
Dimerized Domain V of Beta2-Glycoprotein I Is Sufficient to Upregulate Procoagulant Activity in PMA-Treated U937 Monocytes and Require Intact Residues in Two Phospholipid-Binding Loops

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
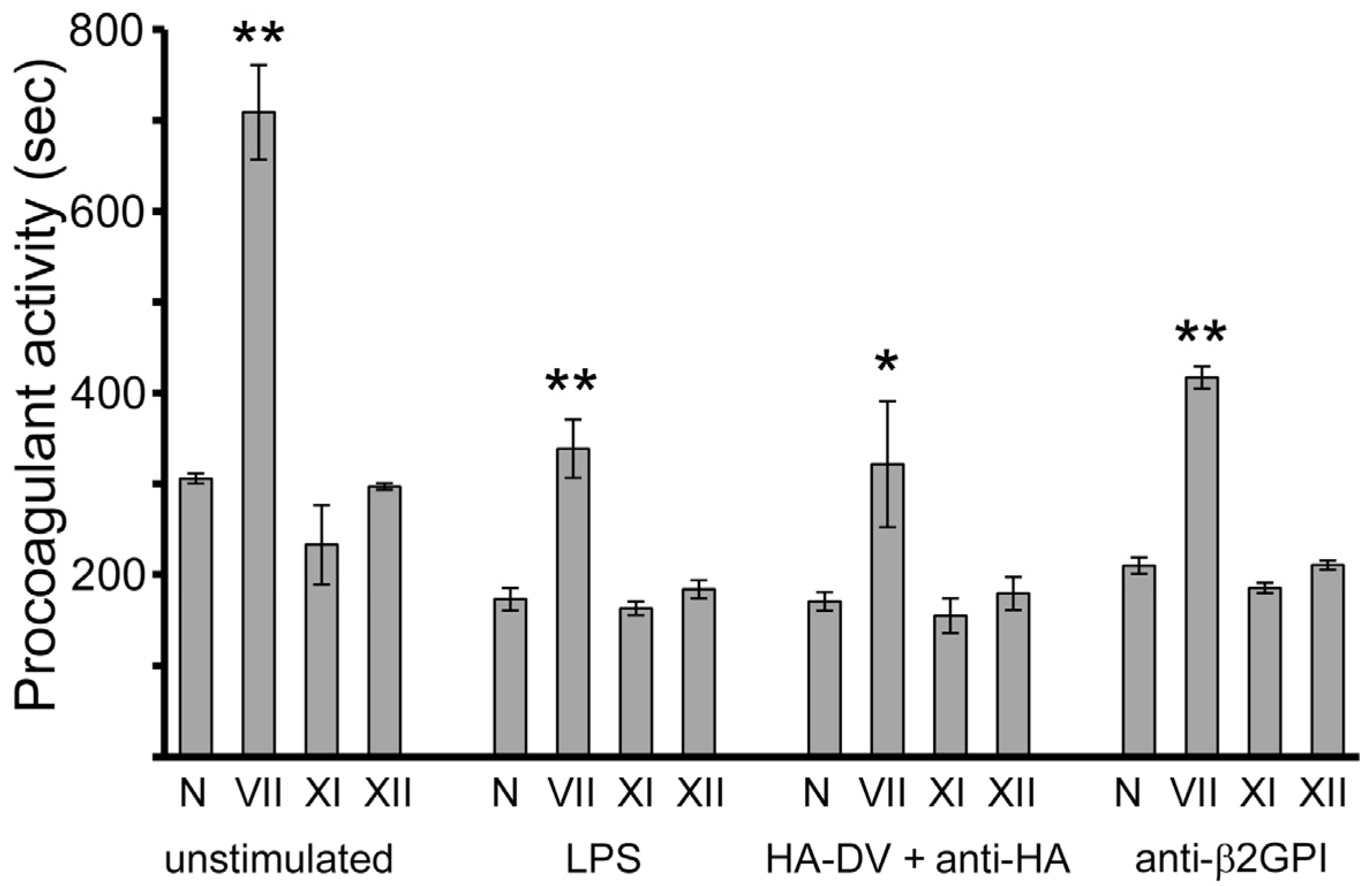
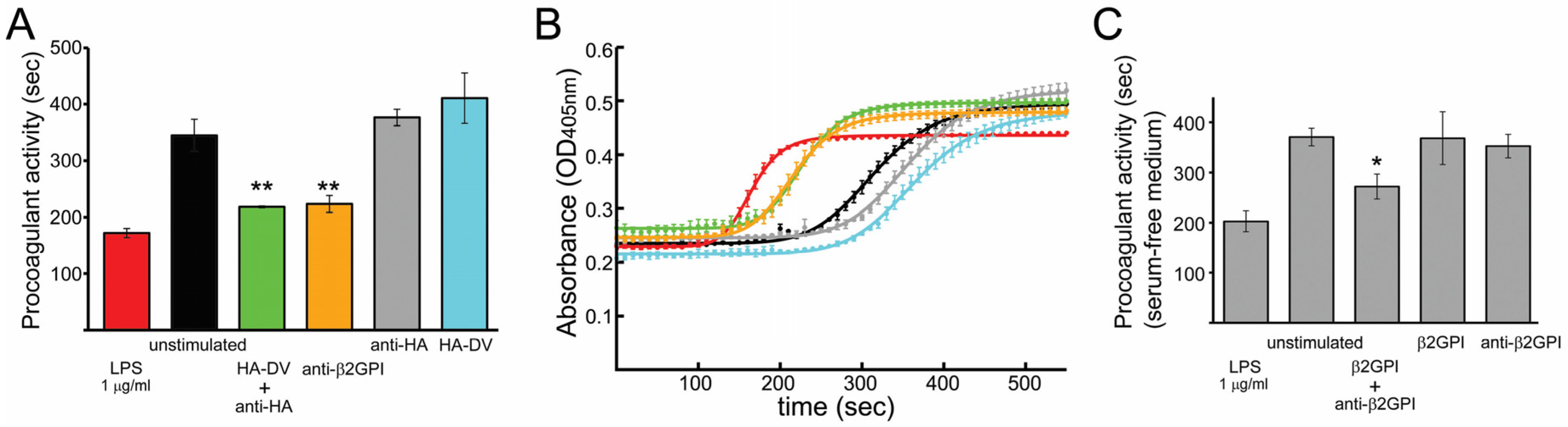
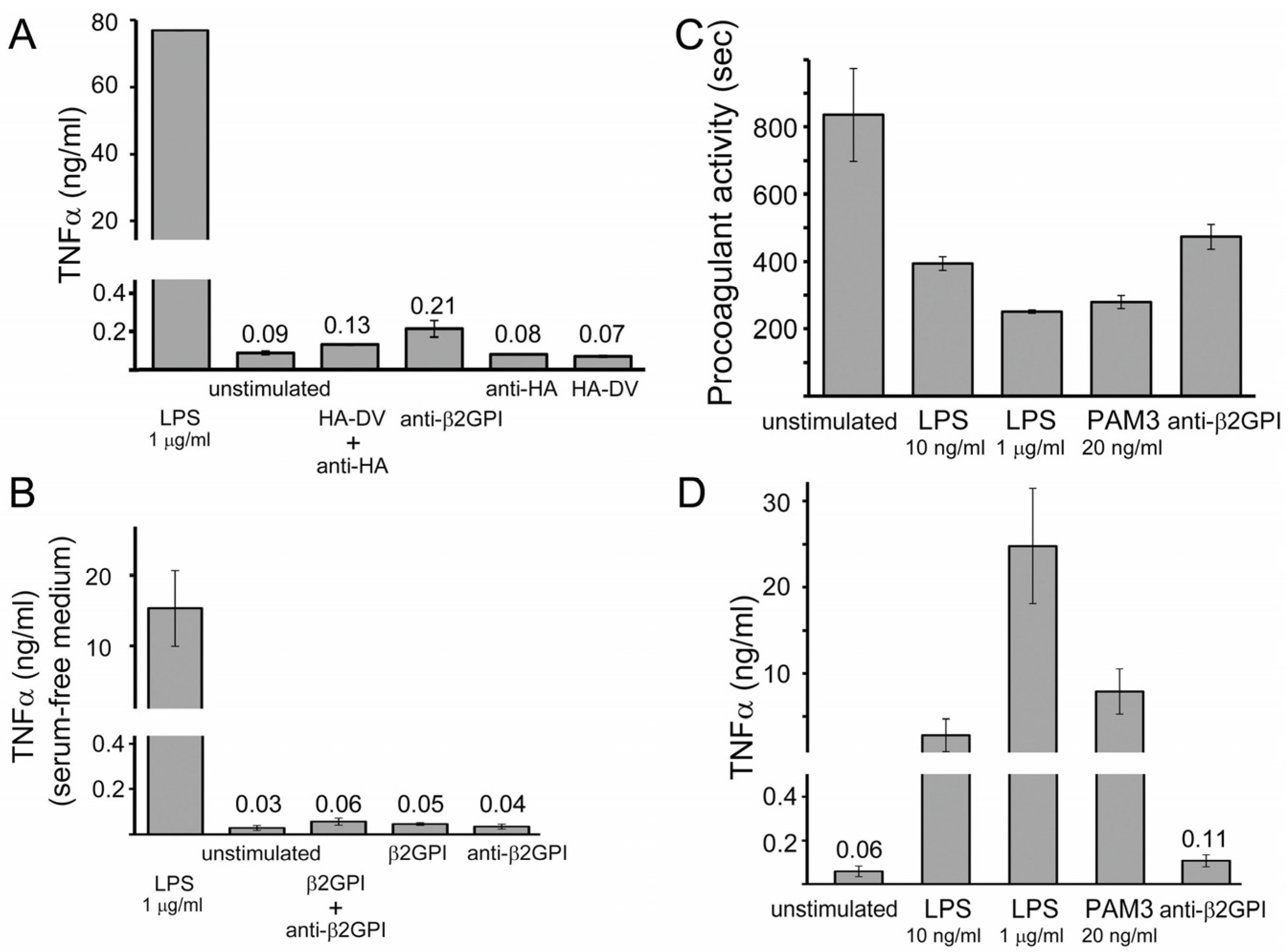
2.1. In the Presence of Dimerizing Antibodies, Domain V of β2GPI Is Sufficient to Stimulate the Procoagulant Activity of PMA-Differentiated U937 Monocytes
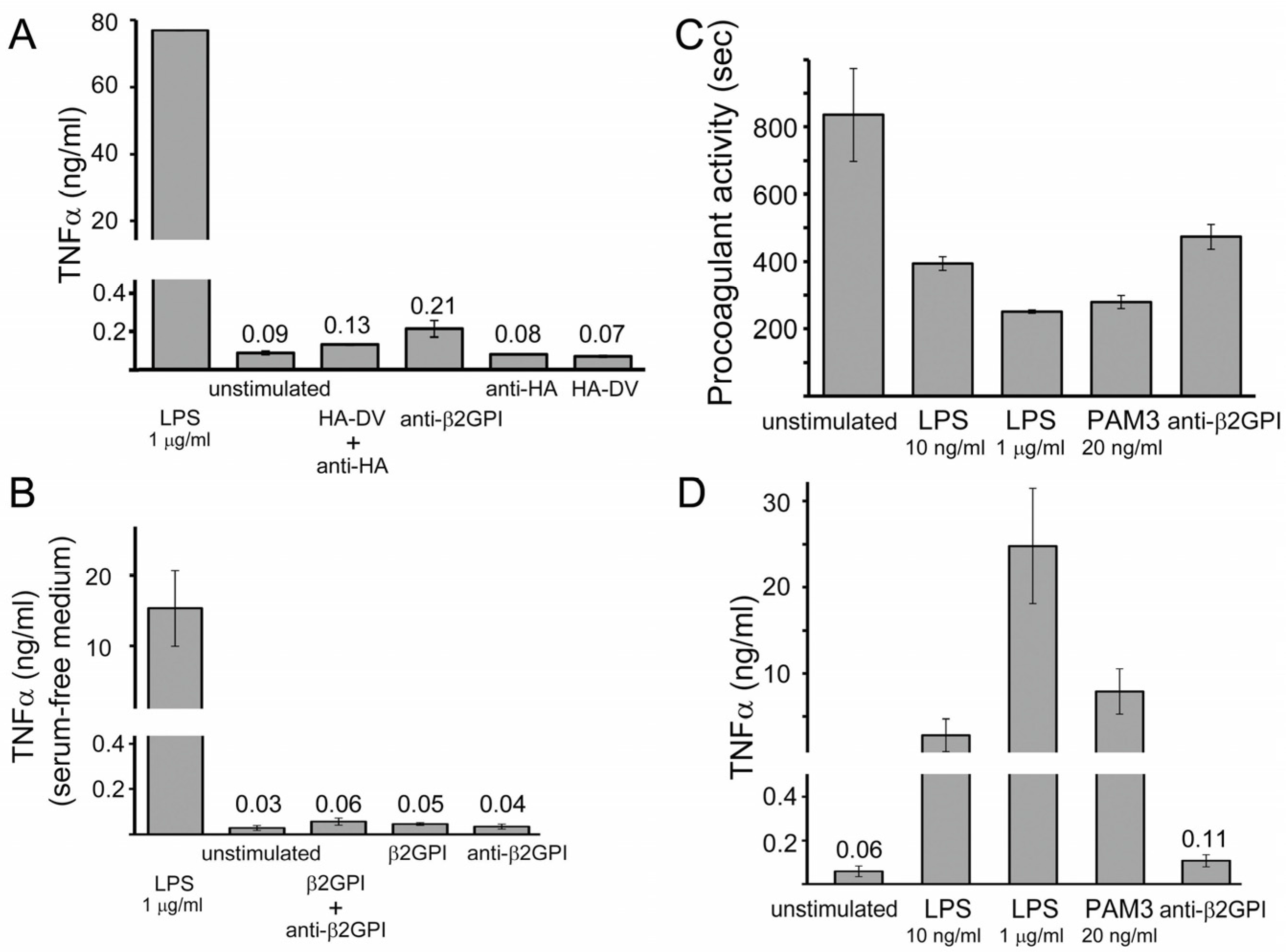
2.2. Measured Procoagulant Activity of U937 Cells Is TF-Dependent
2.3. TNFα Released by U937 Cells Stimulated with either HA-DV/Anti-HA Complexes or Anti-β2GPI Antibodies Was Negligible Compared to TNFα Released by Cells Stimulated with TLR4 and TLR2 Ligands
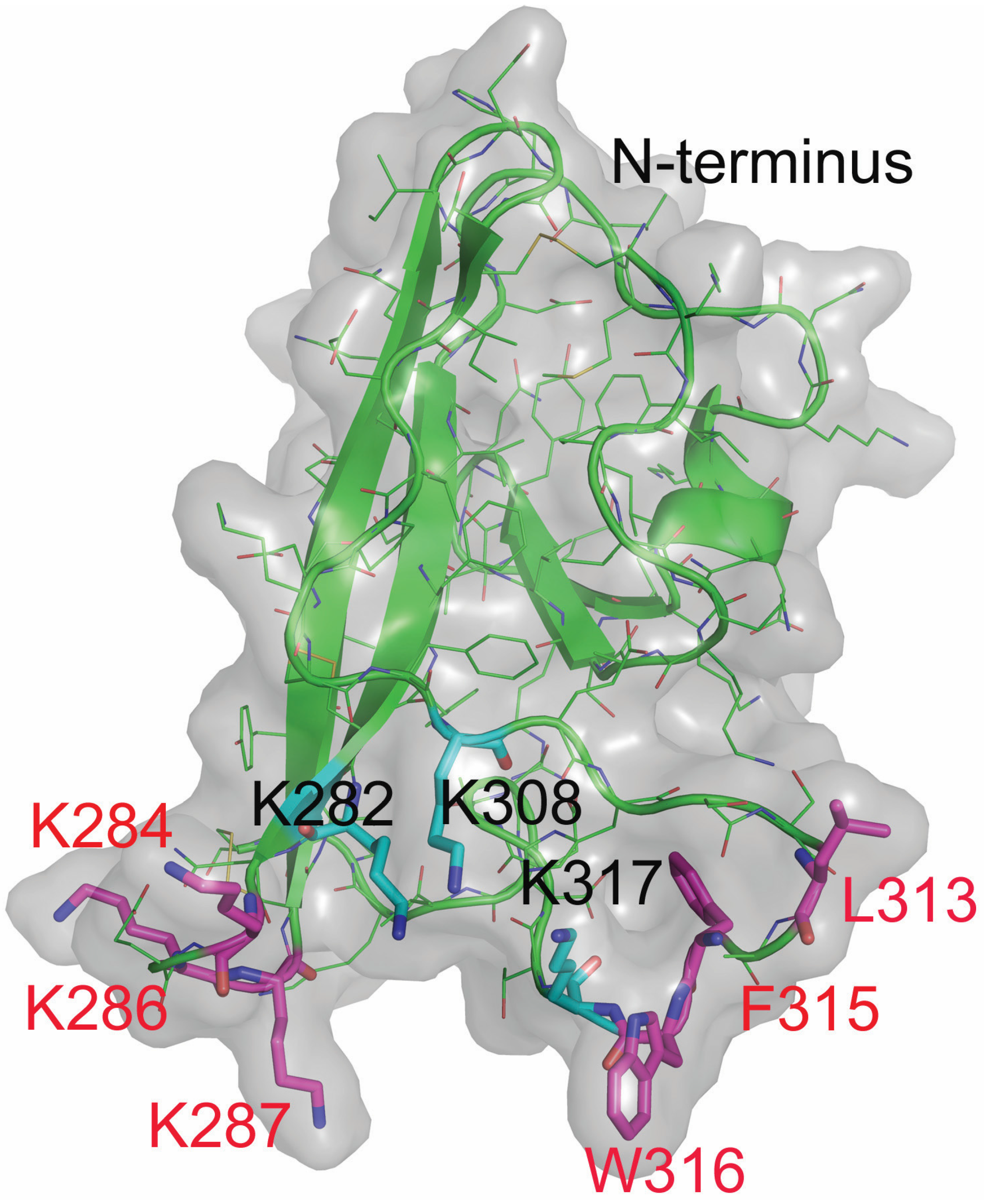
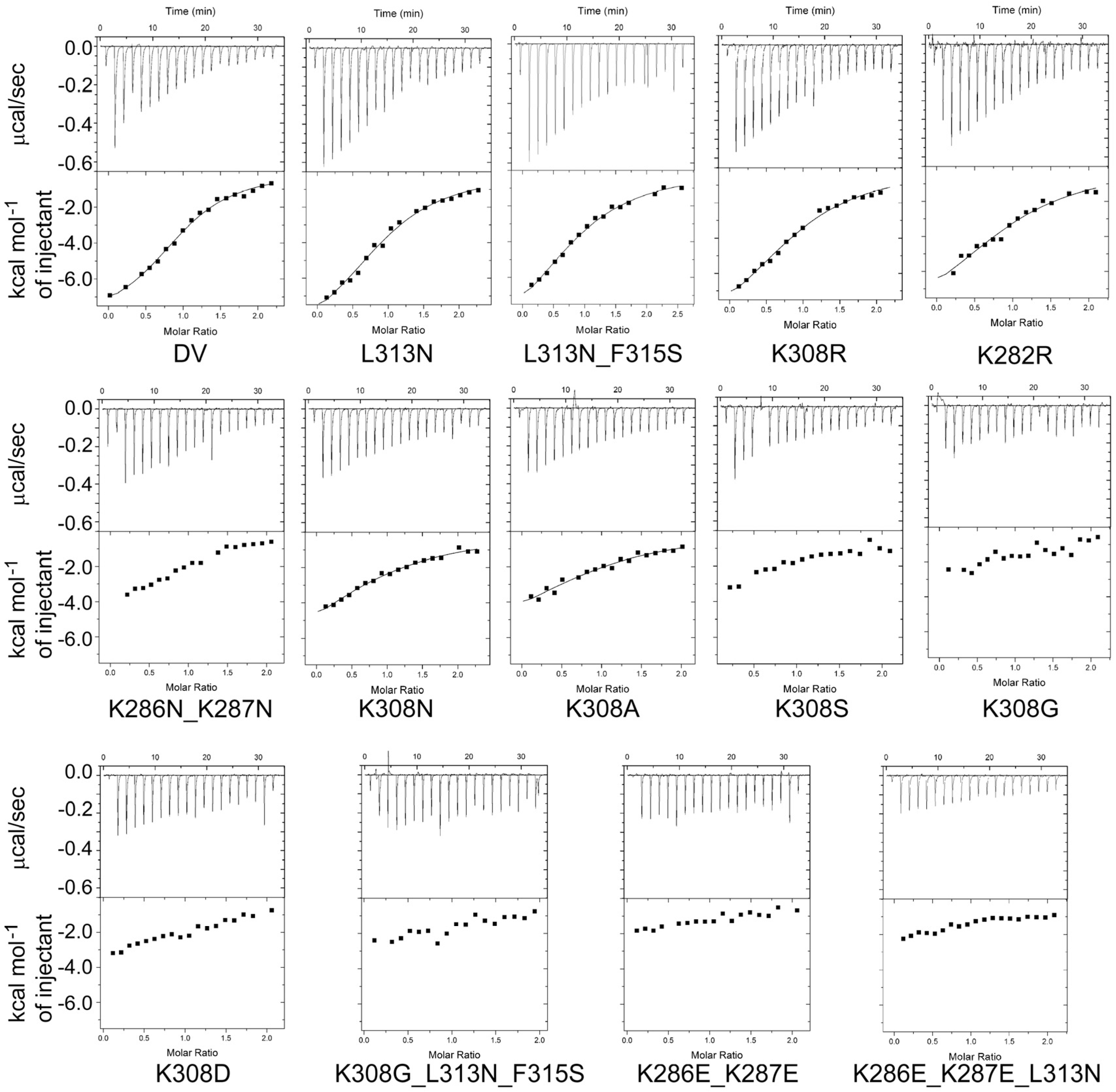
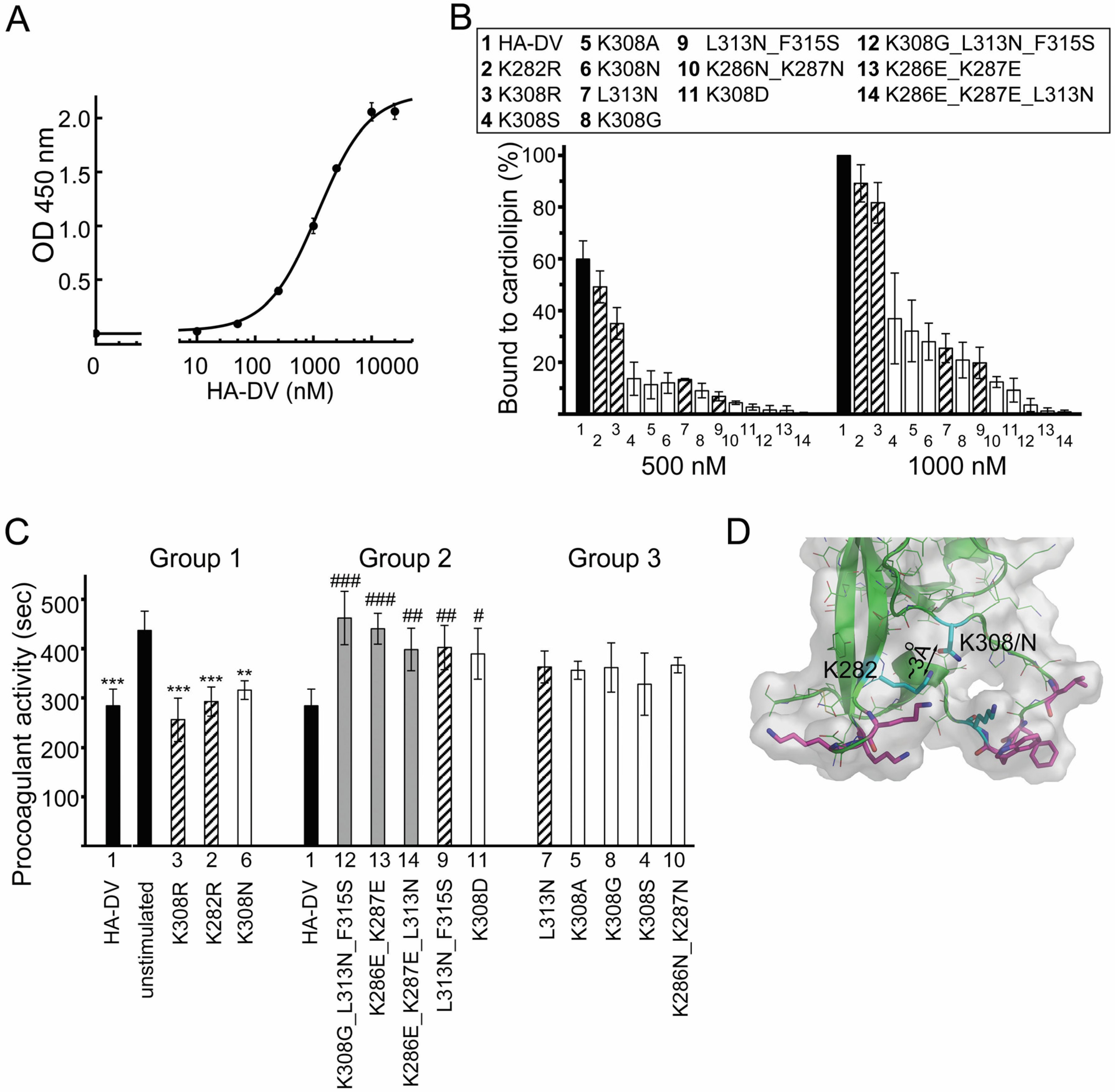
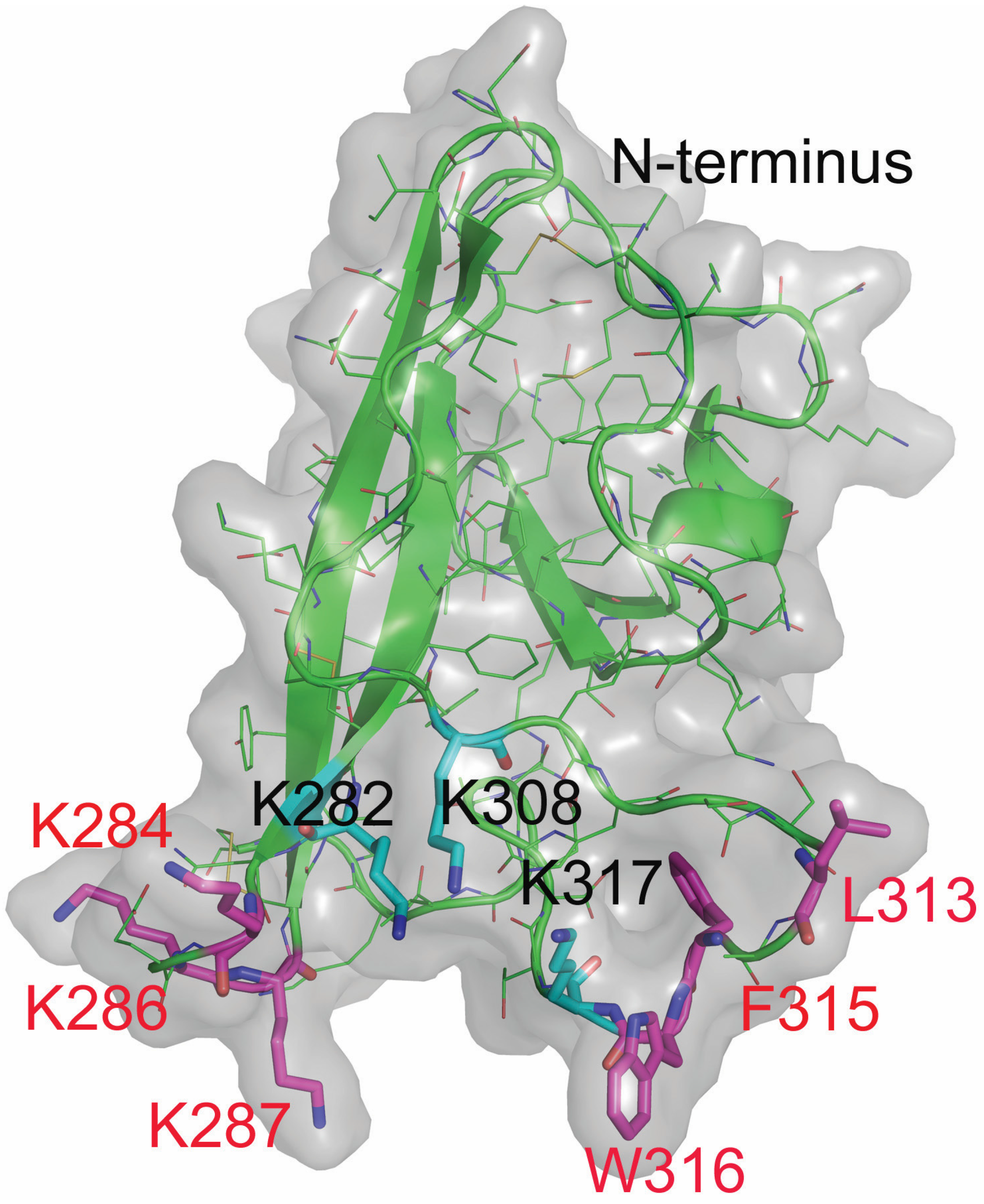
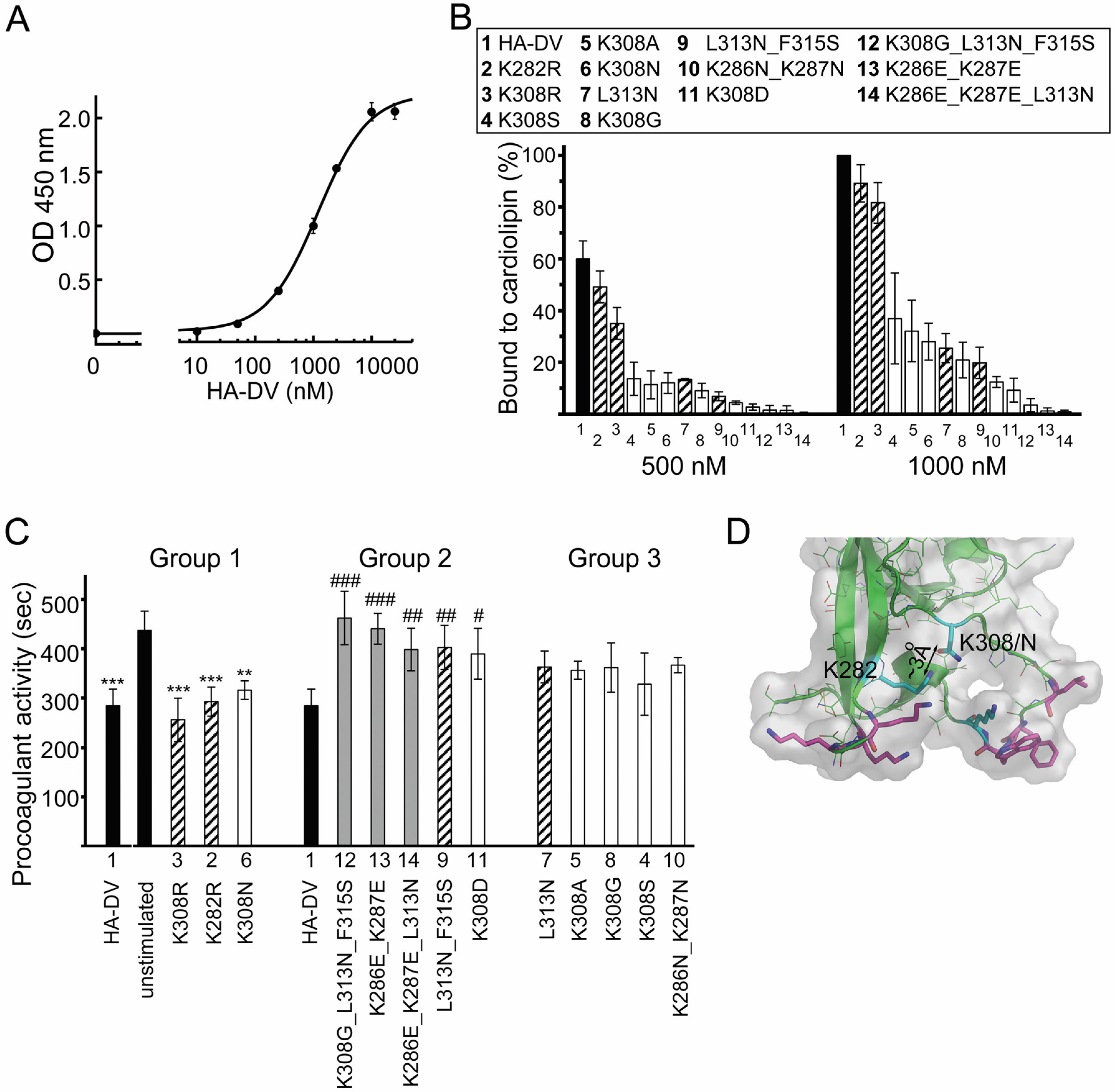
2.4. Design and Characterization of Point Mutants of Domain V of β2GPI (HA-DV)
2.4.1. The Binding of HA-DV Variants to ApoER2
2.4.2. The Binding of HA-DV Variants to Cardiolipin
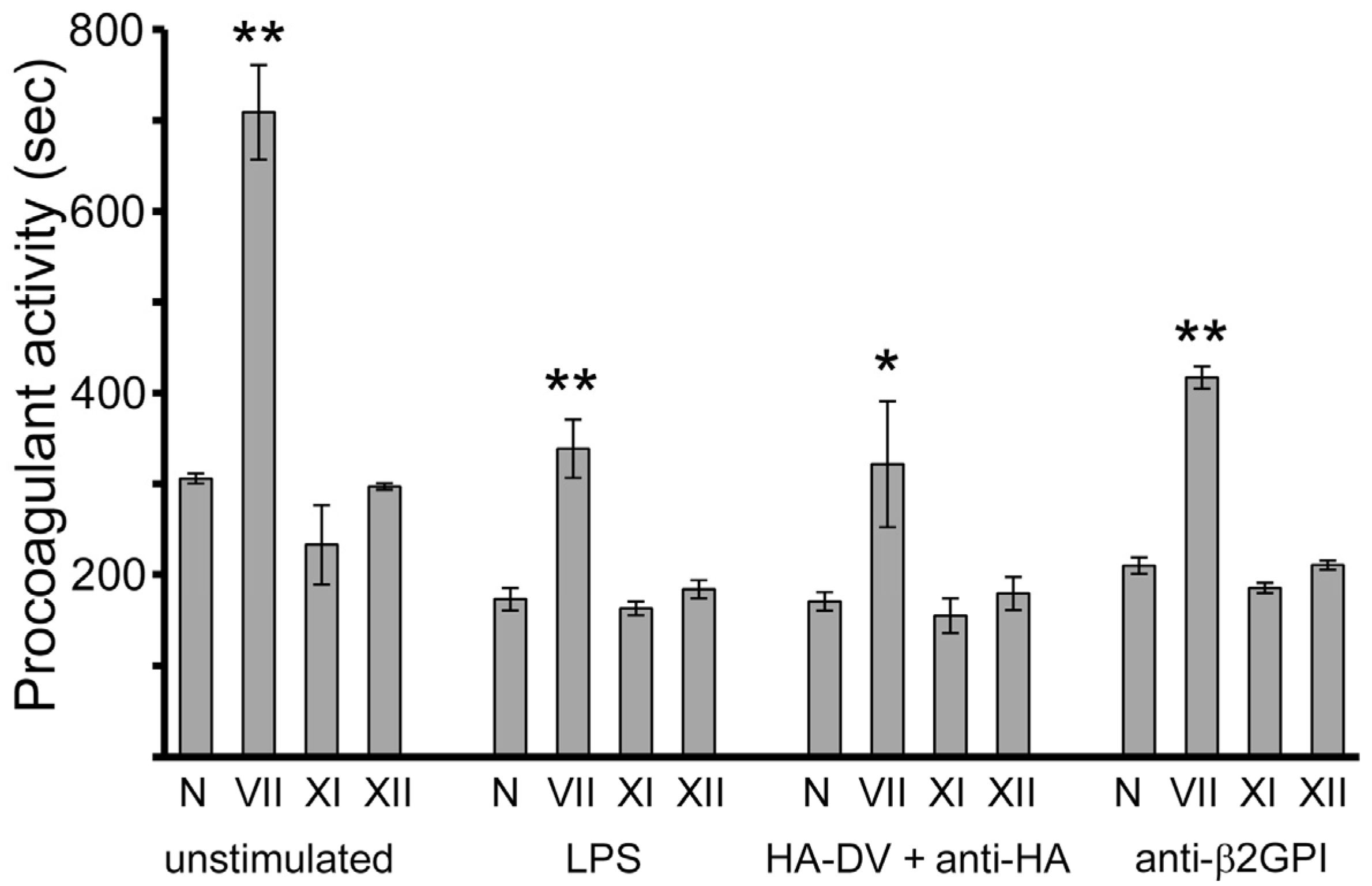
2.5. ApoER2 Does Not Contribute to Upregulation of Procoagulant Activity in U937 Cells
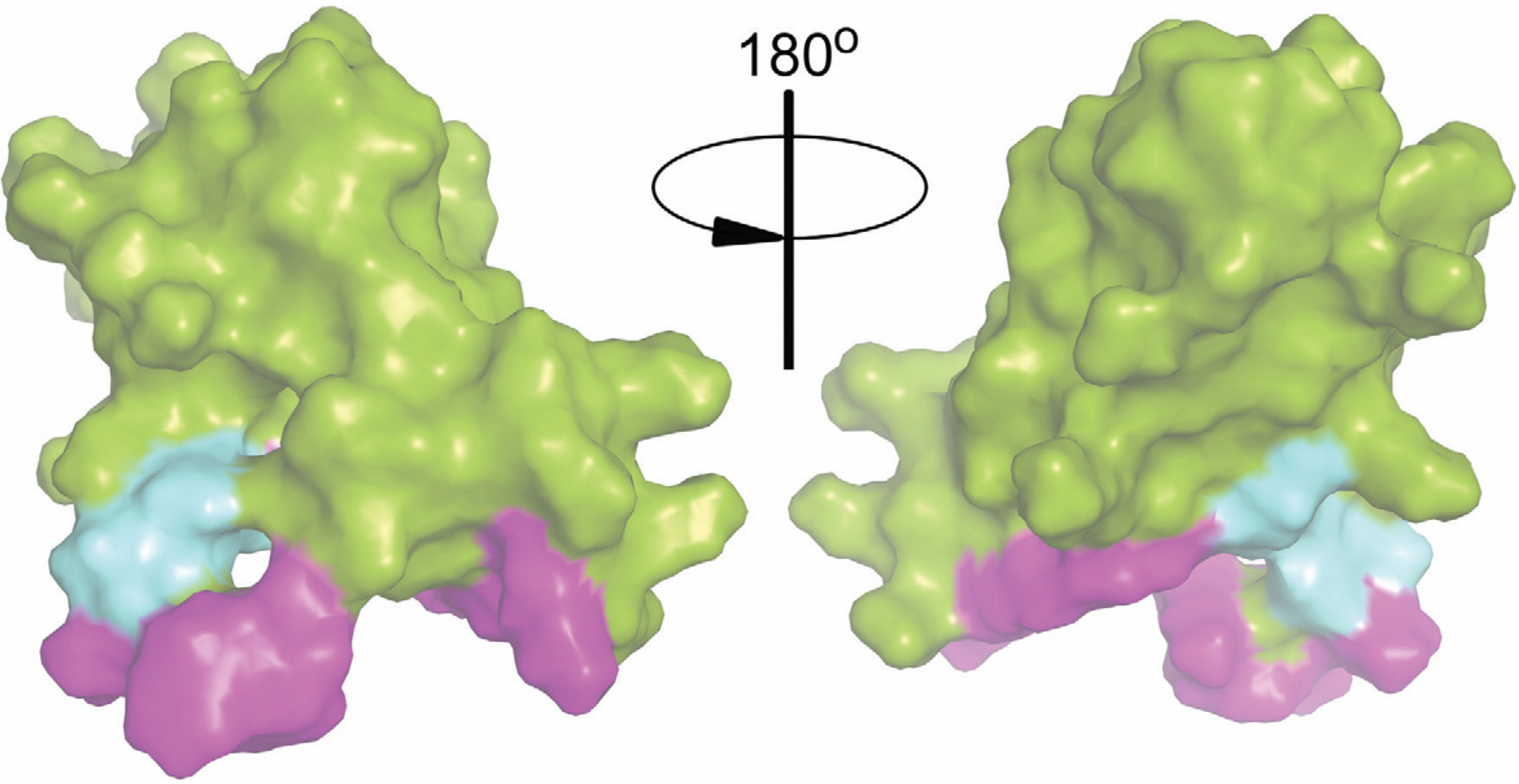
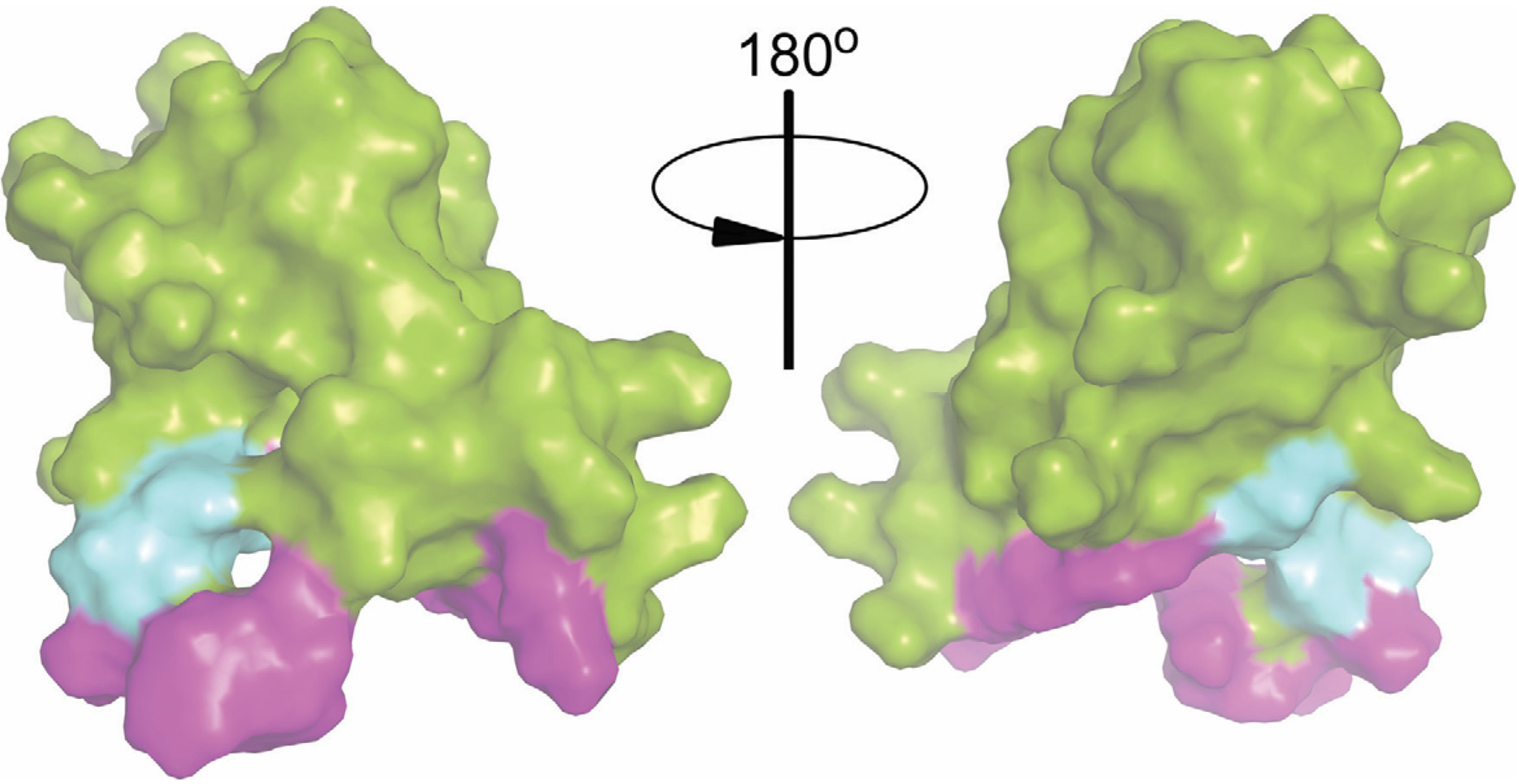
2.6. Intact Residues in the Two Phospholipid-Binding Loops of HA-DV Are Important for the Ability of HA-DV/Anti-HA Complexes to Induce Procoagulant Activity in U937 Cells
3. Discussion
4. Materials and Methods
4.1. Proteins
4.2. Cells and Culture Conditions
4.3. Measurements of the Procoagulant Activity of U937 Cells
4.4. TNFα ELISA
4.5. Isothermal Titration Calorimetry
4.6. Cardiolipin ELISA
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bertolaccini, M.L.; Amengual, O.; Andreoli, L.; Atsumi, T.; Chighizola, C.B.; Forastiero, R.; de Groot, P.; Lakos, G.; Lambert, M.; Meroni, P.; et al. 14th international congress on antiphospholipid antibodies task force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmun. Rev. 2014, 13, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; de Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Giles, I.P.; Isenberg, D.A.; Latchman, D.S.; Rahman, A. How do antiphospholipid antibodies bind beta2-glycoprotein I? Arthritis Rheum. 2003, 48, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Lieby, P.; Soley, A.; Levallois, H.; Hugel, B.; Freyssinet, J.M.; Cerutti, M.; Pasquali, J.L.; Martin, T. The clonal analysis of anticardiolipin antibodies in a single patient with primary antiphospholipid syndrome reveals an extreme antibody heterogeneity. Blood 2001, 97, 3820–3828. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Pittoni, V.; Griggi, T.; Losardo, A.; Leri, O.; Magno, M.S.; Misasi, R.; Valesini, G. Specificity of anti-phospholipid antibodies in infectious mononucleosis: A role for anti-cofactor protein antibodies. Clin. Exp. Immunol. 2000, 120, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Gretler, J.; Aglas, F.; Auer-Grumbach, P.; Rainer, F. Anticardiolipin antibodies in rheumatoid arthritis: Their relation to rheumatoid nodules and cutaneous vascular manifestations. Br. J. Dermatol. 1994, 131, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Forastiero, R.R.; Martinuzzo, M.E.; Kordich, L.C.; Carreras, L.O. Reactivity to beta 2 glycoprotein I clearly differentiates anticardiolipin antibodies from antiphospholipid syndrome and syphilis. Thromb. Haemost. 1996, 75, 717–720. [Google Scholar] [PubMed]
- Manukyan, D.; Muller-Calleja, N.; Jackel, S.; Luchmann, K.; Monnikes, R.; Kiouptsi, K.; Reinhardt, C.; Jurk, K.; Walter, U.; Lackner, K.J. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J. Thromb. Haemost. 2016, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Merashli, M.; Noureldine, M.H.; Uthman, I.; Khamashta, M. Antiphospholipid syndrome: An update. Eur. J. Clin. Investig. 2015, 45, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 2013, 368, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Chighizola, C.B.; Rovelli, F.; Gerosa, M. Antiphospholipid syndrome in 2014: More clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res. Ther. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.R.; Dier, K.J.; Lopez, D.; Merrill, J.T.; Fink, C.A. Anti-beta 2-glycoprotein I and antiphosphatidylserine antibodies are predictors of arterial thrombosis in patients with antiphospholipid syndrome. Am. J. Clin. Pathol. 2004, 121, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Day, H.M.; Thiagarajan, P.; Ahn, C.; Reveille, J.D.; Tinker, K.F.; Arnett, F.C. Autoantibodies to beta2-glycoprotein I in systemic lupus erythematosus and primary antiphospholipid antibody syndrome: Clinical correlations in comparison with other antiphospholipid antibody tests. J. Rheumatol. 1998, 25, 667–674. [Google Scholar] [PubMed]
- Cabiedes, J.; Cabral, A.R.; Alarcon-Segovia, D. Clinical manifestations of the antiphospholipid syndrome in patients with systemic lupus erythematosus associate more strongly with anti-beta 2-glycoprotein-I than with antiphospholipid antibodies. J. Rheumatol. 1995, 22, 1899–1906. [Google Scholar] [PubMed]
- De Laat, B.; Pengo, V.; Pabinger, I.; Musial, J.; Voskuyl, A.E.; Bultink, I.E.; Ruffatti, A.; Rozman, B.; Kveder, T.; de Moerloose, P.; et al. The association between circulating antibodies against domain i of beta2-glycoprotein I and thrombosis: An international multicenter study. J. Thromb. Haemost. 2009, 7, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, Z.; Chen, S.; Li, J.; Wen, X.; Li, L.; Zhang, W.; Zhao, J.; Zhang, F.; Li, Y. Evaluation of the diagnostic potential of antibodies to beta2-glycoprotein I domain I in chinese patients with antiphospholipid syndrome. Sci. Rep. 2016, 6, 23839. [Google Scholar] [CrossRef] [PubMed]
- De Laat, H.B.; Derksen, R.H.; Urbanus, R.T.; Roest, M.; de Groot, P.G. Beta2-glycoprotein I-dependent lupus anticoagulant highly correlates with thrombosis in the antiphospholipid syndrome. Blood 2004, 104, 3598–3602. [Google Scholar] [CrossRef] [PubMed]
- Willis, R.; Pierangeli, S.S. Anti-beta2-glycoprotein I antibodies. Ann. N. Y. Acad. Sci. 2013, 1285, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Arad, A.; Proulle, V.; Furie, R.A.; Furie, B.C.; Furie, B. Beta(2)-glycoprotein-I autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood 2011, 117, 3453–3459. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.; Vreys, I.; Wittevrongel, C.; Boon, D.; Vermylen, J.; Hoylaerts, M.F.; Arnout, J. Thrombogenicity of beta 2-glycoprotein I-dependent antiphospholipid antibodies in a photochemically induced thrombosis model in the hamster. Blood 2003, 101, 157–162. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.G.; Urbanus, R.T. The significance of auto-antibodies against beta2-glycoprotein I. Blood 2012, 120, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, S.S.; Vega-Ostertag, M.E.; Raschi, E.; Liu, X.; Romay-Penabad, Z.; De Micheli, V.; Galli, M.; Moia, M.; Tincani, A.; Borghi, M.O.; et al. Toll-like receptor and antiphospholipid mediated thrombosis: In vivo studies. Ann. Rheum. Dis. 2007, 66, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Romay-Penabad, Z.; Aguilar-Valenzuela, R.; Urbanus, R.T.; Derksen, R.H.; Pennings, M.T.; Papalardo, E.; Shilagard, T.; Vargas, G.; Hwang, Y.; de Groot, P.G.; et al. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood 2011, 117, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Romay-Penabad, Z.; Montiel-Manzano, M.G.; Shilagard, T.; Papalardo, E.; Vargas, G.; Deora, A.B.; Wang, M.; Jacovina, A.T.; Garcia-Latorre, E.; Reyes-Maldonado, E.; et al. Annexin A2 is involved in antiphospholipid antibody-mediated pathogenic effects in vitro and in vivo. Blood 2009, 114, 3074–3083. [Google Scholar] [CrossRef] [PubMed]
- Laplante, P.; Fuentes, R.; Salem, D.; Subang, R.; Gillis, M.A.; Hachem, A.; Farhat, N.; Qureshi, S.T.; Fletcher, C.A.; Roubey, R.A.; et al. Antiphospholipid antibody-mediated effects in an arterial model of thrombosis are dependent on toll-like receptor 4. Lupus 2016, 25, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Brandt, K.J.; Fickentscher, C.; Boehlen, F.; Kruithof, E.K.; de Moerloose, P. NF-kappab is activated from endosomal compartments in antiphospholipid antibodies-treated human monocytes. J. Thromb. Haemost. 2014, 12, 779–791. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.G.; Meijers, J.C. Beta(2)-glycoprotein I: Evolution, structure and function. J. Thromb. Haemost. 2011, 9, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L. Anti-beta-2 glycoprotein I epitope specificity: From experimental models to diagnostic tools. Lupus 2016, 25, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.; Krilis, S. The fifth domain of beta 2-glycoprotein I contains a phospholipid binding site (cys281-cys288) and a region recognized by anticardiolipin antibodies. J. Immunol. 1994, 152, 653–659. [Google Scholar] [PubMed]
- Mehdi, H.; Naqvi, A.; Kamboh, M.I. A hydrophobic sequence at position 313–316 (Leu-Ala-Phe-Trp) in the fifth domain of apolipoprotein H (beta2-glycoprotein I) is crucial for cardiolipin binding. Eur. J. Biochem. 2000, 267, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Pennings, M.T.; Derksen, R.H.; van Lummel, M.; Adelmeijer, J.; VanHoorelbeke, K.; Urbanus, R.T.; Lisman, T.; de Groot, P.G. Platelet adhesion to dimeric beta-glycoprotein I under conditions of flow is mediated by at least two receptors: Glycoprotein Ibalpha and apolipoprotein E receptor 2′. J. Thromb. Haemost. 2007, 5, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Sali, A.; Herzog, H.; Lahnstein, J.; Krilis, S.A. Site-directed mutagenesis of recombinant human beta 2-glycoprotein I identifies a cluster of lysine residues that are critical for phospholipid binding and anti-cardiolipin antibody activity. J. Immunol. 1996, 157, 3744–3751. [Google Scholar] [PubMed]
- Van Lummel, M.; Pennings, M.T.; Derksen, R.H.; Urbanus, R.T.; Lutters, B.C.; Kaldenhoven, N.; de Groot, P.G. The binding site in {beta}2-glycoprotein I for ApoER2′ on platelets is located in domain V. J. Biol. Chem. 2005, 280, 36729–36736. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; De Biasio, A.; Beglova, N. Mode of interaction between beta2GPI and lipoprotein receptors suggests mutually exclusive binding of beta2GPI to the receptors and anionic phospholipids. Structure 2010, 18, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Amengual, O.; Atsumi, T.; Khamashta, M.A. Tissue factor in antiphospholipid syndrome: Shifting the focus from coagulation to endothelium. Rheumatol. Oxf. 2003, 42, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Kinev, A.V.; Roubey, R.A. Tissue factor in the antiphospholipid syndrome. Lupus 2008, 17, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Boles, J.; Mackman, N. Role of tissue factor in thrombosis in antiphospholipid antibody syndrome. Lupus 2010, 19, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, M.J.; Lopez-Pedrera, C.; Khamashta, M.A.; Camps, M.T.; Tinahones, F.; Torres, A.; Hughes, G.R.; Velasco, F. Thrombosis in primary antiphospholipid syndrome: A pivotal role for monocyte tissue factor expression. Arthritis. Rheum. 1997, 40, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Dobado-Berrios, P.M.; Lopez-Pedrera, C.; Velasco, F.; Aguirre, M.A.; Torres, A.; Cuadrado, M.J. Increased levels of tissue factor mrna in mononuclear blood cells of patients with primary antiphospholipid syndrome. Thromb. Haemost. 1999, 82, 1578–1582. [Google Scholar] [PubMed]
- Nojima, J.; Masuda, Y.; Iwatani, Y.; Suehisa, E.; Futsukaichi, Y.; Kuratsune, H.; Watanabe, Y.; Takano, T.; Hidaka, Y.; Kanakura, Y. Tissue factor expression on monocytes induced by anti-phospholipid antibodies as a strong risk factor for thromboembolic complications in sle patients. Biochem. Biophys. Res. Commun. 2008, 365, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Lambrianides, A.; Carroll, C.J.; Pierangeli, S.S.; Pericleous, C.; Branch, W.; Rice, J.; Latchman, D.S.; Townsend, P.; Isenberg, D.A.; Rahman, A.; et al. Effects of polyclonal IgG derived from patients with different clinical types of the antiphospholipid syndrome on monocyte signaling pathways. J. Immunol. 2010, 184, 6622–6628. [Google Scholar] [CrossRef] [PubMed]
- Satta, N.; Kruithof, E.K.; Fickentscher, C.; Dunoyer-Geindre, S.; Boehlen, F.; Reber, G.; Burger, D.; de Moerloose, P. Toll-like receptor 2 mediates the activation of human monocytes and endothelial cells by antiphospholipid antibodies. Blood 2011, 117, 5523–5531. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhou, H.; Wang, H.; Chen, D.; Xia, L.; Wang, T.; Yan, J. Anti-beta(2)GPI/beta(2)GPI induced TF and TNF-alpha expression in monocytes involving both TLR4/MYD88 and TLR4/TRIF signaling pathways. Mol. Immunol. 2013, 53, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Longo, A.; Capozzi, A.; Garofalo, T.; Misasi, R.; Alessandri, C.; Conti, F.; Buttari, B.; Rigano, R.; Ortona, E.; et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis. Rheum. 2007, 56, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, M.; Vaccarino, A.; Stella, S.; Bertero, M.T.; Carignola, R.; Montaruli, B.; Roccatello, D.; Shoenfeld, Y.; Piedmont, A.P.S.C. Thrombotic recurrences and bleeding events in APS vascular patients: A review from the literature and a comparison with the APS piedmont cohort. Autoimmun. Rev. 2013, 12, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Serrano, R.; Pons-Estel, G.J.; Ceberio-Hualde, L.; Shoenfeld, Y.; de Ramon, E.; Buonaiuto, V.; Jacobsen, S.; Zeher, M.M.; Tarr, T.; et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: A multicentre prospective study of 1000 patients. Ann. Rheum. Dis. 2015, 74, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Chighizola, C.B.; Ubiali, T.; Meroni, P.L. Treatment of thrombotic antiphospholipid syndrome: The rationale of current management-an insight into future approaches. J. Immun. Res. 2015, 2015, 951424. [Google Scholar] [CrossRef] [PubMed]
- Pedrinaci, S.; Ruiz-Cabello, F.; Gomez, O.; Collado, A.; Garrido, F. Protein kinase c-mediated regulation of the expression of CD14 and CD11/CD18 in U937 cells. Int. J. Cancer 1990, 45, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Krahling, S.; Smith, D.; Williamson, P.; Schlegel, R.A. Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol. Biol. Cell 2004, 15, 2863–2872. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; McElvaney, N.G.; O’Neill, S.J.; Taggart, C.C. Secretory leucoprotease inhibitor impairs toll-like receptor 2- and 4-mediated responses in monocytic cells. Infect. Immun. 2004, 72, 3684–3687. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hirai, H.; Taniguchi, K.; Shimura, K.; Inaba, T.; Shimazaki, C.; Taniwaki, M.; Imanishi, J. Toll-like receptors (TLRs) are expressed by myeloid leukaemia cell lines, but fail to trigger differentiation in response to the respective TLR ligands. Br. J. Haematol. 2009, 147, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.V.; Banerjee, Y.; Fernandez, J.A.; Deguchi, H.; Xu, X.; Mosnier, L.O.; Urbanus, R.T.; de Groot, P.G.; White-Adams, T.C.; McCarty, O.J.; et al. Activated protein C ligation of ApoER2 (Lrp8) causes DAB1-dependent signaling in U937 cells. Proc. Natl. Acad. Sci. USA 2009, 106, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Kolyada, A.; Karageorgos, I.; Mahlawat, P.; Beglova, N. An A1-A1 mutant with improved binding and inhibition of beta2GPI/antibody complexes in antiphospholipid syndrome. FEBS J. 2015, 282, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Pennings, M.T.; Derksen, R.H.; Urbanus, R.T.; Tekelenburg, W.L.; Hemrika, W.; de Groot, P.G. Platelets express three different splice variants of ApoER2 that are all involved in signaling. J. Thromb. Haemost. 2007, 5, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Urbanus, R.T.; Pennings, M.T.; Derksen, R.H.; de Groot, P.G. Platelet activation by dimeric beta(2)-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2′. J. Thromb. Haemost. 2008, 6, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Agar, C.; van Os, G.M.; Morgelin, M.; Sprenger, R.R.; Marquart, J.A.; Urbanus, R.T.; Derksen, R.H.; Meijers, J.C.; de Groot, P.G. {beta}2-glycoprotein I can exist in two conformations: Implications for our understanding of the antiphospholipid syndrome. Blood 2010, 116, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Roubey, R.A.; Eisenberg, R.A.; Harper, M.F.; Winfield, J.B. “Anticardiolipin” autoantibodies recognize beta 2-glycoprotein I in the absence of phospholipid. Importance of Ag density and bivalent binding. J. Immunol. 1995, 154, 954–960. [Google Scholar] [PubMed]
- Tincani, A.; Spatola, L.; Prati, E.; Allegri, F.; Ferremi, P.; Cattaneo, R.; Meroni, P.; Balestrieri, G. The anti-beta2-glycoprotein I activity in human anti-phospholipid syndrome sera is due to monoreactive low-affinity autoantibodies directed to epitopes located on native beta2-glycoprotein I and preserved during species’ evolution. J. Immunol. 1996, 157, 5732–5738. [Google Scholar] [PubMed]
- Willems, G.M.; Janssen, M.P.; Pelsers, M.M.; Comfurius, P.; Galli, M.; Zwaal, R.F.; Bevers, E.M. Role of divalency in the high-affinity binding of anticardiolipin antibody-beta 2-glycoprotein I complexes to lipid membranes. Biochemistry 1996, 35, 13833–13842. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. The challenge of lipid rafts. J. Lipid Res. 2009, 50, S323–S328. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pedrera, C.; Buendia, P.; Cuadrado, M.J.; Siendones, E.; Aguirre, M.A.; Barbarroja, N.; Montiel-Duarte, C.; Torres, A.; Khamashta, M.; Velasco, F. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappab/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis. Rheum. 2006, 54, 301–311. [Google Scholar] [PubMed]
- Zhou, H.; Wolberg, A.S.; Roubey, R.A. Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood 2004, 104, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Hurst, J.; Lorenz, M.; Prinz, N.; Clemens, N.; Drechsler, M.D.; Bauer, S.; Chapman, J.; Shoenfeld, Y.; Blank, M.; et al. Human antiphospholipid antibodies induce TNFalpha in monocytes via toll-like receptor 8. Immunobiology 2010, 215, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Kolyada, A.; Lee, C.J.; De Biasio, A.; Beglova, N. A novel dimeric inhibitor targeting beta2GPI in beta2GPI/antibody complexes implicated in antiphospholipid syndrome. PLoS ONE 2010, 5, e15345. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolyada, A.; Barrios, D.A.; Beglova, N. Dimerized Domain V of Beta2-Glycoprotein I Is Sufficient to Upregulate Procoagulant Activity in PMA-Treated U937 Monocytes and Require Intact Residues in Two Phospholipid-Binding Loops. Antibodies 2017, 6, 8. https://doi.org/10.3390/antib6020008
Kolyada A, Barrios DA, Beglova N. Dimerized Domain V of Beta2-Glycoprotein I Is Sufficient to Upregulate Procoagulant Activity in PMA-Treated U937 Monocytes and Require Intact Residues in Two Phospholipid-Binding Loops. Antibodies. 2017; 6(2):8. https://doi.org/10.3390/antib6020008
Chicago/Turabian StyleKolyada, Alexey, David A. Barrios, and Natalia Beglova. 2017. "Dimerized Domain V of Beta2-Glycoprotein I Is Sufficient to Upregulate Procoagulant Activity in PMA-Treated U937 Monocytes and Require Intact Residues in Two Phospholipid-Binding Loops" Antibodies 6, no. 2: 8. https://doi.org/10.3390/antib6020008