. These are some of the functions of the anti-GD2 IC hu14.18-IL2 (Figure 1) and its preclinical predecessor ch14.18-IL2. These ICs were constructed by fusing the human IL2 gene to the ch14.18 or hu14.18 IgG1 genes [51]. The Gillies, Reisfeld, and Sondel labs have shown that these ICs activate GD2-specific tumor cell lysis by IL2R+ T cells and NK cells [51,52]. Ch14.18-IL2 induces anti-melanoma activity in a SCID–xenograft model [53] and in conventional mice bearing syngeneic tumors expressing GD2 (B78 melanoma) [54,55], and anti-NBL activity in conventional mice bearing the GD2+ NXS2 NBL [56,57].
Ch14.18-IL2 IC causes dramatically better antitumor effects against localized or metastatic NXS2 NBL than comparable amounts of ch14.18 mAb and IL2 in combination (Table 1) [56,57]. Under these conditions, IC-treated mice show no metastases. The in vivo destruction of NXS2 in mice receiving ch14.18-IL2 is largely NK mediated [56,57], while the antitumor effect against the B78 melanoma involves T cells [54]. This T cell effect demonstrates epitope spread, as ch14.18-IL2 enables C57Bl/6 mice to destroy GD2- B16 melanoma cells, but only if they are a component of mixed tumors, created by co-injection with the GD2+ B78 melanoma cells (Table 1) [55]. Thus ch14.18-IL2 induces far more potent antitumor effects in melanoma or NBL-bearing mice than the combination of ch14.18 mAb + IL2, and functions both as a T cell inducing vaccine and a potent activator of NK-mediated ADCC. More potent tumor eradication is seen in mice with smaller tumor burdens (Figure 2) [39]. Similar results are seen in murine models using the hu14.18-IL2 IC [58]. As a nearly “pure” human protein it is predicted to be less immunogenic in patients than would ch14.18-IL2.
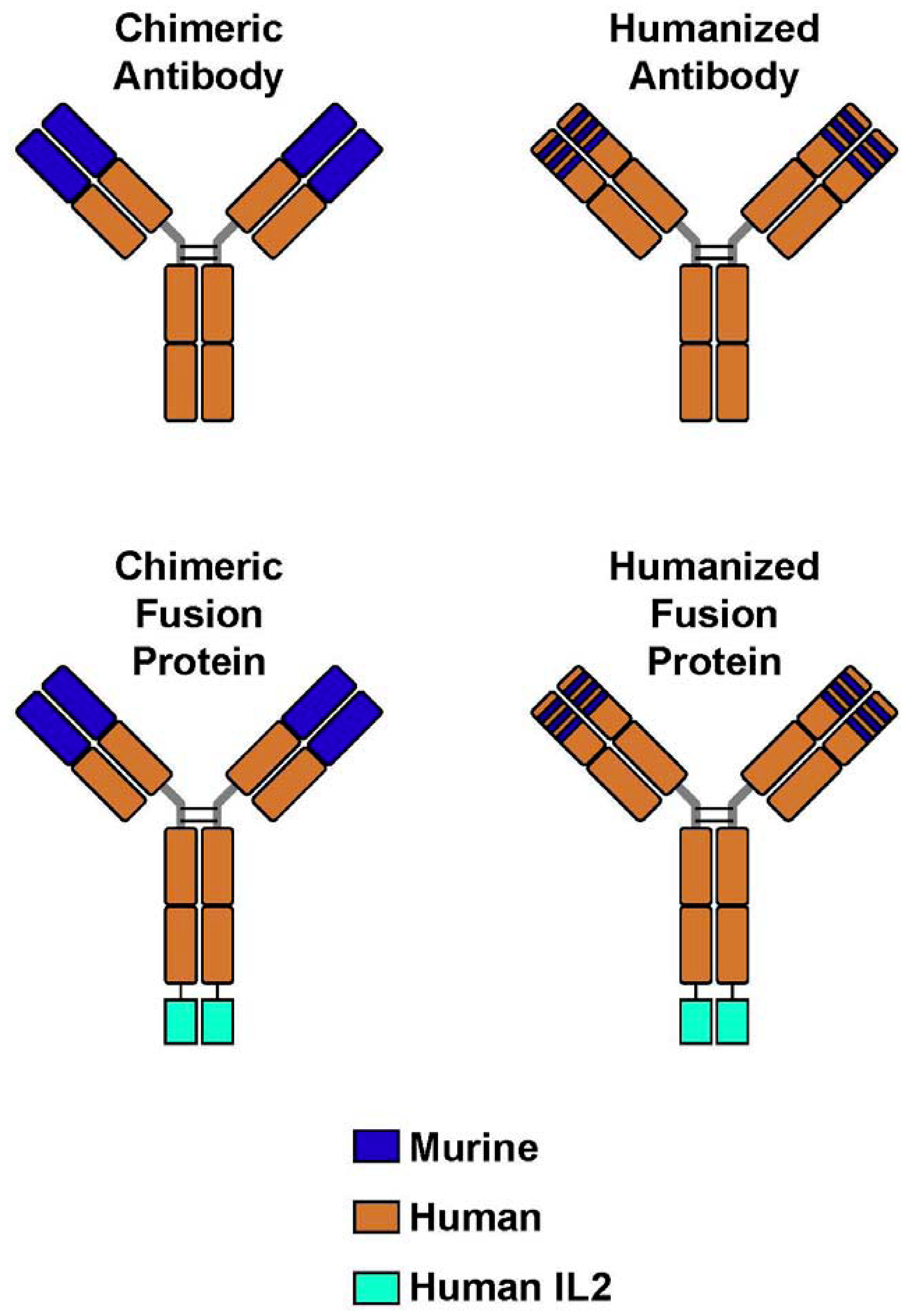
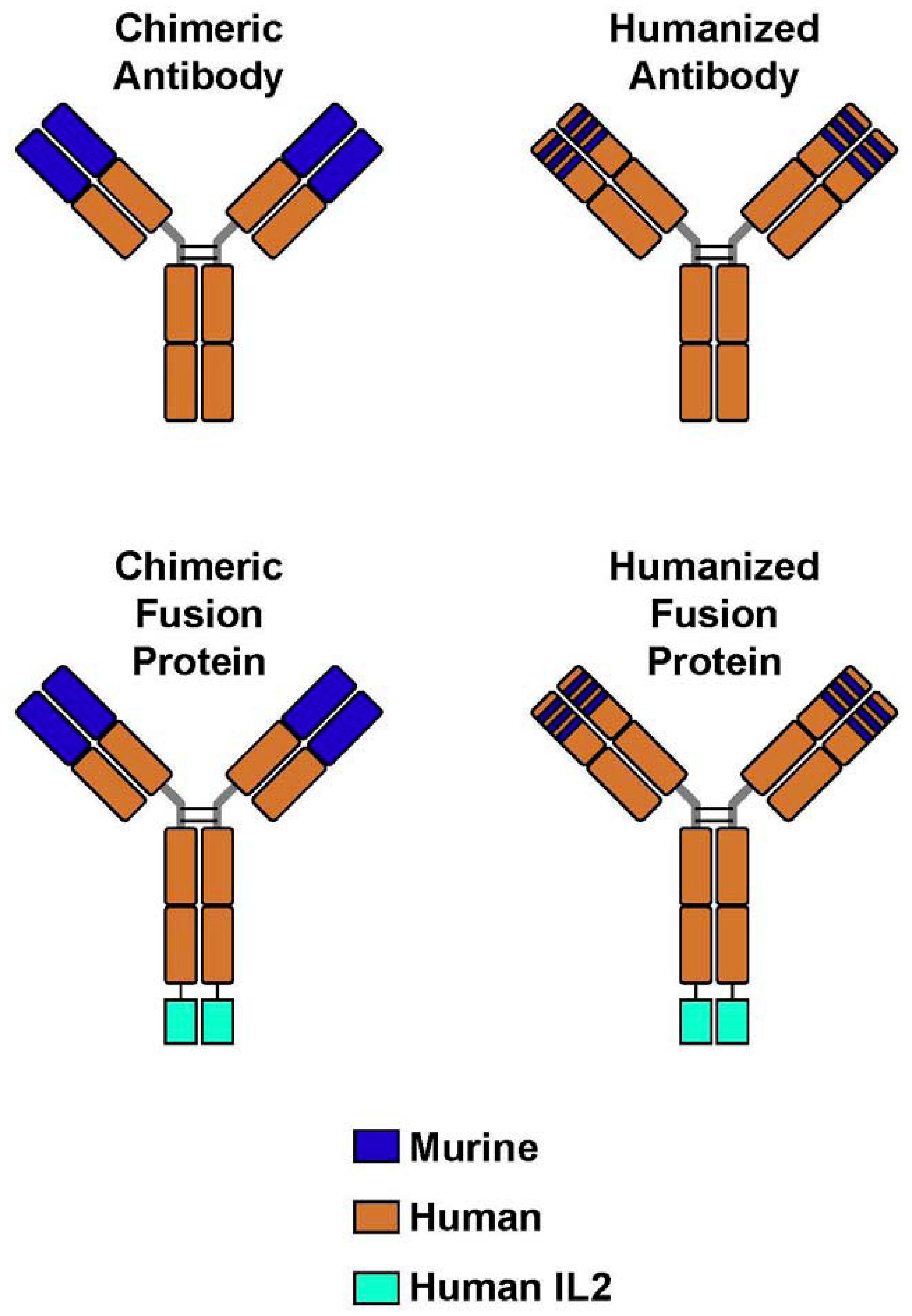
Figure 1.
Monoclonal Antibodies and Immunocytokines. (A) A chimeric monoclonal antibody (mAb) combines the constant region of a human antibody with the variable domain of a murine antibody. The antigen specificity is conferred by the murine variable domain. (B) In the humanized mAb, the murine framework determinants of both the heavy and light chains are replaced with human framework determinants, but the antigen specificity of the original murine mAb is retained. (C,D) Immunocytokines combine the mAb with covalently linked cytokines, such as molecules of interleukin 2 (IL-2), in this case to the end of each of the heavy chains at the C-terminus.
Figure 1.
Monoclonal Antibodies and Immunocytokines. (A) A chimeric monoclonal antibody (mAb) combines the constant region of a human antibody with the variable domain of a murine antibody. The antigen specificity is conferred by the murine variable domain. (B) In the humanized mAb, the murine framework determinants of both the heavy and light chains are replaced with human framework determinants, but the antigen specificity of the original murine mAb is retained. (C,D) Immunocytokines combine the mAb with covalently linked cytokines, such as molecules of interleukin 2 (IL-2), in this case to the end of each of the heavy chains at the C-terminus.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Tumor | Number of Tumor Foci |
|---|---|---|
| PBS | *NXS2 | >250, >250, >250, >250, 240, 115 |
| IL2+ch14.18 | *NXS2 | 174, 134, 105, 102, 91, 83 |
| **ch14.18-IL2 | *NXS2 | 0, 0, 0, 0, 0, 0, |
| ch14.18-IL2 | #B16 | >500, >500, >500, >500, >500, 138, 97 |
| PBS | #B78 +B16 | >500, >500, >500, >500, >500, >500, >500,>500 |
| IL2+ch14.18 | #B78 + B16 | >500, >500, >500, >500, 189, 179, 104 |
| ch14.18-IL2 | #B78 + B16 | ##0, 0, 2, 7, 9, 12, 21, 43 |
* Hepatic metastases (mets) were induced with 106 NXS2 cells IV into AJ mice. On day 1 mice received PBS, 10 mcg ch14.18 mAb + 30,000 IU IL2/d, or 10 mcg of ch14.18-IL2 daily × 6 days. Mets were scored for each of 6 mice on day 21, and were less in the IC group** than the other 2 groups (p < 0.001) [57]; # Pulmonary mets in C57Bl mice were induced by IV injection of 1 × 106 B16 cells (GD2-) alone, or combined with 5 × 106 B78 cells (GD2+). One week post-inoculation, 7 days of PBS, 8 μg ch14.18 + 24,000 IU IL-2, or 8 μg ch14.18-IL2 was initiated, and mets were scored 4 weeks later. ## Mice with mixed tumors treated with IC had fewer mets than all other groups (p ≤ 0.002) [55].
| Receptor | Ligand |
|---|---|
| KIR2DL1 (CD158a) | HLA-C2 (Lys80) |
| KIR2DL2/KIR2DL3 (CD158b) | HLA-C1 (Asp80) |
| KIR3DL1 (CD158e) | HLA-Bw4, HLA-ABw4 |
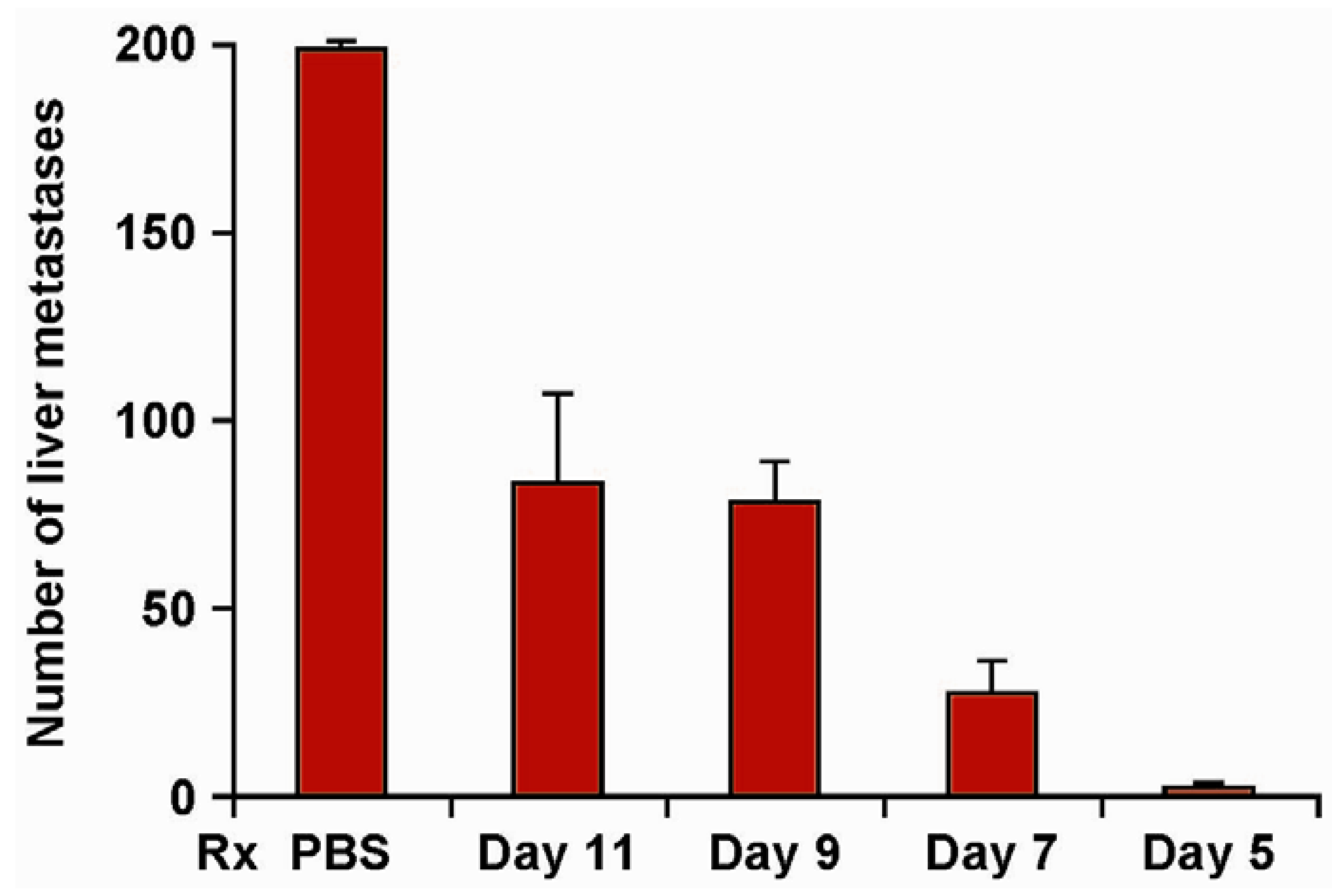
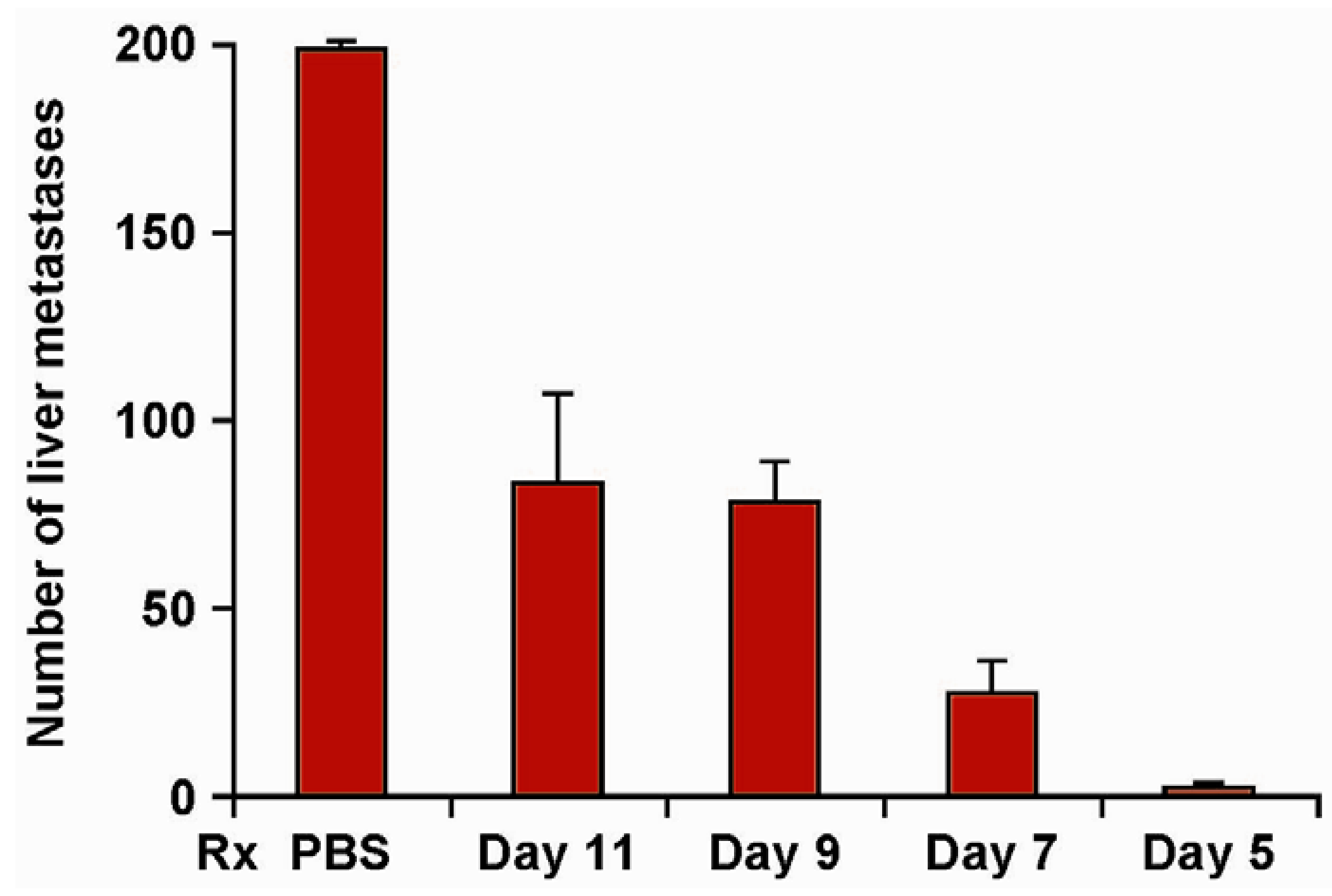
Figure 2.
Efficacy of hu14.18-IL2 therapy is influenced by the timing of its initiation. Groups of 4 mice were treated IV with 5 µg/day × 5 days of hu14.18-IL2 beginning on d 5, 7, 9 or 11 following IV injection of 5 × 105 NXS2 NB cells (or IV PBS). Mice were sacrificed on d28 and the liver metastases were enumerated. Data are mean ± SE (adapted from Neal et al., 2004, [39]).
Figure 2.
Efficacy of hu14.18-IL2 therapy is influenced by the timing of its initiation. Groups of 4 mice were treated IV with 5 µg/day × 5 days of hu14.18-IL2 beginning on d 5, 7, 9 or 11 following IV injection of 5 × 105 NXS2 NB cells (or IV PBS). Mice were sacrificed on d28 and the liver metastases were enumerated. Data are mean ± SE (adapted from Neal et al., 2004, [39]).
4.1. Phase I Clinical Testing of hu14.18-IL2
These results supported the FDA-IND for our “first in human” Phase I study in adults with melanoma at the UWCCC [59], and enabled our team to conduct a Phase I trial of hu14.18-IL2 in children [60], using the same schedule used in our melanoma trial.
Our Phase I trials in melanoma [59], and NBL [60], identified the MTD and immune effects of 3 daily IV doses of hu14.18-IL2. Of the 28 patients with measurable NBL, 3 showed isolated marrow improvement, and 1 showed a non-confirmed CR that was not completely attributable to the IC. Of the 28 melanoma patients with measurable disease, none showed a response. Of the 5 melanoma patients that entered with no evidence of disease (NED) following recent surgical resection of progressive metastases, 3 patients recurred at 1, 6 and 92 months, and 2 remain in remission >74 and >117 months following treatment. Based on the suggestion of IC activity in melanoma patients with NED, the BM-activity seen in our NBL study, and our preclinical murine data documenting better antitumor efficacy for smaller tumors (Figure 2) [39], we proposed that greater antitumor activity would be detected when using hu14.18-IL2 to treat patients with less “bulky” tumors.
4.2. Phase II Clinical Testing of hu14.18-IL2
We thus worked with the COG to conduct a Phase II study of hu14.18-IL2 in Children with Recurrent or Refractory NBL. This Phase II protocol was designed to evaluate the clinical antitumor activity and in vivo biological-immunological effects of hu14.18-IL2, in children with refractory or recurrent NBL, and separately assess patients with bulky disease and patients with minimal evaluable NBL. Patients received 3 daily IV doses of 12.0 mg/m2/day IC in each of 4 monthly courses. Patients with CR or PR could receive 2 more courses [61].
Fifteen patients had disease measurable by standard radiographical criteria (stratum 1) and 24 patients had disease evaluable only by MIBG and/or BM histology (stratum 2). Responses were confirmed by independent radiological review and immunocytochemistry (ICC) evaluation of BM. No responses were seen in the 15 stratum 1 pts. In the 24 stratum 2 pts, 5 showed CR (complete resolution of MIBG avid disease and complete resolution of BM disease both by standard morphology and ICC). Of these 5 pts, 4 relapsed after 8, 12, 18 and 30 months, while 1 remains in CR after 35+ mo. At study entry, all 5 had recurrent-refractory disease following ASCT [61].
Two other stratum-2 patients showed disease improvement suggesting efficacy, but did not quite meet protocol criteria for PR or CR. One pt had BM-CR and a decrease in MIBG-avid disease that was read as a PR by the treating center but not by independent review. A 2nd pt had resolution of MIBG+ disease and BM improvement, but not clearing.
The spectrum and degree of toxicities were similar to that seen in our Phase I studies. Of the 38 patients evaluable for toxicity, 10 did not receive cycle 2 of therapy [2 patients due to protocol defined DLT, 7 because of progressive disease (PD) and 1 due to parental choice]. Of the 28 patients receiving cycle 2, 3 patients had toxicity in cycle 1 (pain, hypotension and hyperbilirubinemia) requiring a reduction in dose for cycle 2 [61].
The clinical response data support the conclusion that this agent and regimen have clinical activity in stratum-2 but not in stratum-1 pts. Although this study was not powered to address whether response may be predicted by entry status, identifying 5 CRs and 2 patients with clear improvement in the 24 patients in stratum-2 vs. 0 responses of 15 patients in stratum-1 suggests there may be a real difference (p = 0.03). As all patients in this study had recurrent/refractory disease following extremely aggressive prior multi-modality therapy, these responses are of interest to pediatric oncologists. COG is currently performing a follow-up feasibility-Phase II trial with this IC in combination with GM-CSF and cis-retinoic acid, focused on high risk NBL patients with non-bulky (stratum-2) disease to confirm/extend these response data to potentially support licensing of this agent. Furthermore, in order to evaluate potential mechanisms of anti-tumor response, we investigated whether the inhibitory NK Killer Immunoglobulin-like receptors (KIR) may be involved in regulating this clinical antitumor activity in patients receiving hu14.18-IL2.
5. The Roles of KIR/KIR-Ligand (KIR-L) and FcR Genotypes in the Responses Induced by hu14.18-IL2
The Killer Immunoglobulin-like Receptor (KIR) genes encode receptors that recognize MHC class I molecules. On a cellular level the expression of KIR and HLA defines the repertoire and responsiveness of NK cells. While there are both activating and inhibitory KIR gene products, most (but not all) clinical attention has been focused on the inhibitory KIR molecules. The inhibitory KIR receptors transmit inhibitory signals to NK cells when they encounter their cognate MHC class I molecules [62,63,64,65,66,67,68,69,70]. The ligand specificity is focused on amino acid position 80 of the HLA class I. As presented in Table 2, HLA-C alleles with Lys80 constitute ligands for KIR2DL1 and those with Asp80 interact with KIR2DL2 and KIR2DL3. Exceptions to this rule have recently been reported [71,72]. KIR3DL1 recognizes HLA-B alleles with the so-called Bw4 motif, also conferred by amino acid positions 77–80. As recently shown, HLA-A alleles with the Bw4 motif can serve as ligands for KIR3DL1 [73]. Ruggeri et al. first reported on the phenomenon of KIR-L incompatibility and response to HLA-haploidentical HSCT, primarily in adult patients with acute myeloid leukemia (AML) [74,75]. According to this analysis, improved leukemia control is seen when there is a difference in HLA between the donor and recipient such that the recipient’s cells lack the ligand specific for the donor KIR, creating a “missing KIR ligand” situation. Leung et al. proposed the principle of missing KIR ligand analysis (designated here as KIR/KIR-L mismatch), in which the HSCT recipient lacks one or greater HLA class-I ligands for the HSCT donor’s inhibitory KIRs (regardless of whether or not the donor’s own HLA provides such ligands) [76]. They found that the response of pediatric patients with AML and acute lymphoid leukemia (ALL) to haploidentical HSCT could be predicted by the presence of this KIR/KIR-ligand mismatch. The KIR/KIR-L mismatch principle also posits that a difference in HLA between the donor and recipient is not necessary for the benefit of KIR-HLA mismatching. This was confirmed when an analysis of results of HLA-identical T cell depleted sibling HSCT also revealed a benefit of KIR/KIR-ligand mismatch [77].
The genes encoding for KIR and HLA class I KIR ligands are polymorphic and inherited independently [78]. As such, individuals differ with respect to the number of KIR genes present in the genome and frequently express KIR receptors that have no corresponding HLA ligands on autologous cells, thus displaying an “autologous” KIR-KIR ligand mismatch. This scenario of autologous KIR/KIR-L mismatch occurs in approximately 60% of the US population and has been implicated as a favorable prognostic factor in pediatric solid tumor patients following ASCT [78,79].
Until 2010 the analyses of KIR/KIR-L mismatch in the setting of cancer treatment had been confined to the clinical setting of allogeneic HSCT [70,71,77], allogeneic adoptive NK infusions [80,81,82] and autologous HSCT [78,79]. We hypothesized that this KIR/KIR-L relationship pertains more to the cellular function of NK cells, independent of allogeneic relationships [83] or autologous HSCT. We also hypothesized that individuals that were KIR/KIR-L mismatched would be more likely to respond favorably to cancer immunotherapy that is NK-mediated. Our preclinical data with the hu14.18-IL2 IC demonstrated that antitumor effects in mice could be mediated by NK cells, in the absence of T cells [39]. Furthermore, we showed that murine NBLs that escape from suboptimal hu14.18-IL2 regimens do so by up-regulating MHC class I, a mechanism that apparently turns off NK cells via the MHC-specific inhibitory Ly 49 receptors on murine NK cells [58]. These observations suggested that circumventing KIR mediated inhibition of NK cells may be helpful in the in vivo efficacy of this form of immunotherapy. Thus we hypothesized that individuals that were “autologous KIR/KIR-L mismatched” would have better NK function against their autologous tumor when treated with the hu14.18-IL2 reagent. Our phase II trial of hu14.18-IL2 treated 38 pts [61]. Five patients showed CR and 2 showed clear clinical benefit, that did not quite meet PR/CR criteria [61]. This provided the opportunity to look at the role of KIR/KIR-L.
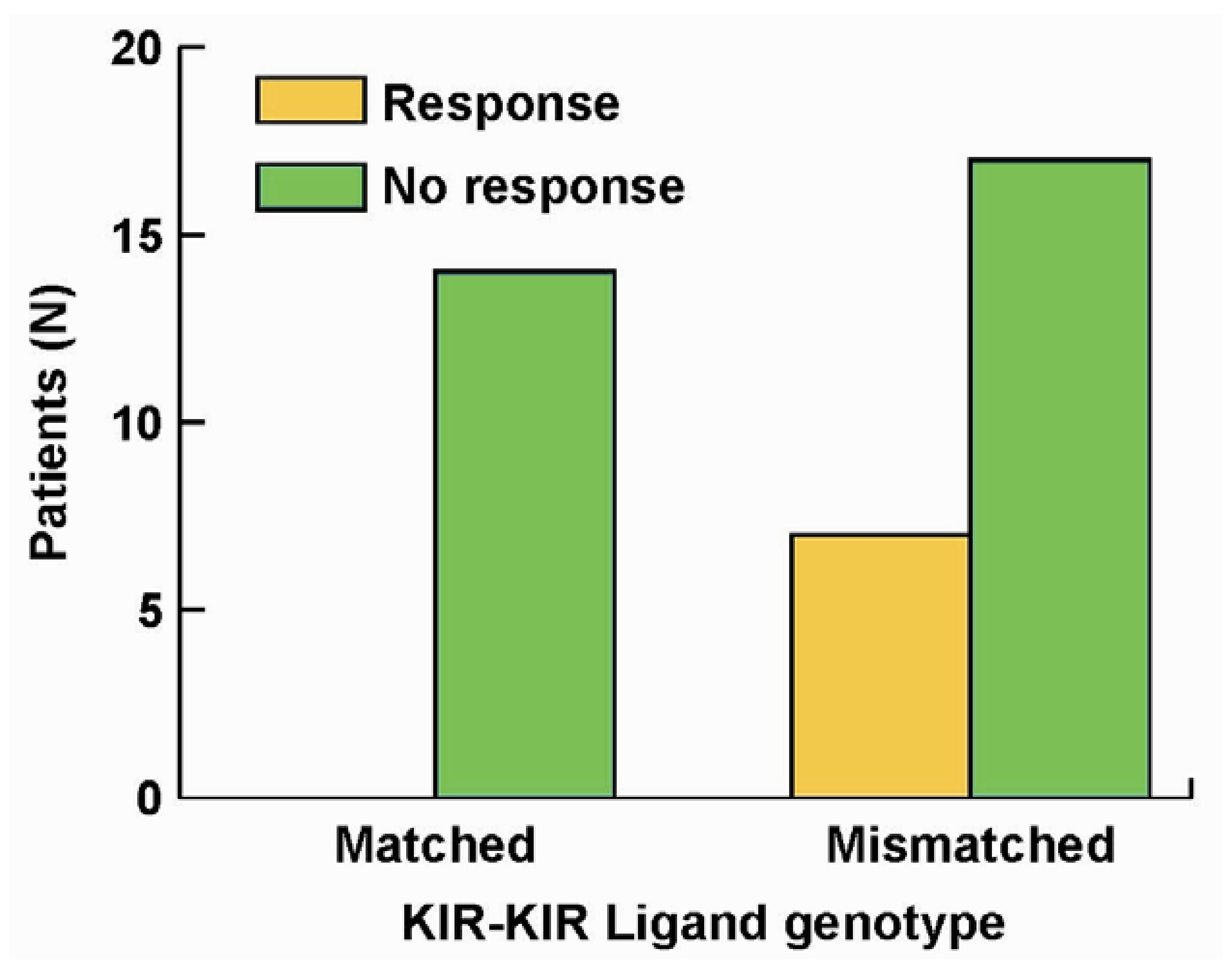
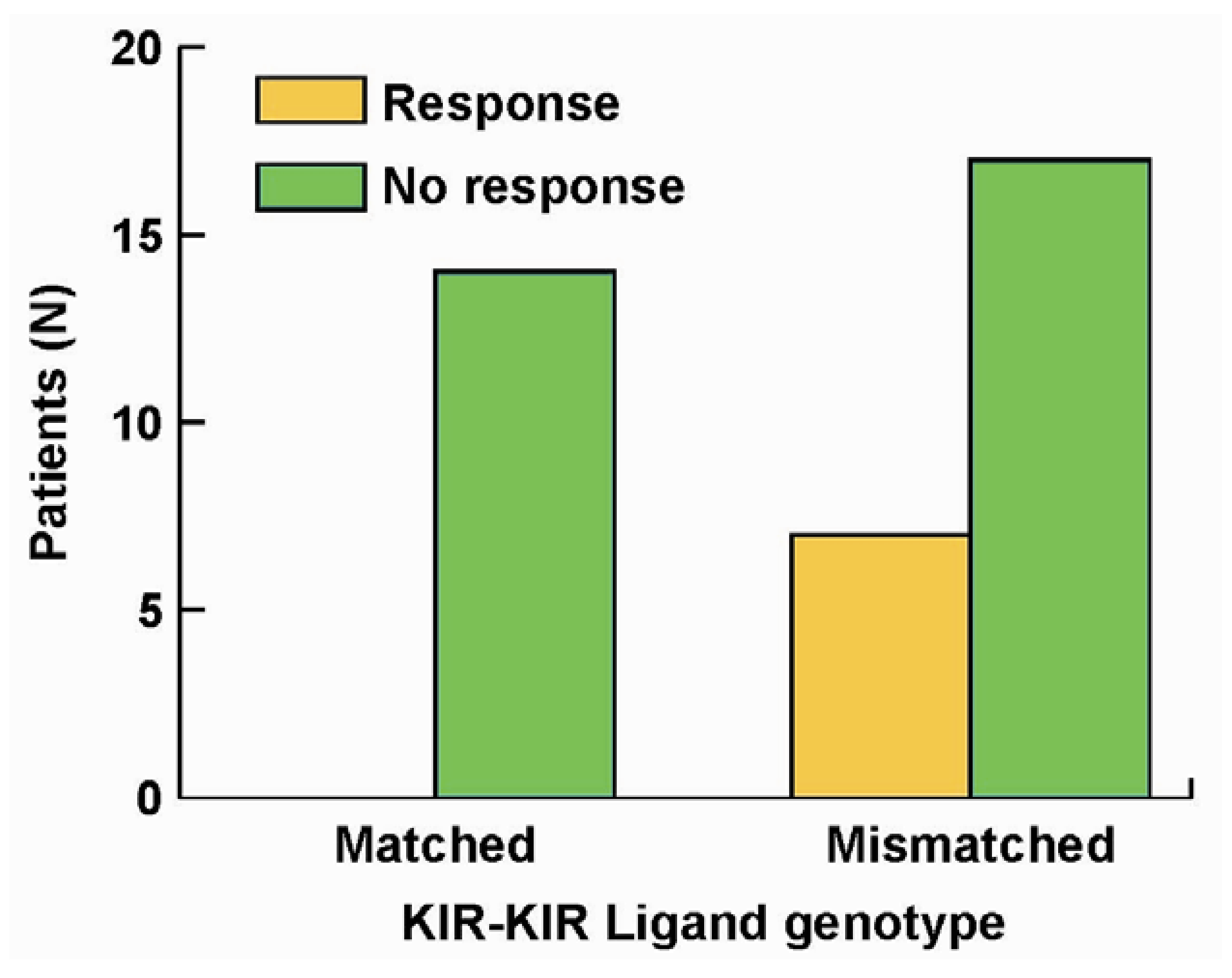
We performed KIR and KIR-L genotyping on the 38 pts. All 7 patients that showed improvement were in the cohort of patients that were KIR/KIR-L mismatched; none of the patients in the KIR/KIR-L matched group showed response or benefit (Figure 3, p = 0.03). Even though our report [84] reflects a study of only 38 pts, this is the first demonstration (to our knowledge), that an individual’s KIR/KIR-L status may predict response to an immunotherapy that does not involve adoptive allogeneic cellular transfer, or autologous HSCT. These results suggest that NK cells may have a significant role in the observed clinical anti-tumor activity.
Figure 3.
Importance of KIR/KIR-Ligand mismatch in clinical response to hu14.18-IL2. Of 38 individuals treated with hu14.18-IL2 in a phase II protocol [61] all of the 7 individuals that showed clinical benefit were from the group of 24 that were KIR/KIR-Ligand mismatched (p = 0.033) [84].
In this same study we also evaluated FcR genotypes. We did find a trend (p = 0.06) for improved likelihood of benefit for those patients that inherited the high affinity genotype (HH) for the FcγR2a receptors on neutrophils and monocytes/macrophages [84]. This suggests that treatment with the hu14.18-IL2 may have been activating these FcγR2a-bearing effector cells to mediate ADCC in vivo. It is notable that there was no correlation between response and FcγR3a genotype [84]. A possible explanation for this is suggested by more recent pre-clinical studies with hu14.18-IL2 and ICs targeting other tumor types.
One such immunocytokine, DI-Leu16-IL2, targets CD20 using the de-immunized form of the murine mAb, Leu16, as the targeting agent. Mouse efficacy studies in SCID mice engrafted with disseminated lymphoma showed very high anti-tumor activity that was more than 50-fold more potent than that of rituximab, or the combination of rituximab and IL2 [85]. This activity was only partly attributable to the standard effector functions of ADCC and CDC because a de-glycosylated form of DI-Leu16-IL2 that lost both of these activities retained most of the anti-tumor activity. Since the primary effector cells in SCID mouse models are expected to be NK cells, and the de-glycosylated form of DI-Leu16-IL2 is not able to bind FcRs, we tested whether other mechanisms were responsible for activating NK killing. The first of these studies were performed with both hu14.18-IL2 and the anti-EpCAM IC, huKS-IL2, and showed that both agents induced target cell conjugation between NK cell lines lacking FcR but constitutively expressing CD25, and the tumor cell expressing the appropriate tumor target (GD2 or EPCAM, respectively) [86]. Furthermore, this conjugate formation led a polarization of CD25 to the immunological synapse and to enhanced target cell lysis [87]. We have named this novel effector function IFCC (immunocytokine-facilitated cellular cytotoxicity). Further studies are needed to assess whether other cell types expressing CD25, besides NK cells, are capable of this novel effector function but in the case of relapsed neuroblastoma, a strong correlation between KIR/KIR-L mismatch certainly implicates NK cells as the primary effectors in this clinical setting.
6. Augmenting Local Antitumor Activity by IT Injection of IC
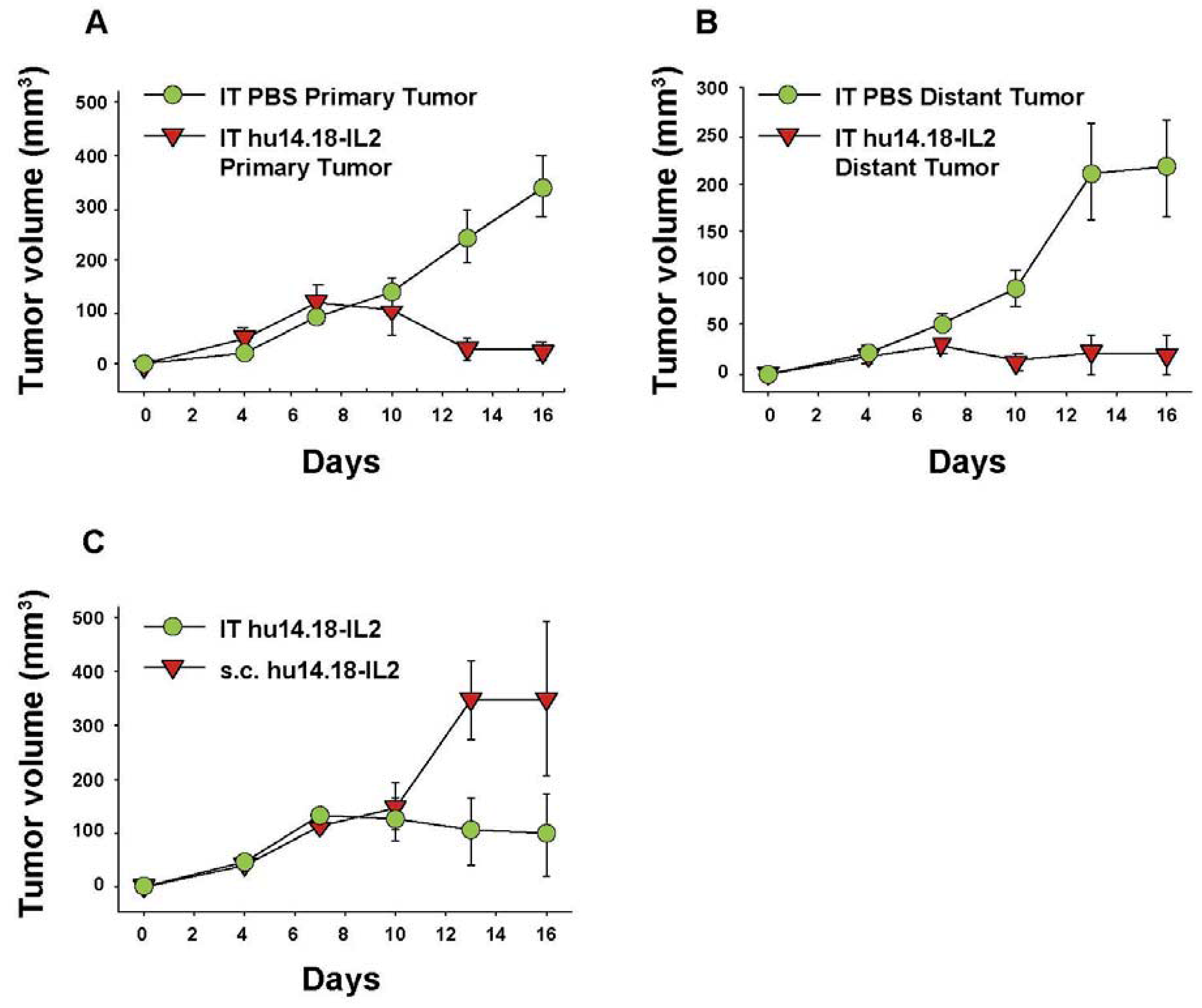
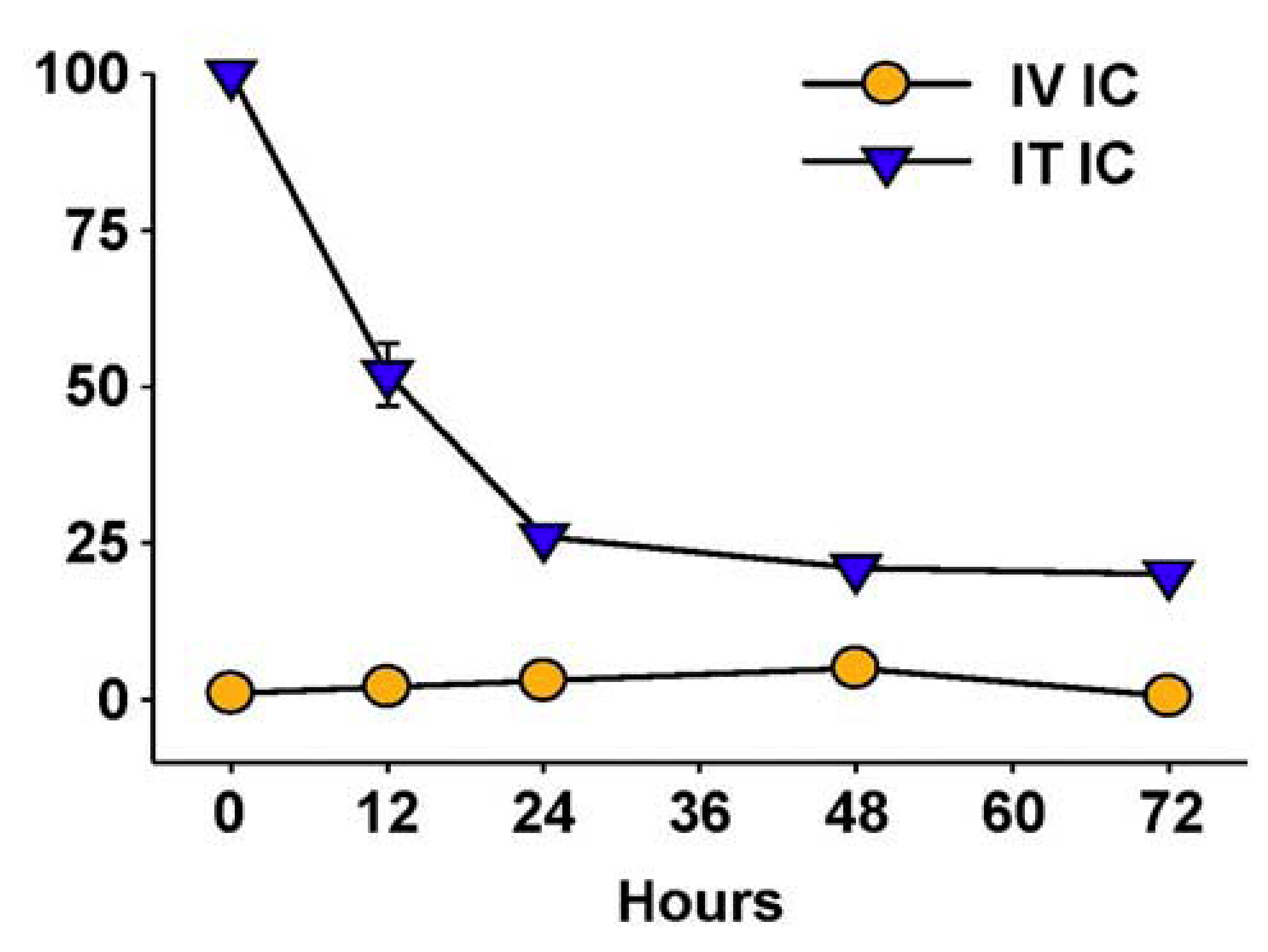
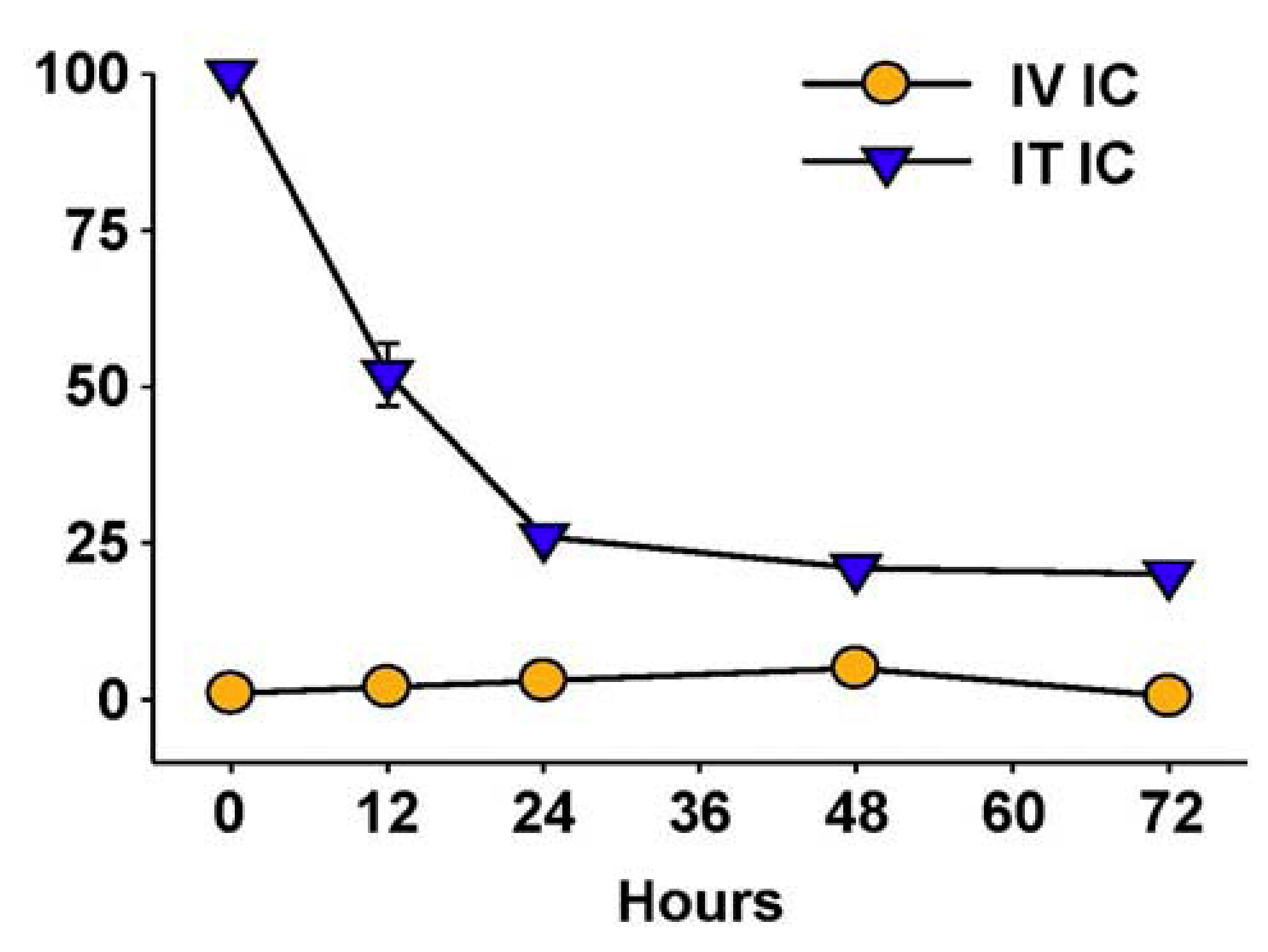
Hu14.18-IL2 administered IV has shown antitumor effects, particularly in mice with MRD, and preliminarily in patients with non-bulky disease [61]. However, its effect against established “bulky” disease in both preclinical and clinical studies has been minimal or transient [39,61,88]. One potential explanation is that following IV injection of IC an inadequate amount of the IC is getting to measurable sites of disease to achieve a clinically significant effect. This is likely due to two major factors. The first is that more than 90% of ICs such as hu14.18-IL2 are lost on first pass clearance by the liver following IV injection [89]. The second is the interstitial pressure of solid tumors that makes it difficult for large proteins to enter from the circulation [90]. We tested this hypothesis by administering hu14.18-IL2 directly into subcutaneously (s.c.) established NXS2 tumors [91]. This IT IC (IT-IC) approach demonstrated significantly greater antitumor effects than IV administration of IC (IV-IC). Only 3 out of 17 mice treated with IV-IC cleared the tumor, while 12 of 17 mice treated with IT-IC cleared tumor (p = 0.002, IT vs. IV groups). IT treatment of a single s.c. tumor (Figure 4A) also prevented the growth of a separate s.c. tumor at a distant site (Figure 4B). Better antitumor effects were seen at distant tumor sites when the IC was injected into the primary tumor, than when the IC was injected s.c. into non-involved skin at an equidistant site (Figure 4C). This argues that injecting IC into the tumor induces a systemic immunologic effect, seen at distant tumor sites. There was also a memory response after IT-IC: most mice that became NXS2-free after IT-IC were able to reject subsequent re-challenges with NXS2 tumor cells, but did not reject an unrelated tumor (YAC-1). Preclinical studies have shown antitumor effects of IT injections of IL2, leading to clinical testing of IT injections of IL2, especially in melanoma [92,93]. IT administration of the IC was significantly more potent at tumor eradication than was IT administration of soluble IL2 (not shown) [91], or of IT injection of a non-specific IC, KS-IL2 (not shown) [91]. Far greater levels of IC are achieved and remain in the tumor site after 24 h following IT vs. IV delivery (25% vs. 4% of injected dose at the tumor, Figure 5) [91].
Figure 4.
IT Immunocytokine effects against primary (injected) and distant tumors. A/J mice (5 per group) received NXS2 (106 s.c. on day 0) in the abdomen (primary tumor). A 2nd NXS2 injection (106 s.c.) was placed on the flank on d4 (distant tumor). The primary tumor received 50 μL PBS or 15 μg hu14.18-IL2 IT on d7-11, while the distant tumor was not treated. (A) Tumor volume of primary tumors (p = 0.001, day 16). Day 0 = implantation of the primary tumor. (B) Tumor volume of distant tumors (p = 0.007, day 16). Day 0 = implantation of the distant tumor. (C) A/J mice (3 per group) received 106 NXS2 cells on d0 in the abdomen. Mice were treated with 15 μg hu14.18-IL2 IT or s.c. into the flank at a site away from the tumor (p = 0.04, days 13–16). (Adapted fromJohnson et al., 2008, [91]).
Figure 4.
IT Immunocytokine effects against primary (injected) and distant tumors. A/J mice (5 per group) received NXS2 (106 s.c. on day 0) in the abdomen (primary tumor). A 2nd NXS2 injection (106 s.c.) was placed on the flank on d4 (distant tumor). The primary tumor received 50 μL PBS or 15 μg hu14.18-IL2 IT on d7-11, while the distant tumor was not treated. (A) Tumor volume of primary tumors (p = 0.001, day 16). Day 0 = implantation of the primary tumor. (B) Tumor volume of distant tumors (p = 0.007, day 16). Day 0 = implantation of the distant tumor. (C) A/J mice (3 per group) received 106 NXS2 cells on d0 in the abdomen. Mice were treated with 15 μg hu14.18-IL2 IT or s.c. into the flank at a site away from the tumor (p = 0.04, days 13–16). (Adapted fromJohnson et al., 2008, [91]).
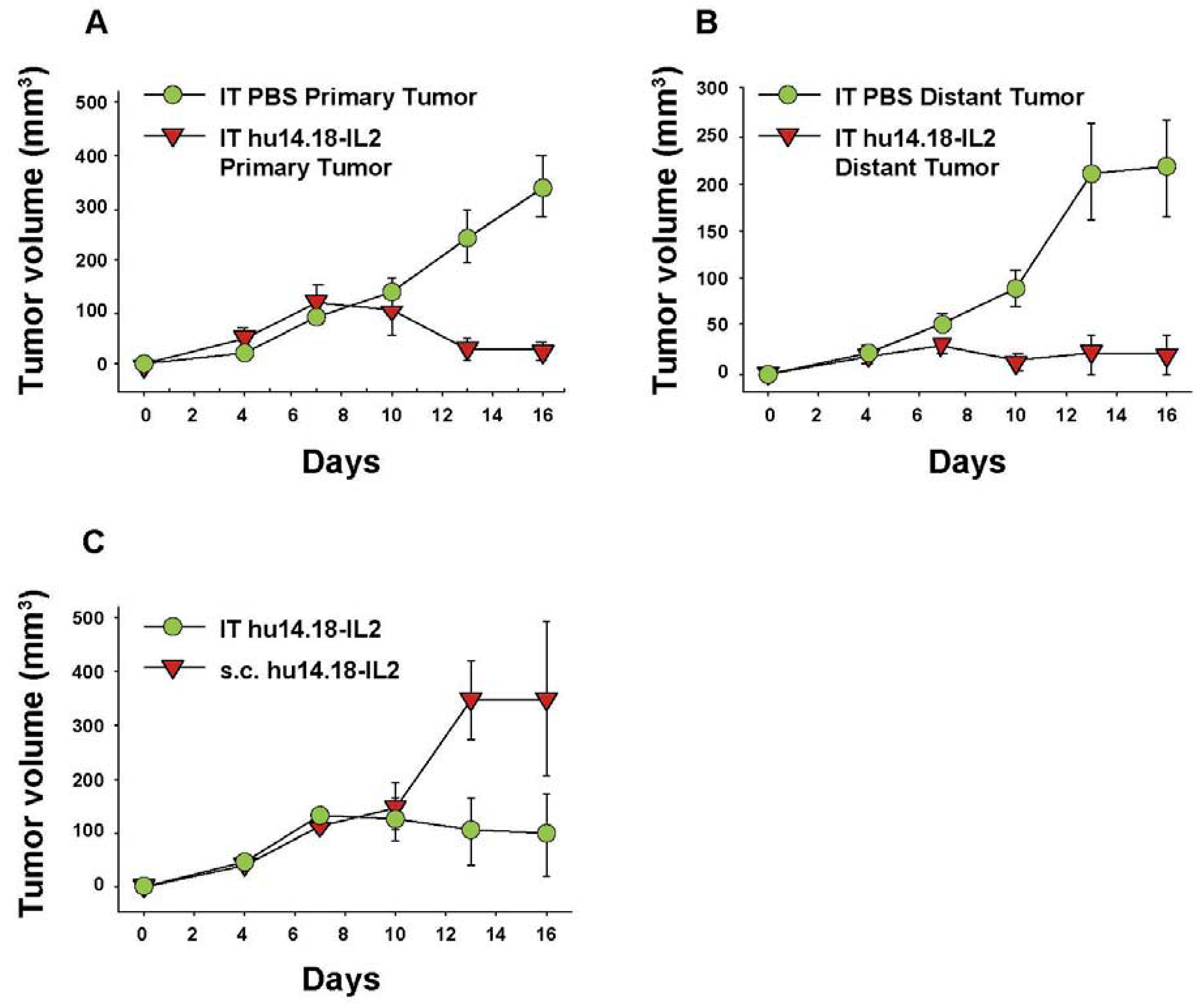
Figure 5.
IT vs. IV delivery results in increased tumor retention of IC. CEA-transgenic C57BL/6 mice bearing d10 s.c. MC-38.CEA tumors were treated with 111In-GcT84.66-IL2 anti-CEA IC. Animals received 25 μg IV (5/grp) or 2.3 µg IT (2/grp). Tumors were harvested serially from the IV-treated mice and the % injected dose (shown on the Y-axis) determined by a γamma counter. IT-treated mice were imaged with a γamma-camera, acquiring % injected dose by gating on the tumor. (Adapted from Johnson et al., 2008, [91]).
Figure 5.
IT vs. IV delivery results in increased tumor retention of IC. CEA-transgenic C57BL/6 mice bearing d10 s.c. MC-38.CEA tumors were treated with 111In-GcT84.66-IL2 anti-CEA IC. Animals received 25 μg IV (5/grp) or 2.3 µg IT (2/grp). Tumors were harvested serially from the IV-treated mice and the % injected dose (shown on the Y-axis) determined by a γamma counter. IT-treated mice were imaged with a γamma-camera, acquiring % injected dose by gating on the tumor. (Adapted from Johnson et al., 2008, [91]).
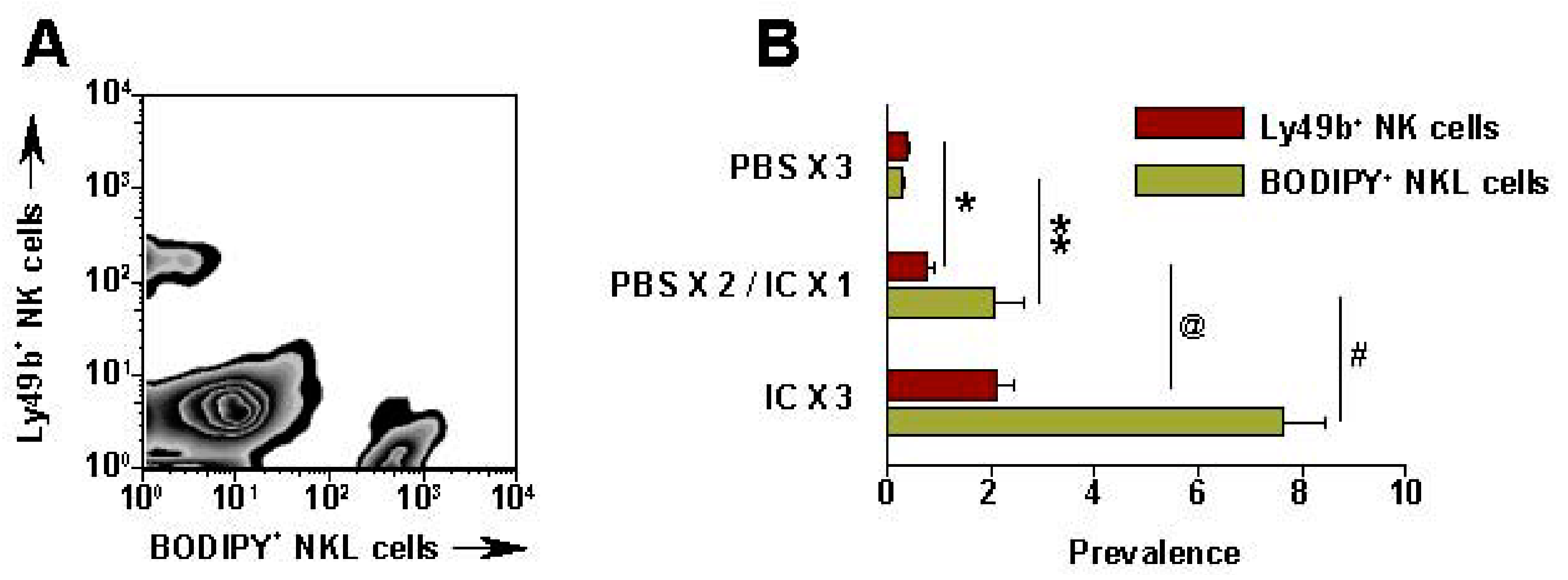
Recent data show that IT administration of IC can also enhance localization of NK cells in the tumor site [87]. CFSE (Carboxyfluorescein succinimidyl ester, a fluorescent stain) labeled human NKL cells were injected IV (5 × 106) into SCID mice bearing human GD2+ M21 melanoma tumors (~1 cm diameter) [87]. After 24 h, the tumors were harvested, disaggregated and evaluated by flow cytometry to determine what % of viable cells represented the CFSE-labeled NKL cells (Figure 6). In PBS treated NXS2 tumors, only 0.06% of the viable cells were CFSE labeled; in contrast, 24 h after IT-IC, 6.9% of the viable cells in the tumor were the CFSE labeled NKL cells [87]. IHC analyses of NXS2 tumors immediately following IT-IC show direct staining by IC and demonstrate NK infiltration 24 h later (data not shown).
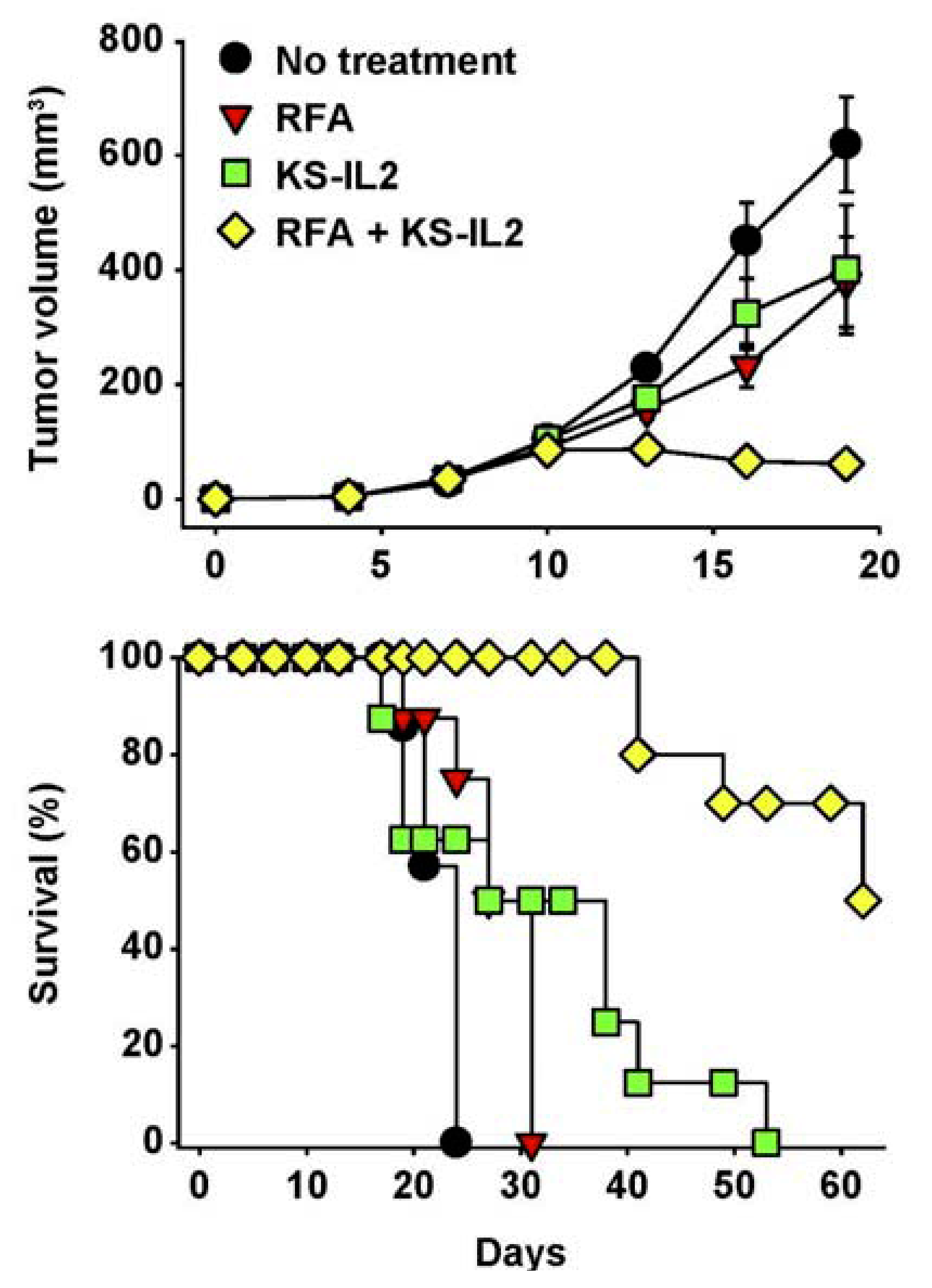
As the localized injection of IC at a tumor site may not only induce ADCC and IFCC, but also serve as an endogenous, autologous tumor, vaccine, we have capitalized on the ability of apoptotic tumor cells to facilitate antigen uptake and presentation for vaccination purposes. In that regard, we have used radio-frequency ablation (RFA) to induce localized tumor cell damage (via thermal tissue injury) in combination with IT-IC treatment [94]. In many of these studies we investigated this in mouse models with the KS-IL2 IC directed against the Epithelial Cell adhesion molecule (EpCam). Dramatic synergy was seen when IT-IC was combined with RFA (Figure 7) [94]. Using a separate IC, synergy has been observed by combining IC treatment with localized radiotherapy, likely inducing similar synergistic mechanisms as the combination of IC + RFA in Figure 7. In this case, an IC targeting necrotic DNA and containing a form of IL2 specific for the high affinity IL2R was administered IV following radiation of s.c. Lewis Lung Carcinoma tumors. This resulted in the rapid regression of tumors in all mice that was preceded by cytotoxic T cell infiltration and followed by the ability of these mice to resist a subsequent tumor challenge [95].
Figure 6.
IT-IC facilitates migration of NK cells into tumor. SCID mice bearing s.c. M21 received IT-PBS (50 μL) or IT-IC (10 μg in 50 μL) on day 27-29. (PBS × 3) = PBS on d 27, 28, 29; (PBS × 2/IC × 1) = PBS on d 27, 28, and IC on d 29; (IC × 3) = IC on d 27, 28, 29. On d 29, right after the last IT- PBS or IC, all mice received 5 × 106 BODIPY-labeled NKL cells IV. 24 h after the NKL cell injection, tumors were harvested, processed, stained and tested by flow for mouse Ly49b+ NK cells and BODIPY+ NKL cells. (A) Representative pattern from a (IC × 3) mouse; numbers are for the 3 (IC × 3) mice (Mean % ± SEM). (B) Data for all groups (represents 2 independent experiments). * p = 0.06; ** p = 0.046; @ p = 0.02; #p = 0.007 (Adapted from Buhtoiarov et al. 2011, [87]).
Figure 6.
IT-IC facilitates migration of NK cells into tumor. SCID mice bearing s.c. M21 received IT-PBS (50 μL) or IT-IC (10 μg in 50 μL) on day 27-29. (PBS × 3) = PBS on d 27, 28, 29; (PBS × 2/IC × 1) = PBS on d 27, 28, and IC on d 29; (IC × 3) = IC on d 27, 28, 29. On d 29, right after the last IT- PBS or IC, all mice received 5 × 106 BODIPY-labeled NKL cells IV. 24 h after the NKL cell injection, tumors were harvested, processed, stained and tested by flow for mouse Ly49b+ NK cells and BODIPY+ NKL cells. (A) Representative pattern from a (IC × 3) mouse; numbers are for the 3 (IC × 3) mice (Mean % ± SEM). (B) Data for all groups (represents 2 independent experiments). * p = 0.06; ** p = 0.046; @ p = 0.02; #p = 0.007 (Adapted from Buhtoiarov et al. 2011, [87]).
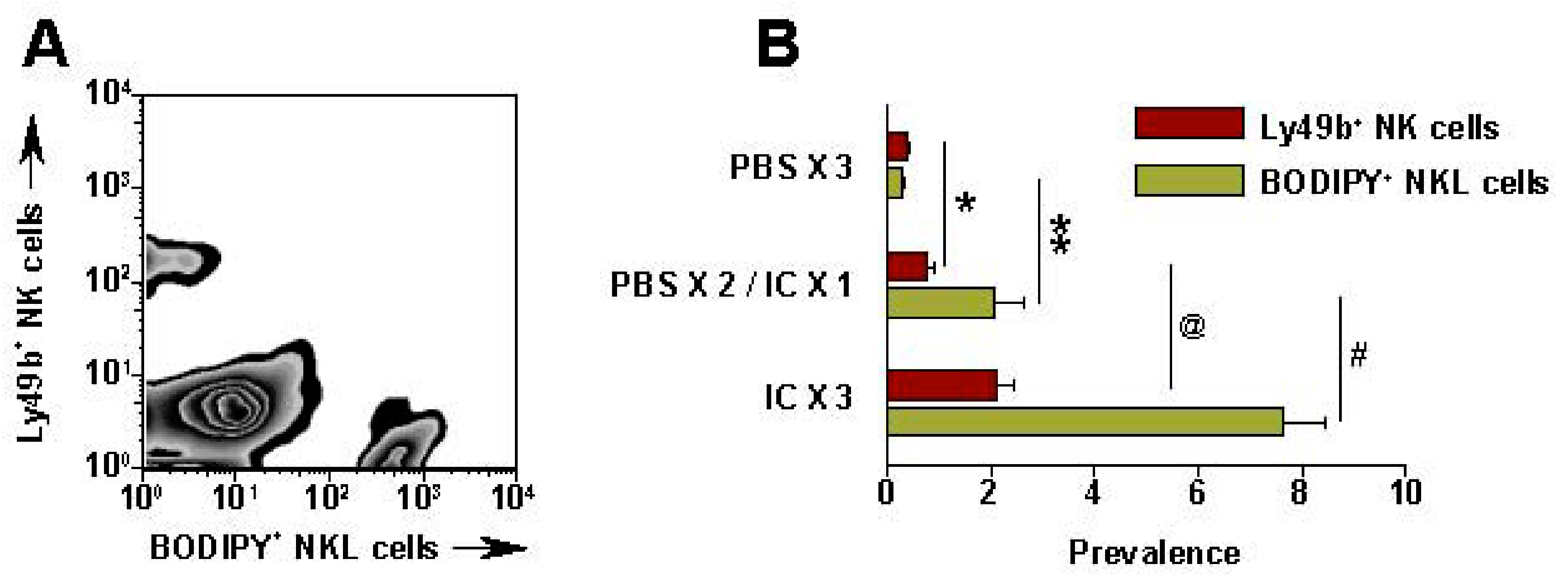
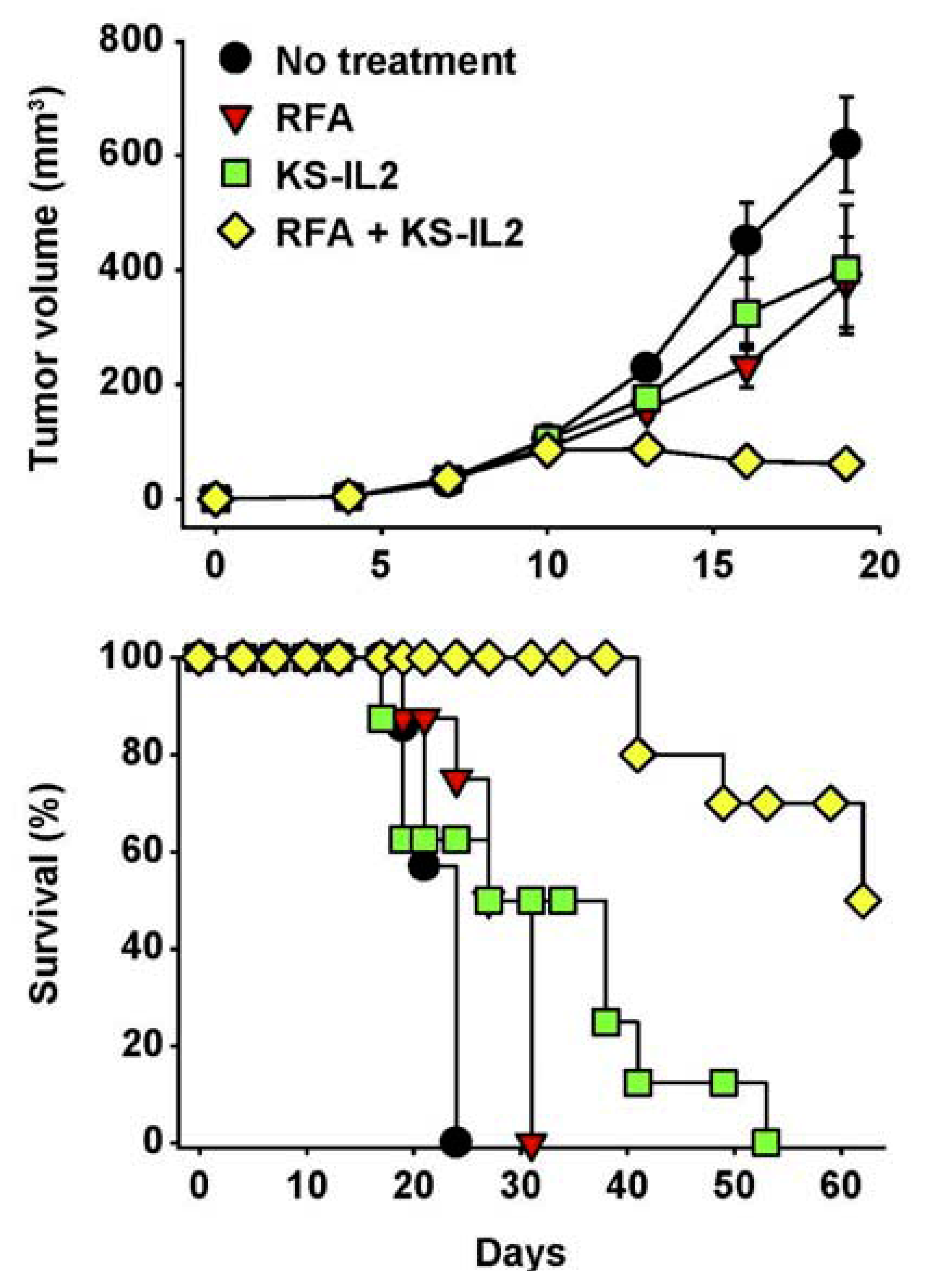
Figure 7.
RFA and huKS-IL2 IC synergize in induction of antitumor effects. Balb/c mice (7–8 mice per group) received 5 × 105 CT26-KS cells s.c. in the abdomen on d0. Mice were treated with partial RFA (25 seconds, d11), KS-IL2 IC (15 μg, d11-15), or both RFA and KS-IL2. Data shown are mean tumor volume ± SEM (A) or survival (B). The results are representative of 3 experiments. (Adapted from Johnson et al., 2008, [91]).
Figure 7.
RFA and huKS-IL2 IC synergize in induction of antitumor effects. Balb/c mice (7–8 mice per group) received 5 × 105 CT26-KS cells s.c. in the abdomen on d0. Mice were treated with partial RFA (25 seconds, d11), KS-IL2 IC (15 μg, d11-15), or both RFA and KS-IL2. Data shown are mean tumor volume ± SEM (A) or survival (B). The results are representative of 3 experiments. (Adapted from Johnson et al., 2008, [91]).
These data support the following hypotheses: (a) IT-IC causes much higher and longer lasting levels of IC in the injected tumor than IV-IC; (b) Greater IC levels in the tumor enhance NK infiltration into the tumor (via FcRs and IL2Rs), leading to greater ADCC and greater tumor destruction, even of larger macroscopic lesions that are unresponsive to IV IC delivery; (c) Some of the IC injected IT circulates systemically (via lymphatics and blood vessels), enabling IC delivery to distant sites as effectively as when IC is given IV (possibly with a better PK profile); (d) The IC-facilitated response within the tumor may attract other effector cells (T cells and macrophages) to the site of necrotic tumor (or to draining lymph nodes), leading to T cell sensitization; (e) The vaccine-like effect resulting in tumor-specific T cell reactivity may impact on distant sites of micrometastatic disease (and prevent subsequent growth upon experimental tumor rechallenge); (f) Combining IC with other treatments that cause localized tumor damage (without local immune suppression), should synergistically augment the antitumor activity of the IC. All data in hand, which were generated with first-generation ICs, hu14.18-IL2 and huKS-IL2, are consistent with, but do not prove, all these hypotheses. Thus, while continued preclinical studies need to better understand the cellular and immune mechanisms of these antitumor effects, the antitumor efficacy of this approach and the relative ease for translating it to the clinical setting justify initial Phase I/II clinical testing of this IT-IC strategy, especially for melanoma.
7. Future Directions for IC Development and Treatment
The data, discussed above, have shown how more effective IT dosing can be for ICs with suboptimal pharmacokinetic properties. While the IT approach can be useful for cancers with easily accessible tumors, the treatment of disseminated, systemic disease might benefit from ICs that have been optimized for tumor targeting and reduced liver clearance. In this regard we reported technical approaches that greatly improved the PK properties of ICs—the genetic removal of the N-linked glycan in the CH2 domain of the H chain (that abrogates FcR binding) and alterations in the fusion junction between the H chain and IL2 that reduce intracellular proteolysis [96,97]. In the first case, blood levels are increased following IV administration, tumor targeting of radio-labeled IC is enhanced, and liver uptake is significantly reduced. In the second case, blood levels are also increased despite retaining FcR binding (and CDC activity), presumably due to more efficient re-cycling of the IC out of cells that have taken it up. In both cases, relatively low doses of the ICs were completely effective at causing the regression of s.c. colon carcinoma tumors in all mice, while the equivalent dose of the first-generation IC (huKS-IL2 in this case) showed only a short growth delay. This improved form of huKS-IL2 is not in clinical development so translation of these results to human therapy is currently not possible. Fortunately, the IC discussed above, DI-Leu16-IL2, was constructed using one of these optimization strategies and is in early clinical trials. In this case, the CH2 domain N-linked glycan was retained (preserving ADCC and CDC activity) but the H chain/IL2 fusion junction was modified for superior PK properties [85]. These changes have created an IC with very potent anti-lymphoma activity, even in the absence of T cells (since data was generated in SCID mice), and that has all effector functions (including IFCC). We are hopeful that DI-Leu16-IL2 studies in lymphoma patients will demonstrate T cell responses as well, since it is known that a high proportion of lymphoma patients have infiltrating T cells in the tumor microenvironment, and that they can be associated with better clinical outcome [98].
Another aspect of these ICs with improved PK properties is their potential for vascular toxicity. It is well known that IL2 toxicity, in large part, is related to Cmax levels in the blood and that this, in turn, is related to the activation of immune cells expressing the intermediate IL2 receptor, CD122 [99]. We have found two ways of greatly reducing the vascular toxicity of ICs based on this hypothesis. The first is through the genetic modification of IL2 at position D20 that interacts with the β chain of CD122 and thereby prevents activation of this receptor. This D20T mutation still allows the high affinity receptor to be activated when the α chain forms the trimeric complex with the β and γ chains [100]. This IC, called NHS-IL2LT (for low toxicity—also called Selectikine) preferentially activates cells expressing CD25, such as effector T cells, over CD122 expressing cells generally associated with vascular toxicity. Studies in both monkeys and cancer patients have shown a dramatic reduction in the IL2 toxicities normally seen with IL2-based ICs administered IV. The second approach to reducing vascular toxicity of IL2 based ICs is to administer them by the s.c. route instead of IV. This avoids the initially high Cmax obtained by IV administration and instead, the IC is taken up by the lymphatics and then slowly released into the bloodstream over a longer period of time. In this way cells expressing CD25 are preferentially activated without the overstimulation of cells expressing CD122. One possible advantage of this approach is that there is still some degree of CD122 activation that can up-regulate the expression of CD25 to increase the effector T cell pool, but still does so in a way that is well tolerated. The first IC that will be compared clinically using IV or s.c. dosing is the anti-CD20 IC, DI-Leu-16-IL2. Pre-clinical studies in monkeys have already confirmed the increased tolerability of s.c. dosing compared to the same doses given IV and studies in patients are underway. Since these studies will be done with patients with CD20-expressing cancers, such as non-Hodgkin’s Lymphoma (NHL), there may be an additional advantage of using s.c. dosing, since the drug is taken up by the compartment in which lymphoma generally resides and thereby enhances tumor targeting. For other cancer types that are not in the lymphatic system, further optimization of ICs may help increase the efficiency of s.c. uptake through the lymphatics and into the blood so that it can effectively target these tumors. We are currently developing such molecules that have longer circulating half lives, better uptake into the blood using s.c. injection and in this way benefit from this well tolerated and convenient route of administration. Further modifications also include modifications that alter the balance between the activation of CD25 and CD122-expressing cells. In this way we hope to find the optimal balance between the effector functions provided by the antibody component of the IC and the potent cytokine activity.
Acknowledgements
The Authors thank Ralph Reisfeld for decades of collaboration, discussions and mentorship. They also thank J. Hank, A. Rakhmilevich, J. Gan, I. Buhtoiarov, Z. Neal, D. Delgado, A. Erbe, K. Alderson, B. Grzywacz, K. Desantes and R. Yang for years of discussion, collaboration, data generation as well as help in creation of figures related to this chapter. The work related to this project performed at The University of Wisconsin was supported by: National Institutes of Health Grants CA032685, CA87025, CA14520, CA166105, GM067386, Department of Defense grant W81XWH-08-1-0559 and grants from the Midwest Athletes for Childhood Cancer Fund, the Crawdaddy Foundation, The Evan Dunbar Foundation, and the University of Wisconsin-Madison Institute for Clinical and Translational Research (ICTR) TL1 Training Grant 1TL1RR025013-01.
References
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kerse, J.; Kolb, H.J.; Rimm, A.A.; Ringden, O.; Rozman, C.; Speck, B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar]
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: Clinical response, cellular immunity, and immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399. [Google Scholar] [CrossRef]
- Krege, S.; Giani, G.; Meyer, R.; Otto, T.; Rubben, H. A randomized multicenter trial of adjuvant therapy in superficial bladder cancer: Transurethral resection only versus transurethral resection plus mitomycin C versus transurethral resection plus bacillus Calmette-Guerin. Participating Clinics. J. Urol. 1996, 156, 962–966. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar]
- Hodi, F.S.; O'Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar]
- Fong, L.; Small, E.J. Anti-cytotoxic T-lymphocyte antigen-4 antibody: The first in an emerging class of immunomodulatory antibodies for cancer treatment. J. Clin. Oncol. 2008, 265, 275–283. [Google Scholar]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.; Chen, H.; Smith, M.; Anderson, B.; Villablanca, J.; Matthay, K.K.; et al. Chimeric Anti-GD2 Antibody with GM-CSF, IL2 and 13-cis Retinoic Acid for High-risk Neuroblastoma: A Children’s Oncology Group (COG) Phase 3 Study. N. Engl. J. Med. 2010, 335, 1324–1334. [Google Scholar]
- Basham, T.Y.; Race, E.R.; Campbell, M.J.; Reid, T.R.; Levy, R.; Merigan, T.C. Synergistic antitumor activity with IFN and monoclonal anti-idiotype for murine B cell lymphoma. Mechanism of action. J. Immunol. 1988, 141, 2855–2860. [Google Scholar]
- Primus, F.J.; Finch, M.D.; Wetzel, S.A.; Masci, A.M.; Schlom, J.; Kashmiri, S.V. Monoclonal antibody gene transfer. Implications for tumor-specific cell-mediated cytotoxicity. Ann. NY Acad. Sci. 1994, 716, 165–166. [Google Scholar]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Jefferis, R. Antibody therapeutics: Isotype and glycoform selection. Expert Opin. Biol. Th. 2007, 7, 1401–1413. [Google Scholar] [CrossRef]
- Farag, S.S.; VanDeusen, J.B.; Fehniger, T.A.; Caligiuri, M.A. Biology and clinical impact of human natural killer cells. Int. J. Hematol. 2003, 78, 7–17. [Google Scholar] [CrossRef]
- Roda, J.M.; Parihar, R.; Magro, C.; Nuovo, G.J.; Tridandapani, S.; Carson, W.E., 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006, 66, 517–526. [Google Scholar]
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725. [Google Scholar] [CrossRef]
- Carlsten, M.; Malmberg, K.J.; Ljunggren, H.G. Natural killer cell-mediated lysis of freshly isolated human tumor cells. Int. J. Cancer 2009, 124, 757–762. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Analyzing antibody-Fc-receptor interactions. Methods Mol. Biol. 2008, 415, 151–162. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Treon, S.P.; Mitsiades, C.; Mitsiades, N.; Young, G.; Doss, D.; Schlossman, R.; Anderson, K.C. Tumor Cell Expression of CD59 Is Associated With Resistance to CD20 Serotherapy in Patients With B-Cell Malignancies. J. Immunother. 2001, 24, 263–271. [Google Scholar] [CrossRef]
- Dzietczenia, J.; Wróbel, T.; Mazur, G.; Poreba, R.; Jaźwiec, B.; Kuliczkowski, K. Expression of complement regulatory proteins: CD46, CD55, and CD59 and response to rituximab in patients with CD20+ non-Hodgkin's lymphoma. Med. Oncol. 2010, 27, 743–746. [Google Scholar] [CrossRef]
- Binyamin, L.; Alpaugh, R.K.; Hughes, T.L.; Lutz, C.T.; Campbell, K.S.; Weiner, L.M. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 2008, 180, 6392–6401. [Google Scholar]
- Wang, S.Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.C.; Vogel, C.W.; St John, W.; Weiner, G.J. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 2009, 114, 5322–5330. [Google Scholar]
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell 2002, 1, 117–123. [Google Scholar] [CrossRef]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frébourg, T.; Michel, P.; et al. Impact of FcgRIIa-FcgRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar]
- Shan, D.; Ledbetter, J.A.; Press, O.W. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood 1998, 91, 1644–1652. [Google Scholar]
- Cheung, N.K.; Sowers, R.; Vickers, A.J.; Cheung, I.Y.; Kushner, B.H.; Gorlick, R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J. Clin. Oncol. 2006, 24, 2885–2890. [Google Scholar]
- Weng, W.K.; Czerwinski, D.; Levy, R. Humoral immune response and immunoglobulin G Fc receptor genotype are associated with better clinical outcome following idiotype vaccination in follicular lymphoma patients regardless of their response to induction chemotherapy. Blood 2007, 109, 951–953. [Google Scholar]
- Wang, B.; Kokhaei, P.; Mellstedt, H.; Liljefors, M. FcγR polymorphisms and clinical outcome in colorectal cancer patients receiving passive or active antibody treatment. Int. J. Oncol. 2010, 37, 1599–1606. [Google Scholar]
- Roda, J.M.; Joshi, T.; Butchar, J.P.; McAlees, J.W.; Lehman, A.; Tridandapani, S.; Carson, W.E., 3rd. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin. Cancer Res. 2007, 13, 6419–6428. [Google Scholar]
- Kloess, S.; Huenecke, S.; Piechulek, D.; Esser, R.; Koch, J.; Brehm, C.; Soerensen, J.; Gardlowski, T.; Brinkmann, A.; Bader, P.; et al. IL-2-activated haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in neuroblastoma patients by scavenging of plasma MICA. Eur. J. Immunol. 2010, 40, 3255–3267. [Google Scholar]
- Hank, J.A.; Kohler, P.C.; Weil-Hillman, G.; Rosenthal, N.; Moore, K.H.; Storer, B.; Minkoff, D.; Bradshaw, J.; Bechhofer, R.; Sondel, P.M. In vivo induction of the lymphokine-activated killer phenomenon: Interleukin 2-dependent human non-major histocompatibility complex-restricted cytotoxicity generated in vivo during administration of human recombinant Interleukin 2. Cancer Res. 1988, 48, 1965–1971. [Google Scholar]
- Hank, J.A.; Robinson, R.R.; Surfus, J.; Mueller, B.M.; Reisfeld, R.A.; Cheung, N.-K.; Sondel, P.M. Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant Interleukin-2. Cancer Res. 1990, 50, 5234–5239. [Google Scholar]
- Sosman, J.A.; Hank, J.A.; Sondel, P.M. In vivo activation of lymphokine-activated killer activity with Interleukin-2: Prospects for combination therapies. Semin. Oncol. 1990, 17, 22–30. [Google Scholar]
- Hank, J.A.; Albertini, M.R.; Schiller, J.; Sondel, P.M. Activation of multiple effector mechanisms to enhance tumor immunotherapy. J. Immunother. 1993, 14, 329–335. [Google Scholar] [CrossRef]
- Mujoo, K.; Kipps, T.J.; Yang, H.M.; Cheresh, D.A.; Wargalla, U.; Sander, D.J.; Reisfeld, R.A. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. 1989, 49, 2857–2861. [Google Scholar]
- Kendra, K.; Malkovska, V.; Allen, M.; Guzman, J.; Albertini, M. In vivo binding and antitumor activity of Ch14.18. J. Immunother. 1999, 5, 23–430. [Google Scholar]
- Neal, Z.C.; Yang, J.C.; Rakhmilevich, A.L.; Buhtoiarov, I.; Lum, H.E.; Imboden, M.; Hank, J.A.; Lode, H.N.; Reisfeld, R.A.; Gillies, S.D.; et al. Enhanced activity of hu14.18-IL2 immunocytokine against the murine NXS2 neuroblastoma when combined with IL2 therapy. Clin. Cancer Res. 2004, 10, 4839–4847. [Google Scholar]
- Hank, J.; Surfus, J.; Gan, J.; Chew, T.-L.; Hong, R.; Tans, K.; Reisfeld, R.; Seeger, R.; Reynolds, C.P.; Bauer, M.; et al. Treatment of neuroblastoma patients with antiganglioside GD2 antibody plus Interleukin-2 induces antibody dependent cellular cytotoxicity against neuroblastoma detected in vitro. J. Immunother. 1994, 15, 29–37. [Google Scholar] [CrossRef]
- Frost, J.D.; Ettinger, L.J.; Hank, J.A.; Cairo, M.S.; Reaman, G.H.; Blazar, B.R.; Frierdich, S.; Krailo, M.; Seeger, R.C.; Matthay, K.; et al. Phase I/IB trial of murine monoclonal anti-GD2 antibody 14.G2a plus IL-2 in children with refractory neuroblastoma: A report of the Children’s Cancer Group. Cancer 1997, 80, 317–333. [Google Scholar] [CrossRef]
- Albertini, M.R.; Hank, J.A.; Schiller, J.H.; Khorsand, M.; Borchert, A.A.; Gan, J.; Bechhofer, R.; Storer, B.; Reisfeld, R.A.; Sondel, P.M. Phase IB trial of chimeric anti-GD2 antibody plus interleukin-2 for melanoma patients. Clin. Cancer Res. 1997, 3, 1277–1288. [Google Scholar]
- Gilman, A.L.; Ozkaynak, F.; Matthay, K.; Krailo, M.; Yu, A.; Gan, J.; Sternberg, A.; Hank, J.; Seeger, R.; Reaman, G.; et al. Phase I Study of ch14.18 with GM-CSF and IL-2 in Children with Neuroblastoma Following Autologous Bone Marrow Transplant or Stem Cell Rescue: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2009, 27, 85–91. [Google Scholar]
- Yu, A.L.; Batova, A.; Alvarado, C.; Rao, V.J.; Castleberry, R.P. Usefulness of a chimeric anti-GD2 (ch14.18) and GM-CSF for refractory neuroblastoma. Proc. Am. Assoc. Clin. Oncol. 1997, 6, 1846. [Google Scholar]
- Kushner, B.H.; Kramer, K.; Cheung, N.K. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J. Clin. Oncol. 2001, 19, 4189–4194. [Google Scholar]
- Arndt, C.A.; Koshkina, N.V.; Inwards, C.Y.; Hawkins, D.S.; Krailo, M.D.; Villaluna, D.; Anderson, P.M.; Goorin, A.M.; Blakely, M.L.; Bernstein, M.; et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: Effects on disease-free survival and immunomodulation. A report from the Children's Oncology Group. Clin. Cancer Res. 2010, 16, 4024–4030. [Google Scholar]
- Simon, T.; Hero, B.; Faldum, A.; Handgretinger, R.; Schrappe, M.; Niethammer, D.; Berthold, F. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J. Clin. Oncol. 2004, 22, 3549–3557. [Google Scholar] [CrossRef]
- Weil-Hillman, G.; Fisch, P.; Prieves, A.F.; Sosman, J.A.; Hank, J.A.; Sondel, P.M. Lymphokine-activated killer activity induced by in vivo interleukin-2 therapy: Predominant role for lymphocytes with increased expression of CD2 and Leul9 antigens but negative expression of CD16 antigens. Cancer Res. 1989, 49, 3680–3688. [Google Scholar]
- Voss, S.D.; Robb, R.J.; Weil-Hillman, G.; Hank, J.A.; Sugamum, K.; Tsudo, M.; Sondel, P.M. Increased expression of the interleukin-2 (IL2) receptor beta chain (p70) on CD56+ natural killer cells after in vivo IL2 therapy: p70 expression does not alone predict the level of intermediate affinity IL2 binding. J. Exp. Med. 1990, 172, 1101–1114. [Google Scholar]
- Sondel, P.M.; Kohler, P.C.; Hank, J.A.; Moore, K.H.; Rosenthal, N.; Sosman, J.; Bechhofer, R.; Storer, B. Clinical and immunological effects of recombinant interleukin-2 given by repetitive weekly cycles to subjects with cancer. Cancer Res. 1988, 48, 2561–2567. [Google Scholar]
- Gillies, S.D.; Reilly, E.B.; Lo, K.-M.; Reisfeld, R.A. Antibody-targeted interleukin 2 stimulates the T-cell killing of autologous tumor cells. Proc. Natl. Acad. Sci. USA 1992, 89, 1428. [Google Scholar]
- Hank, J.A.; Surfus, J.E.; Gan, J.; Jaeger, P.; Gillies, S.; Reisfeld, R.A.; Sondel, P.M. Activation of human effector cells by a tumor reactive recombinant anti-ganglioside GD2/interleukin-2 immunocytokine (ch14.18-IL2). Clin. Cancer Res. 1996, 2, 1951–1959. [Google Scholar]
- Sabzevari, H.; Gillies, S.D.; Mueller, B.M.; Pancook, J.D.; Reisfeld, R.A. A recombinant antibody-interleukin 2 immunocytokine suppresses growth of hepatic human neuroblastoma metastases in severe combined immunodeficiency mice. Proc. Natl. Acad. Sci. USA 1994, 91, 9626. [Google Scholar]
- Becker, J.C.; Pancook, J.D.; Gillies, S.D.; Furukawa, K.; Reisfeld, R.A. T cell mediated eradiation of murine metastatic melanoma induced by targeted interleukin-2 therapy. J. Exp. Med. 1996, 183, 2361. [Google Scholar] [CrossRef]
- Becker, J.C.; Varki, N.; Gillies, S.D.; Furukawa, K.; Reisfeld, R.A. An antibody-interleukin-2 fusion protein overcomes tumor heterogeneity by induction of a cellular immune response. Proc. Natl. Acad. Sci. USA 1996, 93, 7826–7831. [Google Scholar]
- Lode, H.N.; Xiang, R.; Varki, N.M.; Dolman, C.S.; Gillies, S.D.; Reisfeld, R.A. Targeted interleukin-2 therapy of spontaneous neuroblastoma to bone marrow. J. Natl. Cancer Inst. 1997, 89, 1586–1591. [Google Scholar] [CrossRef]
- Lode, H.N.; Xiang, R.; Drier, T.; Varki, N.M.; Gillies, S.D.; Reisfeld, R.A. Natural killer cell mediated eradication of neuroblastoma metastases to bone marrow by targeted IL2 therapy. Blood 1998, 91, 1706–1715. [Google Scholar]
- Neal, Z.C.; Imboden, M.; Rakhmilevich, A.L.; Kim, K.M.; Hank, J.A.; Surfus, J.; Dixon, J.R.; Lode, H.N.; Reisfeld, R.A.; Gillies, S.D. NXS2 murine neuroblastomas express increased levels of MHC class I antigens upon recurrence following NK-dependent immunotherapy. Cancer Immunol Immun. 2003, 53, 41–52. [Google Scholar]
- King, D.M.; Albertini, M.R.; Schalch, H.; Hank, J.A.; Gan, J.; Surfus, J.; Mahvi, D.; Schiller, J.H.; Warner, T.; Kim, K.M.; et al. A Phase I Clinical Trial of the Immunocytokine EMD 273063 (hu14.18-IL2) in Patients with melanoma. J. Clin. Oncol. 2004, 22, 4463–4473. [Google Scholar]
- Osenga, K.L.; Hank, J.A.; Albertini, M.R.; Gan, J.; Sternberg, A.G.; Eickhoff, J.; Seeger, R.C.; Matthay, K.K.; Reynolds, C.P.; Twist, C.; et al. A Phase I Clinical Trial of Hu14.18-IL2 (EMD 273063) as a Treatment for Children with Refractory or Recurrent Neuroblastoma and Melanoma: a Study of the Children’s Oncology Group. Clin. Cancer Res. 2005, 12, 1750–1759. [Google Scholar]
- Shusterman, S.; London, W.B.; Gillies, S.D.; Hank, J.A.; Voss, S.; Seeger, R.C.; Reynolds, C.P.; Kimball, J.; Albertini, M.A.; Wagner, B.; Gan, J.; et al. Anti-tumor activity of hu14.18-IL2 in relapsed/refractory neuroblastoma patients: A Children’s Oncology Group (COG) phase II study. J. Clin. Oncol. 2010, 28, 4969–4975. [Google Scholar]
- Joncker, N.T.; Shifrin, N.; Delebecque, F.; Raulet, D.H. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J. Exp. Med. 2010, 207, 2065–2072. [Google Scholar] [CrossRef]
- Elliott, J.M.; Wahle, J.A.; Yokoyama, W.M. MHC class I-deficient natural killer cells acquire a licensed phenotype after transfer into an MHC class I-sufficient environment. J. Exp. Med. 2010, 207, 2073–2079. [Google Scholar] [CrossRef]
- Sun, J.C. Re-educating natural killer cells. J. Exp. Med. 2010, 207, 2049–2052. [Google Scholar] [CrossRef]
- Orr, M.T.; Lanier, L.L. Natural killer cell education and tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Yawata, M.; Yawata, N.; Draghi, M.; Partheniou, F.; Little, A.M.; Parham, P. MHC class I-specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood 2008, 112, 2369–2380. [Google Scholar] [CrossRef]
- Sola, C.; André, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Bléry, M.; Wagtmann, N.R.; Romagné, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar]
- Sola, C.; André, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Bléry, M.; Wagtmann, N.R.; Romagné, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: Evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar]
- Moesta, A.K.; Norman, P.J.; Yawata, M.; Yawata, N.; Gleimer, M.; Parham, P. Synergistic Polymorphism at Two Positions Distal to the Ligand-Binding Site Makes KIR2DL2 a Stronger Receptor for HLA-C than KIR2DL3. J. Immol. 2008, 180, 3969–3979. [Google Scholar]
- Stern, M.; Ruggeri, L.; Capanni, M.; Mancusi, A.; Velardi, A. Human leukocyte antigens A23, A24, and A32 but not A25 are ligands for KIR3DL1. Blood 2008, 112, 708–710. [Google Scholar]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar]
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Capanni, M.; Carotti, A.; Aloisi, T.; Aversa, F.; Martelli, M.F.; Velardi, A. NK cell alloreactivity and allogeneic hematopoietic stem cell transplantation. Blood Cells Mol. Dis. 2008, 40, 84–90. [Google Scholar] [CrossRef]
- Leung, W.; Iyengar, R.; Turner, V.; Lang, P.; Bader, P.; Conn, P.; Niethammer, D.; Handgretinger, R. Determinants of antileukemia effects of allogeneic NK cells. J. Immunol. 2004, 172, 644–650. [Google Scholar]
- Hsu, K.C.; Keever-Taylor, C.A.; Wilton, A.; Pinto, C.; Heller, G.; Arkun, K.; O'Reilly, R.J.; Horowitz, M.M.; Dupont, B. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood 2005, 105, 4878–4884. [Google Scholar]
- Leung, W.; Handgretinger, R.; Iyengar, R.; Turner, V.; Holladay, M.S.; Hale, G.A. Inhibitory KIR-HLA receptor-ligand mismatch in autologous haematopoietic stem cell transplantation for solid tumour and lymphoma. Br. J.Cancer 2007, 97, 539–542. [Google Scholar] [CrossRef]
- Venstrom, J.M.; Zheng, J.; Noor, N.; Danis, K.E.; Yeh, A.W.; Cheung, I.Y.; Dupont, B.; O'Reilly, R.J.; Cheung, N.K.; Hsu, K.C. KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin. Cancer Res. 2009, 15, 7330–7334. [Google Scholar]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar]
- Bachanova, V.; Burns, L.J.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lindgren, B.R.; Cooley, S.; Weisdorf, D.; Miller, J.S. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol. Immun. 2010, 59, 1739–1744. [Google Scholar]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar]
- Delgado, D.C.; Hank, J.A.; Kolesar, J.; Lorentzen, D.; Gan, J.; Seo, S.; Kim, K.M.; Shusterman, S.; Gillies, S.D.; Reisfeld, R.A.; et al. Genotypes of NK cell KIR receptors, their ligands, and Fcγ receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 2010, 70, 9554–9661. [Google Scholar]
- Gillies, S.D.; Lan, Y.; Williams, S.; Carr, F.; Forman, S.; Raubitschek, A.; Lo, K.M. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood 2005, 105, 3972–3978. [Google Scholar] [CrossRef]
- Gubbels, J.A.; Gadbaw, B.; Buhtoiarov, I.N.; Horibata, S.; Kapur, A.K.; Patel, D.; Hank, J.A.; Gillies, S.D.; Sondel, P.M.; Patankar, M.S.; et al. Ab-IL2 fusion proteins mediate NK cell immune synapse formation by polarizing CD25 to the target cell-effector cell interface. Cancer Immunol. Immun. 2011, 60, 1789–1800. [Google Scholar]
- Buhtoiarov, I.N.; Neal, Z.C.; Gan, J.; Buhtoiarova, T.N.; Patankar, M.S.; Gubbels, J.A.A.; Hank, J.A.; Yamane, B.; Rakhmilevich, A.L.; Reisfeld, R.A.; et al. Differential internalization of hu14.18-IL2 immunocytokine by NK and tumor cells: Impact on conjugation, cytotoxicity and targeting. J. Leuk. Biol. 2011, 89, 625–638. [Google Scholar]
- Albertini, M.R.; Hank, J.A.; Gadbaw, B.; Kostlevy, J.; Haldeman, J.; Schalch, H.; Kim, K.M.; Eickhoff, J.; Gillies, S.D.; Sondel, P.M. Phase II Trial of Hu14.18-IL2 for Patients with Metastatic Melanoma. Cancer Immunol. Immun. 2012, in press.. [Google Scholar]
- Gillies, S.D.; Young, D.; Lo, K.-M.; Roberts, S. Biological activity and in vivo clearance of antitumor antibody/cytokine fusion proteins. Bioconjug. Chem. 1993, 4, 230–235. [Google Scholar] [CrossRef]
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: Clinical implications. CancerRes. 1999, 59, 3776–3782. [Google Scholar]
- Johnson, E.E.; Lum, H.D.; Rakhmilevich, A.L.; Schmidt, B.E.; Furlong, M.; Buhtoiarov, I.N.; Hank, J.A.; Raubitschek, A.; Colcher, D.; Reisfeld, R.A.; et al. Intratumoral Immunocytokine Treatment Results in Enhanced Antitumor Activity. Cancer Immunol. Immun. 2008, 57, 1891–1902. [Google Scholar]
- Weide, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Radny, P.; Zelba, H.; Pföhler, C.; Pawelec, G.; Garbe, C. High response rate after intratumoral treatment with interleukin-2: Results from a phase 2 study in 51 patients with metastasized melanoma. Cancer 2010, 116, 4139–4146. [Google Scholar]
- Weide, B.; Eigentler, T.K.; Pflugfelder, A.; Leiter, U.; Meier, F.; Bauer, J.; Schmidt, D.; Radny, P.; Pföhler, C.; Garbe, C. Survival after intratumoral interleukin-2 treatment of 72 melanoma patients and response upon the first chemotherapy during follow-up. Cancer Immunol. Immun. 2011, 60, 487–493. [Google Scholar]
- Johnson, E.E.; Yamane, B.H.; Lum, H.D.; Buhtoiarov, I.N.; Rakhmilevich, A.L.; Mahvi, D.M.; Gillies, S.D.; Sondel, P.M. Radiofrequency Ablation Combined with KS-IL2 Immunocytokine (EMD 273066) Results in an Enhanced Anti-tumor Effect Against Murine Colon Adenocarcinoma. Clin. Cancer Res. 2009, 15, 4875–4884. [Google Scholar]
- Gillies, S.D. unpublished work, 2012; Provenance Biopharmaceuticals Corp.: Burlington, MA, USA.
- Gillies, S.D.; Lan, Y.; Lo, K.M.; Super, M.; Wesolowski, J. Improving the efficacy of antibody-interleukin 2 fusion proteins by reducing their interaction with Fc receptors. Cancer Res. 1999, 59, 2159–2166. [Google Scholar]
- Gillies, S.D.; Lo, K.M.; Burger, C.; Lan, Y.; Dahl, T.; Wong, W.K. Improved circulating half-life and efficacy of an antibody-interleukin 2 immunocytokine based on reduced intracellular proteolysis. Clin. Cancer Res. 2002, 8, 210–216. [Google Scholar]
- Wahlin, B.E.; Aggarwal, M.; Montes-Moreno, S.; Gonzalez, L.F.; Roncador, G.; Sanchez-Verde, L.; Christensson, B.; Sander, B.; Kimby, E. A unifying microenvironment model in follicular lymphoma: outcome is predicted by programmed death-1—Positive, regulatory, cytotoxic, and helper T cells and macrophages. Clin. Cancer Res. 2010, 16, 637–650. [Google Scholar]
- Shanafelt, A.B.; Lin, Y.; Shanafelt, M.C.; Forte, C.P.; Dubois-Stringfellow, N.; Carter, C.; Gibbons, J.A.; Cheng, S.L.; Delaria, K.A.; Fleischer, R.A. T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat. Biotechnol. 2000, 18, 1197–1202. [Google Scholar]
- Gillies, S.D.; Lan, Y.; Hettmann, T.; Brunkhorst, B.; Sun, Y.; Mueller, S.O.; Lo, K.M. A low-toxicity IL-2-based immunocytokine retains antitumor activity despite its high degree of IL-2 receptor selectivity. Clin. Cancer Res. 2011, 17, 3673–3685. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Sondel, P.M.; Gillies, S.D. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies 2012, 1, 149-171. https://doi.org/10.3390/antib1020149
AMA Style
Sondel PM, Gillies SD. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies. 2012; 1(2):149-171. https://doi.org/10.3390/antib1020149
Chicago/Turabian StyleSondel, Paul M., and Stephen D. Gillies. 2012. "Current and Potential Uses of Immunocytokines as Cancer Immunotherapy" Antibodies 1, no. 2: 149-171. https://doi.org/10.3390/antib1020149