Antibody Drug Conjugates as Cancer Therapeutics

Regeneron Pharmaceuticals, Tarrytown, NY 10591, USA
Antibodies 2013, 2(1), 113-129; https://doi.org/10.3390/antib2010113
Submission received: 24 December 2012
/
Revised: 11 February 2013
/
Accepted: 18 February 2013
/
Published: 27 February 2013
(This article belongs to the Special Issue Antibody-Drug Conjugates)
Abstract
:Monoclonal antibody (MAb) based therapies have achieved considerable success in oncology, primarily when used in combination with cytotoxic drugs. Antibody drug conjugates (ADCs) are a class of therapeutics that harness the antigen-selectivity of MAbs to deliver highly potent cytotoxic drugs to antigen-expressing tumor cells. The use of MAb directed delivery can confer a therapeutic index to highly potent cytotoxic drugs, increasing both the efficacy and safety of therapy. Although simple in concept, to achieve the design goal of improved therapeutic efficacy and reduced toxicity, each of the components of an ADC; the MAb, linker and drug need to considered in the context of the targeted antigen, the selectivity of antigen expression and the biology of the tumor type on which the target antigen is expressed. The characteristics of targets, MAbs, linkers and drugs being used in ADC design are discussed.
1. Introduction
Recent efforts in oncology have focused on identifying drugs with improved selectivity for malignant versus normal cell as a means to improve both the efficacy and tolerability of cancer treatment. One approach to improving selectivity is to identify therapeutic targets with altered levels of expression on malignant versus normal cells and direct therapy against those targets. The introduction of monoclonal antibody (MAb) technology by Kohler and Milstein in 1975 [1] led to intensive efforts to develop MAbs as highly selective antitumor therapeutics, however the immunogenicity of first generation, murine MAbs limited their utility as therapeutics. Several key technology improvements including the development of methods to generate human/mouse chimeric and humanized MAbs substantially reduced immunogenicity and increased MAb half-life in patients. The ability to obtain fully human MAbs from transgenic mice and by phage display has further enhanced the clinical potential of these approaches [2,3,4]. There are currently 9 unconjugated MAbs approved by the FDA as cancer therapeutics. These MAbs include 2 chimeric, 4 humanized and 3 fully human monoclonal antibodies that mediate antitumor activity via blocking ligand/receptor interactions, or induce cell killing by antibody dependent cellular cytotoxicity (ADCC), or complement dependent cytotoxicity (CDC) [5]. Monoclonal antibodies have also been used to selectively deliver radionuclides [6,7], plant and bacterial toxins, [8,9,10] and a variety of cytotoxic drugs [11,12,13,14]. MAb directed delivery of cytotoxic drugs is an area of intense interest and there are currently at least 25 antibody drug conjugates (ADCs) undergoing clinical evaluation in oncology.
2. General Characteristics of ADCs
Antibody drug conjugates consist of a MAb chemically coupled to a linker and cytotoxic drug (Figure 1). Mechanistically, ADCs are designed to be stable in circulation and to effect intracellular drug release following antigen-specific binding and internalization of the ADC. Currently the use of ADCs as therapeutics is focused almost exclusively on the treatment of cancer. In contrast to small molecule cancer agents or function blocking MAbs, the targets for ADCs do not need to be causal in tumor progression. Rather the target antigens need to be differentially expressed on the cell surface of malignant cells relative to normal tissues. The MAbs used in first generation ADCs identified cell surface antigens with varying levels of tumor selectivity and included MAbs that internalized following antigen binding and those that did not. To be effective, non-internalizing ADCs needed to remain intact in the circulation and yet selectively release active drug at the tumor site. Typically these ADCs utilized peptidyl linkers designed to be cleaved by enzymes such as cathepsins and matrix metalloproteinases expressed in the tumor, or linkers that would release drug by hydrolysis at the slightly acidic pH observed in many solid tumors. For the most part, these non-internalizing ADCs did not show significant antigen-specific activity and did not improve the therapeutic index (maximum tolerated dose/active dose) relative to that of the free drug [15,16]. The use of MAbs that internalize following antigen binding has resulted in the design of linkers that are stable in circulation and efficiently release active drug following antigen specific binding, internalization and trafficking to endosomes/lysosomes [17,18,19]. Internalizing ADCs have demonstrated impressive preclinical [20,21,22,23] and clinical [24,25,26,27,28,29] activity.
3. ADC Targets and Mabs
A variety of cell surface antigens expressed on hematologic malignancies and epithelial tumors are being evaluated as ADC targets (Table 1). In general, tumor specific antigens have not been identified and as such ADC’s utilize MAbs directed against tumor-associated antigens [19,20,21,22,23]. These include antigens expressed on malignant cells with limited expression on cells of normal tissues, antigens that are expressed at much higher density on malignant versus normal cells, or antigens that are developmentally expressed. The best ADC targets are selectively and homogeneously expressed at high density on the cell surface of tumor cells. The tumor selectivity and level of expression of a target antigen are critical in the design of safe and efficacious ADCs as the intracellular concentration of drug that can be achieved is determined both by the level of antigen expression and the efficiency of ADC internalization and intracellular trafficking.
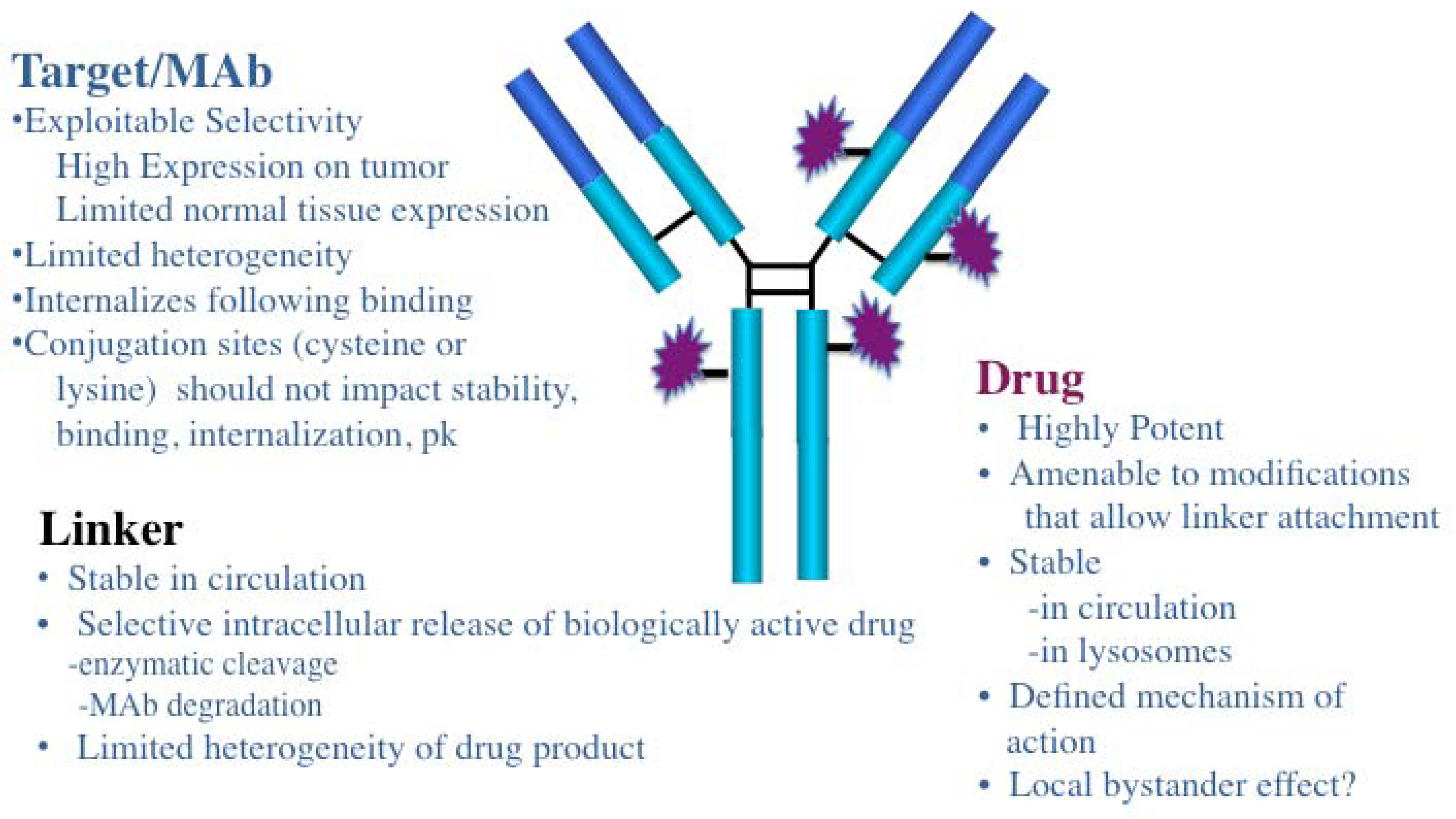
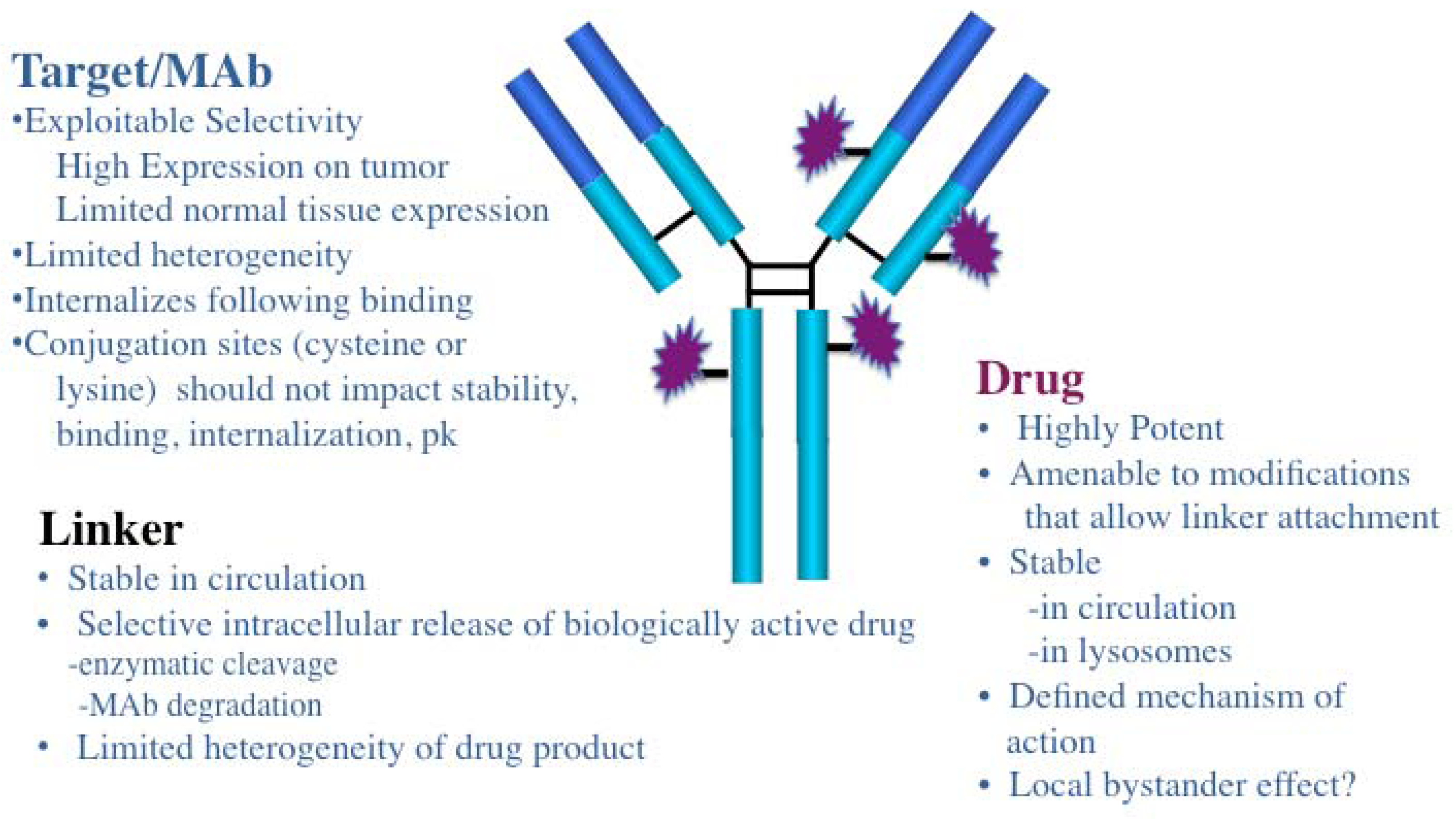
Figure 1.
Schematic of an antibody drug conjugate (ADC): An ADC consists of three components, the MAb, linker, and cytotoxic drug each of which must be optimized to design safe, potent and effective therapeutics.
Figure 1.
Schematic of an antibody drug conjugate (ADC): An ADC consists of three components, the MAb, linker, and cytotoxic drug each of which must be optimized to design safe, potent and effective therapeutics.
In addition to antigens expressed on tumor cells, there are ongoing efforts to develop ADCs to antigens expressed selectively on the tumor vasculature. These approaches seek to exploit differences in the level of antigen expression and/or the higher proliferation rate of endothelial cells of the tumor vasculature relative to that of normal tissues. Several endothelial targets considered to meet these criteria are being evaluated [30]. Among these are prostate-specific membrane antigen (PSMA) which is expressed on prostate cancer cells and also selectively expressed on vascular endothelial cells present in malignant solid tumors but not on endothelial cells of normal vasculature [31,32,33].
As most target antigens used in ADCs are tumor selective rather than tumor specific it is important to select MAbs that cross-react with the corresponding target antigen expressed on cells from rodents and/or non-human primates whenever possible. This enables preclinical toxicology assessments of “on target” toxicities that result from binding of the ADC to antigen expressed on cells of normal tissues. Preclinical pharmacology studies are also facilitated by MAbs that cross-react with rodent species as this allows for an understanding of efficacy in the background of normal tissue expression.
There are currently 19 ADCs in clinical trials (as listed in the database clinicaltrials.gov) for which targets and indications have been disclosed (Table 1). These include 8 ADCs directed against targets expressed on hematologic malignancies and 11 ADCs directed against targets expressed on epithelial tumors. Most ADCs (17/19) in clinical development utilize humanized or fully human MAbs. There are 2 ADCs that incorporate chimeric MAbs, brentuximab vedotin, an anti-CD30 ADC [28,34] and BT062, an anti-CD138 ADC [35]. Brentuximab vedotin recently received accelerated approved for treatment of Hodgkin lymphoma and anaplastic large cell lymphoma [36].

{kind=link}
{kind=link}
{kind=link}
| ADC Designations | Target Antigen | Antibody | Linker | Drug Class | Stage b | Tumor Indication(s) | Developer |
|---|---|---|---|---|---|---|---|
| Brentuximab vedotin | CD30 | Ch IgG1 | Valine-citrulline | Auristatin MMAE | Approved | HL/ALCL | Seattle Genetics |
| Inotuzumab ozogamicin | CD22 | Hz IgG4 | Hydrazone | Calicheamicin | Phase III | NHL | Pfizer |
| Gemtuzumab ozogamicin | CD33 | Hz IgG4 | Hydrazone | Calicheamicin | Phase II | Relapsed AML | Pfizer |
| SAR3419 | CD19 | Hz IgG1 | SPDB | Maytansine DM4 | Phase II | NHL | sanofi |
| BT062 | CD138 | Ch IgG4 | SPDB | Maytansine DM4 | Phase II | MM | Biotest |
| RG7593/DCDT2980S | CD22 | Hz IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | NHL | Genetech/Roche |
| RG-7596 | CD79b | Hz IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | NHL | Genentech/Roche |
| Milatuzumab-doxorubicin | CD74 | HzIgG1 | Hydrazone | Doxorubicin | Phase I | MM | Immunomedics |
| Trastuzumab-emtansine | HER2 | Hz IgG1 | SMCC | Maytansine DM1 | Phase III | Breast Cancer | Genentech/Roche |
| Glembatumomab vedotin | GPNMB | Hu IgG2 | Valine-citrulline | Auristatin MMAE | Phase II | Breast Cancer, Melanoma | Celldex Therapeutics |
| Anti-PSMA ADC | PSMA | Hu IgG1 | Valine-citrulline | Auristatin MMAE | Phase II | Prostate Cancer | Progenics |
| Lorvotuzumab mertansine | CD56 | Hz IgG1 | SPP | Maytansine DM1 | Phase I/II | Solid tumors, MM | Immunogen |
| AGS-5ME | SLC44A4 | Hu IgG2 | Valine-citrulline | Auristatin MMAE | Phase I | Pancreatic, Prostate Cancer | Astellas |
| SAR566658 | CA6 | Hu IgG1 | SPDB | Maytansine DM4 | Phase I | Solid Tumors | Sanofi |
| BAY 79-4620 | CA-IX | Hu IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | Solid Tumors | Bayer |
| BAY 94-9343 | Mesothelin | Hu IgG1 | SPDB | Maytansine DM4 | Phase I | Solid Tumors | Bayer |
| SGN-75 | CD70 | Hz IgG1 | Maleimidocaproyl | Auristatin MMAF | Phase I | RCC, NHL | Seattle Genetics |
| Labestuzumab-SN-38 | CD66e/CEACAM5 | Hz IgG1 | Phenylalanine-lysine | CPT-11 SN38 | Phase I | CRC | Immunomedics |
| ASG-22ME | Nectin-4 | Hu IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | Solid Tumors | Astellas |
Ch: chimeric; Hz: humanized; Hu: fully human; MMAE: monomethyl auristatin E; MMAF: monomethyl auristatin F; GPNMB: Glycoprotein NMB; PSMA: prostate specific membrane antigen; HL: Hodgkin’s Lymphoma; ALCL: Anaplastic Large Cell Lymphoma; NHL: Non-Hodgkin’s Lumphoma; AML: Acute Myelogenous Leukemia; MM: Multiple Myeloma; RCC: Renal Cell Carcinoma; CRC: Colorectal Carcinoma. a Includes ADCs with disclosed targets currently listed on Clinicaltrials.gov bSource: Clinicaltrials.gov: December 2012.
The isotype of the MAb will influence whether the ADC has the potential to kill cells via immune-mediated effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and/or complement dependent cytotoxicity (CDC) in addition to killing mediated by the targeted drug. IgG1 MAbs have the potential to mediate CDC, via binding the complement component C1q, and ADCC, via binding to Fcγ receptors expressed on various effector cells whereas IgG2 and IgG4 MAbs are inefficient in these effector functions [37]. Whether the ADCC and/or CDC activity of a given IgG1 MAb is retained following attachment of the linker/drug is likely to be influenced by both the sequence of the particular MAb, the site of drug conjugation, the drug load, and the drug itself. Notably, Trastuzumab-DM1, an ADC directed to HER-2 currently in Phase III trials, has been shown to retain in vitro ADCC activity [38]. The effector function of the MAb may contribute to the antitumor activity of ADCs. However, the relative contribution of the effector function of IgG1 versus IgG2 and IgG4 ADCs to the potency, selectivity, and off target toxicity of ADCs is not clear and difficult to model in preclinical studies. A comprehensive comparison of the safety profile of ADCs that differ only in MAb isotype has not been published to date. There are currently Mabs of the IgG1, IgG2 and IgG4 isotypes in clinical development, however, the majority (14/19) of current ADCs are IgG1 (Table 1).
4. Drugs and Linkers Used in ADCs
To be effective, an ADC must selectively bind, internalize and deliver an intracellular concentration of drug that is sufficient to result in cell death (Figure 2). While there is general agreement that conjugation strategies used in ADC design should have minimal effects on MAb affinity there are limited data available that can be used to define the optimal, or even the minimal, affinity that is required for an effective ADC. Rather than MAb affinity being the sole driver of ADC efficacy it is likely that the selectivity, efficiency of internalization and intracellular trafficking of a given MAb in composite will define an efficacious and safe ADC. The copy number and heterogeneity of antigen expression must be considered in the selection of drug and linker. This is particularly important for antigens expressed heterogeneously within a tumor where ADCs with local bystander activity [39,40] may be particularly desirable. The linker should be stable in circulation to exploit the long circulating half-life of the MAb and yet release active drug following antigen-mediated internalization. Linkers can be broadly classified by their mechanism of drug release. Cleavable linkers release drug by hydrolysis or enzymatic cleavage following internalization whereas non-cleavable linkers, release drug via degradation of the MAb in lysosomes following antigen-specific internalization [17,41,42,43,44]. In addition to the mechanism of drug release, the site of conjugation, the potency of the drug and the average number of drug molecules per antibody need to be considered in the selection of the linker.
Early ADCs incorporated drugs such as methotrexate [45,46,47], vinblastine [48,49] and doxorubicin [11,18,50,51] each of which had displayed clinical activity as free drugs. In general these ADCs demonstrated antigen-specific activity in vitro and in vivo but required high dose levels of ADC to achieve substantial antitumor activity. A variety of approaches were evaluated to increase the potency of these ADCs including increasing the quantity of drug delivered per MAb. In the case of doxorubicin conjugates, increasing the drug:MAb ratio over a range of 1–25 molecules of drug/MAb was achieved by direct conjugation [52], the use of branched linkers [53,54] or polymeric carriers [55]. The in vitro potency of these ADCs increased as the drug:MAb ratio increased as long as the conjugation did not adversely effect affinity.
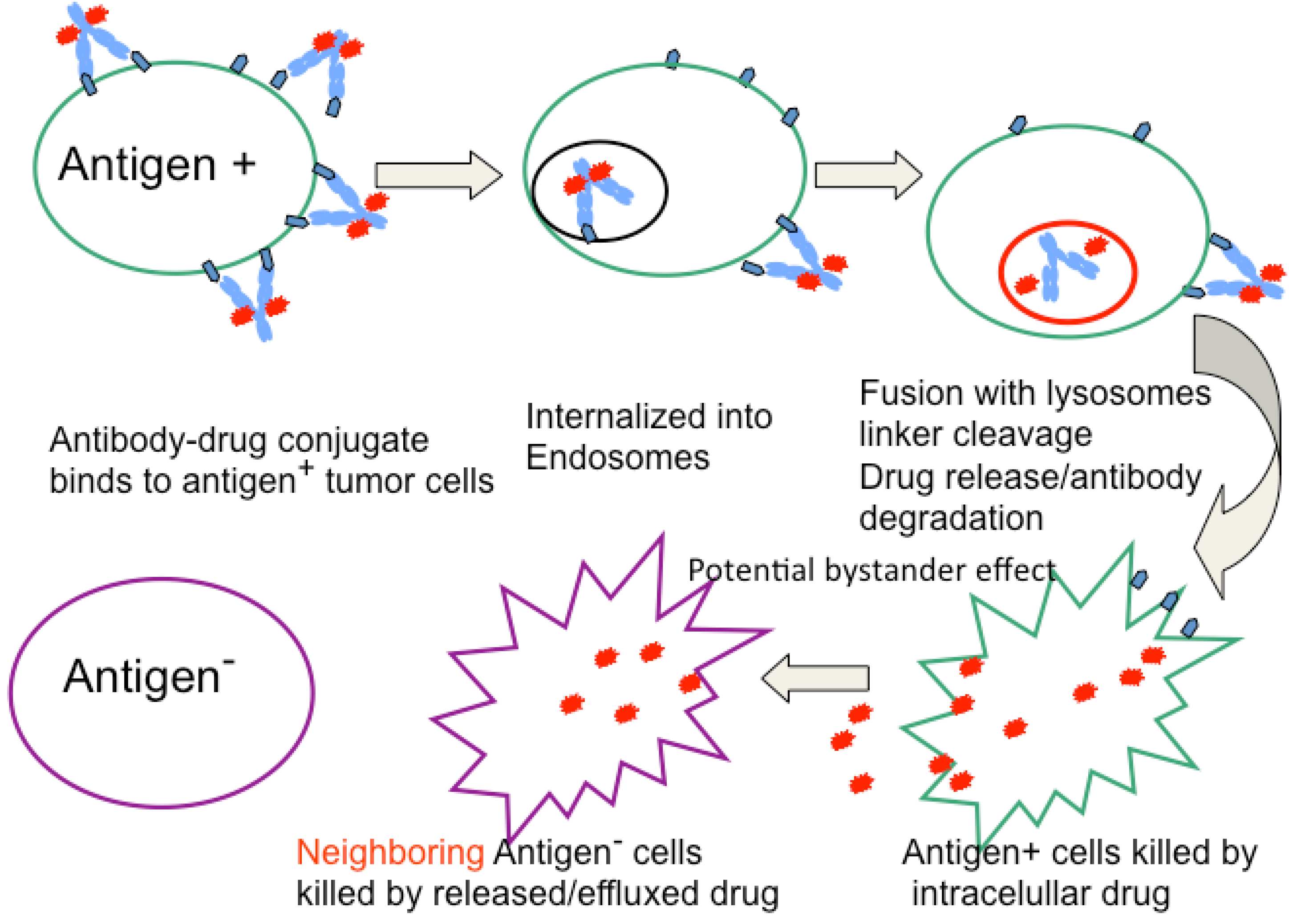
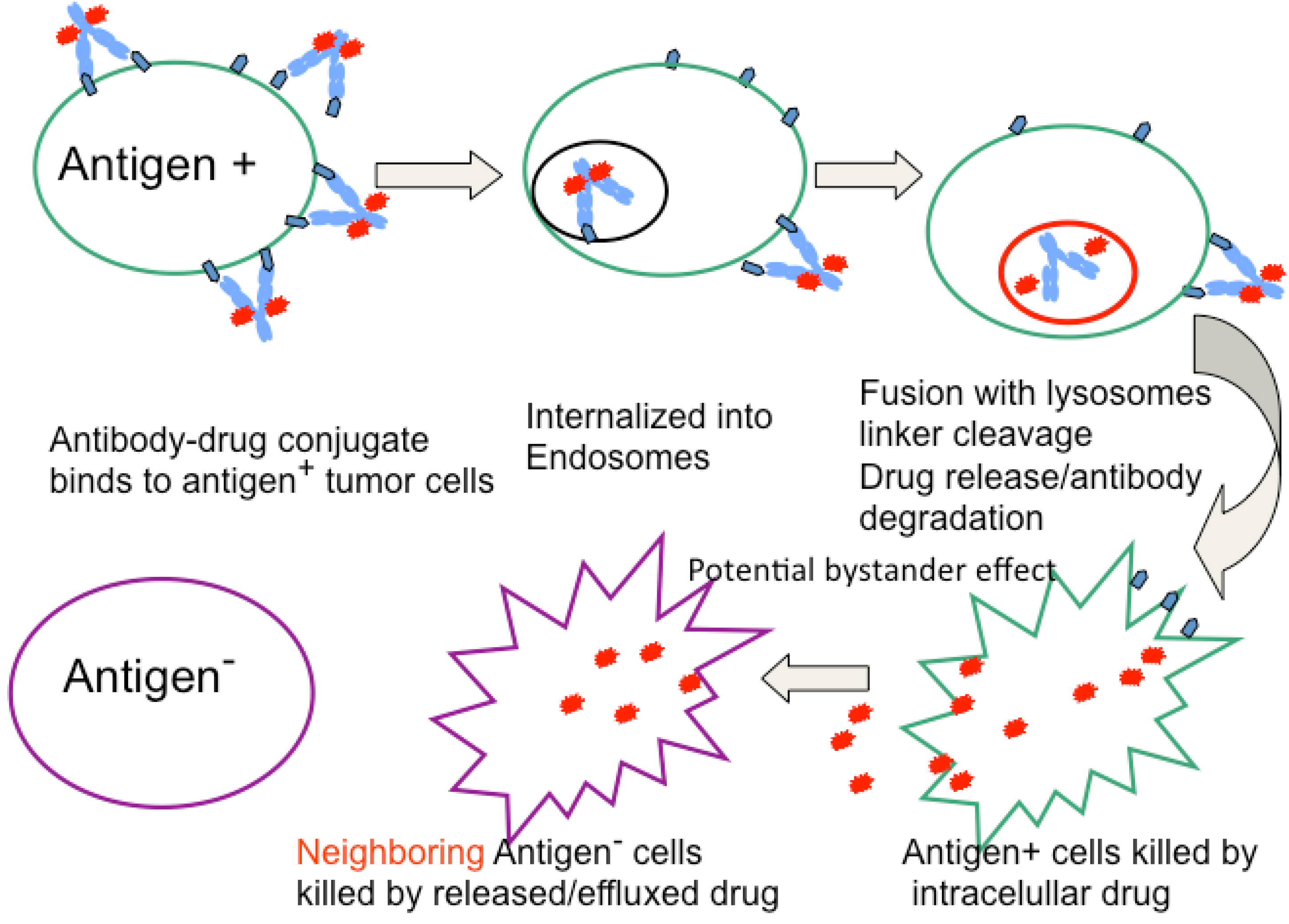
Figure 2.
Mechanism of ADC activity. Following tumor localization, ADCs bind to antigen expressing cells, is internalized into endosomes/lysosomes where biologically active drug is released following enzymatic cleavage or MAb catabolism. The liberated drug enters the cytoplasm where it binds to its molecular target (typically DNA or tubulin) resulting in cell cycle arrest and apoptosis. The drug may also diffuse out of or be released from dying cells and if membrane permeable can enter cells (antigen positive or negative) in close proximity and mediate bystander killing.
Figure 2.
Mechanism of ADC activity. Following tumor localization, ADCs bind to antigen expressing cells, is internalized into endosomes/lysosomes where biologically active drug is released following enzymatic cleavage or MAb catabolism. The liberated drug enters the cytoplasm where it binds to its molecular target (typically DNA or tubulin) resulting in cell cycle arrest and apoptosis. The drug may also diffuse out of or be released from dying cells and if membrane permeable can enter cells (antigen positive or negative) in close proximity and mediate bystander killing.
The importance of drug potency to the efficacy of ADCs was shown in the clinical evaluation of an ADC termed BR96-DOX (BMS-182248) a rapidly internalized chimeric IgG1 anti-Lewisy MAb conjugated to doxorubicin (DOX) via an acid-labile hydrazone linker. The BR96-DOX conjugate produced cures of both DOX sensitive and insensitive human tumors in immune deficient rodents. However, the dose levels of ADC required were high, likely reflecting the low potency of DOX and the sub-optimal half-life of the hydrazone linker [11,18]. The ADC was evaluated in Phase I trials in patients with solid tumors confirmed to express the Lewisy antigen by immunohistochemistry. Tumor biopsies obtained from patients with accessible lesions 48 hours after infusion of the ADC showed tumor localization of both the BR96 MAb and DOX components of the ADC. However, the results of this trial were disappointing, with objective responses seen in only 2 of 66 patients treated every 3 weeks at various dose levels of BR96-DOX [56]. These data suggest the limitations of using a low potency drug such as DOX as the delivery payload for an ADC.
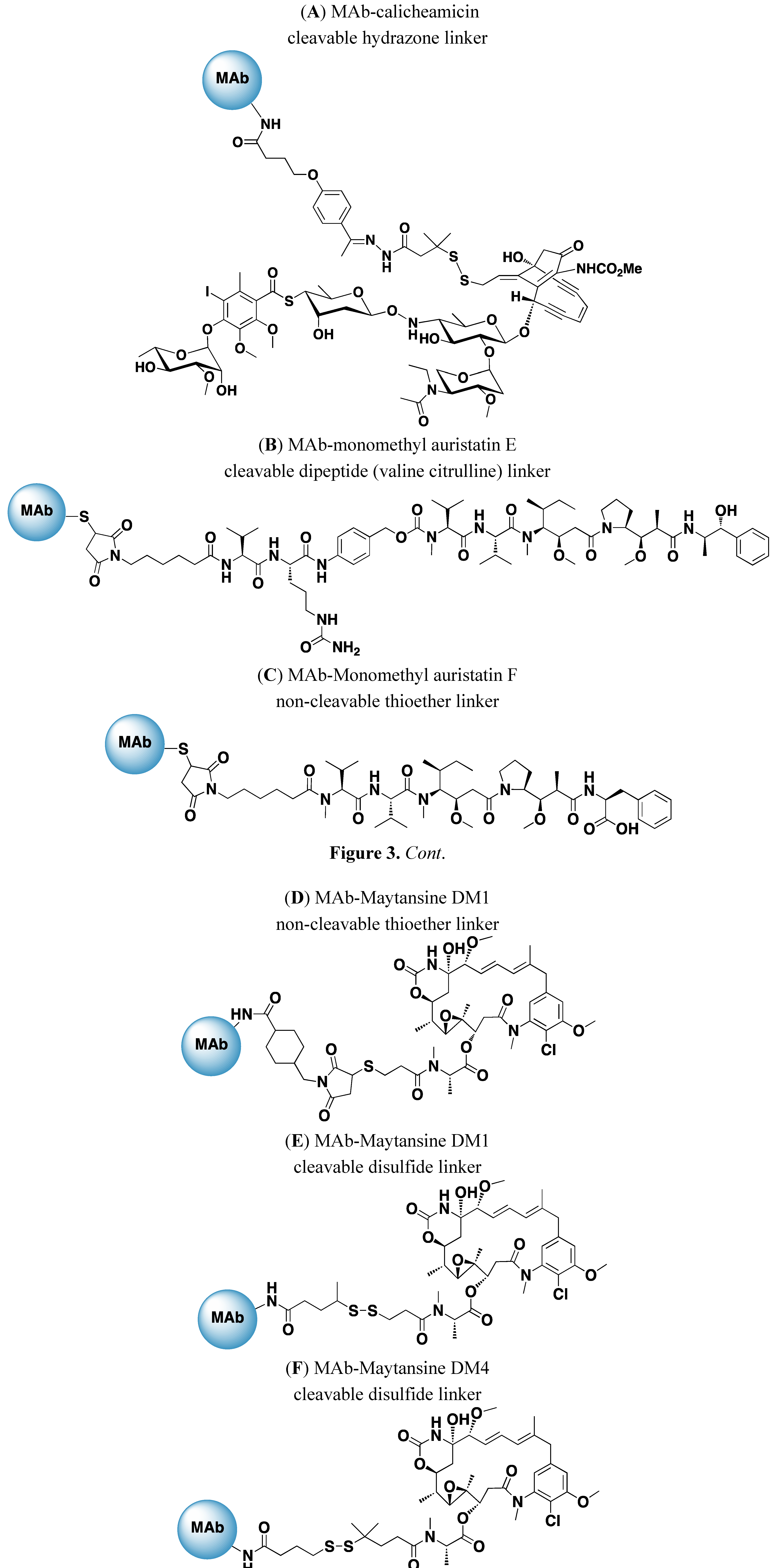
A major advance in ADC optimization resulted from incorporating highly potent cytotoxics such as calicheamicins, maytansinoids, auristatins, and duocarmycins into ADCs. These cytotoxics are 100–1,000 fold more potent than the drugs used in early ADCs. For the most part, these agents had failed clinically as free drugs, as they were very toxic and lacked a therapeutic index. Delivery in the form of an ADC provides a means to clinically exploit the potency of these agents while minimizing their systemic toxicities. The structures of drugs and linkers that are currently being evaluated clinically are shown in Figure 3A–F.
Figure 3.
Structure of drugs and linkers used in ADCs under clinical development.

Several potent DNA-damaging agents, including analogs of calicheamicin [12] and duocarmycin [57] have been evaluated as ADC payloads. The calicheamicins bind to the minor groove of DNA resulting in double strand breaks in DNA and cell death at sub picomolar concentrations. Gemtuzumab ozogamacin is an ADC in which calicheamicin is conjugated to a humanized anti-CD33 MAb utilizing a hydrazone linker (Figure 3A). The conjugate is highly potent and demonstrated antigen-specific activity in preclinical models at doses of ~100 µg/kg [12]. Gemtuzumab ozogamacin was evaluated in 3 open-label single arm Phase II studies and received accelerated approval by the FDA in 2000 for treatment of CD33 positive acute myeloid leukemia (AML) in first relapse in patients over 60 years of age [58,59]. The drug was voluntarily withdrawn from the market in 2010 due to concerns about the relative therapeutic benefit seen in post marketing studies [60]. As shown in Table 1, there are ongoing Phase II and III studies of gemtuzumab ozogamacin in additional indications.
Inotuzumab ozogamacin (CMC-544) is an IgG4 ADC that targets CD22 MAb conjugated to the same calicheamicin analog via a hydrazone linker. The ADC demonstrated potent activity in preclinical models [61]. A Phase I study of inotuzumab ozogamacin was completed in relapsed or refractory CD22 expressing B cell NHL and the ADC demonstrated evidence of clinical activity. At the MTD (1.8 mg/m2 every 4 weeks) 15% of patients with diffuse large B-cell lymphoma (n = 26) and 68% of patients with follicular lymphoma (n = 22) achieved an objective response. The major grade 3–4 toxicity was thrombocytopenia [62]. This may reflect “off target” toxicity of the ADC as a consequence of non-specific release of drug from hydrolysis the hydrazone linker and suggests that improvements in linker stability may be required to fully exploit the potency of calicheamicin based ADCs. Inotuzumab ozogamacin is currently in Phase III studies in NHL (Table 1).
Analogs of duocarmycin [57,63,64,65] have displayed impressive activity in preclinical models. MDX-1203 is a duocarmycin analog directed against CD70 that incorporates a dipeptide linker to release a prodrug of duocarmycin following antigen-specific internalization. MDX-1203 demonstrated potent antigen-specific antitumor activity in vitro and in preclinical tumor models [57,63,64]. A Phase I trial of MDX-1203 in patients with advanced/recurrent renal cell carcinoma or relapsed/refractory Non-Hodgkin’s Lymphoma was initiated, however the trial was suspended (clinicaltrials.gov December 2012).
The auristatins and maytansinoids are two classes of highly potent (pM-nM IC50s), structurally distinct, drugs that bind to the vinca binding site of tubulin and inhibit tubulin polymerization resulting in G2/M arrest and cell death [14,66,67]. The auristatins, monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF) are fully synthetic analogs of the marine natural product dolastatin 10 [14,68]. The maytansinoids DM1 and DM4 are benzoansamacrolides derived semi-synthetically from ansamitocin [67]. The majority of ADCs currently in clinical development (Table 1) are conjugates of auristatin [14,69,70] or maytansine [66,67,71].
There are 9 auristatin-based ADCs with disclosed targets currently in clinical development (Table 1). The majority of the auristatin ADCs use a dipeptide (valine-citrulline) linker conjugated to the auristatin analog MMAE (Figure 3B) via solvent accessible thiols present in MAb cysteines. These conjugates are stable in circulation and the linker is cleaved by lysosomal enzymes, including cathepsin B, following antigen specific binding and internalization [14,17,42]. Biologically active MMAE is released from the ADC following antigen-specific internalization and importantly, due to the membrane permeability of MMAE, the released drug is able to kill cells that are in close proximity, including those that lack antigen expression [40]. The ability of a targeted drug, released in an antigen-specific manner to mediate local bystander killing is an important selection criteria for those ADCs directed against antigens that are heterogeneously expressed in tumors. The most advanced of the vc-MMAE ADCs is an anti-CD30 ADC, termed SGN-35 or brentuximab vedotin, with an average drug:MAb ratio of 4. The ADC produced antigen-specific cell killing in vitro and was active at tolerated doses against human NHL models in athymic mice [23,70]. The conjugate produced impressive activity when evaluated in Phase 1 trials in patients with relapsed CD30-positive lymphomas. Forty-five patients with CD30 positive lymphomas received doses of 0.1–3.6 mg/kg every 3 weeks and objective responses were seen in 17 patients (including 11 complete remissions). The most common adverse events were fatigue, pyrexia, diarrhea, nausea, neutropenia and peripheral neuropathy. An objective tumor response rate of 50% (6/12) was seen in patients treated at the MTD (1.8 mg/kg). The same chimeric anti-CD30 MAb had been evaluated previously in unconjugated form in a similar patient population and had shown minimal efficacy. Taken together these data demonstrate the critical importance of delivery of the cytotoxic drug MMAE to the activity of SGN-35 [28] and provide clear evidence for improving the activity of unconjugated MAbs by incorporating the additional effector mechanism of MAb-mediated drug delivery. Brentuximab vedotin received accelerated approved for treatment of patients with Hodgkin lymphoma or systemic anaplastic large-cell lymphoma (sALCL) in 2011 based on the results of 2 single arm trials. The objective response rate for Hodgkin lymphoma was 73% with a median duration of response of 6.7 months and complete response rate of 32%. In sALCL an objective response rate of 86% with a median response duration of 12.6 months and a complete response rate of 57% [72] was reported.
In addition to the cleavable vc-MMAE ADCs, non-cleavable maleimidocaproyl (mc) linked conjugates of the auristatin analog MMAF (Figure 3C) have shown antigen specific activity in vitro and potent antigen specific activity in human tumor models [69,73,74]. As with vc-MMAE based ADCs, the mc linkers are attached to solvent accessible thiols present in MAb cysteines. For the mc-MMAF conjugates, the intracellular release of drug is believed to be a consequence of degradation of the MAb in lysosomes. In contrast to MMAE, the MMAF analog released following antigen-specific internalization and localization to lysosomes is cell impermeable and as such does not display significant bystander killing activity. An MMAF based ADC termed SGN-75 and directed against CD70 [74] is currently in Phase I evaluation in NHL and renal cell carcinoma (Table 1).
Maytansine-based ADCs represent 6 of 19 currently active clinical programs with disclosed targets (Table 1). There are two maytansine analogs, DM1 (Figure 3D) and DM4 (Figure 3E), being evaluated clinically. These derivatives differ in steric hindrance around the disulfide bridge [21,71]. In each case, maytansine linkers are conjugated through amino groups of MAb lysine residues.
The most advanced of the maytansine based ADCs is trastuzumab emtansine (T-DM1), currently in Phase III clinical trials for treatment of HER2 positive metastatic breast cancer. T-DM1 consists of the clinically approved MAb trastuzumab (humanized IgG1) conjugated to DM-1 via a non-cleavable thioether (SMCC) linker (Figure 3D). HER2, the target of T-DM1 is an excellent ADC target as it is amplified and overexpressed in 20–25% of human breast cancers [75] and is internalized following binding. As T-DM1 utilizes a non-cleavable linker it is anticipated that drug release from the ADC occurs as a result of degradation of the MAb in lysosomes. T-DM1 has an average of 3.5 molecules of DM-1/MAb. In preclinical studies, T-DM1 demonstrated potent antigen-specific activity in vitro and was more active and better tolerated than ADCs prepared with other derivatives of DM1 having varying degrees of steric hindrance around the disulfide. Importantly, T-DM1 demonstrated excellent stability for at least 7 days after administration of the ADC [21]. T-DM1 demonstrated significant activity in a Phase I and Phase II trials in patients with HER2 positive metastatic breast cancer. Results of a 991 patient randomized Phase III trial (EMELIA) that evaluated the activity of T-DM1, administered at a dose of 3.6 mg/kg every 3 weeks, versus lapatinib plus capecitabine in patients with advanced HER2 positive breast cancer previously been treated with unconjugated trastuzumab and a taxane have been recently published [27]. The results of this trial are impressive both in terms of the improvement in activity and safety seen for T-DM1 relative to lapatanib plus capecitabine. T-DM1 significantly increased both progression free survival (9.6 months for T-DM1 versus 6.4 months for lapatinib-capecitabin; p < 0.001) and overall survival (30.9 months for T-DM1 versus 25.1 months for lapatinib-capecitabine; p < 0.001). The objective response rate for T-DM1 was also higher (43.6% for T-DM1 versus 30.8% for lapatinib-capecitabine; p < 0.001). Although adverse events of any grade were similar for both treatment arms, (95.9% and 97.7% for T-DM1 and lapatinib-capecitabine, respectively) the incidence of Grade 3 or 4 events was lower for T-DM1 (40.8%) than was observed for lapatinib-capecitabine (57%). The T-DM1 ADC achieved the primary goals of antibody-directed drug delivery: increased clinical activity and increased safety. Importantly, as these patients had previously progressed when treated with unconjugated trastuzumab the data on T-DM1 demonstrate the critical importance of targeted drug delivery to the clinical activity of the T-DM1 ADC.
Maytansine based ADCs prepared with cleavable linkers (Figure 3E) are also being evaluated clinically (Table 1). The most advanced of these is SAR3419 an ADC that consists of a humanized (IgG1 MAb) conjugated to DM4 using a cleavable hindered disulfide linker. In preclinical models SAR3419 demonstrated activity in several lymphoma models. Clinical activity was seen in Phase I trials when the ADC was administered either every 3 weeks or on a weekly schedule, with objective response seem in 6 of 35 evaluable patients on the every 3 weeks schedule [20]. SAR3419 is currently being evaluated in multiple Phase 2 studies.
5. Optimization of ADCs
Major advances in the efficacy and safety of ADCs have resulted from incorporating highly potent drugs and using stable linkers to better exploit the half-life of the MAb component of the ADC. The recent clinical data from both Adcetris (SGN-35) [76] and Trastuzumab Emtansine (T-DM1) [27] conclusively demonstrate that improvements in both clinical activity and patient safety can be achieved with antibody directed drug delivery. There are ongoing efforts to increase ADC selectivity and efficacy by further optimizing linkers, sites of MAb attachment and cytotoxic payloads [77,78,79]. The selection of suitable ADC targets remains a critical challenge to ADC design. Characteristics of the ADC target such as copy number, heterogeneity and specificity of expression, internalization rate, and intracellular trafficking can be used to guide the selection of linker (cleavable or non-cleavable) and the potency and characteristics of the drug released (cell permeable or impermeable). However, further insight into the optimal design characteristics of effective ADCs will be best gained as additional clinical data become available. There are currently over 25 ADCs in clinical development and evolving clinical data from these trials will provide critical insight into the design of next generation ADCs.
References
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Jakobovits, A. The long-awaited magic bullets: Therapeutic human monoclonal antibodies from transgenic mice. Expert Opin. Investig. Drugs 1998, 7, 607–614. [Google Scholar] [CrossRef]
- Lonberg, N. Human antibodies from transgenic animals. Nat. Biotechnol. 2005, 23, 1117–1125. [Google Scholar] [CrossRef]
- Reichert, J.M.; Dhimolea, E. The future of antibodies as cancer drugs. Drug Discov. Today 2012, 17, 954–963. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Radioactive antibodies: A historical review of selective targeting and treatment of cancer. Hosp. Pract. (Minneap) 2010, 38, 82–93. [Google Scholar] [CrossRef]
- Steiner, M.; Neri, D. Antibody-radionuclide conjugates for cancer therapy: Historical considerations and new trends. Clin. Cancer Res. 2011, 17, 6406–6416. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Lorberboum-Galski, H. Human toxin-based recombinant immunotoxins/chimeric proteins as a drug delivery system for targeted treatment of human diseases. Expert Opin. Drug Deliv. 2011, 8, 605–621. [Google Scholar] [CrossRef]
- Choudhary, S.; Mathew, M.; Verma, R.S. Therapeutic potential of anticancer immunotoxins. Drug Discov. Today 2011, 16, 495–503. [Google Scholar] [CrossRef]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellstrom, I.; Hellstrom, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar]
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar]
- Liu, C.; Chari, R.V. The development of antibody delivery systems to target cancer with highly potent maytansinoids. Expert Opin. Investig. Drugs 1997, 6, 169–172. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Trail, P.A.; Bianchi, A.B. Monoclonal antibody drug conjugates in the treatment of cancer. Curr. Opin. Immunol. 1999, 11, 584–588. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Walker, M.A. Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharmacol. Ther. 1999, 83, 67–123. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar]
- Trail, P.A.; King, H.D.; Dubowchik, G.M. Monoclonal antibody drug immunoconjugates for targeted treatment of cancer. Cancer Immunol. Immunother. 2003, 52, 328–337. [Google Scholar]
- Teicher, B.A. Antibody-drug conjugate targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef]
- Blanc, V.; Bousseau, A.; Caron, A.; Carrez, C.; Lutz, R.J.; Lambert, J.M. SAR3419: An anti-CD19-maytansinoid immunoconjugate for the treatment of B-cell malignancies. Clin. Cancer Res. 2011, 17, 6448–6458. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar]
- Petrul, H.M.; Schatz, C.A.; Kopitz, C.C.; Adnane, L.; McCabe, T.J.; Trail, P.; Ha, S.; Chang, Y.S.; Voznesensky, A.; Ranges, G.; et al. Therapeutic mechanism and efficacy of the antibody-drug conjugate BAY 79-4620 targeting human carbonic anhydrase 9. Mol. Cancer Ther. 2012, 11, 340–349. [Google Scholar] [CrossRef]
- Wahl, A.F.; Klussman, K.; Thompson, J.D.; Chen, J.H.; Francisco, L.V.; Risdon, G.; Chace, D.F.; Siegall, C.B.; Francisco, J.A. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin's disease. Cancer Res. 2002, 62, 3736–3742. [Google Scholar]
- Barginear, M.F.; John, V.; Budman, D.R. Trastuzumab-DM1: A clinical update of the novel antibody-drug conjugate for HER2-overexpressing breast cancer. Mol. Med. 2012, 18, 1473–1479. [Google Scholar] [CrossRef]
- Ogura, M.; Hatake, K.; Ando, K.; Tobinai, K.; Tokushige, K.; Ono, C.; Ishibashi, T.; Vandendries, E. Phase I study of anti-CD22 immunoconjugate inotuzumab ozogamicin plus rituximab in relapsed/refractory B-cell non-Hodgkin lymphoma. Cancer Sci. 2012, 103, 933–938. [Google Scholar] [CrossRef]
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef]
- Younes, A.; Kim, S.; Romaguera, J.; Copeland, A.; Farial Sde, C.; Kwak, L.W.; Fayad, L.; Hagemeister, F.; Fanale, M.; Neelapu, S.; et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J. Clin. Oncol. 2012, 30, 2776–2782. [Google Scholar]
- Gerber, H.P.; Senter, P.D.; Grewal, I.S. Antibody drug-conjugates targeting the tumor vasculature: Current and future developments. MAbs 2009, 1, 247–253. [Google Scholar] [CrossRef]
- Baccala, A.; Sercia, L.; Li, J.; Heston, W.; Zhou, M. Expression of prostate-specific membrane antigen in tumor-associated neovasculature of renal neoplasms. Urology 2007, 70, 385–390. [Google Scholar] [CrossRef]
- Haffner, M.C.; Kronberger, I.E.; Ross, J.S.; Sheehan, C.E.; Zitt, M.; Muhlmann, G.; Ofner, D.; Zelger, B.; Ensinger, C.; Yang, X.J.; et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum. Pathol. 2009, 40, 1754–1761. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86. [Google Scholar]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L.; et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [CrossRef]
- DeFrancesco, L. Seattle genetics rare cancer drug sails through accelerated approval. Nat. Biotechnol. 2011, 29, 851–852. [Google Scholar] [CrossRef]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by antibody-drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef]
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897. [Google Scholar] [CrossRef]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjug. Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Radia, S.; Mastalerz, H.; Walker, M.A.; Firestone, R.A.; Dalton King, H.; Hofstead, S.J.; Willner, D.; Lasch, S.J.; Trail, P.A. Doxorubicin immunoconjugates containing bivalent, lysosomally-cleavable dipeptide linkages. Bioorg. Med. Chem. Lett. 2002, 12, 1529–1532. [Google Scholar]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar]
- Smyth, M.J.; Pietersz, G.A.; McKenzie, I.F. The mode of action of methotrexate-monoclonal antibody conjugates. Immunol. Cell Biol. 1987, 65, 189–200. [Google Scholar] [CrossRef]
- Ghose, T.; Ferrone, S.; Blair, A.H.; Kralovec, Y.; Temponi, M.; Singh, M.; Mammen, M. Regression of human melanoma xenografts in nude mice injected with methotrexate linked to monoclonal antibody 225.28 to human high molecular weight-melanoma associated antigen. Cancer Immunol. Immunother. 1991, 34, 90–96. [Google Scholar] [CrossRef]
- Elias, D.J.; Kline, L.E.; Robbins, B.A.; Johnson, H.C., Jr.; Pekny, K.; Benz, M.; Robb, J.A.; Walker, L.E.; Kosty, M.; Dillman, R.O. Monoclonal antibody KS1/4-methotrexate immunoconjugate studies in non-small cell lung carcinoma. Am. J. Respir. Crit. Care Med. 1994, 150, 1114–1122. [Google Scholar]
- Schrappe, M.; Bumol, T.F.; Apelgren, L.D.; Briggs, S.L.; Koppel, G.A.; Markowitz, D.D.; Mueller, B.M.; Reisfeld, R.A. Long-term growth suppression of human glioma xenografts by chemoimmunoconjugates of 4-desacetylvinblastine-3-carboxyhydrazide and monoclonal antibody 9.2.27. Cancer Res. 1992, 52, 3838–3844. [Google Scholar]
- Petersen, B.H.; DeHerdt, S.V.; Schneck, D.W.; Bumol, T.F. The human immune response to KS1/4-desacetylvinblastine (ly256787) and KS1/4-desacetylvinblastine hydrazide (LY203728) in single and multiple dose clinical studies. Cancer Res. 1991, 51, 2286–2290. [Google Scholar]
- Yang, H.M.; Reisfeld, R.A. Doxorubicin conjugated with a monoclonal antibody directed to a human melanoma-associated proteoglycan suppresses the growth of established tumor xenografts in nude mice. Proc. Natl. Acad. Sci. USA 1988, 85, 1189–1193. [Google Scholar] [CrossRef]
- Shih, L.B.; Goldenberg, D.M.; Xuan, H.; Lu, H.W.; Mattes, M.J.; Hall, T.C. Internalization of an intact doxorubicin immunoconjugate. Cancer Immunol. Immunother. 1994, 38, 92–98. [Google Scholar] [CrossRef]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Greenfield, R.S.; King, D.; Zoeckler, M.E.; Braslawsky, G.R. Antigen-specific activity of carcinoma-reactive BR64-doxorubicin conjugates evaluated in vitro and in human tumor xenograft models. Cancer Res. 1992, 52, 5693–5700. [Google Scholar]
- King, H.D.; Yurgaitis, D.; Willner, D.; Firestone, R.A.; Yang, M.B.; Lasch, S.J.; Hellstrom, K.E.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched linkers: A novel method for increasing the potency of doxorubicin immunoconjugates. Bioconjug. Chem. 1999, 10, 279–288. [Google Scholar] [CrossRef]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar]
- Shih, L.B.; Goldenberg, D.M.; Xuan, H.; Lu, H.; Sharkey, R.M.; Hall, T.C. Anthracycline immunoconjugates prepared by a site-specific linkage via an amino-dextran intermediate carrier. Cancer Res. 1991, 51, 4192–4198. [Google Scholar]
- Saleh, M.N.; LoBuglio, A.F.; Trail, P.A. Immunoconjugate Therapy of Solid Tumors: Studies with BR96-Doxorubicin. In Monoclonal Antibody-Based Therapy of Cancer, 1th ed.; Grossbard, M.L., Ed.; Marcel Dekker, Inc: New York, NY, USA, 1998; Volume 15, pp. 397–416. [Google Scholar]
- Terrett, J.; Gangwar, S.; Rao-Naik, C.; Pan, C.; Guerlavais, V.; Huber, M.; Chong, C.; Green, L.; Cardarelli, P.; King, D.; et al. Single, low dose treatment of lymphoma and renal cancer xenografts with human anti-CD70 antibody-toxin conjugates, results in long term cures. In Proceedings of the 98th Annual Meeting of the American Association for Cancer Research, Los Angeles, CA, USA, 14–18 April 2007.
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Lowenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef]
- Jurcic, J.G. What happened to anti-CD33 therapy for acute myeloid leukemia? Curr. Hematol. Malig. Rep. 2012, 7, 65–73. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Dougher, M.M.; Evans, D.Y.; Zhou, B.B.; Damle, N.K. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother. Pharmacol. 2011, 67, 741–749. [Google Scholar] [CrossRef]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin's lymphoma: Results of a phase I study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar]
- Cardarelli, P.; King, D.; Terrett, J.; Gangwar, S.; Cohen, L.; Pan, C.; Rao, C.; Deshpande, S.; Angipuram, R.; Passmore, D.; et al. Efficacy and safety of a human anti-CD70 antibody-MGBA conjugate. In Proceedings of the 99th Annual Meeting of the American Association for Cancer Research, San Diego, CA, USA, 12-16 April 2008; AACR: Philadelphia, PA, USA, 2008; p. Abstract nr 4061. [Google Scholar]
- King, D.; Terrett, J.; Cardarelli, P.; Pan, C.; Rao, C.; Gangwar, S.; Deshpande, S.; Vangipuram, R.; Passmore, D.; Mirjolet, J.; et al. Mechanism of activation of a human anti-CD70 antibody-mgba conjugate and efficacy in a nude rat model of renal carcinoma. In Proceedings of the 99th Annual Meeting of the American Association for Cancer Research, San Diego, CA, USA, 12-16 April 2008; AACR: Philadelphia, PA, USA, 2008; p. Abstract nr 4057. [Google Scholar]
- Beck, A.; Lambert, J.; Sun, M.; Lin, K. Fourth World Antibody-drug Conjugate Summit: February 29-march 1, 2012, Frankfurt, Germany. MAbs 2012, 4, 637–647. [Google Scholar] [CrossRef]
- Chari, R.V.; Martell, B.A.; Gross, J.L.; Cook, S.B.; Shah, S.A.; Blattler, W.A.; McKenzie, S.J.; Goldmacher, V.S. Immunoconjugates containing novel maytansinoids: Promising anticancer drugs. Cancer Res. 1992, 52, 127–131. [Google Scholar]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 2010, 9, 2700–2713. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Varterasian, M.L.; Almatchy, V.P.; Hannoudi, G.N.; Pettit, G.R.; Al-Katib, A. Successful treatment of human chronic lymphocytic leukemia xenografts with combination biological agents auristatin PE and bryostatin 1. Clin. Cancer Res. 1998, 4, 1337–1343. [Google Scholar]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar]
- de Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. Food and drug administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef]
- Jackson, D.; Gooya, J.; Mao, S.; Kinneer, K.; Xu, L.; Camara, M.; Fazenbaker, C.; Fleming, R.; Swamynathan, S.; Meyer, D.; et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008, 68, 9367–9374. [Google Scholar]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.; Wood, C.G.; Repasky, E.A.; Grewal, I.S.; Law, C.L.; Gerber, H.P. Potent anticarcinoma activity of the humanized anti-CD70 antibody h1F6 conjugated to the tubulin inhibitor auristatin via an uncleavable linker. Clin. Cancer Res. 2008, 14, 6171–6180. [Google Scholar]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar]
- Hartley, J.A.; Hochhauser, D. Small molecule drugs - optimizing DNA damaging agent-based therapeutics. Curr. Opin. Pharmacol. 2012, 12, 398–402. [Google Scholar] [CrossRef]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef]
- Sapra, P.; Hooper, A.T.; O'Donnell, C.J.; Gerber, H.P. Investigational antibody drug conjugates for solid tumors. Expert Opin. Investig. Drugs 2011, 20, 1131–1149. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Trail, P.A. Antibody Drug Conjugates as Cancer Therapeutics. Antibodies 2013, 2, 113-129. https://doi.org/10.3390/antib2010113
AMA Style
Trail PA. Antibody Drug Conjugates as Cancer Therapeutics. Antibodies. 2013; 2(1):113-129. https://doi.org/10.3390/antib2010113
Chicago/Turabian StyleTrail, Pamela A. 2013. "Antibody Drug Conjugates as Cancer Therapeutics" Antibodies 2, no. 1: 113-129. https://doi.org/10.3390/antib2010113