In Vivo Secretion of Bispecific Antibodies Recruiting Lymphocytic Effector Cells

Abstract
:1. Introduction
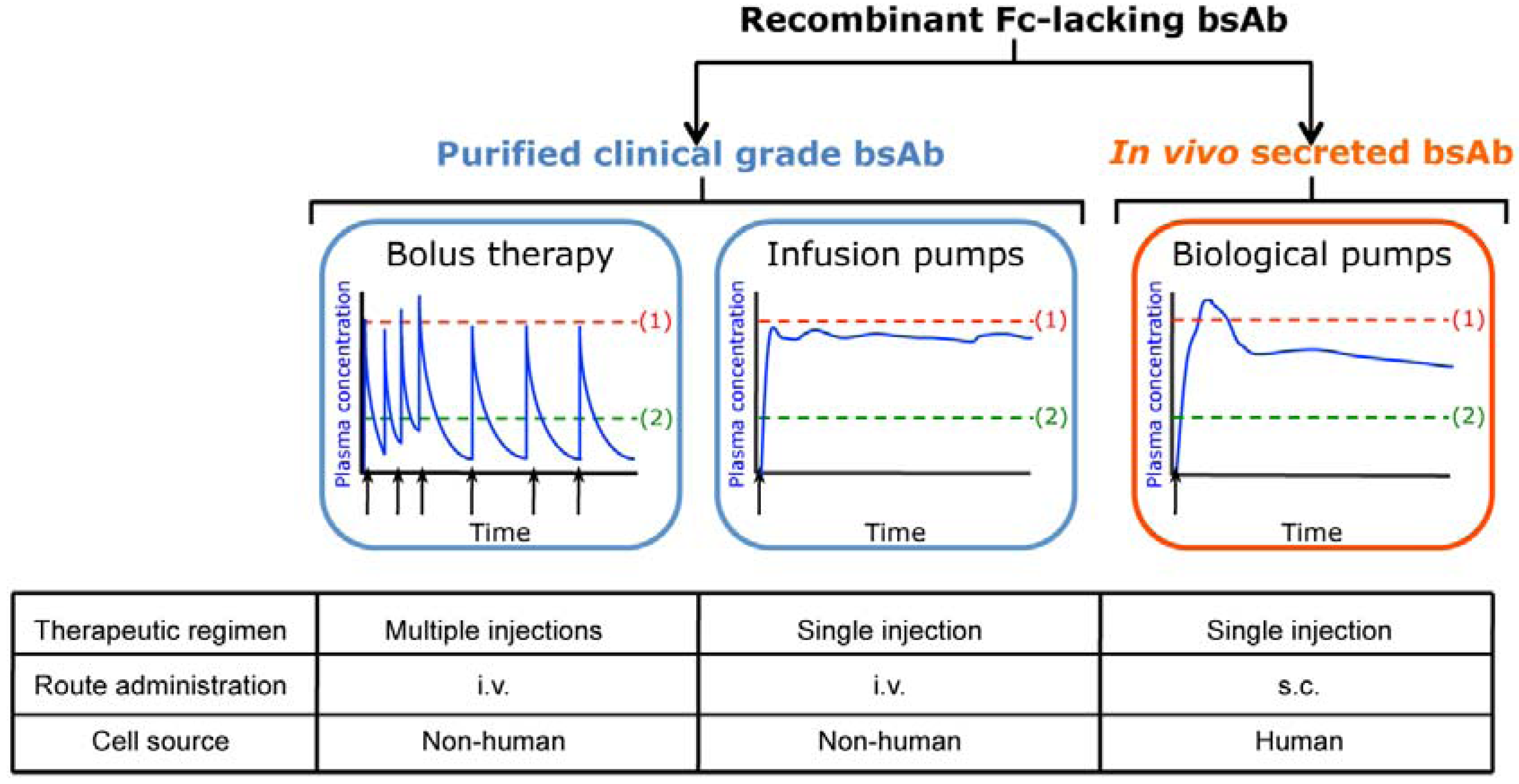
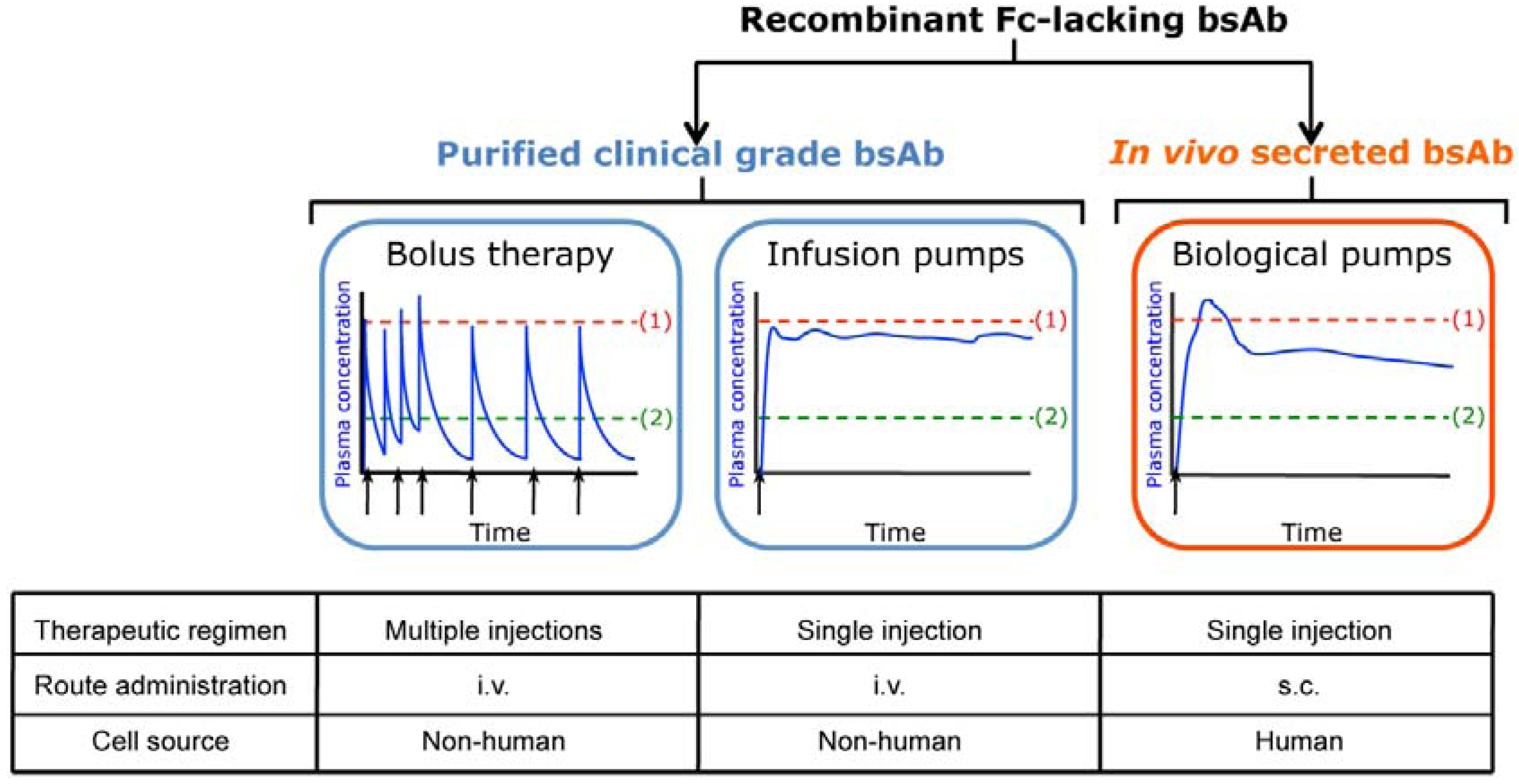
2. In Vivo Secretion of Fc-lacking Bispecific Antibodies

{kind=link}
{kind=link}
| Target antigens | Antibody format | BsAb secreting cells | Effector cells | Disease model | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Cell Vehicle | Transfer Vector | Route of Administration | Cell type | Route of Administration | ||||
| CEA x CD3 | diabody | 293T (h) | plasmid | s.c. co-implant | T cells | i.t. | colon cancer (h) HCT-116 | [23] |
| CEA x CD3 | diabody | T cells (h) | LV | s.c. co-implant | T cells | s.c. co-implant | colon cancer (h) HCT-116 | [26] |
| CEA x CD3 | diabody | MSC (h) | LV | s.c. organoid | T cells | i.v. | colon cancer (h) HCT-116 | [35] |
| HER2 x CD16 | (scFv)2 | MSC-like (m) | RV | i.v. | monocytes | i.p. | breast cancer (h) MDA-MB-453 | [42] |
| CEA x CD3 | diabody | HUVEC | LV | s.c. neovessels | T cells | i.v. | colon cancer (h) HCT-116 | [51] |
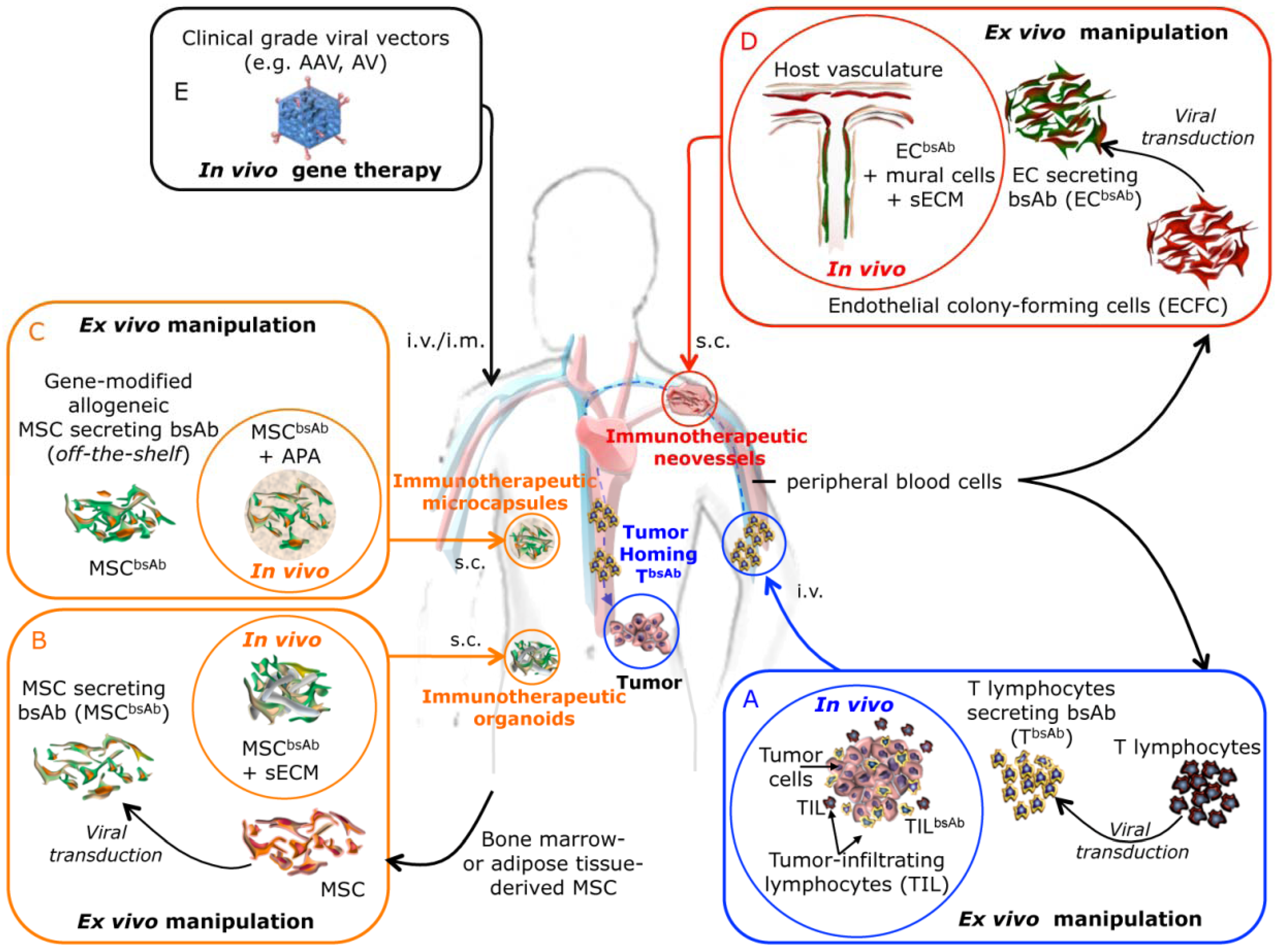
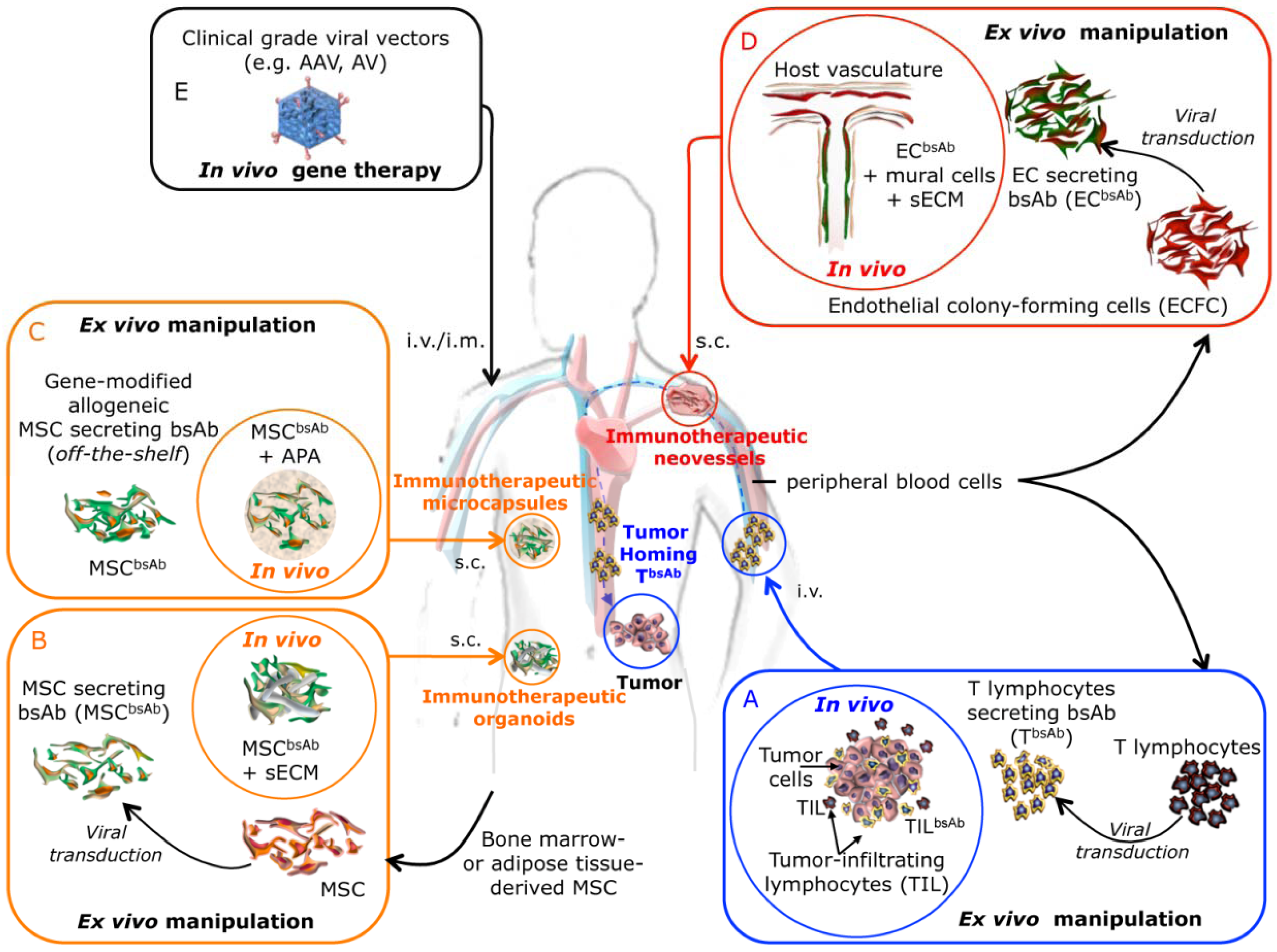
3. Future Prospects
Acknowledgments
Conflict of Interest
References
- Cuesta, A.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Álvarez-Vallina, L. Multivalent antibodies: When design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Álvarez-Vallina, L. Genetic approaches for antigen-selective cell therapy. Curr. Gene Ther. 2001, 1, 385–397. [Google Scholar] [CrossRef]
- Kontermann, R. Dual targeting strategies with bispecific antibodies. MAbs 2012, 4, 182–197. [Google Scholar] [CrossRef]
- Carter, P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 2001, 1, 118–129. [Google Scholar] [CrossRef]
- Muller, D.; Kontermann, R.E. Bispecific antibodies for cancer immunotherapy: Current perspectives. BioDrugs 2010, 24, 89–98. [Google Scholar] [CrossRef]
- Sanz, L.; Cuesta, A.M.; Compte, M.; Álvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648. [Google Scholar] [CrossRef]
- Dhimolea, E.; Reichert, J.M. World Bispecific Antibody Summit, September 27-28, 2011, Boston, MA. MAbs 2012, 4, 4–13. [Google Scholar] [CrossRef]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: clinical development and future directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef]
- Sebastian, M.; Passlick, B.; Friccius-Quecke, H.; Jager, M.; Lindhofer, H.; Kanniess, F.; Wiewrodt, R.; Thiel, E.; Buhl, R.; Schmittel, A. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM x anti-CD3): A phase I study. Cancer Immunol. Immunother. 2007, 56, 1637–1644. [Google Scholar] [CrossRef]
- Ghaderi, D.; Zhang, M.; Hurtado-Ziola, N.; Varki, A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol. Genet. Eng. Rev. 2012, 28, 147–175. [Google Scholar] [CrossRef]
- Mack, M.; Riethmuller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef]
- Holliger, P.; Winter, G. Engineering bispecific antibodies. Curr. Opin. Biotechnol. 1993, 4, 446–449. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Chames, P.; Baty, D. Bispecific antibodies for cancer therapy: The light at the end of the tunnel? MAbs 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Muller, D.; Karle, A.; Meissburger, B.; Hofig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar] [CrossRef]
- Stork, R.; Campigna, E.; Robert, B.; Muller, D.; Kontermann, R.E. Biodistribution of a bispecific single-chain diabody and its half-life extended derivatives. J. Biol. Chem. 2009, 284, 25612–25619. [Google Scholar]
- Libon, C.; Corvaia, N.; Haeuw, J.F.; Nguyen, T.N.; Stahl, S.; Bonnefoy, J.Y.; Andreoni, C. The serum albumin-binding region of streptococcal protein G (BB) potentiates the immunogenicity of the G130-230 RSV-A protein. Vaccine 1999, 17, 406–414. [Google Scholar] [CrossRef]
- Sanz, L.; Blanco, B.; Álvarez-Vallina, L. Antibodies and gene therapy: Teaching old 'magic bullets' new tricks. Trends Immunol. 2004, 25, 85–91. [Google Scholar] [CrossRef]
- Samaranayake, H.; Wirth, T.; Schenkwein, D.; Raty, J.K.; Yla-Herttuala, S. Challenges in monoclonal antibody-based therapies. Ann. Med. 2009, 41, 322–331. [Google Scholar] [CrossRef]
- Sanchez-Martin, D.; Sanz, L.; Álvarez-Vallina, L. Engineering human cells for in vivo secretion of antibody and non-antibody therapeutic proteins. Curr. Opin. Biotechnol. 2011, 22, 924–930. [Google Scholar] [CrossRef]
- Sanz, L.; Compte, M.; Guijarro-Munoz, I.; Álvarez-Vallina, L. Non-hematopoietic stem cells as factories for in vivo therapeutic protein production. Gene Ther. 2012, 19, 1–7. [Google Scholar] [CrossRef]
- Blanco, B.; Holliger, P.; Vile, R.G.; Álvarez-Vallina, L. Induction of human T lymphocyte cytotoxicity and inhibition of tumor growth by tumor-specific diabody-based molecules secreted from gene-modified bystander cells. J. Immunol. 2003, 171, 1070–1077. [Google Scholar]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Compte, M.; Blanco, B.; Serrano, F.; Cuesta, A.M.; Sanz, L.; Bernad, A.; Holliger, P.; Álvarez-Vallina, L. Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA x anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther. 2007, 14, 380–388. [Google Scholar] [CrossRef]
- Shah, K. Mesenchymal stem cells engineered for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 739–748. [Google Scholar] [CrossRef]
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef]
- Aboody, K.S.; Najbauer, J.; Danks, M.K. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008, 15, 739–752. [Google Scholar] [CrossRef]
- Frank, R.T.; Najbauer, J.; Aboody, K.S. Concise review: Stem cells as an emerging platform for antibody therapy of cancer. Stem Cells 2010, 28, 2084–2087. [Google Scholar] [CrossRef]
- Meisel, R.; Zibert, A.; Laryea, M.; Gobel, U.; Daubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef]
- Sato, K.; Ozaki, K.; Oh, I.; Meguro, A.; Hatanaka, K.; Nagai, T.; Muroi, K.; Ozawa, K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007, 109, 228–234. [Google Scholar] [CrossRef]
- Sanz, L.; Santos-Valle, P.; Alonso-Camino, V.; Salas, C.; Serrano, A.; Vicario, J.L.; Cuesta, A.M.; Compte, M.; Sanchez-Martin, D.; Álvarez-Vallina, L. Long-term in vivo imaging of human angiogenesis: critical role of bone marrow-derived mesenchymal stem cells for the generation of durable blood vessels. Microvasc. Res. 2008, 75, 308–314. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Compte, M.; Cuesta, A.M.; Sanchez-Martin, D.; onso-Camino, V.; Vicario, J.L.; Sanz, L.; Álvarez-Vallina, L. Tumor immunotherapy using gene-modified human mesenchymal stem cells loaded into synthetic extracellular matrix scaffolds. Stem Cells 2009, 27, 753–760. [Google Scholar] [CrossRef]
- Descamps, V.; Blumenfeld, N.; Perricaudet, M.; Beuzard, Y.; Kremer, E.J. Organoids direct systemic expression of erythropoietin in mice. Gene Ther. 1995, 2, 411–417. [Google Scholar]
- Eliopoulos, N.; Al-Khaldi, A.; Crosato, M.; Lachapelle, K.; Galipeau, J. A neovascularized organoid derived from retrovirally engineered bone marrow stroma leads to prolonged in vivo systemic delivery of erythropoietin in nonmyeloablated, immunocompetent mice. Gene Ther. 2003, 10, 478–489. [Google Scholar] [CrossRef]
- Stagg, J.; Lejeune, L.; Paquin, A.; Galipeau, J. Marrow stromal cells for interleukin-2 delivery in cancer immunotherapy. Hum. Gene Ther. 2004, 15, 597–608. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Francois, M.; Boivin, M.N.; Martineau, D.; Galipeau, J. Neo-organoid of marrow mesenchymal stromal cells secreting interleukin-12 for breast cancer therapy. Cancer Res. 2008, 68, 4810–4818. [Google Scholar]
- Yokoo, T.; Fukui, A.; Matsumoto, K.; Ohashi, T.; Sado, Y.; Suzuki, H.; Kawamura, T.; Okabe, M.; Hosoya, T.; Kobayashi, E. Generation of a transplantable erythropoietin-producer derived from human mesenchymal stem cells. Transplantation 2008, 85, 1654–1658. [Google Scholar] [CrossRef]
- Wang, N.; Fallavollita, L.; Nguyen, L.; Burnier, J.; Rafei, M.; Galipeau, J.; Yakar, S.; Brodt, P. Autologous bone marrow stromal cells genetically engineered to secrete an igf-I receptor decoy prevent the growth of liver metastases. Mol. Ther. 2009, 17, 1241–1249. [Google Scholar] [CrossRef]
- Kasuya, K.; Shimazu, M.; Suzuki, M.; Itoi, T.; Aoki, T.; Tsuchida, A. Bispecific anti-HER2 and CD16 single-chain antibody production prolongs the use of stem cell-like cell transplantation against HER2-overexpressing cancer. Int. J. Mol. Med. 2010, 25, 209–215. [Google Scholar]
- Squinto, S.P.; Madri, J.A.; Kennedy, S.; Springhorn, J. The ENCEL system: A somatic cell protein delivery system. In Vivo 1994, 8, 771–780. [Google Scholar]
- Wei, Y.; Li, J.; Wagner, T.E. Long-term expression of human growth hormone (hGH) in mice containing allogeneic yolk sac cell derived neovascular implants expressing hGH. Stem Cells 1996, 14, 232–238. [Google Scholar]
- Matsui, H.; Shibata, M.; Brown, B.; Labelle, A.; Hegadorn, C.; Andrews, C.; Hebbel, R.P.; Galipeau, J.; Hough, C.; Lillicrap, D. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells 2007, 25, 2660–2669. [Google Scholar] [CrossRef]
- Koike, N.; Fukumura, D.; Gralla, O.; Au, P.; Schechner, J.S.; Jain, R.K. Tissue engineering: Creation of long-lasting blood vessels. Nature 2004, 428, 138–139. [Google Scholar] [CrossRef]
- Melero-Martin, J.M.; Khan, Z.A.; Picard, A.; Wu, X.; Paruchuri, S.; Bischoff, J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood 2007, 109, 4761–4768. [Google Scholar] [CrossRef]
- Melero-Martin, J.M.; De Obaldia, M.E.; Kang, S.Y.; Khan, Z.A.; Yuan, L.; Oettgen, P.; Bischoff, J. Engineering robust and functional vascular networks in vivo with human adult and cord blood-derived progenitor cells. Circ. Res. 2008, 103, 194–202. [Google Scholar] [CrossRef]
- Au, P.; Tam, J.; Fukumura, D.; Jain, R.K. Bone marrow-derived mesenchymal stem cells facilitate engineering of long-lasting functional vasculature. Blood 2008, 111, 4551–4558. [Google Scholar] [CrossRef]
- Álvarez-Vallina, L.; Sanz, L. The therapeutic potential of engineered human neovessels for cell-based gene therapy. Expert Opin. Biol. Ther. 2011, 11, 67–76. [Google Scholar] [CrossRef]
- Compte, M.; onso-Camino, V.; Santos-Valle, P.; Cuesta, A.M.; Sanchez-Martin, D.; Lopez, M.R.; Vicario, J.L.; Salas, C.; Sanz, L.; Álvarez-Vallina, L. Factory neovessels: Engineered human blood vessels secreting therapeutic proteins as a new drug delivery system. Gene Ther. 2010, 17, 745–751. [Google Scholar] [CrossRef]
- Lin, R.Z.; Dreyzin, A.; Aamodt, K.; Li, D.; Jaminet, S.C.; Dudley, A.C.; Melero-Martin, J.M. Induction of erythropoiesis using human vascular networks genetically engineered for controlled erythropoietin release. Blood 2011, 118, 5420–5428. [Google Scholar] [CrossRef]
- Lohr, M.; Hoffmeyer, A.; Kroger, J.; Freund, M.; Hain, J.; Holle, A.; Karle, P.; Knofel, W.T.; Liebe, S.; Muller, P.; et al. Microencapsulated cell-mediated treatment of inoperable pancreatic carcinoma. Lancet 2001, 357, 1591–1592. [Google Scholar] [CrossRef]
- Cirone, P.; Bourgeois, J.M.; Chang, P.L. Antiangiogenic cancer therapy with microencapsulated cells. Hum. Gene Ther. 2003, 14, 1065–1077. [Google Scholar] [CrossRef]
- Goren, A.; Dahan, N.; Goren, E.; Baruch, L.; Machluf, M. Encapsulated human mesenchymal stem cells: A unique hypoimmunogenic platform for long-term cellular therapy. FASEB J. 2010, 24, 22–31. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Compte, M.; Nuñez-Prado, N.; Sanz, L.; Alvarez-Vallina, L. In Vivo Secretion of Bispecific Antibodies Recruiting Lymphocytic Effector Cells. Antibodies 2013, 2, 415-425. https://doi.org/10.3390/antib2030415
Compte M, Nuñez-Prado N, Sanz L, Alvarez-Vallina L. In Vivo Secretion of Bispecific Antibodies Recruiting Lymphocytic Effector Cells. Antibodies. 2013; 2(3):415-425. https://doi.org/10.3390/antib2030415
Chicago/Turabian StyleCompte, Marta, Natalia Nuñez-Prado, Laura Sanz, and Luis Alvarez-Vallina. 2013. "In Vivo Secretion of Bispecific Antibodies Recruiting Lymphocytic Effector Cells" Antibodies 2, no. 3: 415-425. https://doi.org/10.3390/antib2030415