
TNFR1 and TNFR2 Expression and Induction on Human Treg Cells from Type 1 Diabetic Subjects

Abstract
:1. Introduction
1.1. Supporting Evidence That TNF is Deficient in Many Autoimmune Diseases
1.2. What Could Be The Reason for Low TNF in Autoimmunity?
1.3. Value of Tregs for the Prevention of Autoimmunity
2. Results and Discussion
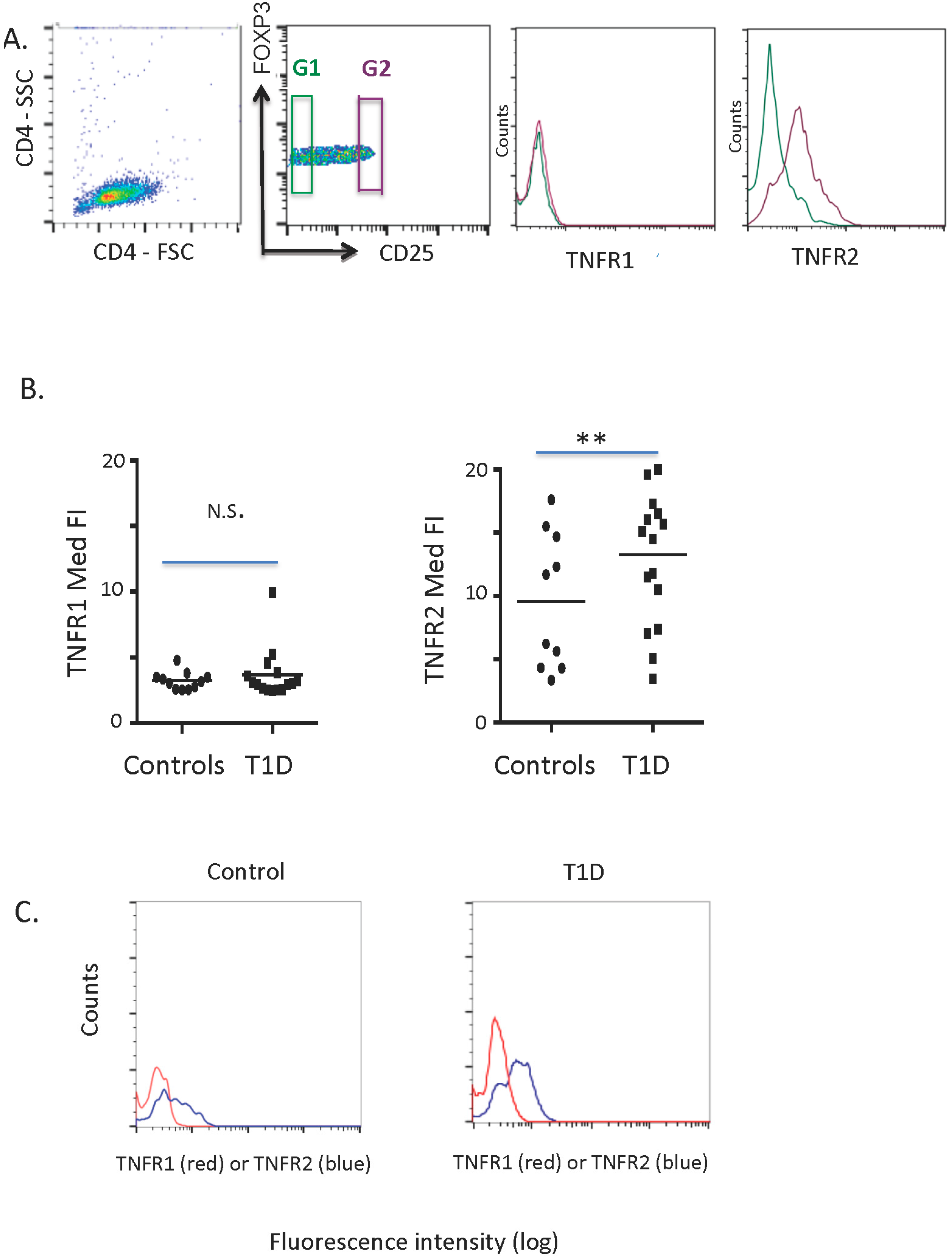
2.1. TNFR1 and TNFR2 Expression on Normal and Diabetic Subject Treg Cells
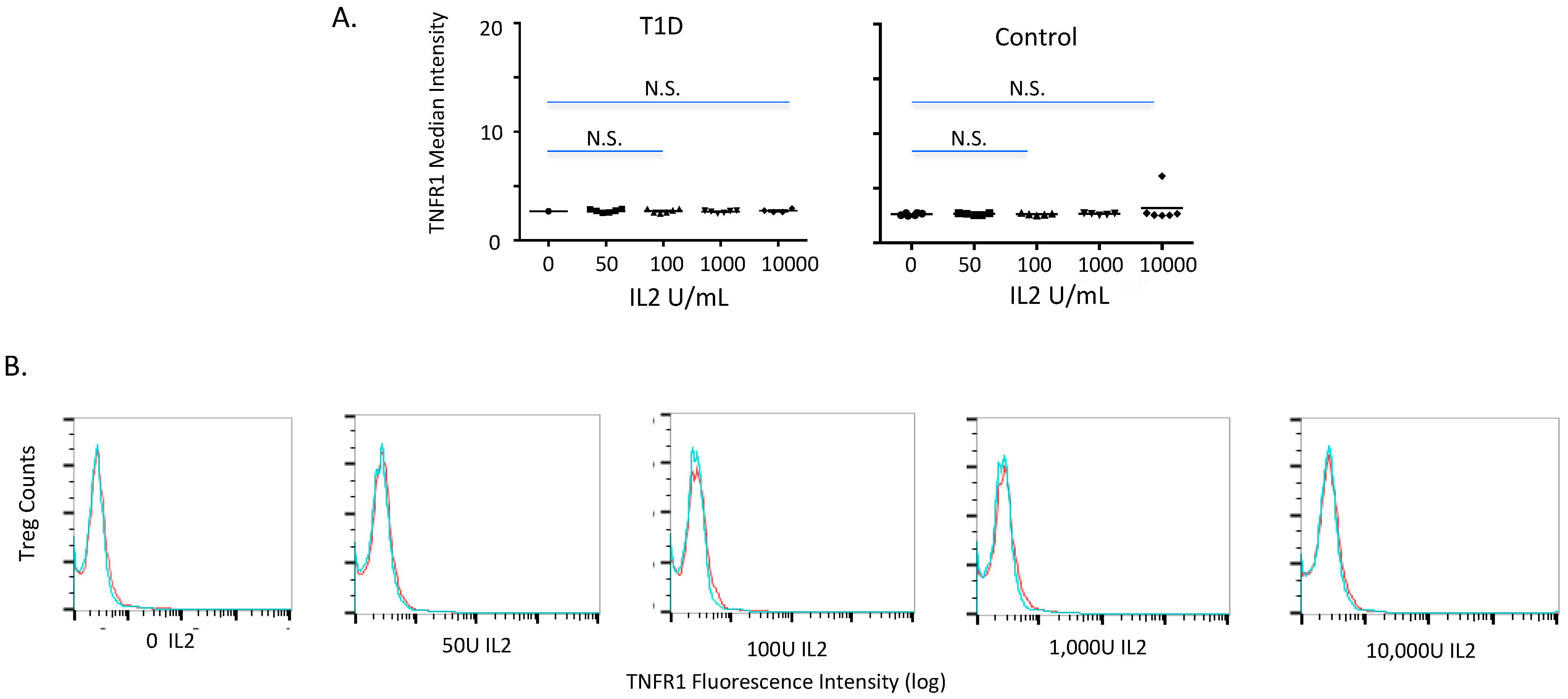
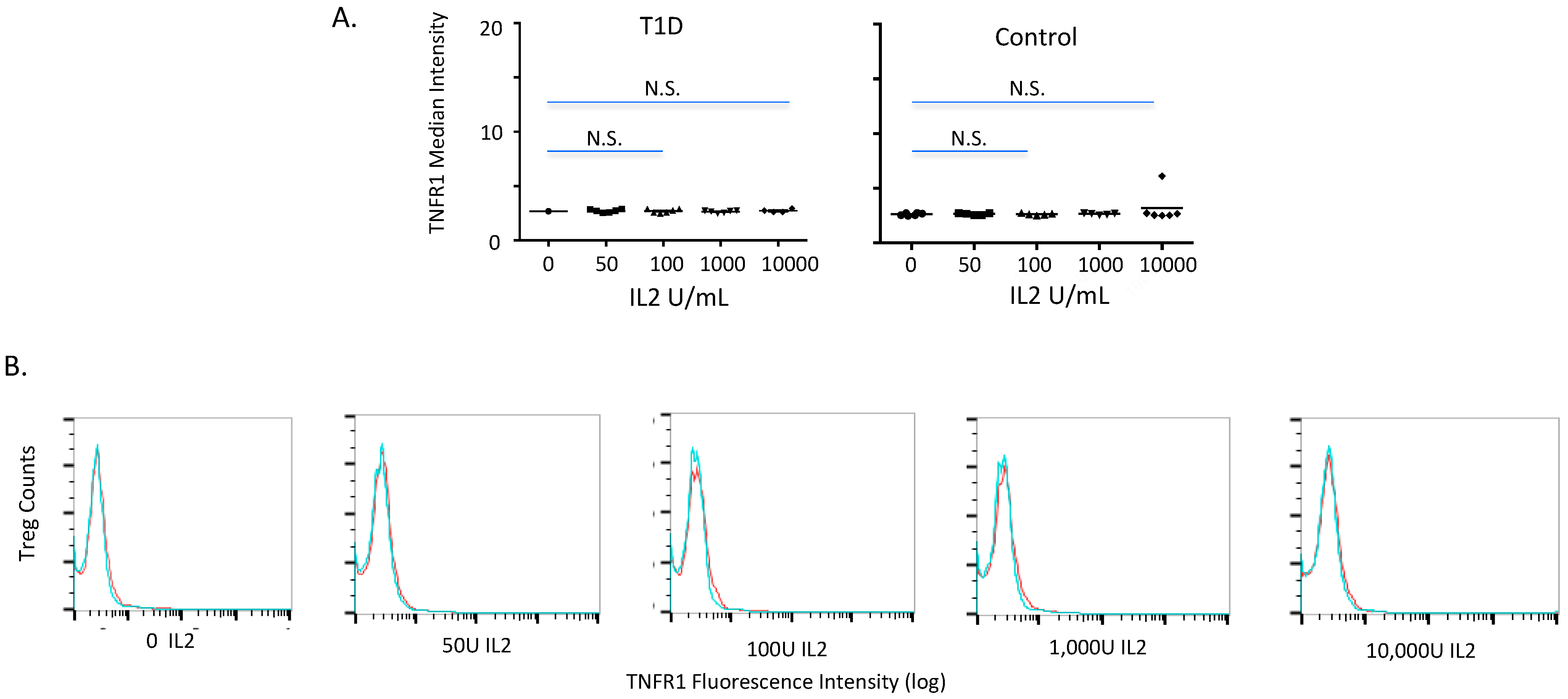
2.2. TNFR1 Expression on Normal and Diabetic Subject Treg Cells after IL2 Exposures
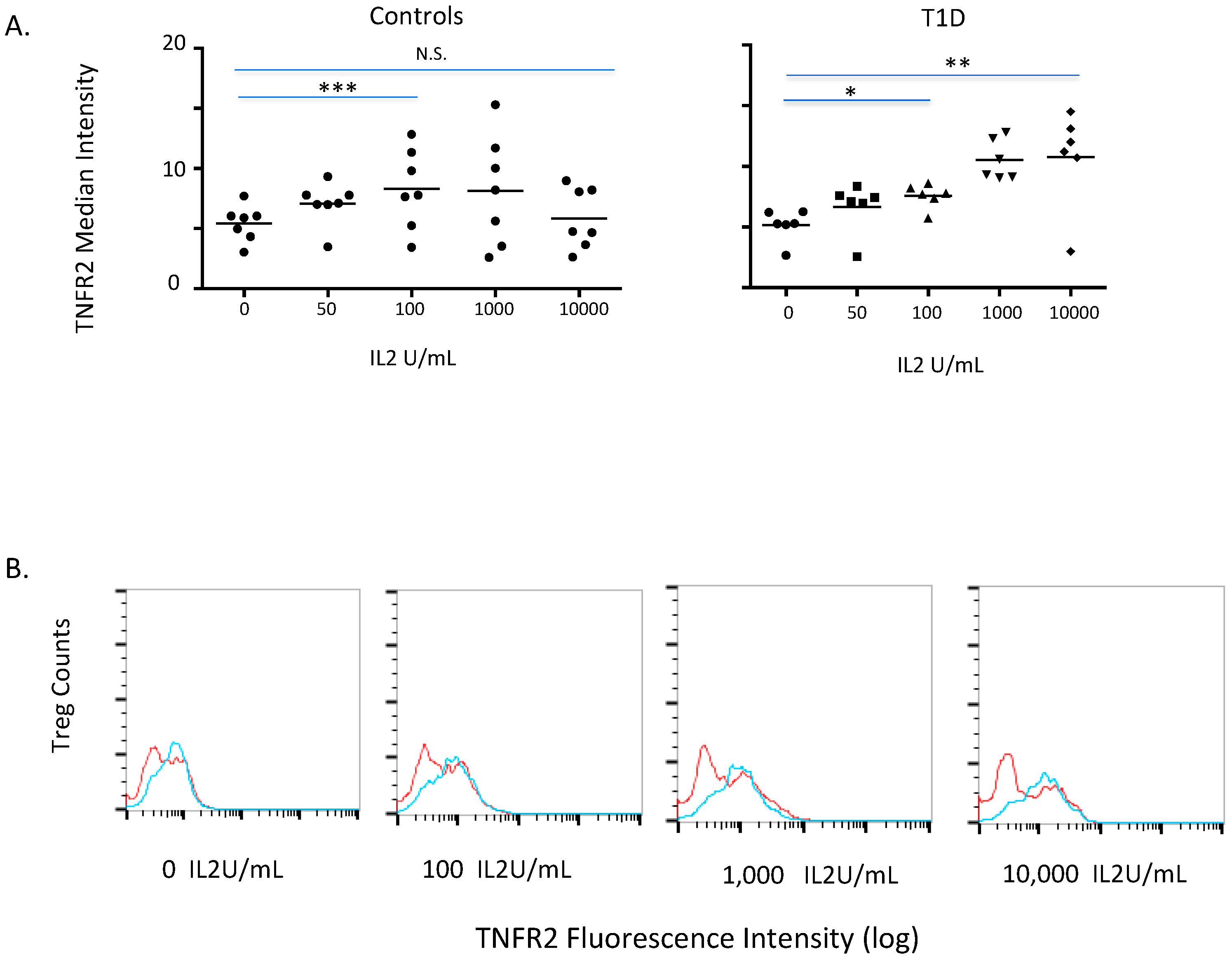
2.3. TNFR2 Expression on Normal and Diabetic Subject Treg Cells after IL2 Exposures
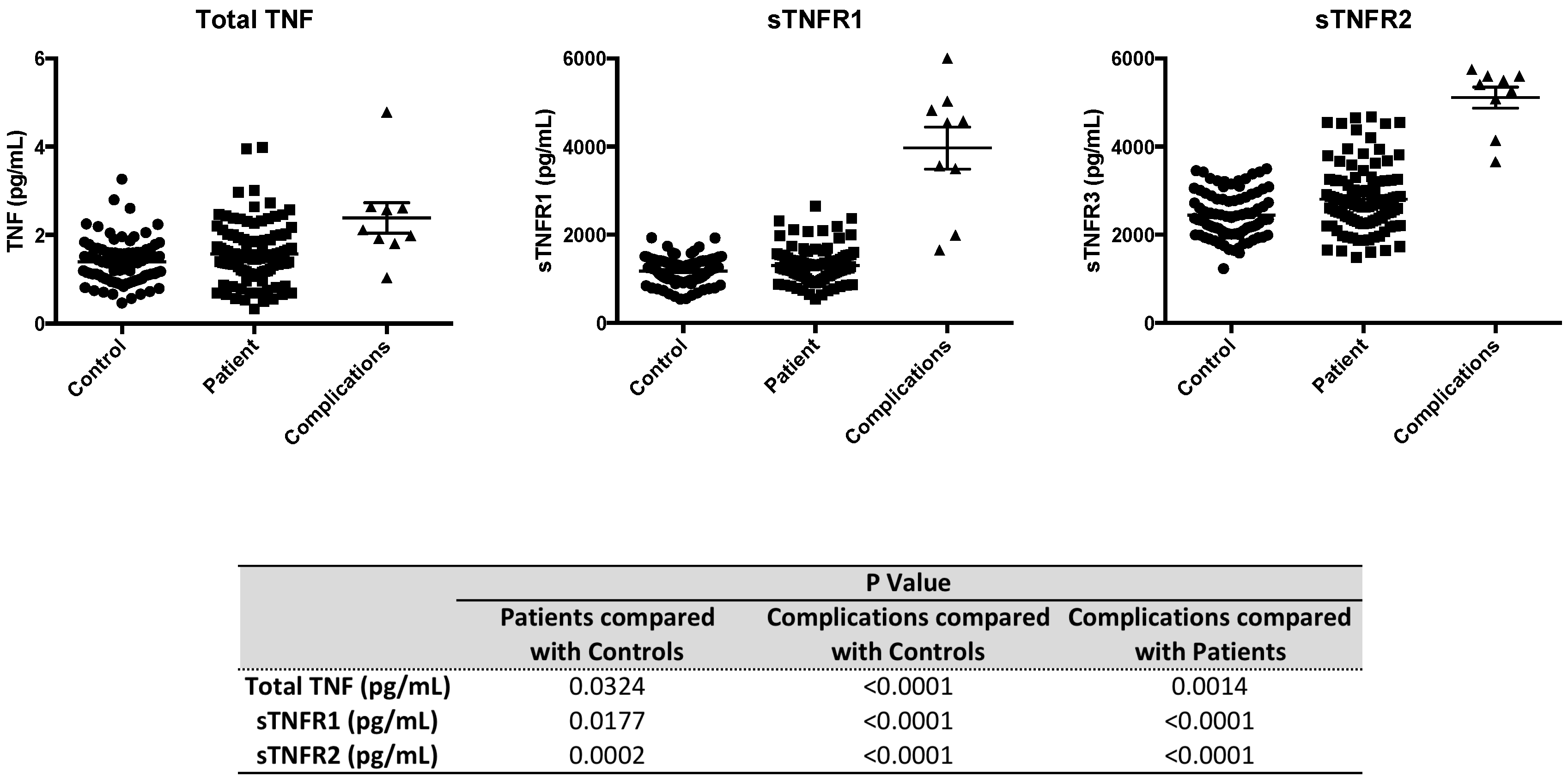
2.4. Serum Levels of sTNFR2 are Significantly Elevated in Type 1 Diabetic Subjects

{kind=link}
{kind=link}
{kind=link}
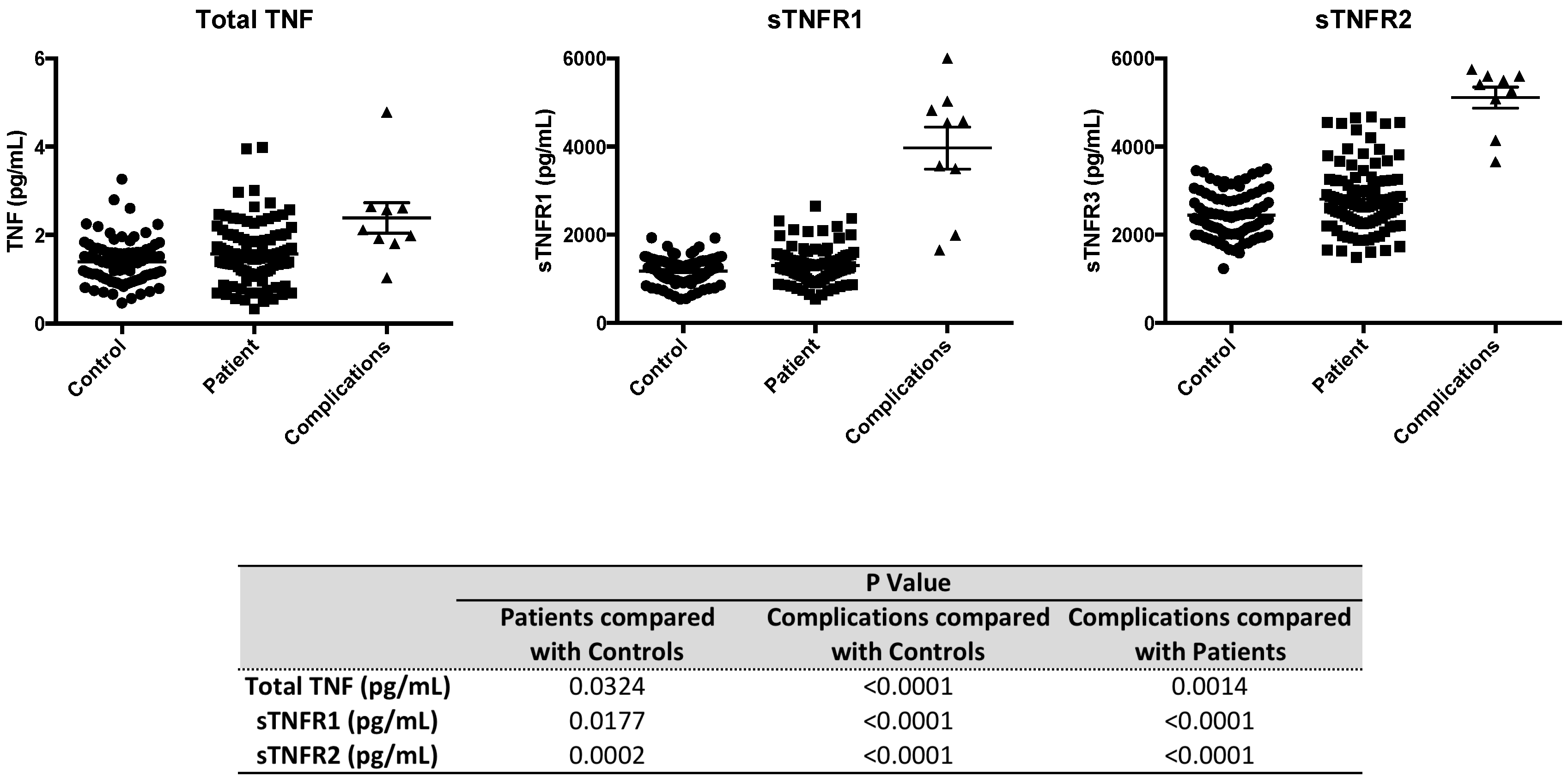{kind=link}
| A. | Study Subjects | ||||||
|---|---|---|---|---|---|---|---|
| Type 1 Diabetics | Controls | ||||||
| N = 15 | N = 12 | ||||||
| Age (yrs) | 36 ± 5 | 46 ± 8 | |||||
| Gender (M/F) | 7/8 | 6/6 | |||||
| Duration (yr) | 20 ± 4 | -- | |||||
| Age of Onset (yr) | 12 ± 5 | -- | |||||
| B. | Study Subjects | ||||||
| Controls | Type 1 Diabetics | Complications | |||||
| N = 63 | N = 63 | N = 6 | |||||
| Age (yrs) | 43 ± 13 | 36 ± 2 | 48 ± 5 | ||||
| Gender (M/F) | 30/33 | 29/34 | 3/3 | ||||
| Duration (yrs) | -- | 17 ± 2 | 34 ± 4 | ||||
| Age of Onset (yrs) | -- | 25 ± 2 | 20 ± 4 | ||||
3. Experimental Section
3.1. Human Subjects and Fresh Blood Procedures
3.2. Blood Preparation and CD4+ T Cell Isolations
3.3. TNFR2, Foxp3 and CD25 Density and Cell Numbers on Human T Regulatory Cells
3.4. Induction Experiments with IL2 to Assess Changes in TNFR1 or TNFR2 Expression on Tregs
3.5. Measurement of Serum TNFR1, Serum TNFR2 and TNF Levels in Human Serum
3.6. Statistical and Analysis Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ban, L.; Zhang, J.; Wang, L.; Kuhtreiber, W.; Burger, D.; Faustman, D.L. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc. Natl. Acad. Sci. USA 2008, 105, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.L.; Davis, M. TNF Receptor 2 and Disease: Autoimmunity and Regenerative Medicine. Front. Immunol. 2013, 4, 478. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Davis, M.; Faustman, D.L. The therapeutic potential of tumor necrosis factor for autoimmune disease: A mechanistically based hypothesis. Cell Mol. Life Sci. 2005, 62, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; von Blomberg, B.M.; Woody, J.N.; Hartung, H.P.; Polman, C.H. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar]
- The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465. [Google Scholar]
- Enayati, P.J.; Papadakis, K.A. Association of Anti-tumor Necrosis Factor Therapy with the Development of Multiple Sclerosis. J. Clin. Gastroenterol. 2005, 39, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.W., Jr.; Weinshenker, B.G.; Sandborn, W.J. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn's disease. Inflamm. Bowel Dis. 2004, 10, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Noman, M.; Van Assche, G.; Baert, F.; Van Steen, K.; Esters, N.; Joossens, S.; Bossuyt, X.; Rutgeerts, P. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn's disease: A prospective cohort study. Gastroenterology 2003, 125, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Klinkhoff, A. Biological agents for rheumatoid arthritis: Targeting both physical function and structural damage. Drugs 2004, 64, 1267–1283. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996, 14, 397–440. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Update on the TNF-alpha blocking agents. Available online: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3930B1_01_B-TNF.briefing.pdf (accessed on 1 August 2009).
- Lipsky, P.E.; van der Heijde, D.M.; St Clair, E.W.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef]
- Moreland, L.W.; Schiff, M.H.; Baumgartner, S.W.; Tindall, E.A.; Fleischmann, R.M.; Bulpitt, K.J.; Weaver, A.L.; Keystone, E.C.; Furst, D.E.; Mease, P.J.; et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann. Intern. Med. 1999, 130, 478–486. [Google Scholar] [CrossRef]
- Bleumink, G.S.; ter Borg, E.J.; Ramselaar, C.G.; Ch Stricker, B.H. Etanercept-induced subacute cutaneous lupus erythematosus. Rheumatology (Oxford) 2001, 40, 1317–1319. [Google Scholar] [CrossRef]
- Cairns, A.P.; Duncan, M.K.; Hinder, A.E.; Taggart, A.J. New onset systemic lupus erythematosus in a patient receiving etanercept for rheumatoid arthritis. Ann. Rheum. Dis. 2002, 61, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Bloom, B.J. Development of diabetes mellitus during etanercept therapy in a child with systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 2000, 43, 2606–2308. [Google Scholar] [CrossRef] [PubMed]
- Aderka, D.; Wysenbeek, A.; Engelmann, H.; Cope, A.P.; Brennan, F.; Molad, Y.; Hornik, V.; Levo, Y.; Maini, R.N.; Feldmann, M.; et al. Correlation between serum levels of soluble tumor necrosis factor receptor and disease activity in systemic lupus erythematosus. Arthritis Rheum. 1993, 36, 1111–1120. [Google Scholar] [CrossRef]
- Gabay, C.; Cakir, N.; Moral, F.; Roux-Lombard, P.; Meyer, O.; Dayer, J.M.; Vischer, T.; Yazici, H.; Guerne, P.A. Circulating levels of tumor necrosis factor soluble receptors in systemic lupus erythematosus are significantly higher than in other rheumatic diseases and correlate with disease activity. J. Rheumatol. 1997, 24, 303–308. [Google Scholar] [PubMed]
- Cope, A.P.; Aderka, D.; Doherty, M.; Engelmann, H.; Gibbons, D.; Jones, A.C.; Brennan, F.M.; Maini, R.N.; Wallach, D.; Feldmann, M. Increased levels of soluble tumor necrosis factor receptors in the sera and synovial fluid of patients with rheumatic diseases. Arthritis Rheum. 1992, 35, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. The genetic association of variants in CD6, TNFRSF1A and IRF8 to multiple sclerosis: A multicenter case-control study. PLoS One 2011, 6, e18813. [Google Scholar]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Wing, K.; Miyara, M. Regulatory T cells—A brief history and perspective. Eur. J. Immunol. 2007, 37 (Suppl. 1), S116–S123. [Google Scholar] [CrossRef]
- Okubo, Y.; Mera, T.; Wang, L.; Faustman, D.L. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci. Rep. 2013, 3, 3153. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Lazzeri, E.; Manetti, R.; Vanini, V.; Romagnani, P.; Maggi, E.; Romagnani, S. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J. Exp. Med. 2002, 196, 379–387. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.J.; Scherer, H.U.; Hameetman, M.; Morgan, M.E.; Flierman, R.; Huizinga, T.W.; Toes, R.E. Cutting edge: TNFR-shedding by CD4+CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J. Immunol. 2008, 180, 2747–2751. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C. Compromised function of regulatroy T cells in rheuymatoid arthritis and reversal by anti-TNF therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, I.; Lindley, K.J.; Londei, M.; Quaratino, S. Anti-TNF therapy increases the nubmer of Foxp3 regulatory T cells in chuildren affected by Crohn's disease. Immunology 2008, 125, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Gambineri, E.; Torgerson, T.R. IPEX, FOXP3 and regulatory T-cells: A model for autoimmunity. Immunol. Res. 2007, 38, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Ottoboni, L.; Frohlich, I.Y.; Lee, M.; Healy, B.C.; Keenan, B.T.; Xia, Z.; Chitnis, T.; Guttmann, C.R.; Khoury, S.J.; Weiner, H.L.; et al. Clinical relevance and functional consequences of the TNFRSF1A multiple sclerosis locus. Neurology 2013, 81, 1891–1899. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinrich, M.; Burger, D.; Wang, L.; Tahhan, G.; Reinhold, P., III; Zhao, M.; Hsu, E.; Warden, S.; Baum, D.; Faustman, D.L. TNFR1 and TNFR2 Expression and Induction on Human Treg Cells from Type 1 Diabetic Subjects. Antibodies 2015, 4, 34-47. https://doi.org/10.3390/antib4010034
Heinrich M, Burger D, Wang L, Tahhan G, Reinhold P III, Zhao M, Hsu E, Warden S, Baum D, Faustman DL. TNFR1 and TNFR2 Expression and Induction on Human Treg Cells from Type 1 Diabetic Subjects. Antibodies. 2015; 4(1):34-47. https://doi.org/10.3390/antib4010034
Chicago/Turabian StyleHeinrich, Melanie, Douglas Burger, Limei Wang, Georges Tahhan, Peter Reinhold, III, Menghan Zhao, Elise Hsu, Sarah Warden, Danielle Baum, and Denise L Faustman. 2015. "TNFR1 and TNFR2 Expression and Induction on Human Treg Cells from Type 1 Diabetic Subjects" Antibodies 4, no. 1: 34-47. https://doi.org/10.3390/antib4010034