
Both Tumor Necrosis Factor Receptor Signaling Pathways Contribute to Mortality but not to Splenomegaly in Generalized Lymphoproliferative Disorder


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
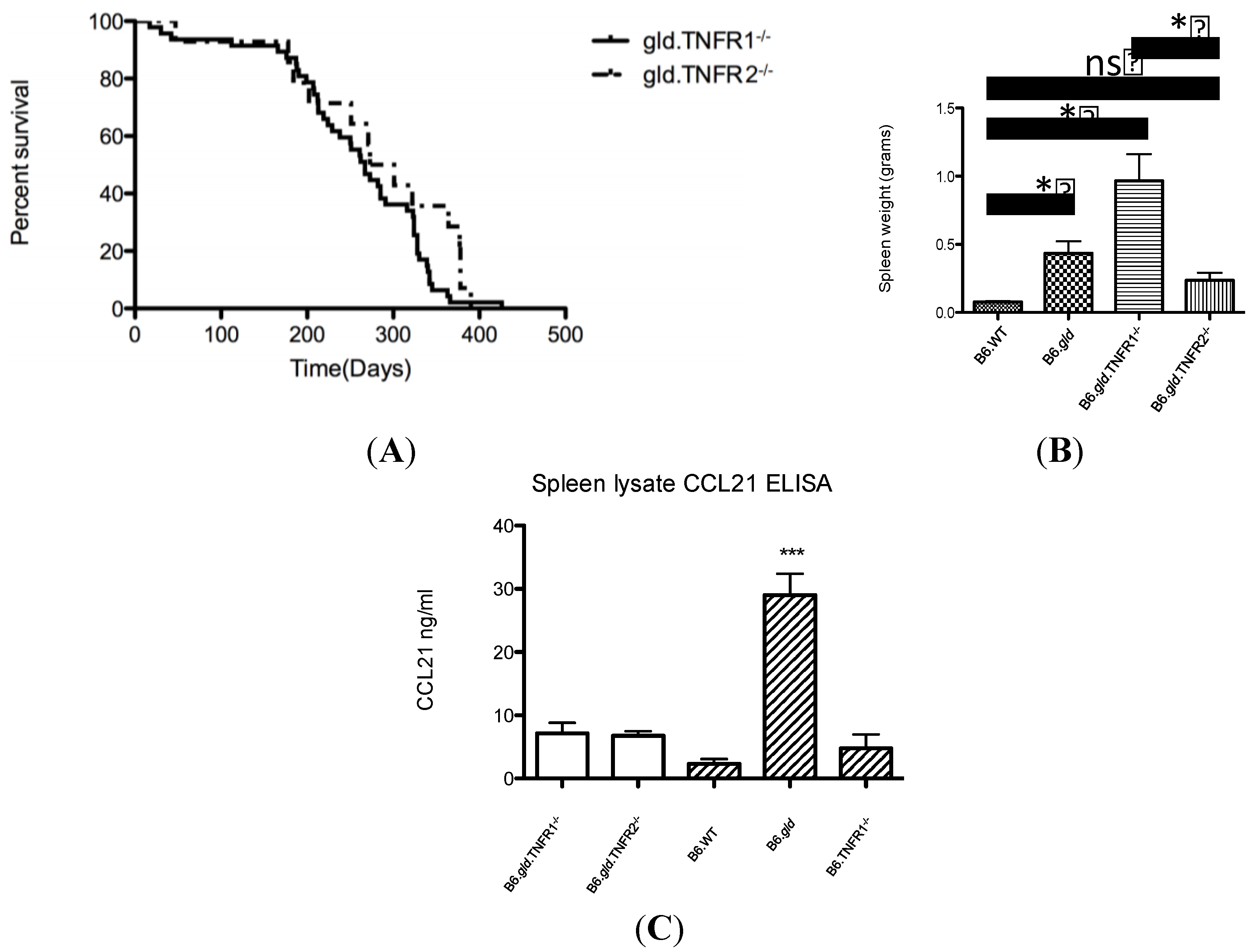
2.1. Comparison of the Overall Phenotype of B6.gld.TNFR1−/− and B6.gld.TNFR2−/− Mice

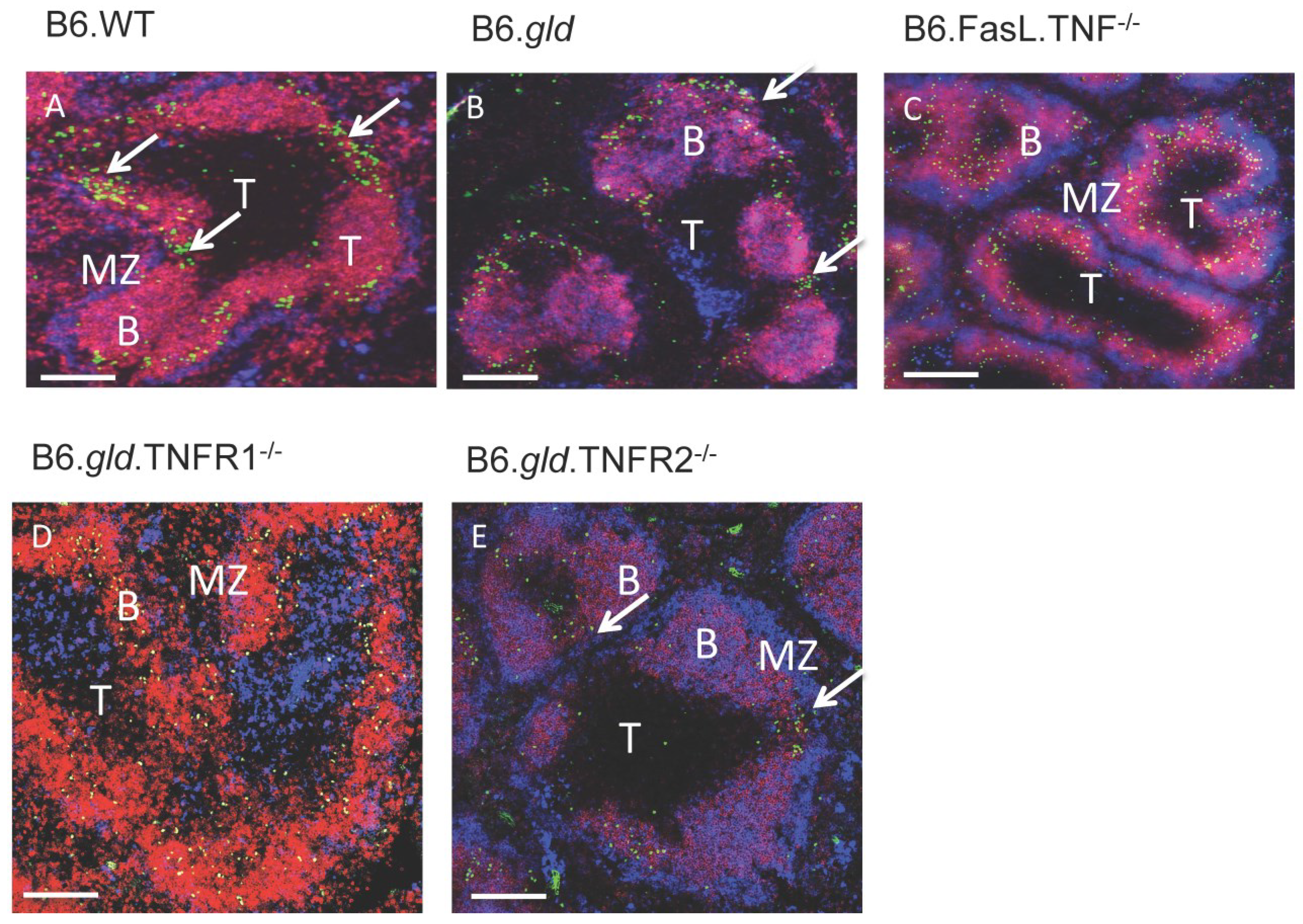
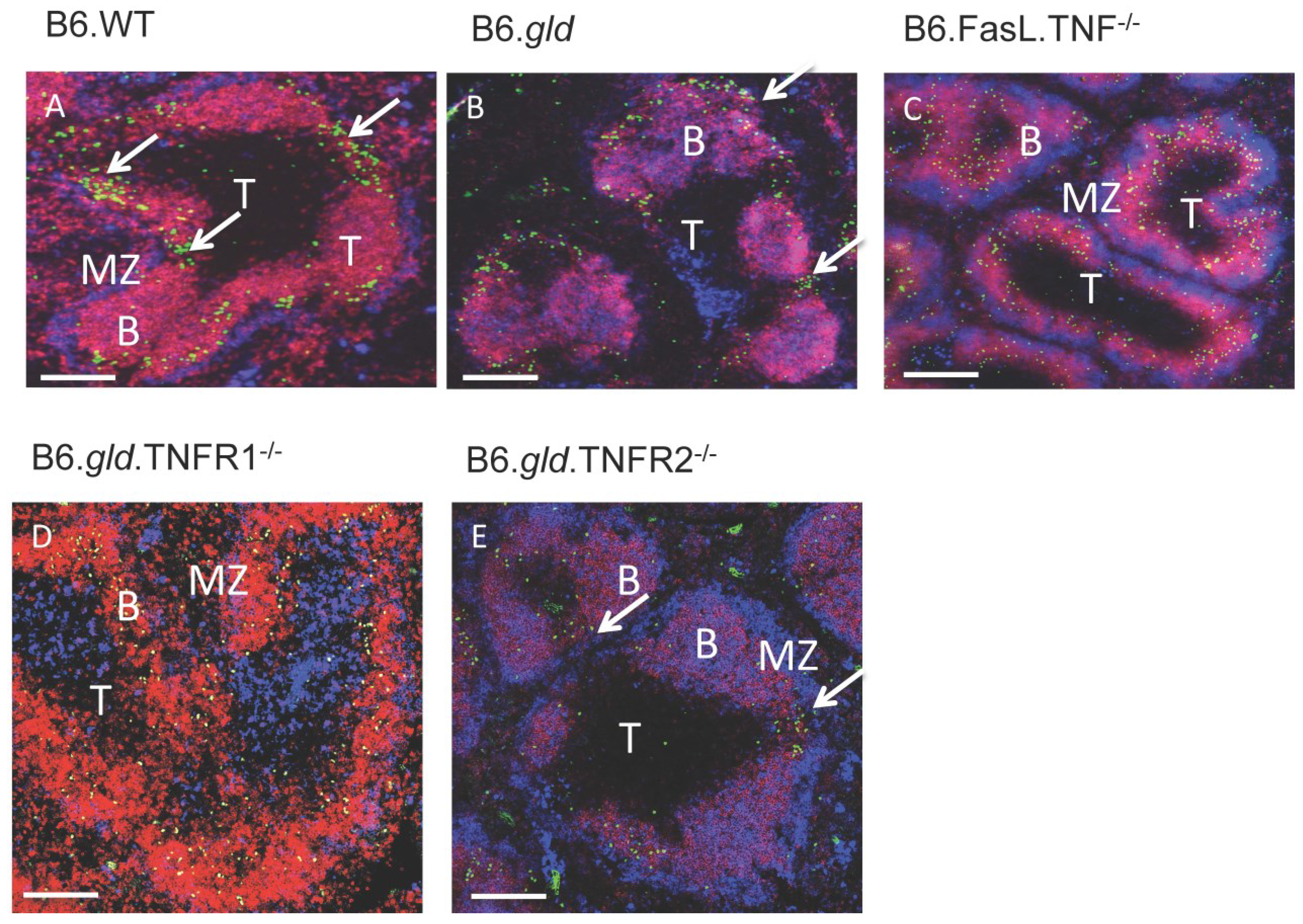
2.2. Transfer of CD4−/CD8− DN T Cells into TNFR Double Knockouts Follows the Respective TNFR Phenotype and Results in Migration to the Bridging Channels or Ends in the B Cell Area
3. Experimental Section
3.1. Mouse Strains
3.2. Ethics Statement
3.3. CCL21 Elisa
3.4. Fluorescence Immunohistology
3.5. Flow Cytometry
3.6. Adoptive Cell Transfer
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Strasser, A.; Jost, P.J.; Nagata, S. The Many Roles of FAS Receptor Signaling in the Immune System. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Thompson, C.B. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 2002, 109, S97–S107. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Eisenberg, R.A. The lpr and gld genes in systemic autoimmunity: Life and death in the Fas lane. Immunol. Today 1992, 13, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.H.; Ramsdell, F.; Alderson, M.R. Fas and FasL in the homeostatic regulation of immune responses. Immunol. Today 1995, 16, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Fas and Fas ligand: A death factor and its receptor. Adv. Immunol. 1994, 57, 129–144. [Google Scholar] [PubMed]
- Pisetsky, D.S.; Caster, S.A.; Roths, J.B.; Murphy, E.D. Ipr gene control of the anti-DNA antibody response. J. Immunol. 1982, 128, 2322–2325. [Google Scholar] [PubMed]
- Roths, J.B.; Murphy, E.D.; Eicher, E.M. A new mutation, gld, that produces lymphoproliferation and autoimmunity in C3H/HeJ mice. J. Exp. Med. 1984, 159, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Rothe, M.; Hu, Y.F.; Goeddel, D.V. Tumor necrosis factor’s cytotoxic activity is signaled by the p55 TNF receptor. Cell 1993, 73, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Edwards, C.K., 3rd; Yang, P.; Wang, Z.; Bluethmann, H.; Mountz, J.D. Greatly accelerated lymphadenopathy and autoimmune disease in lpr mice lacking tumor necrosis factor receptor I. J. Immunol. 1996, 156, 2661–2665. [Google Scholar] [PubMed]
- Körner, H.; Cretney, E.; Wilhelm, P.; Kelly, J.M.; Röllinghoff, M.; Sedgwick, J.D.; Smyth, M.J. Tumor necrosis factor sustains the generalized lymphoproliferative disorder (gld) phenotype. J. Exp. Med. 2000, 191, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Vana, K.; Sedger, L.M.; Lechner, A.; Körner, H. TNF-dependent overexpression of CCL21 is an underlying cause of progressive lymphoaccumulation in generalized lymphoproliferative disorder. Eur. J. Immunol. 2007, 37, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Roomberg, A.; Cretney, E.; Lechner, A.; Fromm, P.; Wren, L.; Smyth, M.J.; Korner, H. Age-dependent, polyclonal hyperactivation of T cells is reduced in TNF-negative gld/gld mice. J. Leukoc. Biol. 2009, 85, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J.; Torrance, D.S.; Stocking, K.L.; Glaccum, M.B.; Otten, C.; Willis, C.R.; Charrier, K.; Morrissey, P.J.; Ware, C.B.; Mohler, K.M. TNF Receptor-Deficient Mice Reveal Divergent Roles for p55 and p75 in Several Models of Inflammation. J. Immunol. 1998, 160, 943–952. [Google Scholar] [PubMed]
- Davidson, W.F.; Calkins, C.; Hugins, A.; Giese, T.; Holmes, K.L. Cytokine secretion by C3H-lpr and -gld T cells. Hypersecretion of IFN-gamma and tumor necrosis factor-alpha by stimulated CD4+ T cells. J. Immunol. 1991, 146, 4138–4148. [Google Scholar] [PubMed]
- Körner, H.; McMorran, B.; Schluter, D.; Fromm, P. The role of TNF in parasitic diseases: Still more questions than answers. Int. J. Parasitol. 2010, 40, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Efimov, G.A.; Kruglov, A.A.; Tillib, S.V.; Kuprash, D.V.; Nedospasov, S.A. Tumor Necrosis Factor and the consequences of its ablation in vivo. Mol. Immunol. 2009, 47, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. TNF-alpha: An activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr. Dir. Autoimmun. 2010, 11, 119–134. [Google Scholar] [PubMed]
- Andrews, B.S.; Eisenberg, R.A.; Theofilopoulos, A.N.; Izui, S.; Wilson, C.B.; McConahey, P.J.; Murphy, E.D.; Roths, J.B.; Dixon, F.J. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978, 148, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Dixon, F.J.; Andrews, B.S.; Eisenberg, R.A.; McConahey, P.J.; Theofilopoulos, A.N.; Wilson, C.B. Etiology and pathogenesis of a spontaneous lupus-like syndrome in mice. Arthrits Rheum. 1978, 21, S64–S67. [Google Scholar] [CrossRef]
- Teh, H.-S.; Seebaran, A.; Teh, S.-J. TNF Receptor 2-Deficient CD8 T Cells Are Resistant to Fas/Fas Ligand-Induced Cell Death. J. Immunol. 2000, 165, 4814–4821. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Baltimore, D. TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-kappaB activation. Genes Dev. 1996, 10, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar]
- Körner, H.; Cook, M.; Riminton, D.S.; Lemckert, F.A.; Hoek, R.M.; Ledermann, B.; Köntgen, F.; Fazekas de St Groth, B.; Sedgwick, J.D. Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 1997, 27, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Alexopoulou, L.; Grell, M.; Pfizenmaier, K.; Bluethmann, H.; Kollias, G. Peyer’s patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 6319–6323. [Google Scholar] [CrossRef] [PubMed]
- Körner, H.; Winkler, T.H.; Sedgwick, J.D.; Röllinghoff, M.; Basten, A.; Cook, M.C. Recirculating and marginal zone B cell populations can be established and maintained independently of primary and secondary follicles. Immunol. Cell Biol. 2001, 79, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Tomooka, S.; Serushago, B.A.; Himeno, K.; Nomoto, K. A new T cell subset expressing B220 and CD4 in lpr mice: Defects in the response to mitogens and in the production of IL-2. Clin. Exp. Immunol. 1988, 74, 36–40. [Google Scholar] [PubMed]
- Ansel, K.M.; McHeyzer-Williams, L.J.; Ngo, V.N.; McHeyzer-Williams, M.G.; Cyster, J.G. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J. Exp. Med. 1999, 1123–1134. [Google Scholar]
- Hoek, R.C.; Kortekaas, M.C.; Sedgwick, J.D. Allele-specific PCR analysis for detection of the gld Fas-ligand point mutation. J. Immunol. Methods 1997, 210, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Ritter, U.; Wiede, F.; Mielenz, D.; Kiafard, Z.; Zwirner, J.; Körner, H. Analysis of the CCR7 expression on murine bone marrow-derived and spleen dendritic cells. J. Leukoc. Biol. 2004, 76, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, P.D.; Lee, J.H.; Lyons, A.B. B cell differentiation and isotype switching is related to division cycle number. J. Exp. Med. 1996, 184, 277–281. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiede, F.; Roomberg, A.; Darby, J.; Gollan, R.; Körner, H. Both Tumor Necrosis Factor Receptor Signaling Pathways Contribute to Mortality but not to Splenomegaly in Generalized Lymphoproliferative Disorder. Antibodies 2015, 4, 1-10. https://doi.org/10.3390/antib4010001
Wiede F, Roomberg A, Darby J, Gollan R, Körner H. Both Tumor Necrosis Factor Receptor Signaling Pathways Contribute to Mortality but not to Splenomegaly in Generalized Lymphoproliferative Disorder. Antibodies. 2015; 4(1):1-10. https://doi.org/10.3390/antib4010001
Chicago/Turabian StyleWiede, Florian, Alicia Roomberg, Jocelyn Darby, Rene Gollan, and Heinrich Körner. 2015. "Both Tumor Necrosis Factor Receptor Signaling Pathways Contribute to Mortality but not to Splenomegaly in Generalized Lymphoproliferative Disorder" Antibodies 4, no. 1: 1-10. https://doi.org/10.3390/antib4010001