Asymmetric Organocatalytic Reactions of α,β-Unsaturated Cyclic Ketones

1
Department of Chemistry, University of Calabria, Ponte Bucci cubo 12/c, I-87030 Arcavacata di Rende (Cs), Italy
2
Department of Organic Chemistry “A. Mangini”, University of Bologna, viale Risorgimento 4, I-40136 Bologna, Italy
*
Author to whom correspondence should be addressed.
Symmetry 2011, 3(1), 84-125; https://doi.org/10.3390/sym3010084
Submission received: 18 February 2011
/
Revised: 8 March 2011
/
Accepted: 9 March 2011
/
Published: 22 March 2011
(This article belongs to the Special Issue Asymmetric Organocatalysis)
Abstract
:The 1,4-conjugate addition of nucleophiles to α,β-unsaturated carbonyl compounds represents one fundamental bond-forming reaction in organic synthesis. The development of effective organocatalysts for the enantioselective conjugate addition of malonate, nitroalkane and other carbon and heteroatom nucleophiles to cycloenones constitutes an important research field and has been explored in recent years. At the same time, asymmetric Diels-Alder reactions have been developed and often a mechanism has been demonstrated to be a double addition rather than synchronous. This review aims to cover literature up to the end of 2010, describing all the different organocatalytic asymmetric 1,4-conjugate additions even if they are listed as transfer hydrogenation, cycloadditions or desymmetrization of aromatic compounds.
Contents
- Introduction
- Transfer Hydrogenations
- 1,4-Conjugate Additions of Carbon Nucleophiles
- 3.1
- Malonates and related 1,3-dicarbonyls
- 3.2
- Nitroalkanes
- 3.3
- Other carbon nucleophiles
- 1,4-Conjugate Additions of Heteroatom Nucleophiles
- Diels-Alder
- Desymmetrization of cyclohexadienones
- Conclusions
- Abbreviations
- References
Abbreviations
| Ac | Acetyl (MeCO) |
| Bn | benzyl (PhCH2) |
| Boc | tert-butoxycarbonyl (Me3COC=O) |
| DCE | 1,2-dichloroethane |
| DMF | dimethylformammide |
| MS | molecular sieves |
| NMM | N-methylmorpholine |
| PMP | 4-methoyphenyl (4-MeOC6H4) |
| rt | room temperature |
| TBDPS | tert-butyldiphenylsilyl |
| TBAI | tetrabutylammonium iodide |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TRIP | 3,3′-Bis(2,4,6-triisopropylphenyl)-1,1′-bi-2-naphthol cyclic monophosphate |
| TSA | p-toluenesulfonic acid (4-MeC6H4SO3H) |
| Ts | p-tosyl (4-MeC6H4SO2) |
1. Introduction
1,4-Conjugate addition of nucleophiles to the β-position of α,β-unsaturated carbonyl compounds is one of the most frequently used reactions for bond formation. Originally [1], this reaction was restricted to the conjugate addition of an enolate to an α,β−unsaturated carbonyl (Michael reaction). Michael donors that contain active methylene centers can be directly applied, whereas simple carbonyl compounds had generally to be activated into more reactive species such as enolates or enamines. Now “Michael reaction” is often referred to the 1,4-addition of every nucleophile and a prefix indicating the nucleophilic species is generally added (e.g., oxa-, thia- or aza- for oxygen, sulfur and nitrogen nucleophiles, respectively). However 1,4-conjugate addition reaction is the most correct name.
Since stereogenic centers can be created in the course of the 1,4-conjugate addition reaction, much effort has been made to develop efficient catalytic stereoselective methods [2,3,4,5,6,7,8,9]. Carbocycles with more than one stereocenter are among the most broadly represented synthons found in natural products and medicinal agents. Moreover, the effect of conformational constraints in cyclic compounds can enhance the selectivity of the reaction, making cyclic enones especially effective in controlling the stereochemistry. Thus, the asymmetric 1,4-conjugate addition has been widely studied onto cyclic compounds.
Moreover, atom-economic chemical transformations that avoid additional reagents, waste, and working time and metal-free processes are highly desirable. In this environmentally friendly fashion, asymmetric organocatalysis that employs small chiral organic molecules to accelerate asymmetric reactions has become very attractive in recent years [10,11,12,13]. Organocatalysts provide a chiral environment to the process activating the nucleophile, the electrophile or both reagents through weak interactions, such as ion pairing (Scheme 1, Eq. 1), by deprotonation with a chiral base or by employing a chiral phase-transfer catalyst, or hydrogen bonding with molecules that coordinate the unsaturated substrate (Michael acceptor) decreasing its electron density, thus activating it toward nucleophilic attack by the Michael donor (Scheme 1, Eq. 2).
On the other hand, amino-catalysis has gained considerable attention among the strong interactions through covalent bonding strategies. In fact, the presence of a carbonyl group on the Michael acceptor allows the formation of an iminium species (Scheme 1, Eq. 3) or, alternatively for carbonyl-derived Michael donors, they can be activated via formation of enamines (Scheme 1, Eq. 4).
Often a proton source, either present in the molecule or externally added, is crucial to achieve elevated yields because it increases catalyst turnover, speeding up the hydrolysis rate of the functionalized iminium or enamine.
Finally, some organocatalyzed Diels-Alder reactions of cyclic enones have been found to occur by a stepwise mechanism; the first one being a 1,4-conjugate addition. These unconventional Diels-Alder reactions obtained by cascade reaction will also be described in this review and compared with classical synchronous reactions.
This review intends to cover literature on the organocatalyzed 1,4-conjugate additions of cyclic enones until the end of 2010. The surveyed literature covers papers specifically devoted to this topic; many papers indicate cyclic enones among the examples of conjugate addition to α,β−unsaturated carbonyl compounds, but they are reviewed only when they brought a prominent enhancement to chemical knowledge.
2. Transfer Hydrogenations
The simplest nucleophile that can be added to an α,β-unsaturated carbonyl compound is the hydride ion. Actually, the enantioselective transfer hydrogenation of a wide range of β-substituted carbocycles was carried out in 2006 by MacMillan et al. [14] and List et al. [15] (Scheme 2).
They both used Hantzsch esters (2) as the hydrogen transfer reagent but different organocatalysts, the (2S,5S)-5-benzyl-3-methyl-2-(5-methyl-2-furyl)-4-oxoimidazolidinium trichloroacetate (4) by MacMillan et al. and a chiral binaphthol derived phosphate of l-valine (5) by List et al., that is a primary amine to reduce steric requirements in the iminium ion of the ketone with an enantiopure counterion. The reaction conditions are slightly different, MacMillan et al. reported 0 °C in diethyl ether and 20 mol % of catalyst 4; List et al. butyl ether at 60 °C but only 5 mol % of catalyst 5. Yields and enantiomeric excesses of reduced product 3 are comparable (66-89% yield, 88-96% ee for MacMillan et al. with respect to 68-99% yield, 84-98% ee for List et al.), but opposite enantiomers of the product 3 are obtained.
The possibility to accommodate severe steric constraints on the ketone component, and the scarce influence on the enantiocontrol with variation in its electronic nature, are among the advantages of MacMillan group’s reaction. Moreover, the structure of the dihydropyridine reagent seemed to have an important effect on the selectivity of the process, since improved enantiocontrol of the reaction was observed as the steric demand of the ester moiety increased. Finally, the stable iminium adduct of 3-benzyloxy-2-cyclopentenyl system is a suitable substrate for this reduction, since it generally undergoes β-benzyloxy elimination.
On the other hand, when List et al. used the opposite enantiomeric counteranion in the catalyst, interestingly, they obtained the same enantiomeric product, but with much lower enantioselectivity, illustrating a dramatic case of a matched/mismatched catalyst-ion pair combination. At the same time, List et al. affirmed that MacMillan catalyst worked under his reaction conditions with low conversion (40%) and enantioselectivity (50% ee).
From a mechanistic point of view, MacMillan group’s stereoselectivity hypothesis was later supported by DFT calculations on the energies of the species involved in the different possible conformation and configuration of the iminium ion. Only one face of the more stable (E)-iminium intermediate is accessible to attack by the Hantzsch ester (Figure 1) [16].
List group’s hypothesis, instead, was not followed by further investigations. However, he assumed the reaction proceeds via an iminium-phosphate ion pair that may be stabilized by hydrogen bonding interactions. In addition, the phosphate counteranion may also interact with the Hantzsch ester via an additional hydrogen bond.
3. 1,4-Conjugate Additions of Carbon Nucleophiles
In this section, the 1,4-conjugate addition of carbon nucleophiles to unsaturated cyclic ketones carried out by means of organocatalysis will be compiled, including cascade reactions.
3.1. Malonates and Related 1,3-Dicarbonyls
The Michael addition of a malonate nucleophile to cyclic enones is considered a worthwhile subject in asymmetric catalysis of cyclic enones. Yamaguchi group’s pioneering work in the 1990s introduced l-proline rubidium salt as a catalyst for the addition of diisopropyl and di-tert-butyl malonates to cyclic enones with low to good enantioselectivities (35-88% ee) [17,18]. Then List and co-workers treated a mixture of acetone and l-proline (35 mol %) in DMSO with cyclohex-2-en-1-one [19]. The expected Michael adduct was actually formed, but the enantioselectivity was unsatisfactory (15% yield, 20% ee).
During the last decade, many papers appeared on the Michael addition of a malonate nucleophile to cyclic enones reaching high yields and enantioselectivity; many catalysts were suggested and are listed in Table 1. The particular features of these reactions will be highlighted below.
The fundamental mechanistic idea is that malonate must be deprotonated (pKa diethyl malonate = 13) and the anion can attack the iminium ion. Thus, piperidine was added to the reaction mixture for this purpose (entry 1, Table 1) [20,21]. As reported below, nitroalkanes (pKa MeNO2 = 10) need weaker bases. Then l-proline functionalized mesoporous silica was employed as the catalyst under the same reaction conditions (i.e., piperidine in dichloromethane) in the asymmetric diethyl malonate addition reaction to cyclohex-2-en-1-one: 7.5% l-proline vs. silica molar ratio was found to give the best result (61% conversion, 70% ee) at 40 °C. Interestingly, the L-proline functionalized mesoporous silica could be reused many times without losing significant stereoselectivity and can be easily recovered being in heterogeneous phase [29].
Most recently, catalysts have been designed with further intramolecular amino functions thus avoiding the addition of the co-catalyst. Thus, guanidine or diamine moieties are bound to the asymmetric core of the catalyst.
Among them, the conjugate addition of malonate to cyclic enones by modified cinchona alkaloid catalysis (entry 4, Table 1) was found to be strictly dependent on the presence of both the primary amine on diaminocyclohexane and thiourea moieties. Their absence causes in fact a drastic decrease in enantioselectivity. Also, stereochemistry is controlled by 1,2-diaminocyclohexane motif; in fact if (1R,2R)-1,2-diaminocyclohexane was replaced with (1S,2S)-1,2-diaminocyclohexane the configuration of the adduct is reversed. Comparing data in Table 1, this reaction appears the most fruitful for the R-enantiomer [24].
Interestingly, although restricted to only a few instances, guanidine catalysts (entries 2, 5, 6, Table 1) have been studied in detail. In fact, the pKa of guanidinium ion (pKa ≈ 13) is well suited for deprotonation of the malonate. Modification of the guanidine moiety with enantiopure frameworks were scarcely successful (entry 2, Table 1), although the chosen substrate was surely the worst for testing the enantioselectivity of the catalyst (see below).
Better results were obtained when a guanidine motif was linked to l-proline. Moreover, in these cases, the ability of guanidinium ion to efficiently complex malonate ion by both electrostatic interaction and two-directional hydrogen bonds is suitable for the efficient syn-orientation with respect to the pyrrolidine substituent of nucleophilic attack of malonate (Figure 2).
The enantioselectivity of the reaction was then found to be dependent on dilution and a concomitant decrease in catalyst loading that improved the enantioselectivity, but at the expense of the yield. This behavior was observed both in dichloromethane and 1,2-dichloroethane, but, in the latter case, decreasing the catalyst loading was less detrimental to the overall yield. The reason for the effect of dilution on the enantioselection was hypothesized, supposing that at higher reaction concentrations, deprotonation of the malonate is faster than iminium ion formation and therefore results in a direct conjugate addition of malonate anion to cyclohex-2-en-1-one (background reaction). The poor enantioselectivity observed in malonate as the reaction medium, supports this proposal. Lowering the concentration of malonate, the rate of malonate deprotonation by free catalyst is reduced to allow iminium ion formation [25].
Moreover, the use of carboxylic acid salts of the catalysts was explored to render faster iminium formation and to allow a carboxylate assisted deprotonation of malonate. Pivalic acid was found to be very efficient owing to the basicity of its counteranion [26].
It should be noted also that acyclic guanidine catalyst is very efficient with diethylmalonate, when other catalysts need bulky alkoxy frameworks to give high enantioselectivity [26].
The Yamaguchi group’s idea of aminoacid salts used as the catalyst, has been recently resumed by Yoshida and co-workers. They observed that lipophilic O-tert-butyldiphenylsilyl-l-serine lithium salt is an effective catalyst in non-polar solvents (Table 1, entry 7). Authors supposed that, in the imine intermediate, the relatively bulky methylene group comes to the less-hindered side rather than the vinyl group and the Lewis acidic lithium ion coordinates with the nitrogen atom both to reduce the electron density of the β-carbon and to hold the side chain of the amino acid on the Re-face (Figure 3). Thus adduct 8 has the S-configuration. This procedure appears, until now, the simplest available protocol in order to obtain the S-isomer of 8 [27].
All malonates and cyclic enones react generally smoothly to generate adducts with high yields and excellent enantioselectivities, except 2-cyclopentenone, which gives 88%, 64%, 47% yield and 30%, 63%, 55% ee under Ley [21], Ye [24], and Yoshida [27] conditions respectively. The reason is mainly that the conformational control of the iminium intermediate derived from cyclopent-2-en-1-one is more difficult than when other enones are employed as substrates. Calculations of the energy difference between iminium intermediate A and B optimized at the HF/6-31G(d) level of theory, showed no clear energy difference (ΔE = 0.8 kcal/mol, Figure 4) [30].
This difficulty has been overcome using a chiral diamine–acid combination catalyst in methanol [23]. At the HF/6-31G(d) level of theory, the transition states C has a minimum of energy over transition states, where methanol or malonate are not involved in hydrogen bonding with the catalyst. The hydrogen bonding system in C forms a proton relay system to activate the malonate nucleophile with the aid of the methanol molecule that could indirectly catalyze the Michael reaction. Actually, this flexible catalytic system behaves the opposite of the classical catalyst. In fact, worse enantioselectivities are observed with larger cyclic systems (Table 1, entry 8).
Many other 1,3-dicarbonyl compounds are strictly related to malonates, but these substrates are less studied.
For example, Tan and co-workers found that efficiency of guanidinium catalyst 9 (Scheme 3) suffers from the acidity of the CH2 group, and the reaction with dimethylmalonate was relatively slow, giving the Michael adduct only a 20% yield after 96 h and only with 47% ee. Thus, they employed catalyst 9 at the more acidic dithiomalonates or β-ketothioesters and they obtained significant improvements in both yields (85-99%) and ee values (90-98%) [31]. This reaction also represents the first example of the organocatalytic enantioselective Michael addition of 1,3-dicarbonyl donors to lactones: a very difficult reaction due to the labile nature of lactones under strongly basic and Lewis acidic conditions. In fact the reaction with 2(5H)-furanone (10) with the hindered dithiomalonate 11b, yielded 85% of the adduct 12e with 96% ee [31].
The asymmetric Michael addition of 4-hydroxycoumarin (14), a cyclic 1,3-dicarbonyl compound, to cyclohept-2-en-1-one has been reported to occur with 78% yield and 94% ee using 9-amino-9-deoxyepiquinine TFA salt (13, Scheme 4) among the examples of a more general report on the conjugate addition of coumarin derivatives to unsaturated compounds [32].
3.2. Nitroalkanes
The base-catalyzed addition of nitroalkanes to cyclic enones is another fundamentally important preparative route to β-substituted cycloalkanones. In fact, the resulting nitroalkyl appendage and the resident carbonyl group can be chemically transformed into a variety of other functionally useful moieties.
The first example of a useful organocatalytic asymmetric version of this reaction was once more proposed by Yamaguchi and co-workers. [34] They succeeded in the addition of 2-nitropropane to cyclohex-2-en-1-one (59% ee) and cyclohept-2-en-1-one (79% ee) in the presence of rubidium l-prolinate, whereas enantioselectivity was substantially lower with nitromethane and 2-cyclohex-2-en-1-one (45% ee).
Then Hanessian’s group has done a great piece of work on this topic followed by other research groups over the years (Table 2).
Some features can be deduced from these studies. Above all, a base additive is often required. In Scheme 6, a plausible mechanism is depicted, where the catalytic cycle is based on l-proline as the catalyst, but can be extended to other catalysts with little modification, so it can be considered general. The high stereo-differentiation of the two enantiotopic faces arises from an approach of an ammonium nitronate 23, from interaction between the additive 21 and nitro derivative 20, from the less hindered Re-face of the double bond in the iminium ion 22. Notably, NMR studies showed that 2-nitropropane remained unchanged in the presence of piperazine, indicating that the equilibrium is not in favor of nitronate 23, but, evidently, enough quantities of nitronate 23 are however present during the catalytic cycle.
The presence of further non-covalent interactions, such as hydrogen bond between the iminium ion and the nitronate can invert this steric-directed stereoselection.
It should be noted that in virtue of its basicity, the additive can rapidly attack on the carbonyl group to give the corresponding iminium ions 24, thus catalyzing an asymmetric background reaction especially in very polar solvents [35].
Thus iminium ion 24 must be highly disfavored due to severe strain or steric effects with an accurate choice of the basic additive, rendering its population in the equilibrium negligible in comparison to the chiral iminium ion 22 from the organocatalyst [46].
Only trans-2,5-dimethylpiperazine, piperazine, piperidine, and morpholine, among the possible amines, provide this quality together with the basicity and the quasi-“optimal” spatial requirement as a countercation in the transition state; thus leading to the adduct with higher enantiomeric excess.
Another feature arising from the basic medium is the scarcely satisfactory results with nitromethane. The low yield of the nitromethane adduct can be likely attributed to a second addition reaction with another molecule of cycloenone [39,40]. However, increasing steric congestion of the substrate, this drawback can be overcome, as demonstrated by the reaction of 3-methylcyclohex-2-en-1-one with nitromethane under diethyl (2R)-tetrahydropyrro-2-ylphosphonate catalysis [43].
Finally, no stereocontrol was generally achieved at the exocyclic stereocentre, leading to very poor diastereomeric ratios when two stereocenters are formed. This poor selectivity is due to the acidity of the C-position near to the nitro group that undergoes epimerization under the basic conditions typically required for these reactions.
Comparison of the results for the nitroalkanes demonstrates the importance of steric effects for the reaction yields in many cases. The larger nucleophiles react slowly with the activated enone. The highest yield was therefore observed for nitromethane, but the highest selectivity with bulky nitroalkanes.
As in the case of malonates with Ye’s catalyst (entry 8, Table 2), the insertion of 1,2-diaminocyclohexane motif in the catalyst avoids the necessity of a co-catalyst and the configuration of the adduct depends on diaminocyclohexane stereochemistry [44]. Remarkably, 3-substituted cyclohex-2-enones endow adducts with 90-95% ee and 70-82% isolated yield. Notwithstanding the fact that a quaternary chiral carbon centre was produced, it is generally difficult to achieve high levels of selectivity. However, diastereoselectivity is not elevated owing to the cited epimerization by-process [44].
Under Pansare group’s conditions (entry 9, Table 2), the trend of nitroalkane addition in stereoselection was less predictable than with malonate, since lowering the catalyst loading, and increasing dilution, did not always increase enantioselectivity [25]. However, only in this reaction, nitromethane adduct does not react further, suggesting that these reaction conditions are mild enough to avoid further deprotonation of the substrate. Finally, products have the (S) configuration whereas most of l-proline derived catalysts provide the (R) products, owing to hydrogen bonding between nitronate and the protonated guanidine, as in the case of malonate (Figure 2).
A quite different approach was proposed by Maruoka and co-workers, who employed silylnitronates instead of nitroalkanes and N-spiro chiral quaternary ammonium bifluoride as the catalyst (Scheme 7). Both the (R,R)- [47] and the (S,S)-enantiomer [48] of the catalyst were employed with very good results. Scheme 7 shows results of the (R,R)-catalyst 27 [47], but very similar yields (94-99%), dr (from 64-36 to >95:5) and ee (81-93%) are reported for the (S,S)-catalyst 27’ [48].
Cyclopent-2-en-1-one gave moderate stereoselectivity, while high levels of catalytic efficiency and stereocontrol was observed with various silyl nitronates regardless of the length of carbon chain or the presence of additional oxygen-containing functional group. The most interesting feature is the high-obtained diastereoisomeric ratio, since the acidic reaction conditions did not allow the epimerization of the carbon bearing the nitro group. This served as a stimulus to attempt the construction of three consecutive stereo-defined carbon centers. The silyl enol ether 28, in fact, could react with electrophiles and the already present stereo-defined groups could address the diastereoselection. Actually, (R,R)-catalyst 27’ allowed the formation of compound 28 (n = 1, R = Et) in particularly high diastereo- and enantio-selectivity. Then the addition of N-bromosuccinimide, fluorotetraphenylbismuth or paraformaldehyde (mediated by dimethylaluminum chloride) allowed production of prevalent cis-α-bromo-, exclusive trans-α-phenyl- and α-hydroxymethyl- (as a single diastereoisomer, the geometry of which was not elucidated) cyclohexanone, respectively.
Recently α,β’-unsaturated β-ketoesters (29) have also been found to undergo addition of nitroalkanes in a quinine mediated transformation, where multiple stereocentres are formed in good stereoselectivity. For instance, significant results were achieved by employing 1-nitropropane, where essentially only one of the eight potential stereoisomers was isolated. Both the enol and keto form of compound 31 are monitored, but, very interestingly, the keto form is always a single diastereoisomer (with trans relationship between protons on the ring). Moreover, the interesting control of the selectivity in the α-position with respect to the nitro group has been ascribed to the use of quinine which is not a strong base, thus preventing the stereocentre epimerization (Scheme 8) [49].
As expected, employing quinidine (the “pseudo”-enantiomer of quinine) the major product is the opposite enantiomer of compounds as well as employing the quinine-based thiourea (32). Bromonitromethane (30e) led to the cyclopropyl derivative (see below). The desired adduct is obtained in good yields but in moderate enantioselectivity (52% ee).
Finally, the authors successfully achieved the addition of an electrophile, such as methyl vinyl ketone, on the Michael adduct intermediates (Scheme 9) [49]. Surprisingly, the resultant adduct from the nitromethane addition spontaneously cyclized to a bicyclic compound 33.
The examples proposed by Maruoka and Bella groups are clever applications of cascade reactions in this field. Actually, asymmetric organocatalytic conjugate addition of nucleophiles, followed by loss of a leaving group with cascade intramolecular ring closure, provides a highly efficient strategy for the synthesis of chiral cyclopropanes. These compounds are important structural motifs in many drugs and natural products and useful synthetic intermediates for organic synthesis. In particular, nitrocyclopropanes are a class of interesting cyclopropane compounds that found application as intermediates in many syntheses.
Historically, the first one-step enantioselective nitrocyclopropanation is through the phase transfer catalyzed reaction of α-bromocyclopentenone with nitromethane, but the yield (50%) and the enantioselectivity (62%) are moderate [50]. Arai et al. attempted also the de-nitro reduction to cyclopropane but with poor yield (26%). Other details of this reaction will be described later. Now most of the instances of cyclopropanation in order to form bicyclo[n.1.0]alkanes are carried out by bromonitromethane (29).
Scheme 10 describes the proposed mechanism. The formation of iminium cation 35 with the primary amine is generally promoted by an acid. On the other hand, a base deprotonates compound 30e forming the nitronate anion. The additional nitrogen atoms on the catalyst generate hydrogen bond(s) that put the bromonitroalkane anion over the cyclic double bond of iminium ion, owing to the spatial arrangement of the reported catalysts, and then an attack occurs from the Re-face of the cycle. Tautomerization of the imine intermediate results in the formation of nucleophilic enamine 36. Subsequent intramolecular substitution provides iminium ion 37, and, finally, the release of the catalyst by hydrolysis furnishes the nitrocyclopropane derivative 38 and allows the catalytic loop to re-start.
Based on this mechanism some catalyst-cocatalyst combinations have been proposed and they are summarized in Table 3.
From these reports, some interesting features can be seen. Tetrazole catalysis seems to suffer from both electronic and steric factors depending on the ring magnitude (entry 1, Table 3). In fact, 3-methoxycyclohex-2-en-1-one gives no reaction, whereas 3-methylcyclohex-2-en-1-one works well. On the other hand, methyl substitution in the 2- or 3-position on the cyclopent-2-en-1-one leads to the worst results under these conditions. Finally, 5-methyl-6-nitrobicyclo[3.1.0]hexan-2-one is the only substrate obtained as a 2:1 mixture of two diastereoisomers [51].
Cyclopent-2-en-1-one provided bycyclo compound 38 with good enantioselectivity, but in poor yield with thiourea derived catalyst (entry 4, Table 3), since the initial conjugate addition products of bromonitromethane with enones were recovered along with the nitrocyclopropane. [52] Conversely, only moderate enantioselectivity, but good yield was achieved with the very similar sulfonamide (entry 3, Table 3). Under these conditions, moreover, 3,5,5-trimethylcyclohex-2-en-1-one is unreactive probably due to its bulkiness [53].
Interestingly, the racemic 4-tert-amylcyclohex-2-en-1-one gave a single isomer with high enantioselectivity (up to >99% ee) under Cinchona alkaloid derived catalysis. Thus, these conditions were attempted to be applied in the kinetic resolution of 4-substituted cyclohex-2-en-1-ones. Actually, kinetic resolution efficiency depends on the bulkiness of the 4-substitutent. In fact, selectivity in the asymmetric nitrocyclopropanation decreases with smaller groups [55].
3.3. Other Carbon Nucleophiles
Malonates and nitroalkanes have been the most used compounds in the conjugate addition of nucleophiles to cyclic alkenones, since they are easily deprotonated. However, other examples of carbon nucleophiles are described in the literature and they are conveyed in this section.
For instance, cyclohex-2-en-1-one and cyclohept-2-en-1-one can enantioselectively dimerize in the 3-position in 90% and 80% yield with 88% ee and 87% ee, respectively, by using 3,4,5-tribenzyloxybenzyl cinchoninium bromide (39), as the catalyst (Scheme 11, for cyclohex-2-en-1-one) [56].
Interestingly, under these conditions only the most stable γ-anion is formed, as demonstrated by the non-reactivity of 4,4-dimethylcyclohex-2-enone. However, this anion has two reactive nucleophilic positions, but only the α-position reacts and the isomer arising from alkylation of the γ-position is never found. The only exception is represented by 3-methylcyclopent-2-enone, where the exocyclic γ-position partially overcomes the crowding of the formed quaternary centre.
β-Ketosulfones are very interesting compounds. For instance, Jørgensen and co-workers introduced benzothiazolyl-β-ketosulfones (41) as valuable starting material for the asymmetric synthesis of these compounds, using 9-amino-9-deoxyepiquinine TFA salt (42) as the catalyst in the first stage of the reaction [57]. In fact, they can be easily converted into enantioenriched alkenyl (44), alkynyl (45), and ketone derivatives (46) [57] as well as 6-substituted bicyclo[2.2.2]oct-5-en-2-ones (47) (Scheme 12) [58].
As observed in other reactions, cyclopent-2-en-1-one is responsible for the lowest enantioselectivities in the synthesis of 44 and 45, whereas the best results are obtained with cyclohept-2-en-1-one. As expected, the “pseudo”-enantiomer of the catalyst furnishes the products with virtually the same enantioselectivity and opposite configuration in this reaction, as well as in the previous described dimerization reaction.
Arai and co-workers used cyanomethylsulfone (49b) as well as nitromethane (49a) and cyanoacetate (49c) as the precursor for the synthesis of bicyclo[n.1.0] compounds under phase transfer catalysis (Scheme 13). In 1999, only pioneering works on organocatalysis were published and the governing parameters had not been discovered at that time. Thus, the better yield and selectivity often did not match each other, and were obtained under different reaction conditions for every tested compound [50].
α-Cyanosulfones (52) were later used in the reaction with acyclic and cyclic α,β-unsaturated ketones catalyzed by Cinchona alkaloid 53 [59]. In particular, the reaction of cyclic ketones is of interest for this review. (Scheme 14) Two stereocentres were simultaneously created in 58-81% yield, from 65:35 to 90:10 dr and 60-80% ee. The stereoselectivity control at the carbon ring as well as reactivity slightly decreased for cyclohept-2-en-1-one.
Χ-Ray analysis of crystals of compound 54 (Ar=Ph) allowed to unequivocally assign the (S,S) configurations for the two stereocentres and the same configuration was assigned to the other compounds. The same compound was submitted to the Baeyer-Villiger reaction that yielded the corresponding lactone with no erosion of the enantiomeric ratio.
The indole framework represents a privileged structure motif in a large number of natural products and therapeutic agents. In this regard, the development of effective asymmetric routes to indole architectures has attracted much attention. The pyrrole electron rich π-system of indole can undergo Friedel—Crafts alkylations or, from the downside, the C-3 position can act as a nucleophile and Michael-type-Friedel—Crafts reactions of these compounds are well known and has recently been reviewed [60,61].
Thus the reaction of indole with cyclohex-2-en-1-one was attempted but both stereoselectivity (29% ee) and chemical yield (56%) were decreased for this type of substrate with respect to acyclic substrates with 2 mol % of H8-BINOL phosphate as the catalyst in dichloromethane at room temperature. [62] On the other hand, Melchiorre and co-workers found that 9-amino(9-deoxy)epi-hydroquinine·2 d-N-Boc phenylglycine (10 mol%) yielded 65% of the product of the same reaction in 78% ee [63].
2-Oxoindoles, as well as benzofuranones, represent challenging substrates for conjugate additions and, owing to the abundance of indole and benzofuranone derived natural products bearing a quaternary stereocentre in the 3-position of the heterocycle, the asymmetric synthesis is very important. The 3-position of these heterocycles is acid enough to be easy deprotonated leading to nucleophiles that can attack unsaturated compounds. In particular, with cyclic enones they represent the access to functionalized molecules that contain vicinal quaternary and tertiary stereocentres. Recently the highly stereoselective addition of oxoindole (benzofuranone) to cyclic enones, using a readily available chiral primary Cinchona alkaloid amine catalyst (56) has been reported (Scheme 15) [64].
The most significant features of this reaction are the high degree of stereocontrol but decreased reactivity with 5-dimethylcyclohex-2-en-1-one, the classical and remarkably lower yield and enantioselectivity with cyclopent-2-en-1-one, and the almost perfect enantiocontrol with 3-phenyl-2-oxindole, albeit at the expense of the lowest diastereoselection. It should be noted that the “pseudo”-enantiomer 56’ allows the access to the other antipode of the products 57 in almost identical diastereo- and enantio-selectivity. Longer reaction times (72 h vs. 24 h) and higher catalyst loading (20 mol% vs. 10 mol%) are necessary for the reaction of benzofuranone.
4. 1,4-Conjugate Additions of Heteroatom Nucleophiles
In this section, the 1,4-conjugate additions, where sulfur, nitrogen, oxygen and selenium are the nucleophilic centers, are surveyed.
Among various types of this reaction, asymmetrical 1,4-conjugate addition of sulfur nucleophiles provides for example direct access to optically active sulfides that are versatile precursors for the synthesis of biologically interesting compounds.
The first asymmetric organocatalyzed (quinine 0.8 mol%) conjugate addition of a sulfur nucleophile (thiophenol derivatives and benzyl mercaptan) to cyclohex-2-en-1-one was achieved in 1977 by Wynberg et al. [65]. Very good yields (82-95%) and enantiomeric excesses of up to 46% were obtained. Various studies followed these preliminary results in order to improve the reaction with respect to the mechanism and reaction scope. These are listed in Table 4.
Wynberg himself performed a detailed mechanistic study of the cinchonidine-catalyzed addition of thiophenols to cyclic enones (Scheme 16). [66]
A bifunctional activation mode of the catalyst was proposed. The basic catalyst is supposed to deprotonate the thiol (58) as well as the electrophile to hydrogen bond to the catalyst β-hydroxy group and a ternary complex, formed by the protonated catalytic base, the thiolate anion and the enone, accounts for the transition state (60). The thiolate adds from the less hindered face that is the Re-face in the case of quinine depicted in Scheme 16. The authors also demonstrated that the addition of the thiophenolate anion to the β-carbon atom of the electrophile was indeed the rate and chirality-determining step. Reaction conditions and ee’s are reported in entry 2, Table 4.
However, also iminium ion catalysis can be employed in this reaction as demonstrated by catalysts reported in entries 6 and 9, Table 4.
The necessity of providing activation of the unsaturated carbonyl derivative by asymmetric catalysis as well as activation of the sulfur nucleophile envisaged to test the use of ionic liquid as the solvent. In fact, in these solvents nucleophilicity and dissociation constants are higher than in organic solvents.
Unfortunately, neat ionic liquids greatly improve the background addition of thiophenols to unsaturated ketones, so 15 organocatalysts were unsuccessful in addressing enantioselection [77].
The reaction has been found to be dependent on pressure; at 0.9 GPa yields were almost quantitative, but ee’s are lower than at atmospheric pressure (0.1 MPa) and their values are very poor (entry 3, Table 4) [69].
It should be noted that in the (thio)urea derived catalyst depicted in entries 5 [71] and 8 [75] (Table 4) water plays a very different role. In fact on the former reaction, molecular sieves have to be added as water scavenger, while in the latter the addition of 4 Å MS has a negative effect on the enantioselectivity of the reaction. The role of water on the enantioselectivity enhancement is not clear, but probably it plays a crucial role in non- or stabilizing the transition state via weak hydrogen bonding.
A quite different catalyst is the calyx[4]arene, proposed by Shimizu (entry 7, Table 4): an inherently chiral compound with no chiral residue. [74] The design of this catalyst contemplates an amino group and a hydroxy group to activate thiophenol by acid-base interaction and cyclohex-2-en-1-one by hydrogen bonding respectively, in order to obtain a transition state very similar to 60 and finally a bulky substituent (the 3,5-dimethylphenyl group) has selectively to block one of the enantio-face of cyclohex-2-en-1-one. However, the enantio selectivity of the reaction is unsatisfactory.
Most of the reactions reported in Table 4 used arylthiols as the nucleophiles. However, these compounds have little synthetic significance, since they are too stable to be converted into other useful organic motifs. In order to increase the synthetic value of the unsymmetrical addition of thiols to cyclic enones other sulfur nucleophiles were tested.
For instance the addition of benzylmercaptan is synthetically very interesting since benzyl group can be easily removed leading to free SH function. Unfortunately, Mukaiyama [64], Deng [70], and Shimizu [74], reported 1% ee (entry 1, Table 4) 21% (entry 4, Table 4) and 0% ee (entry 7, Table 4) respectively for the reaction of benzylmercaptan with cyclohex-2-en-1-one. Very recently, however, the addition of arylmethylmercaptans to cyclic enones has been successfully accomplished by a simple and commercially available catalyst: S-triphenylmethyl-l-cysteine (entry 9, Table 4) [76].
In 1981, Wynberg et al. reported the addition of thiocarboxylic S-acids (acetic and benzoic) to cyclohex-2-en- 1-one and 5,5-dimethylcyclohex-2-en-1-one in the presence of catalytic amounts of cinchonine and cinchonidine for the (S) and (R)-isomers respectively. The reaction was carried out in benzene, at ambient temperature for about 4 h yielding almost quantitatively products in about 50% ee. Authors claimed the possibility to convert the product for other interesting organic functionalities without affecting ee’s, but examples are not reported [78].
Also, for the addition of thiols, a cascade reaction has been proposed. In fact as described in Scheme 10 an enamine intermediate like 36, is formed under iminium ion catalysis. This compound acts as a nucleophile on any electrophilic motif present on the molecule. With this design in his mind, Cordova envisioned a simple catalytic route to the synthesis of tetrahydrothioxanthenones (65, X=S, Scheme 17) via an asymmetric domino reaction between 2-mercaptobenzaldehyde (62, X=S) and cyclic enones [79]. The reaction was carried out in dimethylformamide at −20 °C with good yields (70-78%) and moderate ee’s (38-62%).
The same reaction was then applied to the synthesis of tetrahydroxanthenones (65, X = O, n = 2) with very little modifications i.e., acetonitrile as the solvent, a reaction temperature of 40 °C, and the necessity of an acid co-catalyst such as 2-nitrobenzoic acid. [80] Moreover in this second paper, Cordova also substituted salicylic aldehydes, recovering 65 in moderate yields (51-56%) but with good ee’s (85-89%).
More recently for the same reaction, Xu et al. introduced the couple pyrrolidine-amino acid in order to obtain cooperative catalysis [81]. The idea was that the highly modular nature of the assembly should allow a more convenient optimization of the catalyst in order to achieve high enantioselectivity. Both partners are enantiopure but, notably, all the synthesized 65 showed the same R configuration, independent of the stereochemistry of the amino acid, implying that the pyrrolidine module plays the leading role in the enantioselectivity of the oxa-1,4-conjugate addition step (Figure 5).
Nevertheless, the best yields (86-95%) and enantioselectivities (80-98% ee) of this reaction are obtained with the couple 66/67 and they are much higher than Cordova’s, providing a very valuable entry to compounds 65, considering also the easy availability of the catalyst.
A very particular oxygen nucleophile is hydrogen peroxide. Its addition to cyclic enones allows the epoxidation of the double bond. Actually, upon treatment with hydrogen peroxide and a catalytic amount of a chiral primary amine salt oxabicyclo[n.1.0], compounds are obtained with 33-84% yields and 92-99% ee [82]. Diamine salts, owing to their bifunctional mode of activation, can activate both the enone substrate via iminium ion formation and hydrogen peroxide via general base catalysis. The best results were achieved with (1R,2R)-1,2-diphenylethane-1,2-diamine mono-(S)-TRIP salt (68) and 9-amino-9-deoxyepiquinine bis-TFA salt (13) for the two possible enantiomers (Scheme 18). α-Substituted enones were unreactive under the reaction conditions.
Only one instance has been carried out on the organocatalytic asymmetric conjugate reaction of selenium nucleophiles: the addition of selenophenols to cyclohex-2-en-1-ones catalyzed by cinchonidine to afford products in very low enantioselectivities (up to 43% ee) reported by Wynberg et al. in 1979 (Scheme 19) [83].
The asymmetric 1,4-conjugate addition of nitrogen nucleophiles to α,β-unsaturated carbonyl compounds allows the preparation of optically active β-amino carbonyl compounds, the key intermediate in the synthesis of many biologically important compounds [7].
Hydrazine derivatives are interesting nucleophiles, since they are suitable substrates for further modifications and posses enhanced nucleophilicity. On the other hand, they could also be competitors to amine-based catalysts because they are basic and react quickly with carbonyl groups. However, hydrazones (69) can represent the best compromise in this dilemma [84].
In fact, they add stereoselectively to cycloenones in the presence of Cinchona alkaloid derivative (70) as the catalyst (Scheme 20). The enantioselectivity is very dependent on the ring size and substitution pattern of the ring, whereas the yields are comparable in all the reported examples. Once more, the worst enantioselectivity is obtained for cyclopent-2-en-1-one, and in the six-membered ring systems, steric hindering in position 4 does not allow the reaction. The racemic 5-isopropylcyclohex-2-en-1-one gives a mixture of diastereoisomers (71 cis/trans 1:2) each one with good enantiomeric excess.
Enantiomerically pure aziridine is another essential motif of biologically interesting compounds, as well as a valuable chiral synthon for the stereoselective preparation of useful amine derivatives. A smart strategy is using a sequential approach based on the use of a suitable compound that should first act as an N-centred nucleophile, and then should become electrophilic to facilitate an intramolecular cyclization step in a similar manner to that already used for nitroalkanes.
An O-tosylated hydroxycarbamate (72) was found to be a valuable compound with the iminium–enamine tandem sequence necessary for this reaction. Moreover, the catalyst for the reaction is an amino acid-Cinchona alkaloid salt and both the pseudo-enantiomers of the catalyst (73 and 73’, the same as 56 with different counterions) are available, so both optical antipodes of the aziridine can be prepared (Scheme 21) [85,86].
Interestingly, the sense of asymmetric induction is mainly driven by the chiral primary amine on the Cinchona partner, but, conversely from what has previously been observed [81], the chiral cocatalyst is important to improve the general efficiency of the aziridination. In fact, mismatched combinations of the partners gave lower reactivity and slightly decreased stereoselectivity.
Under the optimum conditions, aziridines are obtained in 33-98% yields and 61-99% ee’s with comparable results for the two enantiomers. The only limitation is the α-substituted cyclic enones that proved to be unreactive, as was observed in the previously reported epoxidation reaction [82].
5. Diels-Alder
The Diels–Alder reaction is one of the most powerful pericyclic reactions for the construction of six-membered functionalized cyclic frameworks. Great efforts have been devoted to the development of organocatalytic enantioselective versions of this reaction, as it allows in principle the formation of four contiguous asymmetric centers.
The first reported instance for cyclic enones dates from 2002, when MacMillan and co-workers used their catalyst 4 (Scheme 2) for the enantioselective addition of cyclopentadiene to cyclic enones (Scheme 22). [87] In that reaction, small rings were found to exhibit the lowest selectivities. Authors envisaged that the inherent conformational restrictions did not exclude participation of both cis- and trans-iminium isomers in the Diels-Alder event. With larger ring size dienophiles, the enhanced torsional freedom around the N=C-alkyl bond should increasingly shield the trans-iminium Re-face, thus improving asymmetric induction.
An example of asymmetric [3 + 2] dipolar cycloaddition has also been reported. A covalent-bonding (the amino group), and a hydrogen-bonding (the remote hydroxy) motif were necessary to ensure the best selectivity, since they allow synergistic interaction between the organocatalyst and the two reactants (Scheme 23). Under optimal conditions, better results were obtained with catalysis by 70 [88].
Excellent diastereoselectivity (dr > 99:1) was noted in all reactions and very similar enantioselectivity was obtained for the optical antipode when the “pseudo”-enantiomer of 74 was used. Substrates 75 with an electron-donating substituent displayed higher reactivity.
Further examples of Diels–Alder products under organocatalytic conditions have also been obtained using the cyclic enone as the diene. These reactions take advantage of either the keto-enol tautomerism enhanced by hydrogen-bonding catalysts, or the iminium-enamine tautomerism for covalent-bonding catalysts. Moreover, two mechanisms are described for each type of catalyst. In Scheme 24, they are pictured for covalent-bonding catalysis, but, with little modifications, it accounts also for hydrogen-bonding catalysis. The first hypothesis of mechanism is a true cycloaddition, where the catalyst forms a chiral enamine 77 with the cycloenone that undergoes [4 + 2]pericyclization by the dienophile, leading to formation of the corresponding enamine 78. Hydrolysis by water finally affords the desired Diels–Alder adduct 79 and releases the catalyst (Scheme 24, Eq. a). Alternatively, a stepwise mechanism has been proposed: addition of the enamine 77 to the activated “dienophile”, followed by intramolecular 1,4-conjugate addition onto the intermediate 80, and, finally, hydrolysis (Scheme 24, Eq. b).
During this review, an unconventional Diels-Alder obtained by cascade reaction as depicted in Scheme 24, Eq. B, was already described in Section 3.3 (Scheme 10) [58].
According to these mechanisms, a series of dienophiles was tested. For instance, nitrostyrenes were found to be good dienophiles in this reaction under l-proline derived catalysts. At first Cordova demonstrated that 64 coupled with 2,4-dinitrosulfonic acid (30 mol %) catalyzes this asymmetric Diels–Alder reaction in 65-95% yields and with high stereoselectivity [65-86 % ee of the major (>25:1) diastereomer] [89].
In another report, the same reaction was performed affording 55-98% yields and 83-96% with the catalytic couple 81 (from catalyst 66, coupled with trifluoromethylbenzoic acid, Scheme 25). [90] It should be noted that a curious solvent, East China Sea water, was used for this reaction. The fact that seawater significantly enhances the performance of the catalyst in this Diels–Alder reaction was explained by assuming that NaCl plays other roles in the process besides the salt effect, which might stabilize the transition state through a hydrogen-bonding interaction with the acid, and favor the hydrolysis step to finish the catalytic cycle. Actually, the reaction carried out in brine accomplished products in comparable yields and ee’s.
A formal [4 + 2] cycloaddition between the enolic forms of both cyclic enones and aldehydes has been recently reported (“Formal”-TS, Figure 6). Actually, the true reaction mechanism is a combination of conjugate addition of an enamine (from asymmetric catalyst and arylacetaldehydes) to the cyclic enone (TS-1, Figure 6), followed by a classical aldol condensation of the intermediate (TS-2, Figure 6) [91].
The authors claim the most interesting feature of this reaction is that, conversely from most of the reactions described in this review, the catalytic couple is not formed by an amine and an acid, but by two amines. A couple between a Cinchona alkaloid derivative and an aminoacid has already been used by other research groups and examples have also been reported in this review, too [63,81,85,86]. However, in previous reports, Cinchona alkaloid derivatives act as the catalysts and drive the enantioselectivity, whereas aminoacid sometimes improves the general efficiency of the catalyst trough matched/mismatched combinations. In the present reaction, the amine motif of the aminoacid plays the pivotal role to enhance enantioselectivity, whereas Cinchona alkaloid derivative plays only a synergic role.
The yields were unsatisfactory (20-55%); dr (10:1) and ee (72-90%) are good for arylacetalaldehydes, but not for β-methylarylcetalaldehyde [3(71% ee):1(80% ee)].
As well as Diels–Alder reaction being one of the most powerful C-C bond-forming reactions, its aza-modification enables the preparation of nitrogen-containing heterocycles. The dienophiles are imines or nitroso derivatives. In the latter version, the bicyclic compounds are valuable intermediates for the synthesis of enantio enriched amino diols.
The accepted mechanism for these formal “aza-Diels-Alder” reactions is actually a two-step Mannich-1,4-conjugate addition resembling Eq. 2, Scheme 22.
Some reactions are based on the Brønsted acid catalyzed activation of the imine by a chiral acid such as H8-BINOL (entry 1, Table 5) [92] or BINOL phosphate (entries 2 and 3, Table 5) [93] and of the ketone. This second activation step could also be performed by an achiral acid, if both activation processes behave cooperatively. Generally better results are obtained with more sterically shielding aryl groups on the BINOL catalyst [93].
The reaction proposed by Gong et al. was attempted in a one-pot, three-component, asymmetric manner by reaction of cyclohex-2-en-1-one with p-methoxyaniline and aromatic aldehydes, and furnished the products in good yields with enantioselectivities similar to those observed for the corresponding reactions with preformed aldimines [92].
The same approach was envisaged by Cordova and co-workers (entry 4, Table 5) [94], for the reaction of formaldehyde and the appropriate aniline derivative. This is the sole reaction applied to cyclic enones other than cyclohex-2-en-1-one. In fact it has been applied to 4,4-dimethylcyclohex-2-en-1-one and cyclohept-2-en-1-one with good results.
However, 3-substituted cyclohex-2-en-1-one failed to undergo the second 1,4-conjugate step and only the α,β-unsaturated Mannich adduct recovered 40% yield and 94% ee. Moreover, the reaction with cyclopent-2-en-1-one only furnished trace amounts of the aza-Diels–Alder adduct.
The p-dodecylphenylsulfonamide-modified l-proline catalyst, proposed by Carter et al., (85 entry 5, Table 5) which is readily available also in its enantiomeric form, allows the formal aza-Diels-Alder process to be obtained with a completely reversed endo/exo selectivity favoring exclusively the exo product [95]. However, the yields are very unsatisfactory, especially with ortho-substituted arylimines (19%).
Very interestingly, the same reaction, extended to include aliphatic imines structures, led to a divergent reaction pathway. In fact it did not result in the formation of the corresponding azabicyclo[2.2.2]octane 84, but of the bicyclo[2.2.2]octane 86 (Scheme 26) [95].
In addition, the nitroso Diels-Alder-type reaction is actually a sequential nitroso aldol-1,4-conjugate addition, rather than a concerted reaction.
Nitroso compounds are ambident electrophiles and only an appropriate amine framework of enamine from cyclic enone, and a proper choice of acidity of Brønsted acid catalyst, play contributing roles in O/N regioselectivities in the nitroso aldol reaction. Chiral alcohols exhibit complete N-selection and a high enantioselection with piperidine or morpholine enamines. In contrast, l-proline derivative catalysts exhibit high O selectivity [96]. Thus the reaction of morpholino diene (87) derived from 4,4-dimethylcylohex-2-en-1-one, conducted in the presence of the BINOL catalyst 88 at −78 °C, in a mixture with pentane/dichloromethane as the solvent, led to the synthesis of 2-oxa-3-azabicyclo[2.2.2]octan-5-one 90 in moderate to good yields and enantioselectivity (Scheme 25). [97] The corresponding 4,4-diphenyl derivative failed to undergo the second reaction step and only nitroso aldol adduct was recovered.
On the other hand, 3-oxa-2-aza bicycloketones were obtained with pyrrolidinyl tetrazole, at 40 °C, with MeCN as the solvent. When cyclohept-2-en-1-one was used as diene precursor, l-proline provided a better yield than pyrrolidinyl tetrazole, but 4,4-diphenylcyclohex-2-en-1-one is reactive only with pyrrolidinyl tetrazole as the catalyst. Yields are generally moderate, but enantioselectivity are very high (Scheme 27) [97,98].
6. Desymmetrization of Cyclohexadienones
Dearomatization of aromatic compounds provides a useful cyclic synthetic building block due to its high economy and simple elegance. When coupled with a desymmetrization process it affords enantioenriched material from commonly available precursors in rapid fashion. This second step (desymmetrization) has been recently performed by means of organocatalysis.
The requisite substrates are generally accessible from the corresponding phenols, but their preparation is out of topic in this review, and readers are invited to refer to the original papers to have more details for their synthesis.
The first report on this reaction was the synthesis of bicyclo[4.3.0]nonenones (94). This skeleton was synthesized from an achiral precursor (92), via asymmetric intramolecular 1,4-conjugate reaction with creation of three contiguous asymmetric centers in a single step using a cysteine derivative as the catalyst. Diastereo and enantioselectivity yields are very good (Scheme 28) [99].
The asymmetric intramolecular Stetter reaction on substrate 95 proceeds with very good enantioselectivities and yields leading to hydrobenzofurans 97 using a chiral triazolium salt as the catalyst (Scheme 29) [100]. With all substrates, the reaction showed the same 95/5 diastereoselectivity.
The reaction was then extended to 2,4,6-trisubstituted phenols as the precursors and it provided an almost exclusively enantiopure single isomer of hexahydrobenzofuranone in 62-86% yields. Moreover, 3,4,5-trimethylphenol provided the corresponding 97 analogue in 64% yield as a 95/5 mixture of diastereomers the most abundant of which was almost enantiopure. Finally, the carbocyle analogue of 97, which is a hydrindane, is isolated in 60% yield and 90% ee of the major diastereomer (dr > 95/5).
The reactions described until now require the isolation of the meso-cyclohexadienone (92 or 95), but recently Gaunt’s group envisaged the possibility of a one-pot reaction by the realization of a method to oxidize the phenol ring without affecting the sensitive aldehyde function. The optimum reagent was found to be hypervalent iodine reagent PhI(OAc)2 (Scheme 30) [101].
The reaction was extended to biarylphenol to obtain comparable results to benzo-fused structures like 100. When MeCN was used as the nucleophile, the corresponding amide was recovered through a Ritter-type reaction. Finally, the stereochemistry of the products can be rationalized on the basis of a transition state that involves an endo-like attack onto the Si-face of the meso-cyclohexadienone (Scheme 30, red figure). The unnatural prolinol derivative 99 prevalently led to the diastereomer with trans relationship between the incoming nucleophile and exocyclic CHO group.
Recently chiral phosphoric acid 102 was found to catalyze the enantioselective oxo-1,4-conjugate reaction, of cyclohexa-2,5-dien-1-ones (101) leading to highly enantioenriched 1,4-dioxane derivatives (103, Scheme 31) [102].
The alkyl group at the 4-position of the cyclohexadienone has great influence on the enantioselectivity and reactivity, both decreasing with bulkier groups. This reaction was also applied to the synthesis of cleroindicins C, D, and F, natural products isolated from Clerodendum indicum, where the R group in compound 101 is interestingly a peroxo group (104).
A borderline reaction with respect to the topic of this review, is the addition of aldehydes [103] and β-ketoesters [104] to 1,4-quinones using organocatalysts. Actually, it is a “formal” 1,4-conjugate addition to unsaturated cyclic enones, but the products are substituted aromatic compounds with α-stereocentres. This reaction allows an easy-to-control one-pot synthesis of complicated polycyclic and spiro chiral compounds (Scheme 32).
7. Conclusions
In this review, we wish to focus reader attention on the development of effective catalysts for the enantioselective conjugate additions of malonate, nitroalkane and other carbon and heteroatom nucleophiles, to cycloenones and to cycloaddition reactions, which constitutes an important research field in synthetic organic chemistry. This research has become more important in recent years as most of the products are either building blocks or key intermediates in the synthesis of biologically and naturally occurring compounds, as well as drugs. Much work can still be carried out in order to increase enantioselectivity and yields, as the reader will have noted, most of the reactions showed satisfactory results only in one of the two termini of the dilemma of asymmetric synthesis.
References
- Michael, A. Ueber die Addition von Natriumacetessig- und Natriummalonsäureäthern zu den Aethern ungesättigter Säuren. J. Prakt. Chem. 1887, 35, 349–356. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael Additions to Nitroalkenes. Eur. J. Org. Chem. 2002, 1877–1894. [Google Scholar] [CrossRef]
- Almasi, D.; Alonso, D.A.; Nájera, C. Organocatalytic Asymmetric Conjugate Additions. Tetrahedron: Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 1701–1716. [Google Scholar] [CrossRef]
- Enders, D.; Wang, C.; Bats, J.W. Organocatalytic Asymmetric Domino Reactions: A Cascade Consisting of a Michael Addition and an Aldehyde α-Alkylation. Angew. Chem. Int. Ed. 2008, 47, 7539–7542. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.P.; Sunoj, R.B. The Role of Noninnocent Solvent Molecules in Organocatalyzed Asymmetric Michael Addition Reactions. Chem. Eur. J. 2008, 14, 10472–10485. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Wang, C.; Liebich, J.X. Organocatalytic Asymmetric Aza-Michael Additions. Chem. Eur. J. 2009, 15, 11058–11076. [Google Scholar] [CrossRef]
- Janecki, T.; Kedzia, J.; Wasek, T. Michael Additions to Activated Vinylphosphonates. Synthesis 2009, 1227–1254. [Google Scholar] [CrossRef]
- Li, N.; Xi, G.H.; Wu, Q.H.; Liu, W.H.; Ma, J.J.; Wang, C. Organocatalytic Asymmetric Michael Additions. Chin. J. Org. Chem. 2009, 29, 1018–1038. [Google Scholar]
- Bertelsen, S.; Jørgensen, K.A. Organocatalysis-After the Gold Rush. Chem. Soc. Rev. 2009, 38, 2178–2189. [Google Scholar] [CrossRef]
- Amarante, G.W.; Coelho, F. Organocatalysis Reactions with Chiral Amines. Mechanistic Aspects and Use on Organic Synthesis. Quim. Nova 2009, 32, 469–481. [Google Scholar] [CrossRef]
- Xu, L.W.; Luo, J.; Lu, Y.X. Asymmetric Catalysis with Chiral Primary Amine-based Organocatalysts. Chem. Commun. 2009, 1807–1821. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Lopez, R. Asymmetric Organocatalysis by Chiral Brønsted Bases: Implications and Applications. Chem. Soc. Rev. 2009, 38, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, J.B.; Ouellet, S.G.; MacMillan, D.W.C. Organocatalytic Transfer Hydrogenation of Cyclic Enones. J. Am. Chem. Soc. 2006, 128, 12662–12663. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.J.A.; List, B. Highly Enantioselective Transfer Hydrogenation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2006, 128, 13368–13369. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.; Iafe, R.G.; Houk, K.N. Origin of Stereoselectivity in the Imidazolidinone-Catalyzed Reductions of Cyclic α,β-Unsaturated Ketones. Org. Lett. 2009, 11, 4298–4301. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Shiraishi, T.; Hirama, M. A Catalytic Enantioselective Michael Addition of a Simple Malonate to Prochiral α,β-Unsaturated Ketoses and Aldehydes. Angew. Chem. Int. Ed. 1993, 32, 1176–1178. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Shiraishi, T.; Hirama, M. Asymmetric Michael Addition of Malonate Anions to Prochiral Acceptors Catalyzed by L-Proline Rubidium Salt. J. Org. Chem. 1996, 61, 3520–3530. [Google Scholar] [CrossRef]
- List, B. Asymmetric Aminocatalysis. Synlett 2001, 1675–1686. [Google Scholar] [CrossRef]
- Knudsen, K.R.; Mitchell, C.E.T.; Ley, S.V. Asymmetric Organocatalytic Conjugate Addition of Malonates to Enones Using a Proline Tetrazole Catalyst. Chem. Commun. 2006, 66–68. [Google Scholar] [CrossRef]
- Wascholowski, V.; Knudsen, K.R.; Mitchell, C.E.T.; Ley, S.V. A General Organocatalytic Enantioselective Malonate Addition to α,β-Unsaturated Enones. Chem. Eur. J. 2008, 14, 6155–6165. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Ebine, K.; Endo, M.; Araki, Y.; Fushimi, Y.; Miyamoto, I.; Ishikawa, T.; Isobe, T.; Fukuda, K. Guanidine-catalyzed Asymmetric Addition Reactions: Michael Reaction of Cyclopentenone with Dibenzyl Malonates and Epoxidation of Chalcone. Hetrocycles 2005, 66, 347–359. [Google Scholar]
- Yang, Y.-Q.; Zhao, G. Organocatalyzed Highly Enantioselective Michael Additions of Malonates to Enones by Using Novel Primary–Secondary Diamine Catalysts. Chem. Eur. J. 2008, 14, 10888–10891. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wen, S.; Yu, F.; Liu, Q.; Li, W.; Wang, Y.; Liang, X.; Ye, J. Enantioselective Organocatalytic Michael Addition of Malonates to α,β-Unsaturated Ketones. Org. Lett. 2009, 11, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Pansare, S.V.; Lingampally, R. Synthesis and Evaluation of Guanidinyl Pyrrolidines as Bifunctional Catalysts for Enantioselective Conjugate Additions to Cyclic Enones. Org. Biomol. Chem. 2009, 7, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Riguet, E. Novel Guanidinyl Pyrrolidine Salt-based Bifunctional Organocatalysts: Application in Asymmetric Conjugate Addition of Malonates to Enones. Tetrahedron Lett. 2009, 50, 4283–4285. [Google Scholar] [CrossRef]
- Yoshida, M.; Narita, M.; Hirama, K.; Hara, S. Asymmetric Michael Addition of Malonates to Enones Catalyzed by a Siloxy Amino Acid Lithium Salt. Tetrahedron Lett. 2009, 50, 7297–7299. [Google Scholar] [CrossRef]
- Mase, N.; Fukasawa, M.; Kitagawa, N.; Shibagaki, F.; Noshiro, N.; Takabe, K. Organocatalytic Enantioselective Michael Additions of Malonates to 2-Cyclopentenone. Synlett 2010, 2340–2344. [Google Scholar] [CrossRef]
- Adi Prasetyanto, E.; Lee, S.-C.; Jeong, S.-M.; Park, S.-E. Chiral Enhancement in Diethyl Malonate Addition by Morphosynthesized L-proline Mesoporous Silica. Chem. Commun. 2008, 1995–1997. [Google Scholar] [CrossRef]
- Dinér, P.; Nielsen, M.; Marigo, M.; Jørgensen, K.A. Enantioselective Organocatalytic Conjugate Addition of N-Heterocycles to α,β-Unsaturated Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 1983–1987. [Google Scholar] [CrossRef]
- Ye, W.; Jiang, Z.; Zhao, Y.; Goh, S.L.M.; Leow, D.; Soh, Y.-T.; Tan, C.-H. Chiral Bicyclic Guanidine as a Versatile Brønsted Base Catalyst for the Enantioselective Michael Reactions of Dithiomalonates and β-Keto Thioesters. Adv. Synth. Catal. 2007, 349, 2454–2458. [Google Scholar] [CrossRef]
- Xie, J.-W.; Yue, L.; Chen, W.; Du, W.; Zhu, J.; Deng, J.-G.; Chen, Y.-C. Highly Enantioselective Michael Addition of Cyclic 1,3-Dicarbonyl Compounds to α,β-Unsaturated Ketones. Org. Lett. 2007, 9, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Li, H.; Hong, R.; Deng, L. Construction of Quaternary Stereocenters by Efficient and Practical Conjugate Additions to α,β-Unsaturated Ketones with a Chiral Organic Catalyst. Angew. Chem. Int. Ed. 2006, 45, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Shiraishi, T.; Igarashi, Y.; Hirama, M. Catalytic Asymmetric Michael Addition of Nitroalkane to Enone and Enal. Tetrahedron Lett. 1994, 35, 8233–8236. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Jagtap, S.B.; Ardemasova, Z.A.; Kalikhevich, V.N. Trends in Asymmetric Michael Reactions Catalysed by Tripeptides in Combination with an Achiral Additive in Different Solvents. Eur. J. Org. Chem. 2004, 4014–4019. [Google Scholar] [CrossRef]
- Hanessian, S.; Pham, V. Catalytic Asymmetric Conjugate Addition of Nitroalkanes to Cycloalkenones. Org. Lett. 2000, 2, 2975–2978. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Jagtap, S.B.; Ardemasova, Z.A. 4-trans-Amino-proline Based Di- and Tetrapeptides as Organic Catalysts for Asymmetric C-C Bond Formation Reactions. Tetrahedron: Asymmetry 2006, 17, 989–992. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Jagtap, S.B. Dual Catalyst Control in the Chiral Diamine-Dipeptide-Catalyzed Asymmetric Michael Addition. Synlett 2004, 2624–2626. [Google Scholar] [CrossRef]
- Mitchell, C.E.T.; Brenner, S.E.; Garcia-Fortanet, J.; Ley, S.V. An Efficient, Asymmetric Organocatalyst-mediated Conjugate Addition of Nitroalkanes to Unsaturated Cyclic and Acyclic Ketones. Org. Biomol. Chem. 2006, 4, 2039–2049. [Google Scholar] [CrossRef]
- Mitchell, C.E.T.; Brenner, S.E.; Ley, S.V. A Versatile Organocatalyst for the Asymmetric Conjugate Addition of Nitroalkanes to Enones. Chem. Commun. 2005, 5346–5348. [Google Scholar] [CrossRef]
- Prieto, A.; Halland, N.; Jørgensen, K.A. Novel Imidazolidine-Tetrazole Organocatalyst for Asymmetric Conjugate Addition of Nitroalkanes. Org. Lett. 2005, 7, 3897–3900. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Shao, Z.; Warrier, J.S. Optimization of the Catalytic Asymmetric Addition of Nitroalkanes to Cyclic Enones with trans-4,5-Methano-L-proline. Org. Lett. 2006, 8, 4787–4790. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, M.; Granander, J.; Amedjkouh, M. Asymmetric Conjugate Addition of Nitroalkanes to Enones with a Chiral α-Aminophosphonate Catalyst. Tetrahedron: Asymmetry 2008, 19, 1934–1940. [Google Scholar] [CrossRef]
- Li, P.; Wang, Y.; Liang, X.; Ye, J. Asymmetric multifunctional organocatalytic Michael addition of nitroalkanes to α,β-unsaturated ketones. Chem. Commun. 2008, 3302–3304. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lu, Y. Primary Amine/(+)-CSA Salt-Promoted Organocatalytic Conjugate Addition of Nitro Esters to Enones. Org. Lett. 2010, 12, 2278–2281. [Google Scholar] [CrossRef]
- Hanessian, S.; Govindan, S.; Warrier, J.S. Probing the “Additive Effect” in the Proline and Proline Hydroxamic Acid Catalyzed Asymmetric Addition of Nitroalkanes to Cyclic Enones. Chirality 2005, 17, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Doda, K.; Takada, S.; Maruoka, K. Asymmetric Michael Addition of Silyl Nitronates to Cyclic α,β-Unsaturated Ketones Catalyzed by Chiral Quaternary Ammonium Bifluorides: Isolation and Selective Functionalization of Enol Silyl Ethers of Optically Active γ-Nitro Ketones. Tetrahedron Lett. 2006, 47, 145–148. [Google Scholar] [CrossRef]
- Ooi, T.; Takada, S.; Fujioka, S.; Maruoka, K. N-Spiro Chiral Quaternary Ammonium Bromide Catalyzed Diastereo- and Enantioselective Conjugate Addition of Nitroalkanes to Cyclic α,β-Unsaturated Ketones under Phase-Transfer Conditions. Org. Lett. 2005, 7, 5143–5146. [Google Scholar] [CrossRef]
- Piovesana, S.; Schietroma, D.M.S.; Tulli, L.G.; Monaco, M.R.; Bella, M. Unsaturated β-Ketoesters as Versatile Electrophiles in Organocatalysis. Chem. Commun. 2010, 46, 5160–5162. [Google Scholar] [CrossRef]
- Arai, S.; Nakayama, K.; Ishida, T.; Shioiri, T. Asymmetric Cyclopropanation Reaction under Phase-transfer Catalyzed Conditions. Tetrahedron Lett. 1999, 40, 4215–4218. [Google Scholar] [CrossRef]
- Wascholowski, V.; Hansen, H.M.; Longbottom, D.A.; Ley, S.V. A General Organocatalytic Enantioselective Nitrocyclopropanation Reaction. Synthesis 2008, 1269–1275. [Google Scholar]
- Dong, L.-T.; Du, Q.-S.; Lou, C.-L.; Zhang, J.-M.; Lu, R.-J.; Yan, M. Asymmetric Synthesis of Nitrocyclopropanes Catalyzed by Chiral Primary Amines. Synlett 2010, 266–270. [Google Scholar]
- Du, Q.-S.; Dong, L.-T.; Wang, J.-J.; Lu, R.-J.; Yan, M. Asymmetric Conjugate Addition of Bromonitromethane to Cyclic Enones Catalyzed by Chiral Monosulfonated Diamines. Arkivoc 2009, XIV, 191–199. [Google Scholar]
- Hansen, H.M.; Longbottom, D.A.; Ley, S.V. A New Asymmetric Organocatalytic Nitrocyclopropanation Reaction. Chem. Commun. 2006, 4838–4840. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, J.; Lin, Z.; Wang, Y. Enantioselective Synthesis of Functionalized Nitrocyclopropanes by Organocatalytic Conjugate Addition of Bromonitroalkanes to α,β-Unsaturated Enones. Chem. Eur. J. 2009, 15, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, R.; Insogna, S.; Bella, M. Organocatalytic Regioselective Michael Additions of Cyclic Enones via Asymmetric Phase Transfer Catalysis. Org. Biomol. Chem. 2006, 4, 4281–4284. [Google Scholar] [CrossRef]
- Paixão, M.W.; Holub, N.; Vila, C.; Nielsen, M.; Jørgensen, K.A. Trends in Organocatalytic Conjugate Addition to Enones: An Efficient Approach to Optically Active Alkynyl, Alkenyl, and Ketone Products. Angew. Chem. Int. Ed. 2009, 48, 7338–7342. [Google Scholar] [CrossRef]
- Holub, N.; Jiang, H.; Paixão, M.W.; Tiberi, C.; Jørgensen, K.A. An Unexpected Michael–Aldol–Smiles Rearrangement Sequence for the Synthesis of Versatile Optically Active Bicyclic Structures by Using Asymmetric Organocatalysis. Chem. Eur. J. 2010, 16, 4337–4346. [Google Scholar] [CrossRef]
- Cid, M.B.; Lopez-Cantarero, J.; Duce, S.; Ruano, J.L.G. Enantioselective Organocatalytic Approach to the Synthesis of α,α-Disubstituted Cyanosulfones. J. Org. Chem. 2009, 74, 431–434. [Google Scholar] [CrossRef]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic Strategies for the Asymmetric Functionalization of Indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A. Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed. 2009, 48, 9608–9644. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.-Y.; Lu, A.-D.; Zhou, Z.-H.; Zhao, G.-F.; He, L.-N.; Tang, C.-C. Chiral Phosphoric Acid Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles with Simple α,β-Unsaturated Aromatic Ketones. Eur. J. Org. Chem. 2008, 1406–1410. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Carlone, A.; Pesciaioli, F.; Sambri, L.; Melchiorre, P. Organocatalytic Asymmetric Friedel-Crafts Alkylation of Indoles with Simple α,β-Unsaturated Ketones. Org. Lett. 2007, 9, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Pesciaioli, F.; Tian, X.; Bencivenni, G.; Bartoli, G.; Melchiorre, P. Organocatalytic Asymmetric Conjugate Additions of Oxindoles and Benzofuranones to Cyclic Enones. Synlett 2010, 1704–1708. [Google Scholar]
- Helder, R.; Arends, R.; Bolt, W.; Hiemstra, H.; Wynberg, H. Alkaloid Catalyzed Asymmetric Synthesis III the Addition of Mercaptans to 2-Cyclohexene-1-one; Determination of Enantiomeric Excess Using 13C NMR. Tetrahedron Lett. 1977, 18, 2181–2182. [Google Scholar] [CrossRef]
- Hiemstra, H.; Wynberg, H. Addition of Aromatic Thiols to Conjugated Cycloalkenones, Catalyzed by Chiral β-Hydroxy Amines. A Mechanistic Study of Homogeneous Catalytic Asymmetric Synthesis. J. Am. Chem. Soc. 1981, 103, 417–430. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Ikegawa, A.; Suzuki, K. Highly Enantioselective Michael Addition of Thiols to 2-Cyclohexenone by Using (2S,4S)-2-(Anilinomethyl)-1-ethyl-4-hydroxypyrrolidine as a Chiral Catalyst. Chem. Lett. 1981, 10, 165–168. [Google Scholar] [CrossRef]
- Suzuki, K.; Ikegawa, A.; Mukaiyama, T. The Enantioselective Michael Addition of Thiols to Cycloalkenones by Using (2S, 4S)-2-Anilinomethyl-1-ethyl-4-hydroxypyrrolidine as Chiral Catalyst. Bull. Chem. Soc. J. 1982, 55, 3277–3282. [Google Scholar] [CrossRef]
- Sera, A.; Takagi, K.; Katayama, H.; Yamada, H.; Matsumoto, K. High-pressure Asymmetric Michael Additions of Thiols, Nitromethane, and Methyl Oxoindancarboxylate to Enones. J. Org. Chem. 1988, 53, 1157–1161. [Google Scholar] [CrossRef]
- McDaid, P.; Chen, Y.; Deng, L. A Highly Enantioselective and General Conjugate Addition of Thiols to Cyclic Enones with an Organic Catalyst. Angew. Chem. Int. Ed. 2002, 41, 338–340. [Google Scholar] [CrossRef]
- Li, B.-J.; Jiang, L.; Liu, M.; Chen, Y.-C.; Ding, L.-S.; Wu, Y. Asymmetric Michael Addition of Arylthiols to α,β-Unsaturated Carbonyl Compounds Catalyzed by Bifunctional Organocatalysts. Synlett 2005, 603–606. [Google Scholar] [CrossRef]
- Kumar, A. Akanksha Amino Acid Catalyzed Thio-Michael Addition Reactions. Tetrahedron 2007, 63, 11086–11092. [Google Scholar] [CrossRef]
- Shirakawa, S.; Moriyama, A.; Shimizu, S. Design of a Novel Inherently Chiral Calix[4]arene for Chiral Molecular Recognition. Org. Lett. 2007, 9, 3117–3119. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Kimura, T.; Murata, S.-I.; Shimizu, S. Synthesis and Resolution of a Multifunctional Inherently Chiral Calix[4]arene with an ABCD Substitution Pattern at the Wide Rim: The Effect of a Multifunctional Structure in the Organocatalyst on Enantioselectivity in Asymmetric Reactions. J. Org. Chem. 2009, 74, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Rana, N.K.; Selvakumar, S.; Singh, V.K. Highly Enantioselective Organocatalytic Sulfa-Michael Addition to α,β-Unsaturated Ketones. J. Org. Chem. 2010, 75, 2089–2091. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ohno, Y.; Hara, S. Organocatalytic Asymmetric Thio-Michael Addition of Arylmethyl Mercaptans to Cyclic Enones by a Primary Amino Acid. Tetrahedron Lett. 2010, 51, 5134–5136. [Google Scholar] [CrossRef]
- Meciarova, M.; Toma, S.; Kotrusz, P. Michael Addition of Thiols to α-Enones in Ionic Liquids with and without Organocatalysts. Org. Biomol. Chem. 2006, 4, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Gawronski, J.; Gawronska, K.; Wynberg, H. Alkaloid-catalysed Asymmetric Addition of Thiocarboxylic S-Acids to Cyclohex-2-en-1-ones. Absolute Configuration of the Adducts by O,S-Dibenzoate Cotton Effect. J. Chem. Soc., Chem. Commun. 1981, 307–308. [Google Scholar] [CrossRef]
- Rios, R.; Sundén, H.; Ibrahem, I.; Zhao, G.-L.; Córdova, A. A One-pot Organocatalytic Asymmetric Entry to Tetrahydrothioxanthenones. Tetrahedron Lett. 2006, 47, 8679–8682. [Google Scholar] [CrossRef]
- Rios, R.; Sundén, H.; Ibrahem, I.; Córdova, A. A Simple and Concise Catalytic Asymmetric Entry to Tetrahydroxanthenones. Tetrahedron Lett. 2007, 48, 2181–2184. [Google Scholar] [CrossRef]
- Xia, A.-B.; Xu, D.-Q.; Luo, S.-P.; Jiang, J.-R.; Tang, J.; Wang, Y.-F.; Xu, Z.-Y. Dual Organocatalytic Ion-Pair Assemblies: A Highly Efficient Approach for the Enantioselective Oxa-Michael–Mannich Reaction of Salicylic Aldehydes with Cyclohexenones. Chem. Eur. J. 2010, 16, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Reisinger, C.M.; List, B. Catalytic Asymmetric Epoxidation of Cyclic Enones. J. Am. Chem. Soc. 2008, 130, 6070–6071. [Google Scholar] [CrossRef] [PubMed]
- Pluim, H.; Wynberg, H. Alkaloid Catalysed Asymmetric Synthesis. The Addition of Selenophenols to 2-Cyclohexen-1-ones and Conversion to Optically Active Allylic Alcohols. Tetrahedron Lett. 1979, 20, 1251–1254. [Google Scholar] [CrossRef]
- Perdicchia, D.; Jørgensen, K.A. Asymmetric Aza-Michael Reactions Catalyzed by Cinchona Alkaloids. J. Org. Chem. 2007, 72, 3565–3568. [Google Scholar] [CrossRef] [PubMed]
- Pesciaioli, F.; de Vincentiis, F.; Galzerano, P.; Bencivenni, G.; Bartoli, G.; Mazzanti, A.; Melchiorre, P. Organocatalytic Asymmetric Aziridination of Enones. Angew. Chem. Int. Ed. 2008, 47, 8703–8706. [Google Scholar] [CrossRef]
- de Vincentiis, F.; Bencivenni, G.; Pesciaioli, F.; Mazzanti, A.; Bartoli, G.; Galzerano, P.; Melchiorre, P. Asymmetric Catalytic Aziridination of Cyclic Enones. Chem. Asian J. 2010, 5, 1652–1656. [Google Scholar] [CrossRef] [PubMed]
- Northrup, A.B.; MacMillan, D.W.C. The First General Enantioselective Catalytic Diels-Alder Reaction with Simple α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2002, 124, 2458–2460. [Google Scholar] [CrossRef]
- Chen, W.; Du, W.; Duan, Y.-Z.; Wu, Y.; Yang, S.-Y.; Chen, Y.-C. Enantioselective 1,3-Dipolar Cycloaddition of Cyclic Enones Catalyzed by Multifunctional Primary Amines: Beneficial Effects of Hydrogen Bonding. Angew. Chem. Int. Ed. 2007, 46, 7667–7670. [Google Scholar] [CrossRef]
- Sundén, H.; Rios, R.; Xu, Y.; Eriksson, L.; Córdova, A. Direct Enantioselective Synthesis of Bicyclic Diels–Alder Products. Adv. Synth. Catal. 2007, 349, 2549–2555. [Google Scholar] [CrossRef]
- Xu, D.-Q.; Xia, A.-B.; Luo, S.-P.; Tang, J.; Zhang, S.; Jiang, J.-R.; Xu, Z.-Y. In Situ Enamine Activation in Aqueous Salt Solutions: Highly Efficient Asymmetric Organocatalytic Diels–Alder Reaction of Cyclohexenones with Nitroolefins. Angew. Chem. Int. Ed. 2009, 48, 3821–3824. [Google Scholar] [CrossRef]
- Bella, M.; Schietroma, D.M.S.; Cusella, P.P.; Gasperi, T.; Visca, V. Synergic Asymmetric Organocatalysis (SAOc) of Cinchona alkaloids and Secondary Amines in the Synthesis of Bicyclo[2.2.2]octan-2-ones. Chem. Commun. 2009, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. Enantioselective Direct Aza Hetero-Diels-Alder Reaction Catalyzed by Chiral Brønsted Acids. Org. Lett. 2006, 8, 6023–6026. [Google Scholar] [CrossRef]
- Rueping, M.; Azap, C. Cooperative Coexistence: Effective Interplay of Two Brønsted Acids in the Asymmetric Synthesis of Isoquinuclidines. Angew. Chem. Int. Ed. 2006, 45, 7832–7835. [Google Scholar] [CrossRef] [PubMed]
- Sundén, H.; Ibrahem, I.; Eriksson, L.; Córdova, A. Direct Catalytic Enantioselective Aza-Diels–Alder Reactions. Angew. Chem. Int. Ed. 2005, 44, 4877–4880. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Carter, R.G. Asymmetric Construction of Nitrogen-Containing [2.2.2] Bicyclic Scaffolds Using N-(p-Dodecylphenylsulfonyl)-2-pyrrolidinecarboxamide. J. Org. Chem. 2009, 74, 5151–5156. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, H. Brønsted Acid Catalysis of Achiral Enamine for Regio- and Enantioselective Nitroso Aldol Synthesis. J. Am. Chem. Soc. 1081, 127, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, Y.; Yamamoto, H. Diastereo- and Enantioselective Synthesis of Nitroso Diels-Alder-Type Bicycloketones Using Dienamine: Mechanistic Insight into Sequential Nitroso Aldol/Michael Reaction and Application for Optically Pure 1-Amino-3,4-diol Synthesis. J. Am. Chem. Soc. 2007, 129, 1190–1195. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Momiyama, N.; Yamamoto, H. Enantioselective Tandem O-Nitroso Aldol/Michael Reaction. J. Am. Chem. Soc. 2004, 126, 5962–5963. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Tamura, T.; Yamaguchi, H.; Masui, R.; Shoji, M. Cysteine-Derived Organocatalyst in a Highly Enantioselective Intramolecular Michael Reaction. J. Am. Chem. Soc. 2005, 127, 16028–16029. [Google Scholar] [CrossRef]
- Liu, Q.; Rovis, T. Asymmetric Synthesis of Hydrobenzofuranones via Desymmetrization of Cyclohexadienones Using the Intramolecular Stetter Reaction. J. Am. Chem. Soc. 2006, 128, 2552–2553. [Google Scholar] [CrossRef]
- Vo, N.T.; Pace, R.D.M.; O’Har, F.; Gaunt, M.J. An Enantioselective Organocatalytic Oxidative Dearomatization Strategy. J. Am. Chem. Soc. 2007, 130, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Rong, Z.-Q.; Zheng, C.; You, S.-L. Desymmetrization of Cyclohexadienones via Brønsted Acid-Catalyzed Enantioselective Oxo-Michael Reaction. J. Am. Chem. Soc. 2010, 132, 4056–4057. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S.; Maerten, E.; Overgaard, J.; Jørgensen, K.A. Asymmetric Organocatalytic α-Arylation of Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 5520–5523. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Richter, B.; Jørgensen, K.A. Organocatalytic Highly Enantioselective α-Arylation of β-Ketoesters. Angew. Chem. Int. Ed. 2007, 46, 5515–5519. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Main organocatalytic pathways: Michael acceptor (red), Michael donor (blue), asymmetric catalyst (magenta): (Eq. 1) ion pairing mechanism; (Eq. 2) Hydrogen bonding mechanism; (Eq. 3) Iminium ion (acceptor) mechanism; (Eq. 4) Enamine (donor) mechanism.
Scheme 1.
Main organocatalytic pathways: Michael acceptor (red), Michael donor (blue), asymmetric catalyst (magenta): (Eq. 1) ion pairing mechanism; (Eq. 2) Hydrogen bonding mechanism; (Eq. 3) Iminium ion (acceptor) mechanism; (Eq. 4) Enamine (donor) mechanism.

Scheme 2.
MacMillan (top) and List (bottom) transfer hydrogenation reaction.

Figure 1.
Calculated minimum energy transition state for MacMillan’s catalyst.

Figure 2.
Envisaged transition state for the addition of malonates to cyclohex-2-en-1-one with guanidine catalysts.
Figure 2.
Envisaged transition state for the addition of malonates to cyclohex-2-en-1-one with guanidine catalysts.

Figure 3.
Envisaged transition state for the addition of malonates to cyclohex-2-en-1-one with amino acid lithium salt catalyst.
Figure 3.
Envisaged transition state for the addition of malonates to cyclohex-2-en-1-one with amino acid lithium salt catalyst.

Figure 4.
Calculated most stable configurations of iminium ions of cyclopent-2-en-1-one (A, B) and of transition state (C) with involvement of methanol.
Figure 4.
Calculated most stable configurations of iminium ions of cyclopent-2-en-1-one (A, B) and of transition state (C) with involvement of methanol.

Scheme 3.
Asymmetric addition of dithiomalonates or β-ketothioesters to cyclic enones.

Scheme 4.
Asymmetric addition of 4-hydroxycoumarin to cyclohept-2-en-1-one.

Scheme 5.
Asymmetric addition of cyclic α-substituted ketoesters to cyclic enones.

Scheme 6.
Envisaged mechanism for the addition of nitroalkane mediated by piperazine.

Scheme 7.
Asymmetric addition of silylnitronates to cyclic enones and its use in cascade reactions.

Scheme 8.
Asymmetric addition of nitroalkenes to α,β’-unsaturated β-ketoesters.

Scheme 9.
Asymmetric cascade reaction to α,β’-unsaturated β-ketoesters.

Scheme 10.
Envisaged mechanism for the cascade cyclopropanation reaction.

Scheme 11.
Asymmetric dimerization of cyclohex-2-en-1-one.

Scheme 12.
Asymmetric addition of β-ketosulfones to cyclic enones and its use in synthesis.

Scheme 13.
Asymmetric synthesis of bicyclo[n.1.0] compounds.

Scheme 14.
Asymmetric addition of α-cyanosulfones to cyclic enones.

Scheme 15.
Conjugate addition of 2-oxoindoles or benzofuranones to cyclic enones.

Scheme 16.
Proposed mechanism for the quinine-catalyzed addition of thiols to α,β-unsaturated cyclic ketones.
Scheme 16.
Proposed mechanism for the quinine-catalyzed addition of thiols to α,β-unsaturated cyclic ketones.

Scheme 17.
Cascade reaction between 2-substituted benzaldehydes and α,β-unsaturated cyclic ketones: General reaction (top) and envisaged mechanism (bottom).
Scheme 17.
Cascade reaction between 2-substituted benzaldehydes and α,β-unsaturated cyclic ketones: General reaction (top) and envisaged mechanism (bottom).

Figure 5.
Best catalytic couple (left) and envisaged transition state by Xu et al. (right).

Scheme 18.
Enantioselective epoxidation of cyclic enones.

Scheme 19.
Asymmetric addition of selenides to cyclohex-2-en-1-ones.

Scheme 20.
Reaction (left), catalyst (right), envisaged transition states for cyclohex-2-en-1-one (bottom) for the addition of hydrazones to cyclic enones.
Scheme 20.
Reaction (left), catalyst (right), envisaged transition states for cyclohex-2-en-1-one (bottom) for the addition of hydrazones to cyclic enones.

Scheme 21.
Enantioselective aziridination of cyclic enones.

Scheme 22.
Enantioselective addition of cyclopentadiene to cyclic enones.

Scheme 23.
Asymmetric [3 + 2] dipolar cycloaddition to cyclic enones.

Scheme 24.
Cycloaddition (Eq. 1) and step mechanism (Eq. 2) for the formal organocatalytic Diels-Alder addition under covalent-bonding conditions.
Scheme 24.
Cycloaddition (Eq. 1) and step mechanism (Eq. 2) for the formal organocatalytic Diels-Alder addition under covalent-bonding conditions.

Scheme 25.
Asymmetric “pseudo” Diels-Alder reaction of nitrostyrenes and cyclic enones.

Figure 6.
Formal [4 + 2] cycloaddition between the enolic forms of both cyclic enones and aldehydes (left). True transition states of the stepwise mechanism (center and right).
Figure 6.
Formal [4 + 2] cycloaddition between the enolic forms of both cyclic enones and aldehydes (left). True transition states of the stepwise mechanism (center and right).

Scheme 26.
Formal Diels Alder reaction of aliphatic imines.

Scheme 27.
Formal nitroso Diels-Alder reaction: N-selectivity (top), O-selectivity (bottom).

Scheme 28.
Intramolecular 1,4-conjugate addition with creation of three asymmetric centers.

Scheme 29.
Asymmetric intramolecular Stetter reaction.

Scheme 30.
Reaction scheme (black), catalyst (magenta), envisaged transition state (red).

Scheme 31.
Enantioselective oxo-1,4-conjugate addition leading to 1,4-dioxane derivatives.

Scheme 32.
“Formal” 1,4-conjugate addition to quinines.


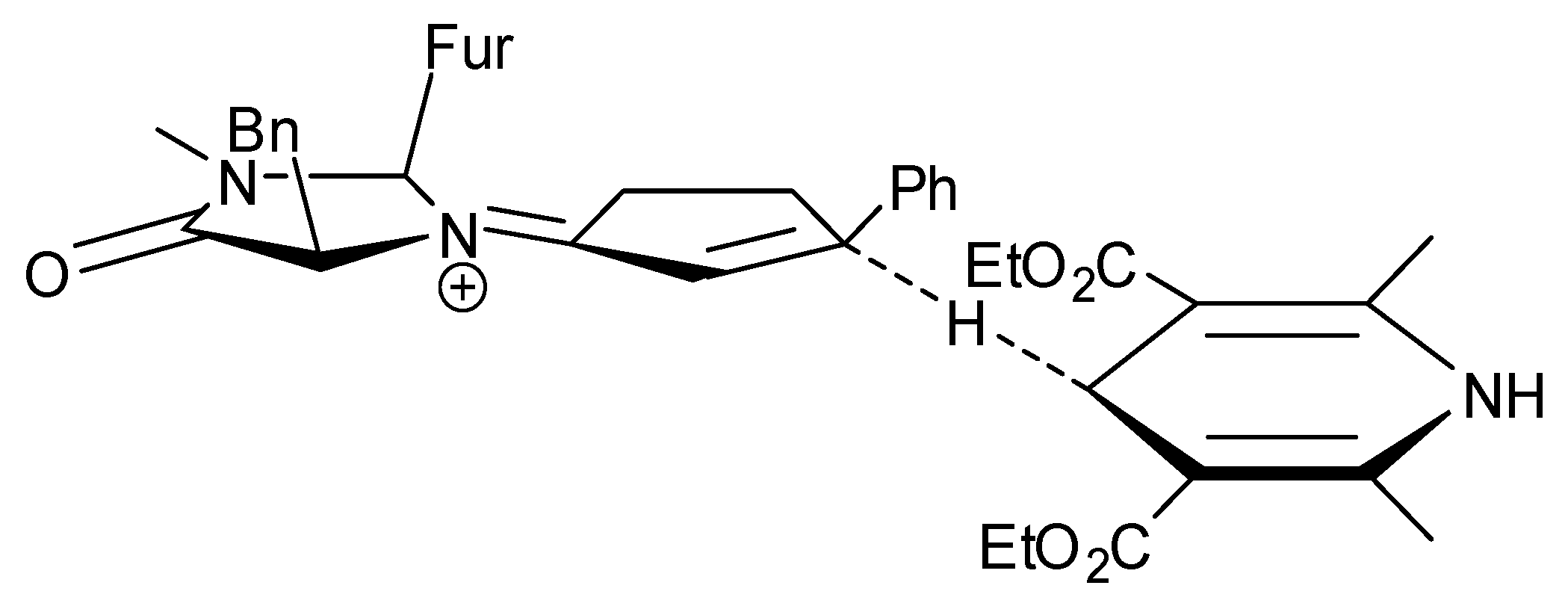{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
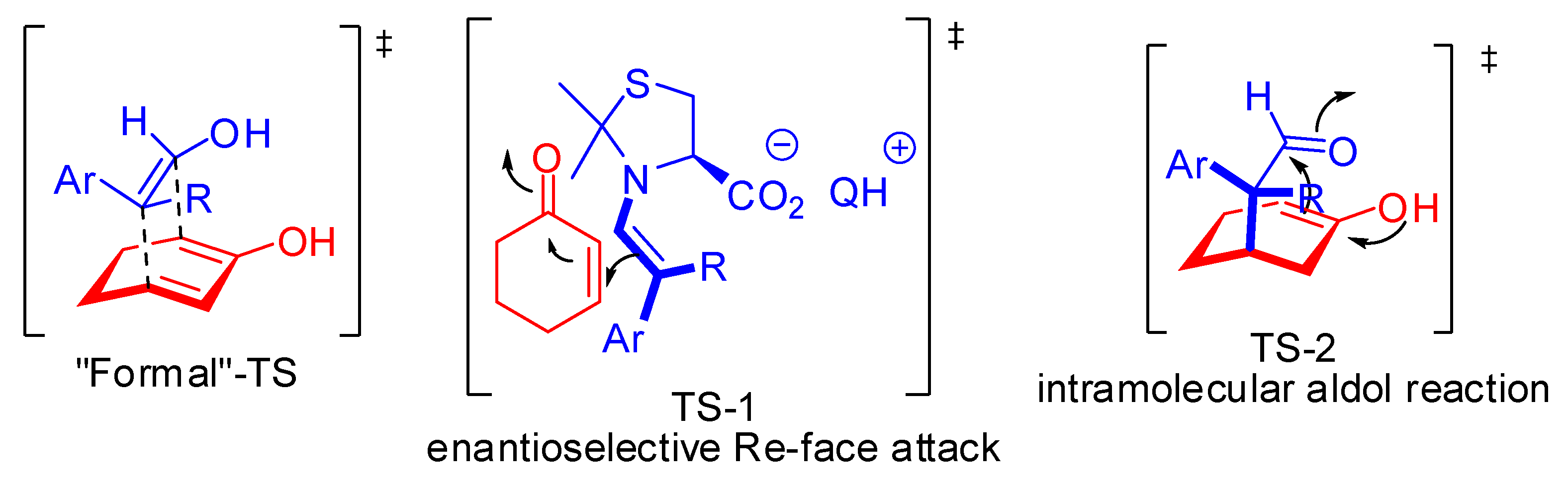{kind=link}
{kind=link}
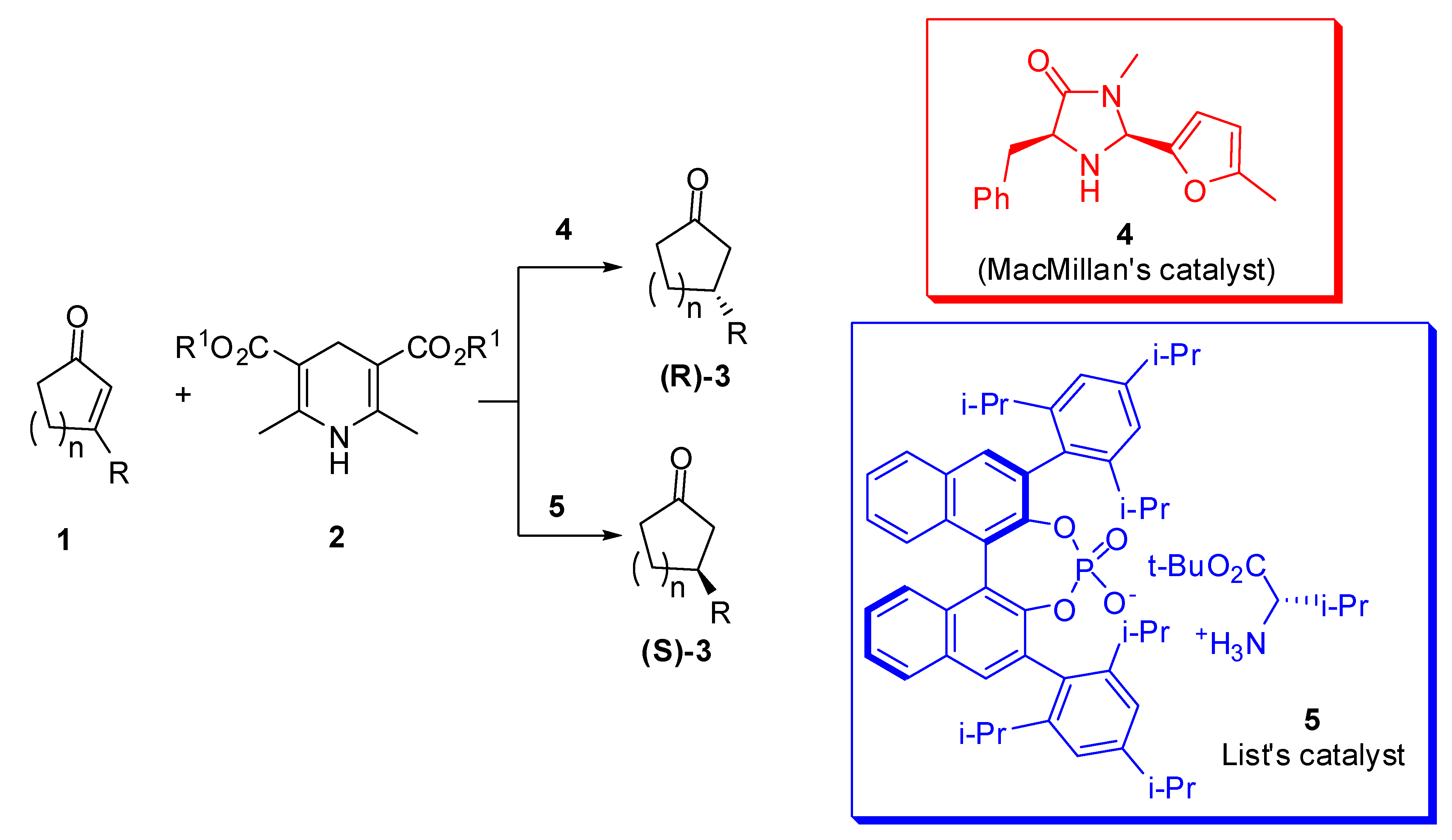{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
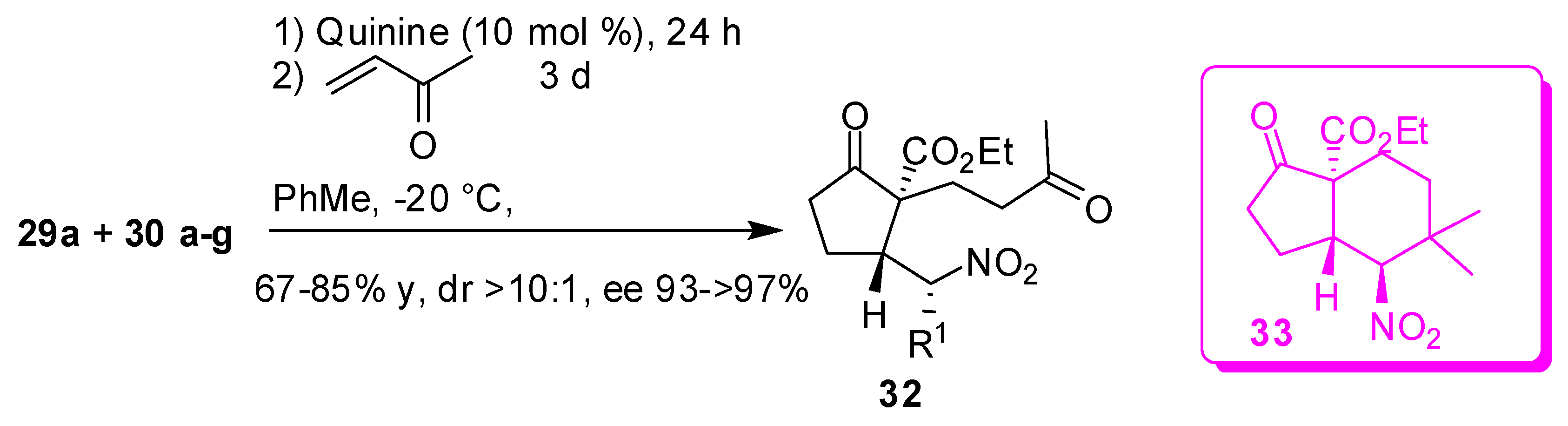{kind=link}
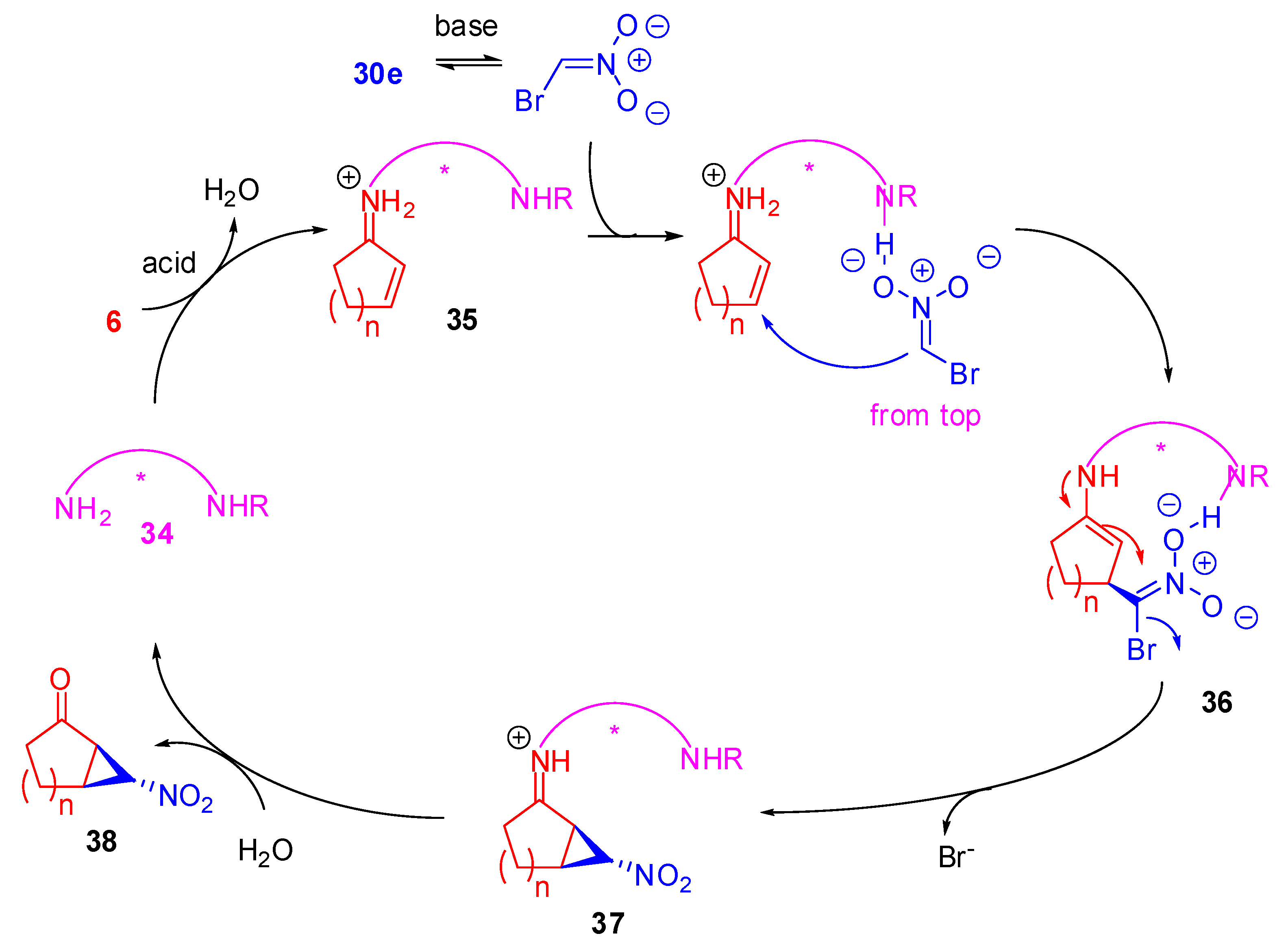{kind=link}
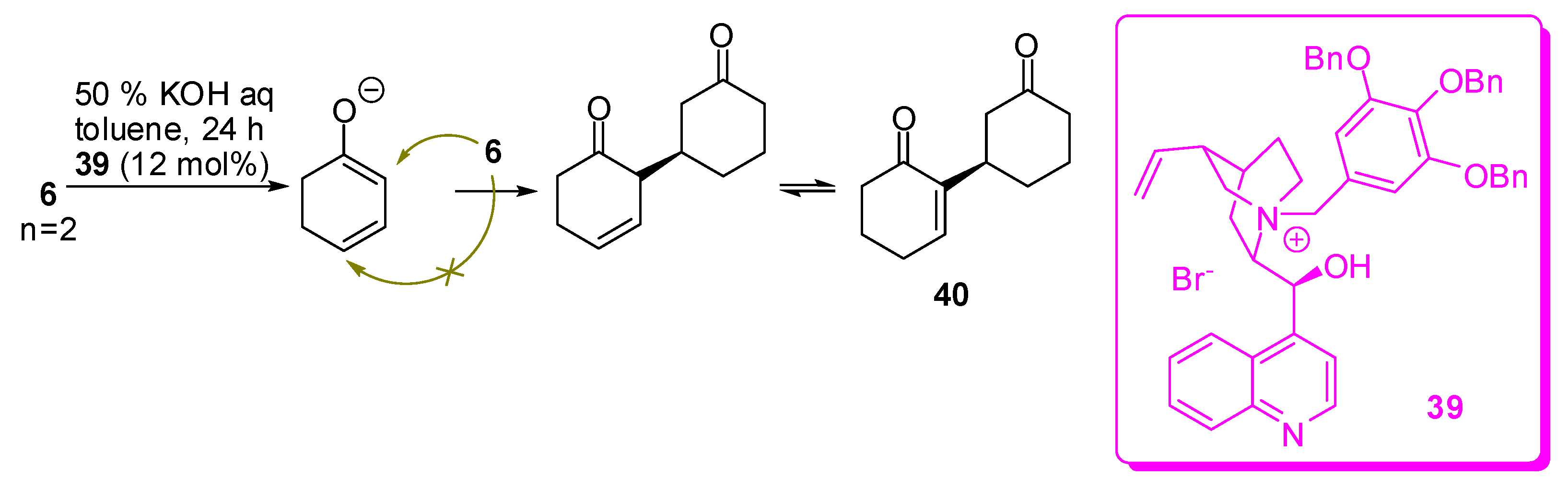{kind=link}
{kind=link}
{kind=link}
{kind=link}
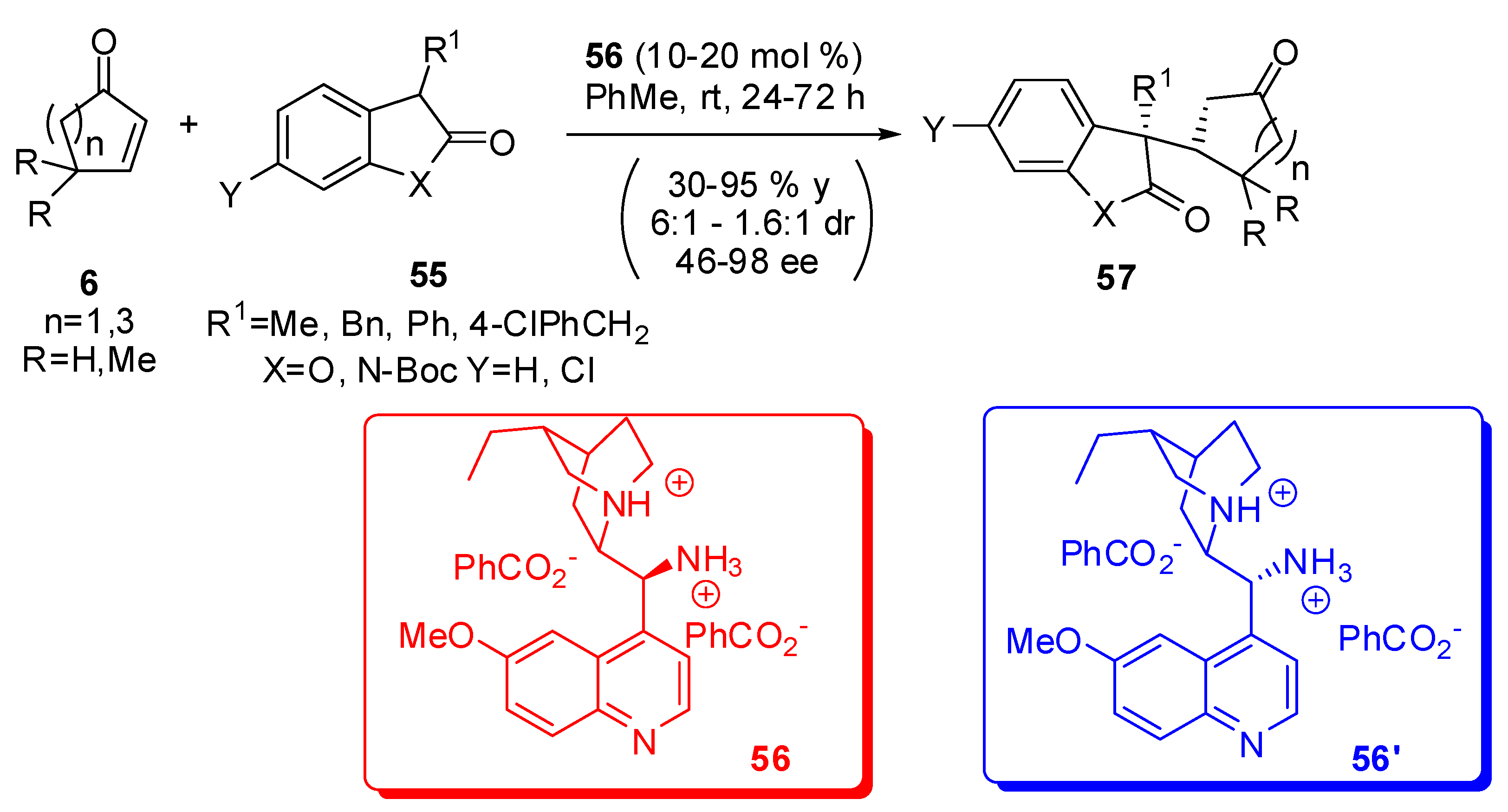{kind=link}
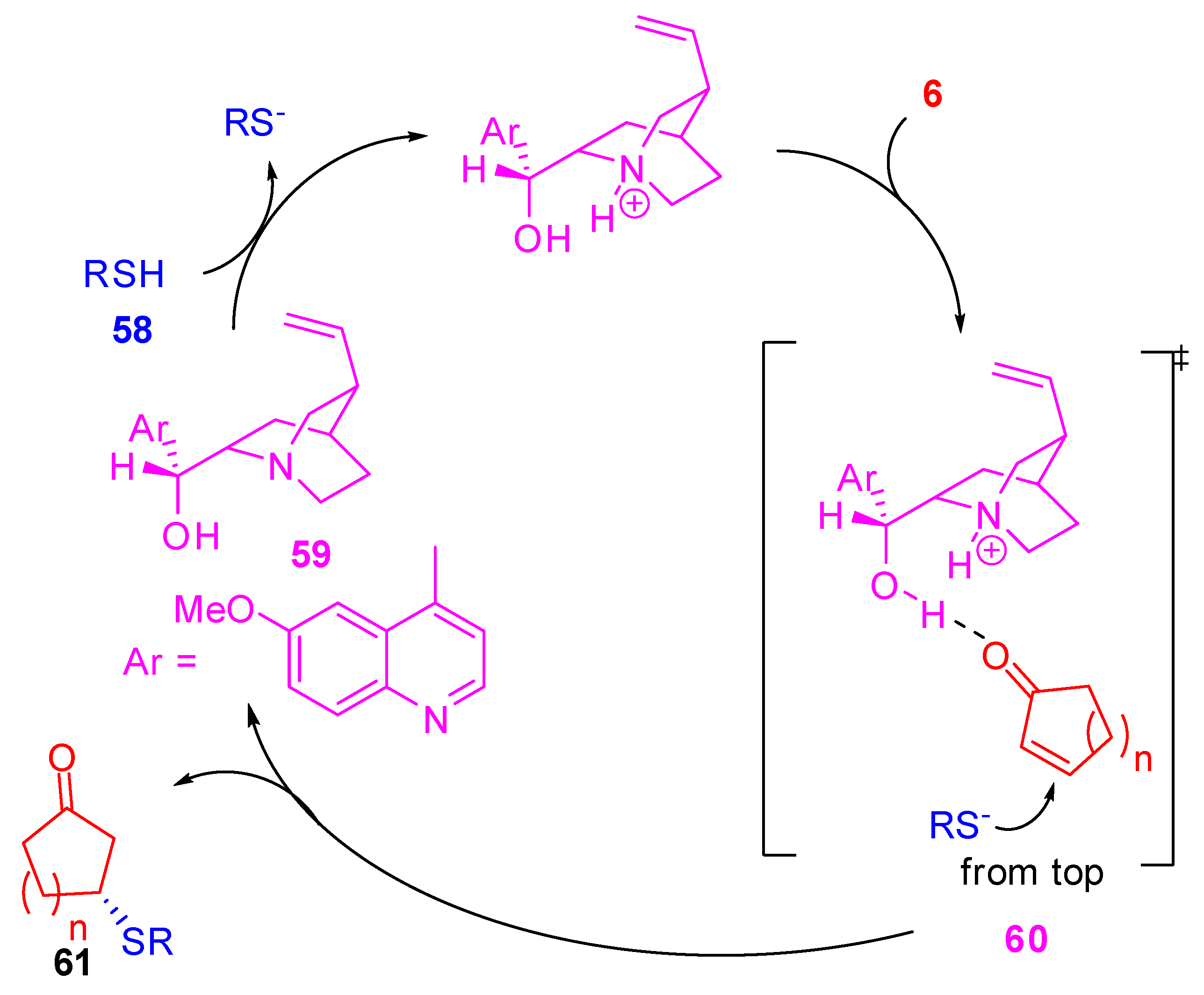{kind=link}
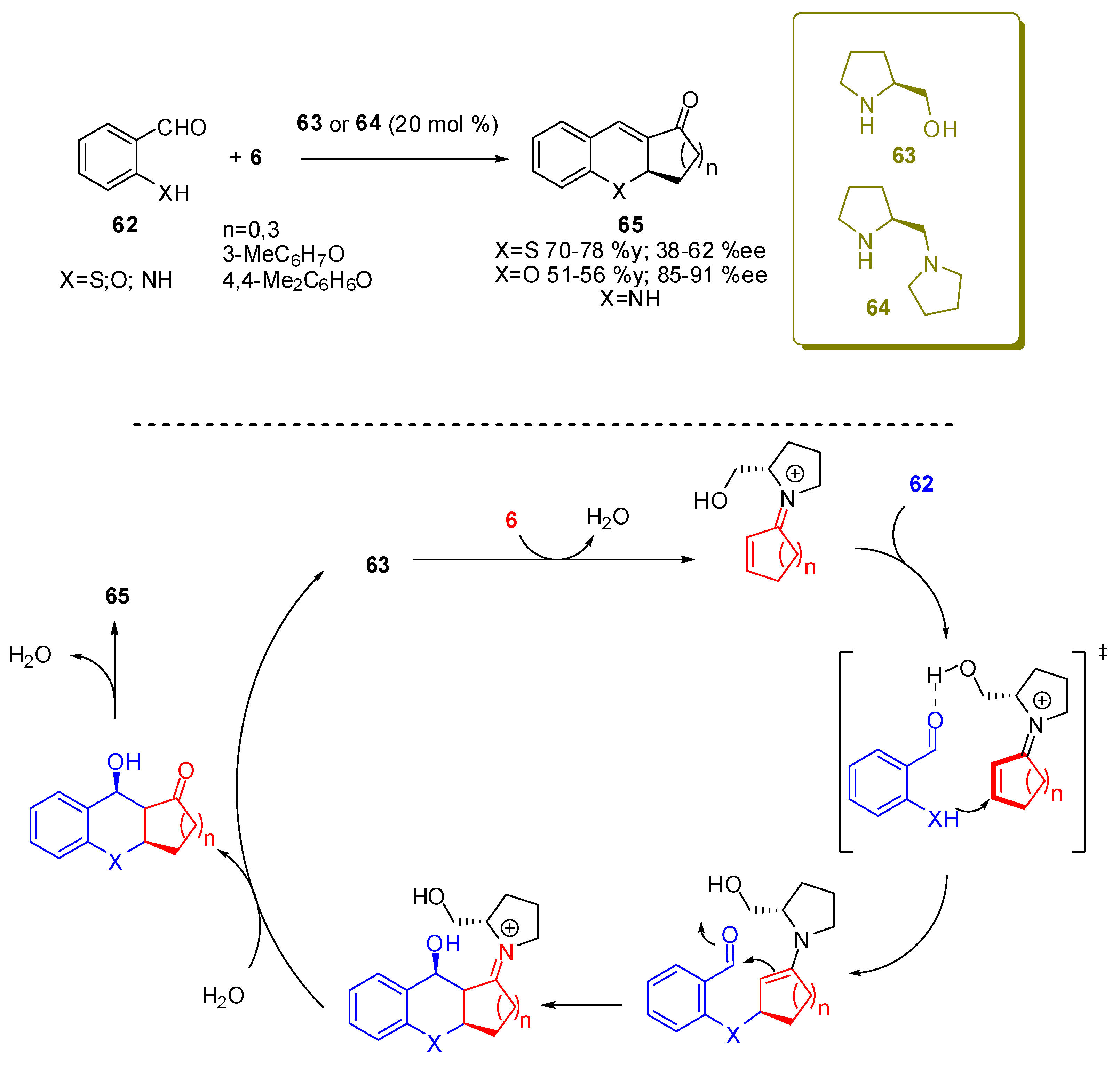{kind=link}
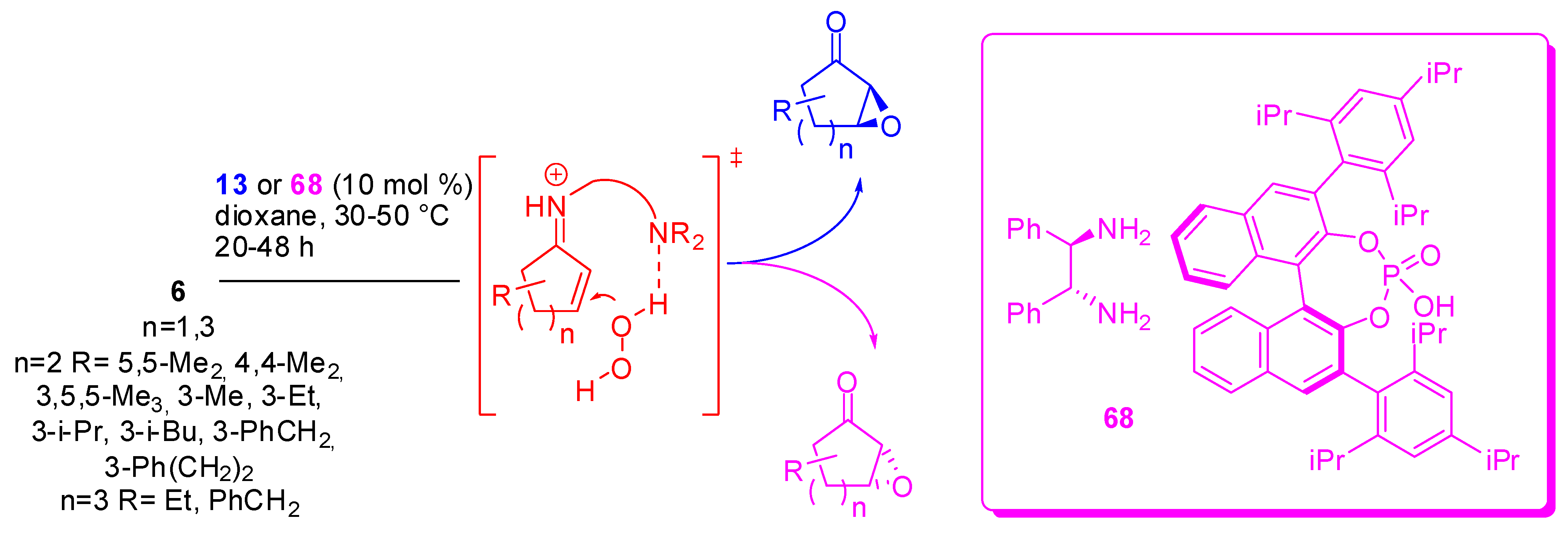{kind=link}
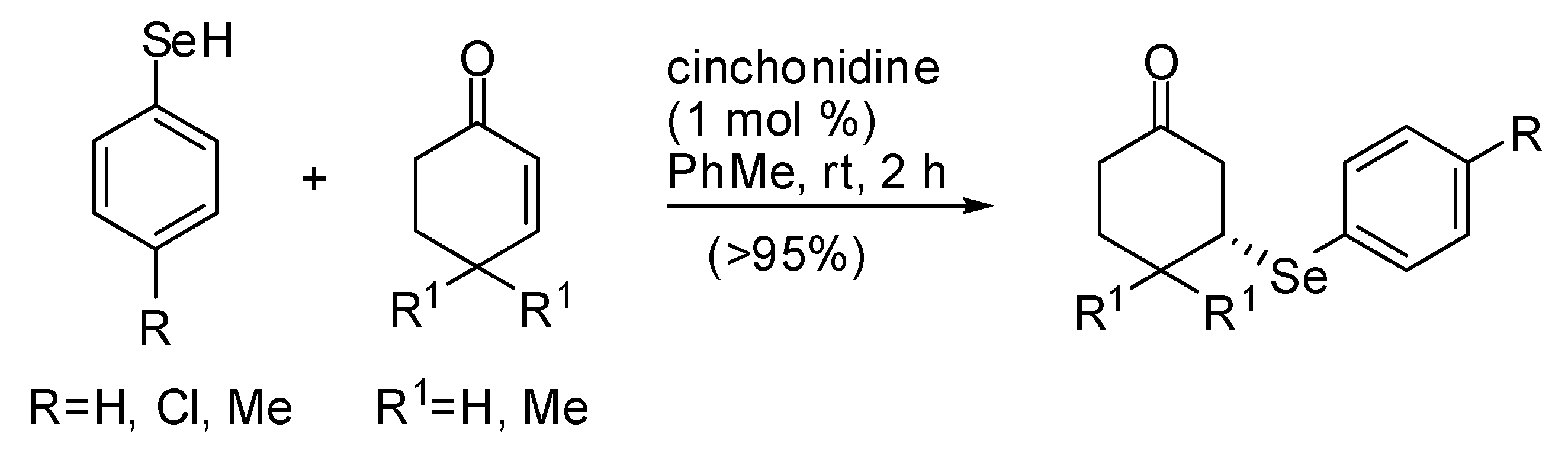{kind=link}
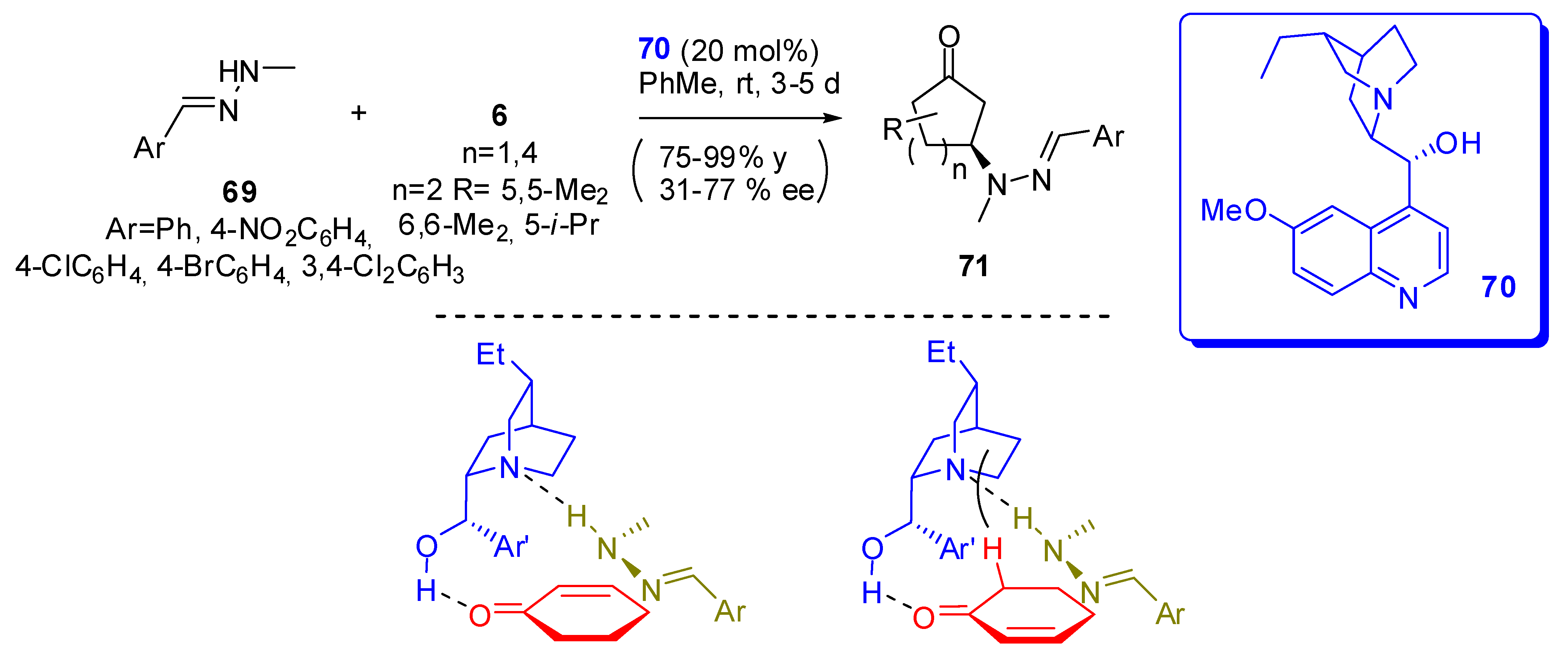{kind=link}
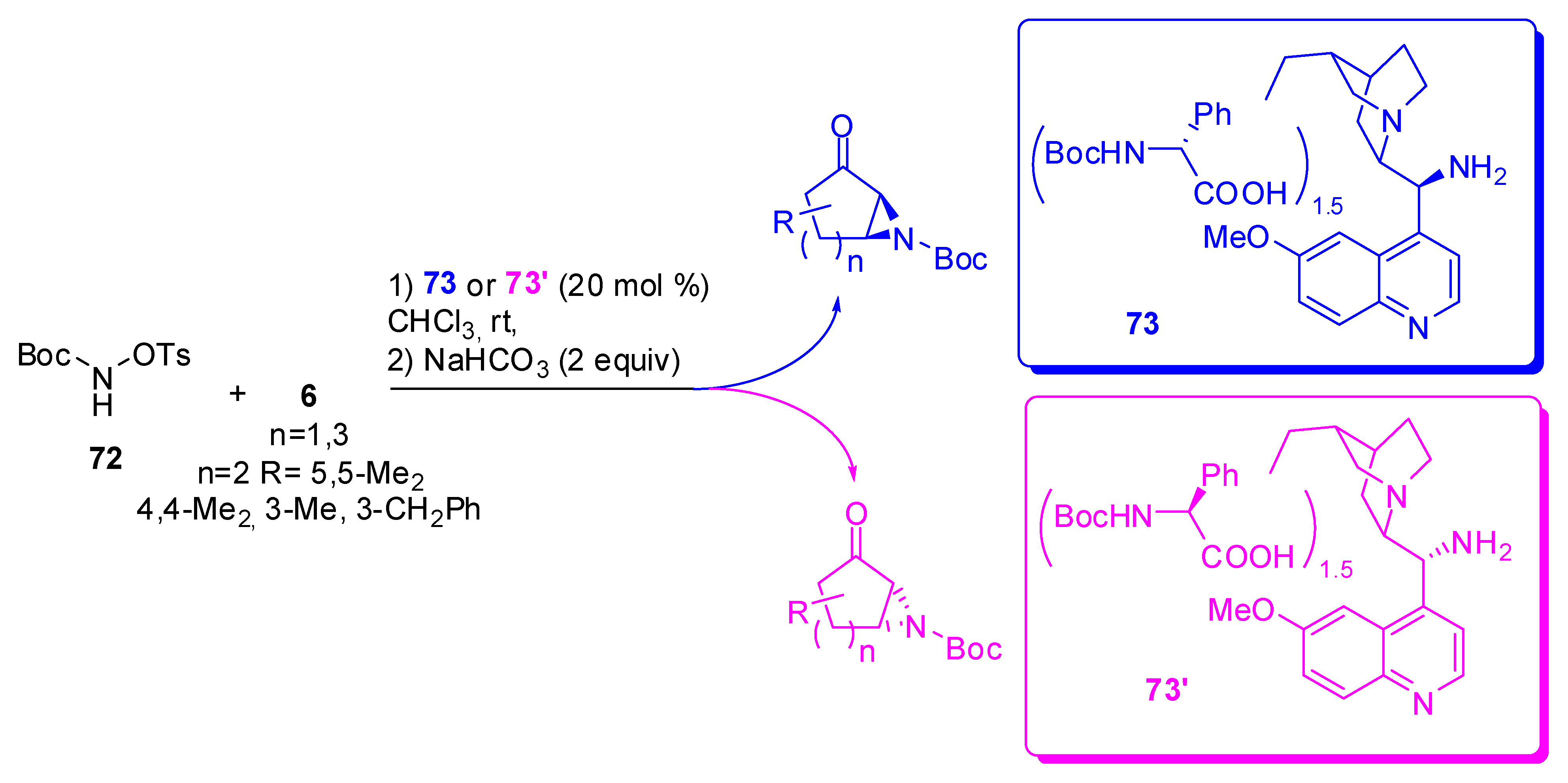{kind=link}
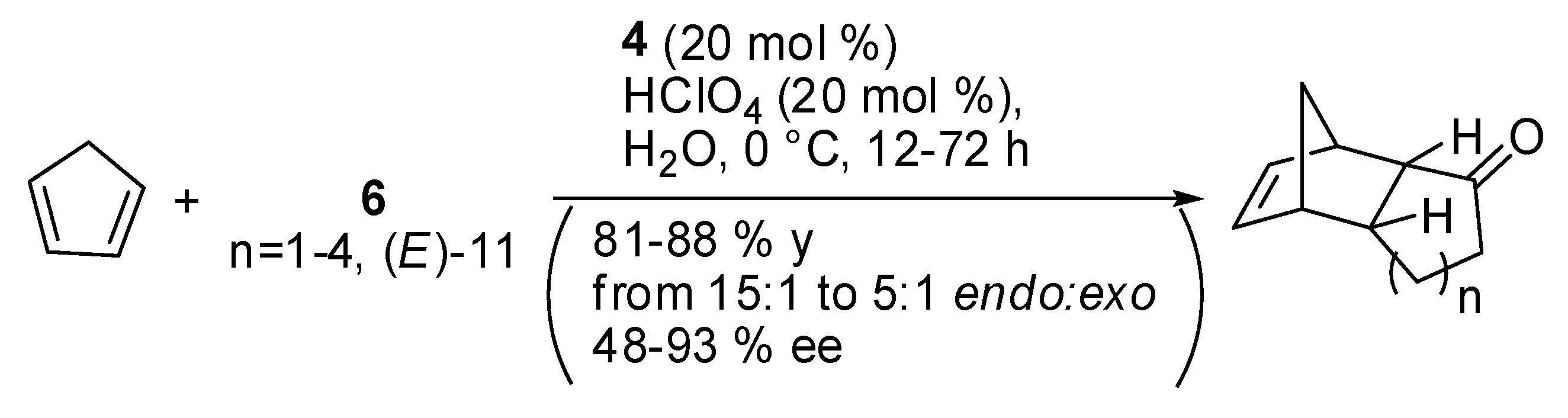{kind=link}
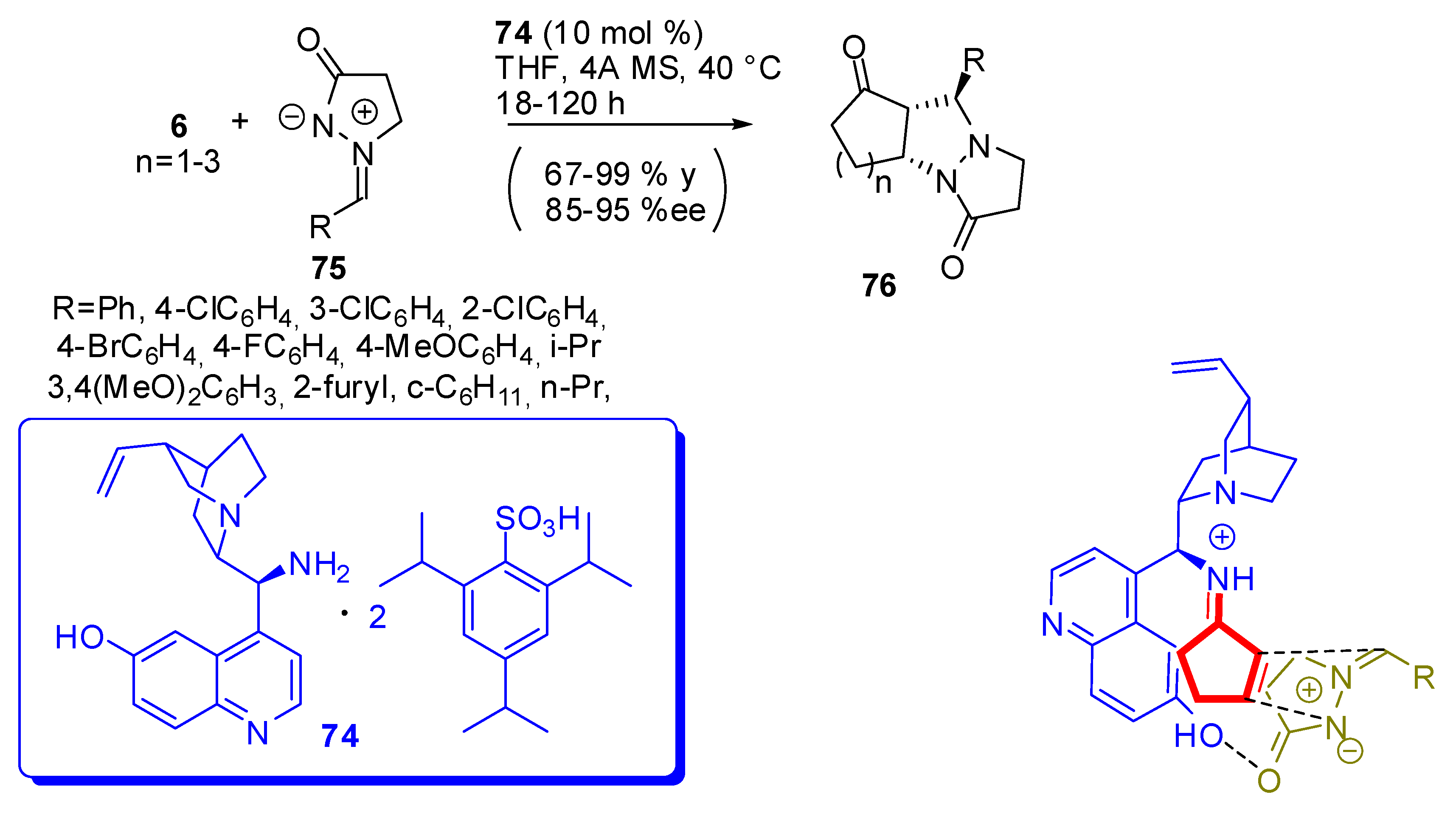{kind=link}
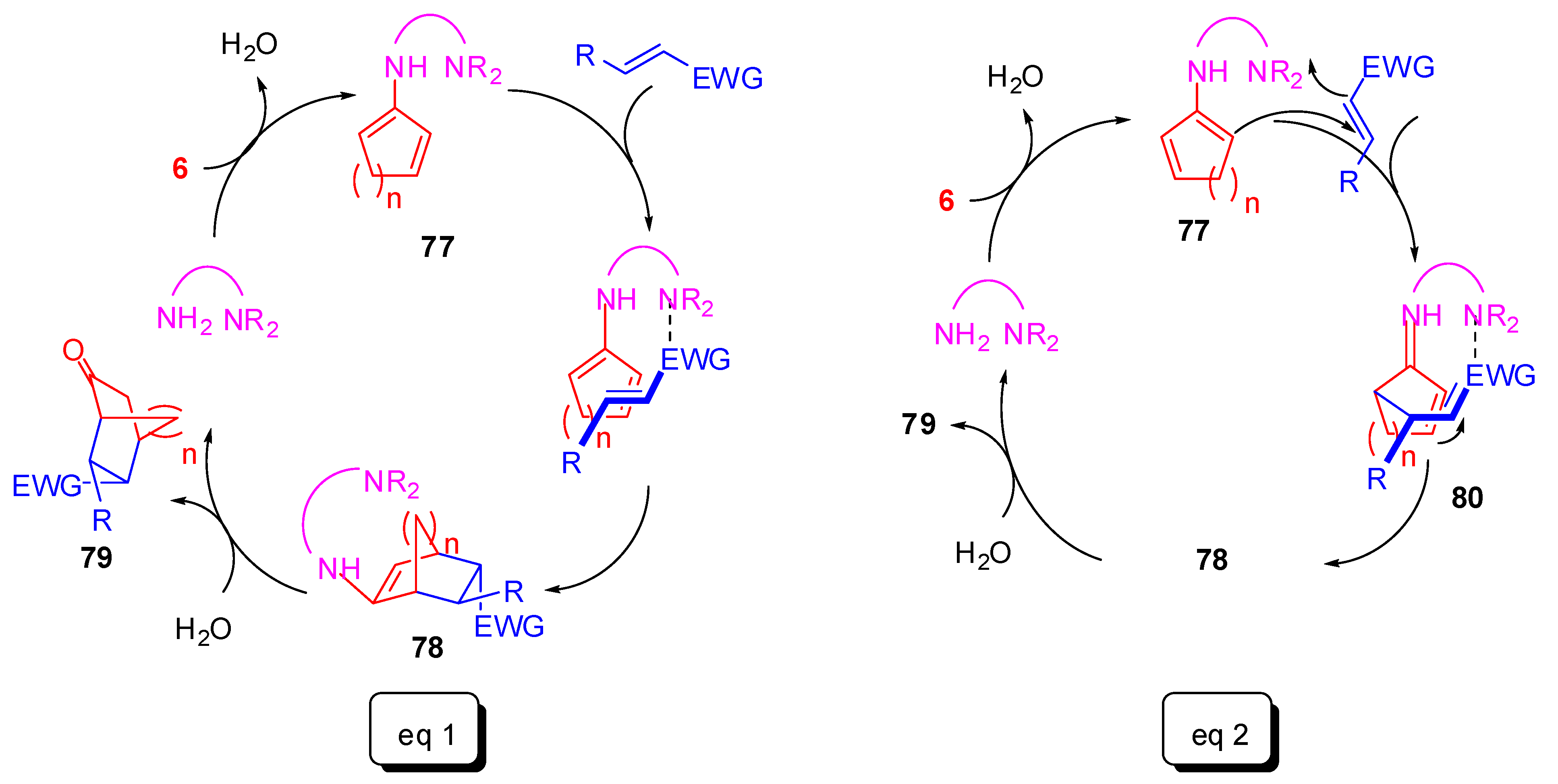{kind=link}
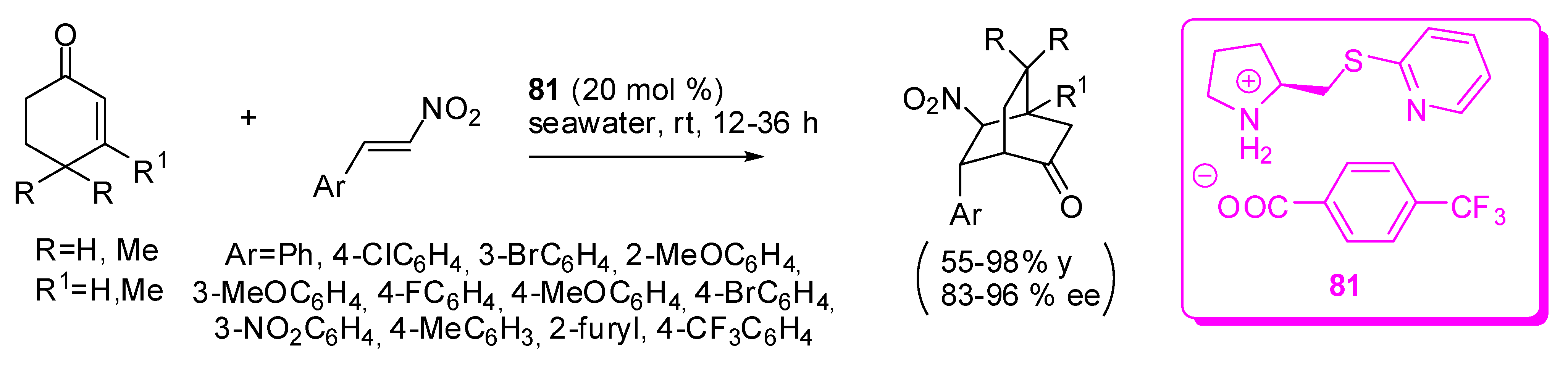{kind=link}
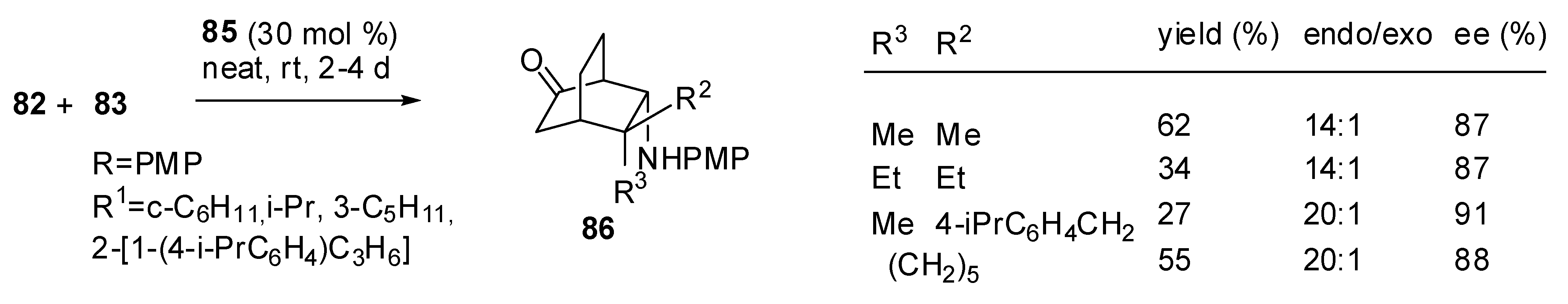{kind=link}
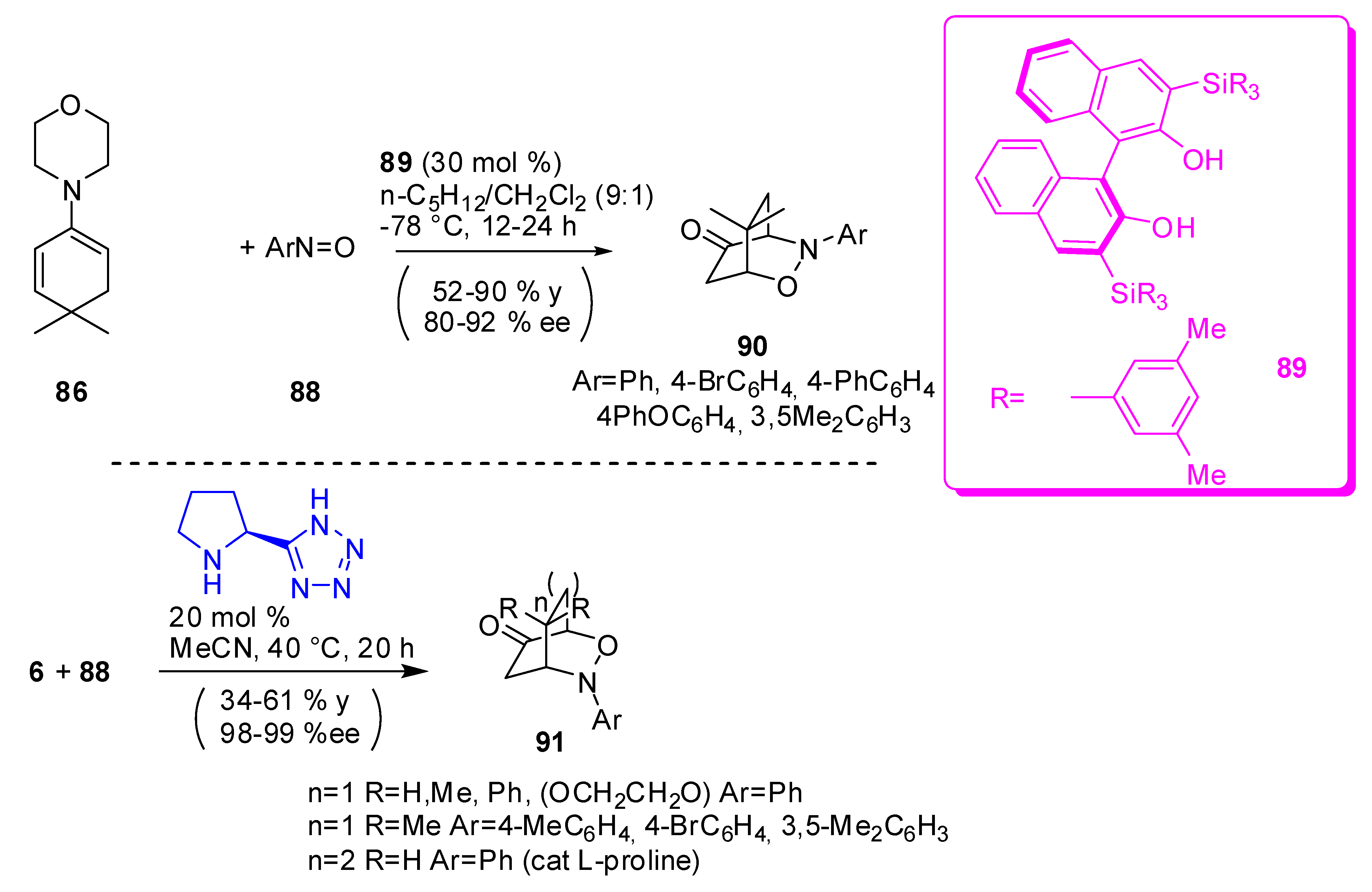{kind=link}
{kind=link}
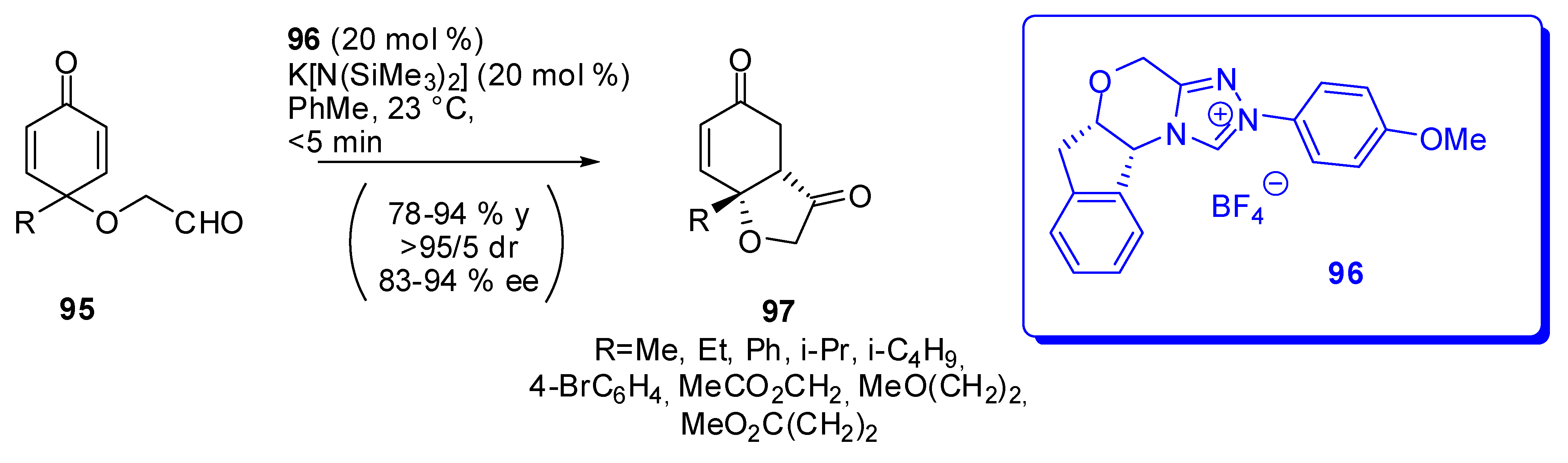{kind=link}
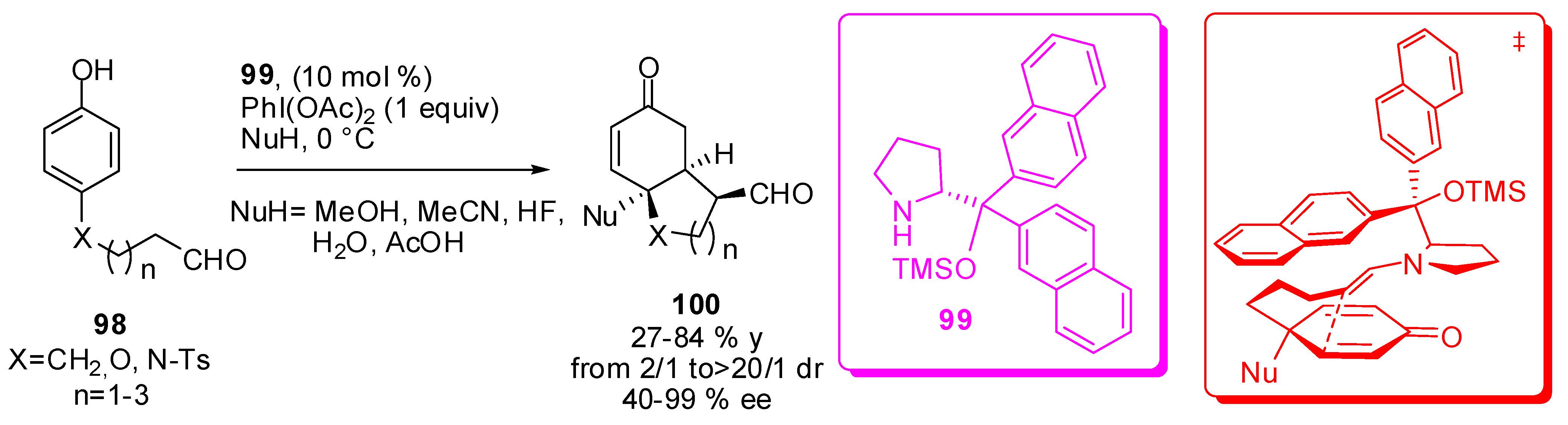{kind=link}
{kind=link}
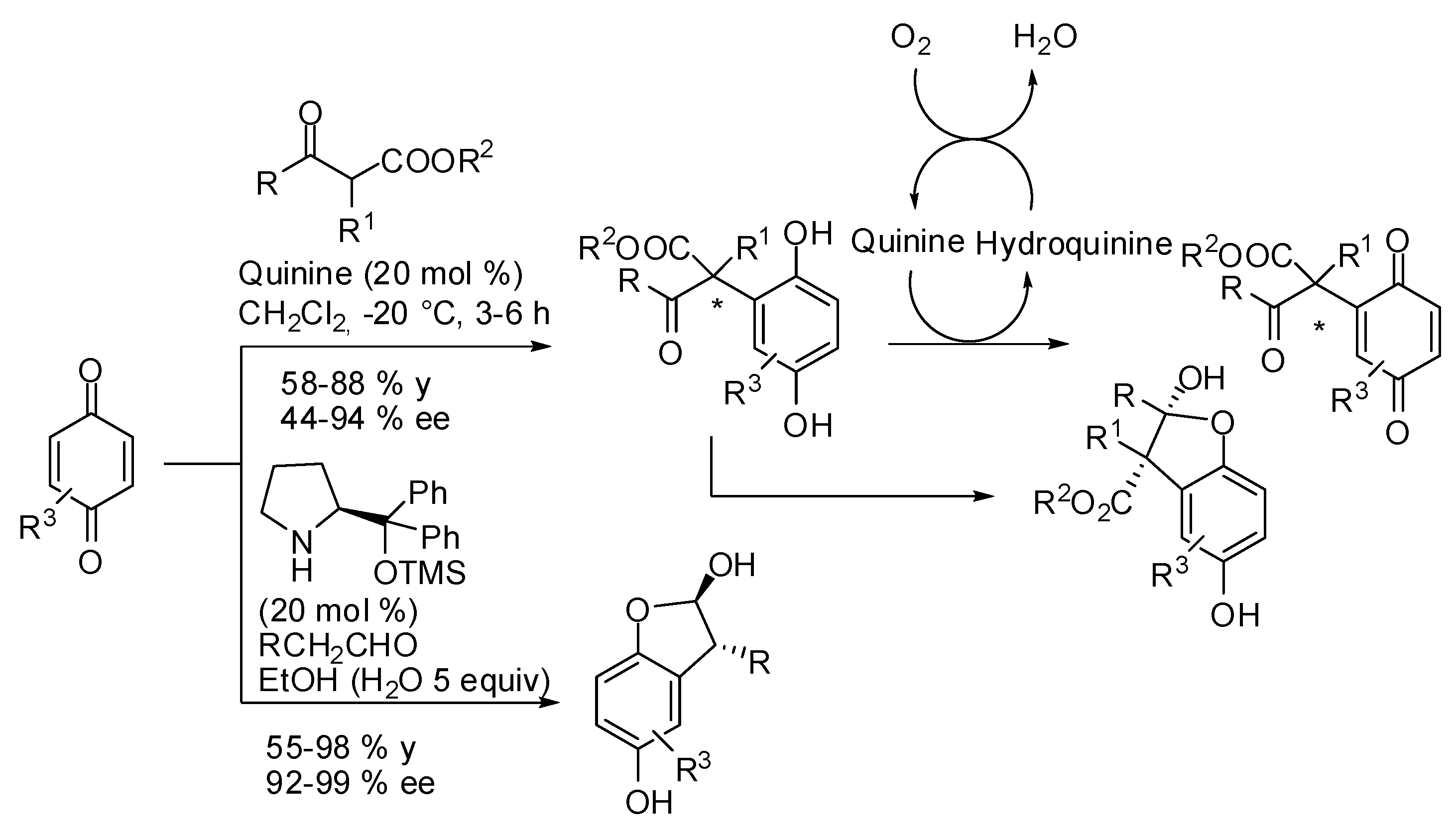{kind=link}
Table 1.
Malonate addition to cycloalkenones under various conditions and catalysts.

| Entry | Examples | Catalyst | Yield range (%) | ee range (%) a | Ref. |
|---|---|---|---|---|---|
| 1 | 3 |  15 mol %, piperidine100 mol% | 69-94 | 30-93 (R) | [20,21] |
| 2 | 2 |  10 mol% | 77-82 b | 30-40 (R) | [22] |
| 3 | 1 |  20 mol% | 71 | 90 (R) | [23] |
| 4 | 9 |  0.5-10 mol% | 64-95 | 63-96 (R) | [24] |
| 5 | 1 |  10 mol% | 64 | 86 (S) | [25] |
| 6 | 1 |  Ar = 2,6-i-Pr2 C6H3, 10 mol % | 89 | 90 (S) | [26] |
| 7 | 10 |  30 mol% | 47-96 | 55-87 (S) | [27] |
| 8 | 7 |  10 mol% | 23-98 | 69-94 (S) | [28] |
a (R) and (S) descriptors assigned according to priority in formula 8; b Yields and ee’s refer to dibenzyl malonate and dibenzyl methylmalonate, respectively.
Table 2.
Nitroalkane addition to cycloalkenones under various conditions and catalysts.
| Entry | Examples | Catalyst | Yield range (%) | ee range a (%) | Ref. |
|---|---|---|---|---|---|
| 1 | 15 | l-proline (3-7 mol%), 21 (100 mol%) | 30-88 | 63-93 (R)b | [36] |
| 2 | 7 |  m = 2-4 m = 2-4(2 mol%), 21 (100 mol%) | 24-99 | 56-88 (R) | [35,37] |
| 3 | 1 | H-Leu-His-OH (30 mol%), (R,R)-1,2-diphenylethylenediamine (30 mol%) | 86 | 75 (R) | [38] |
| 4 | 10 |  (15 mol%), (15 mol%), 21 (100 mol%) | 47-84 | 80-98(R) c | [39,40] |
| 5 | 2 |  (20 mol%) (20 mol%) | 70-75 | 61-77 (S) | [41] |
| 6 | 5 |  (10 mol%), (10 mol%),21 (100 mol%) | 50-94 | 60-99 (S) d | [42] |
| 7 | 12 |  (10 mol%) (10 mol%)21 (100 mol%) | 15-85 | 42-82 (S) e | [43] |
| 8 | 10 |  (2-10 mol%) | 25-92 | 80-98 (R) f | [44] |
| 9 | 6 |  (10-15 mol%) | 31-88 | 26-72 (S) | [25] |
| 10 | 2 |  (10 mol%) (10 mol%) | 83-91 | 96-99 (R) c | [45] |
a Configuration of compound 26 depicted in Scheme 6 is assumed (R); b dr from 2:1 to 1:1; c dr about 1:1; d dr from 2:3 to 1:2; e dr from about 1:1 to about 7:3; f dr from 4:6 to 7:3.
Table 3.
Synthesis of nitrocyclopropanes 34 under various conditions and catalysts.
| Entry | Examples | Catalyst | Yield range (%) | ee range (%) | Ref. |
|---|---|---|---|---|---|
| 1 | 8 |  (15 mol%), NaI (1.5 mol %), morpholine (300 mol%) (15 mol%), NaI (1.5 mol %), morpholine (300 mol%) | 11-99 | 14-90 | [51,54] |
| 2 | 8a |  (20 mol %), (20 mol %), NMM (100 mol%) | 83-99 | 83-99 | [55] |
| 3 | 6 |  (20 mol%), NMM (100 mol%) | 70-95 | 63-96 | [53] |
| 4 | 4 |  (20 mol%), NMM (100 mol %) (20 mol%), NMM (100 mol %) | 31-98 | 88-99 | [52] |
a 1-bromonitroethane was also used.
Table 4.
Addition of thiols to cyclic enones under various conditions and catalysts.
| Entry | Examples | Catalyst | Yield range (%) | ee range a (%) | Ref. |
|---|---|---|---|---|---|
| 1 | 9 |  2 mol% | 22-84 | 1-88 (R) | [67,68] |
| 2 | 15 | Cinchonidine (1 mol%) | n.d.b | 5-75 (R) | [66] |
| 3 | 8 | Quinine or quinidine (0.8 mol%) | > 99 | 18-52c | [69] |
| 4 | 12 |  1.0-1.6 mol% | 55-91 | 21-99 (R) | [70] |
| 5 | 9 |  10 mol %, 4Å MS, | 95-99 | 63-85 (S) | [71] |
| 6 | 3 | l-proline (30 mol%) | 89-94 | n.d.b | [72] |
| 7 | 9 |  1 mol% | 18-99 | 0-31 (R) | [73,74] |
| 8 | 32 |  0.1 mol% | 93->99 | 80->99(S) | [75] |
| 9 | 9 |  10 mol% | 44-86 | 8-58 (S) | [76] |
a Configuration of compound 61 depicted in Scheme 16 is assumed (R); b n.d. = not determined. c (R) for quinine, (S) for quinidine.
Table 5.
Formal aza-Diels Alder reaction for the synthesis of isoquinuclidines (84).

| Entry | Examples | Catalyst | Yield range (%) | Endo/exo | ee range a (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | 13 |  5 mol% 5 mol% | 70-82 | 4/1-6/1 | 76-87 | [92] |
| 2 | 10 |  10 mol %, AcOH (20 mol %) | Ar = 4-PhC6H4, | [93] | ||
| 51-76 | 4/1-9/1 | 76-86 | ||||
| 3 | 10 | Ar = 2-naphthyl | ||||
| 54-84 | 3/1-8/1 | 82-88 | ||||
| 4 | 13 | lproline (30 mol%) | 20-90 | 94-99 | [94] | |
| 5 | 11 |  30 mol% | 19-61 | <1/99 | 80-99 | [95] |
a of the major isomer
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Asymmetric Organocatalytic Reactions of α,β-Unsaturated Cyclic Ketones. Symmetry 2011, 3, 84-125. https://doi.org/10.3390/sym3010084
AMA Style
Dalpozzo R, Bartoli G, Bencivenni G. Asymmetric Organocatalytic Reactions of α,β-Unsaturated Cyclic Ketones. Symmetry. 2011; 3(1):84-125. https://doi.org/10.3390/sym3010084
Chicago/Turabian StyleDalpozzo, Renato, Giuseppe Bartoli, and Giorgio Bencivenni. 2011. "Asymmetric Organocatalytic Reactions of α,β-Unsaturated Cyclic Ketones" Symmetry 3, no. 1: 84-125. https://doi.org/10.3390/sym3010084