Enantiomeric Separation of Tramadol and Its Metabolites: Method Validation and Application to Environmental Samples

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Standards and Reagents
2.2. Instrumental Conditions
2.3. Chromatographic Conditions
2.4. Sample Preparation Procedure
2.5. Method Validation
2.6. Application of Developed HPLC-FD Method in WWTP Samples
3. Results and Discussion
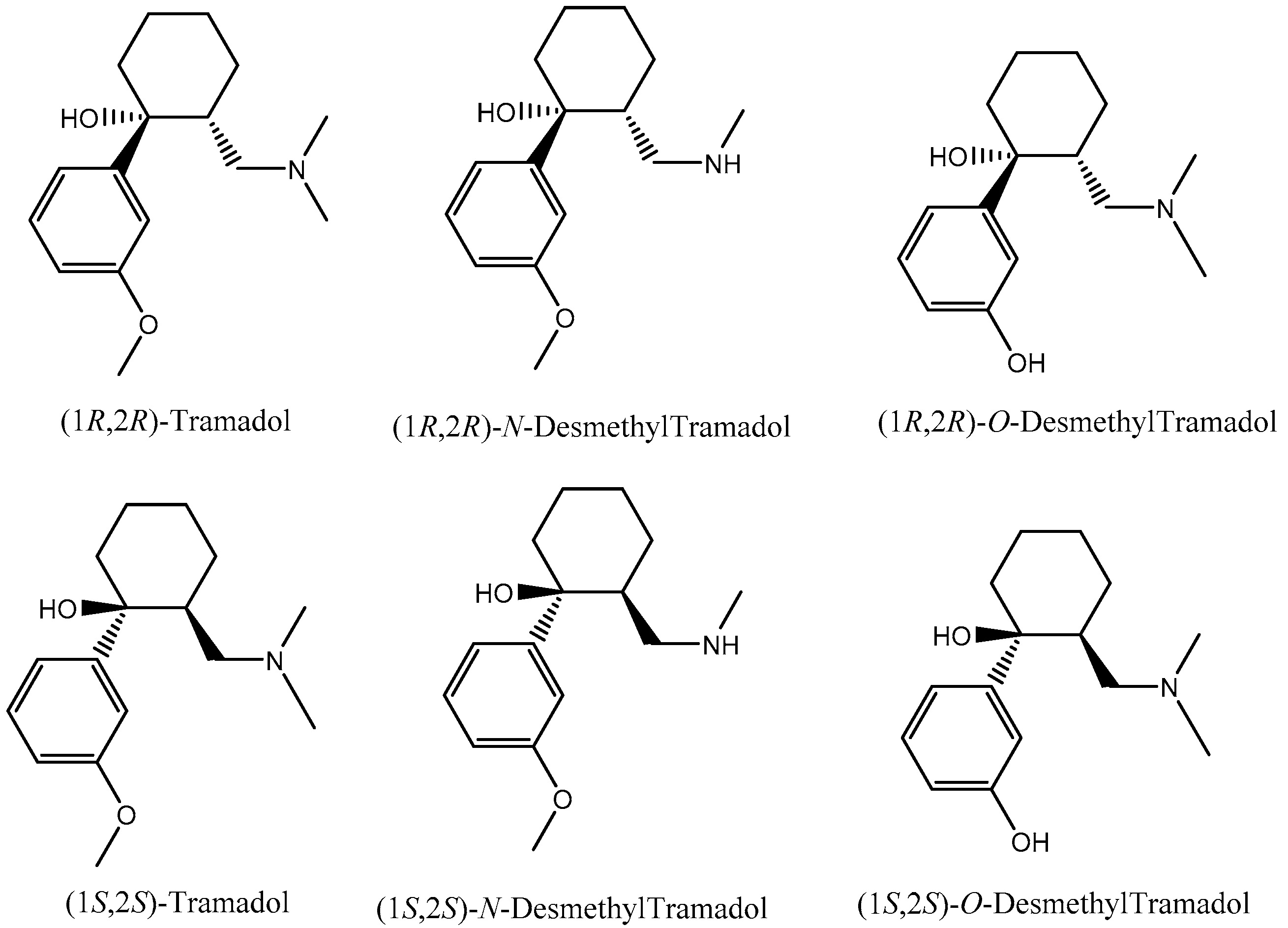
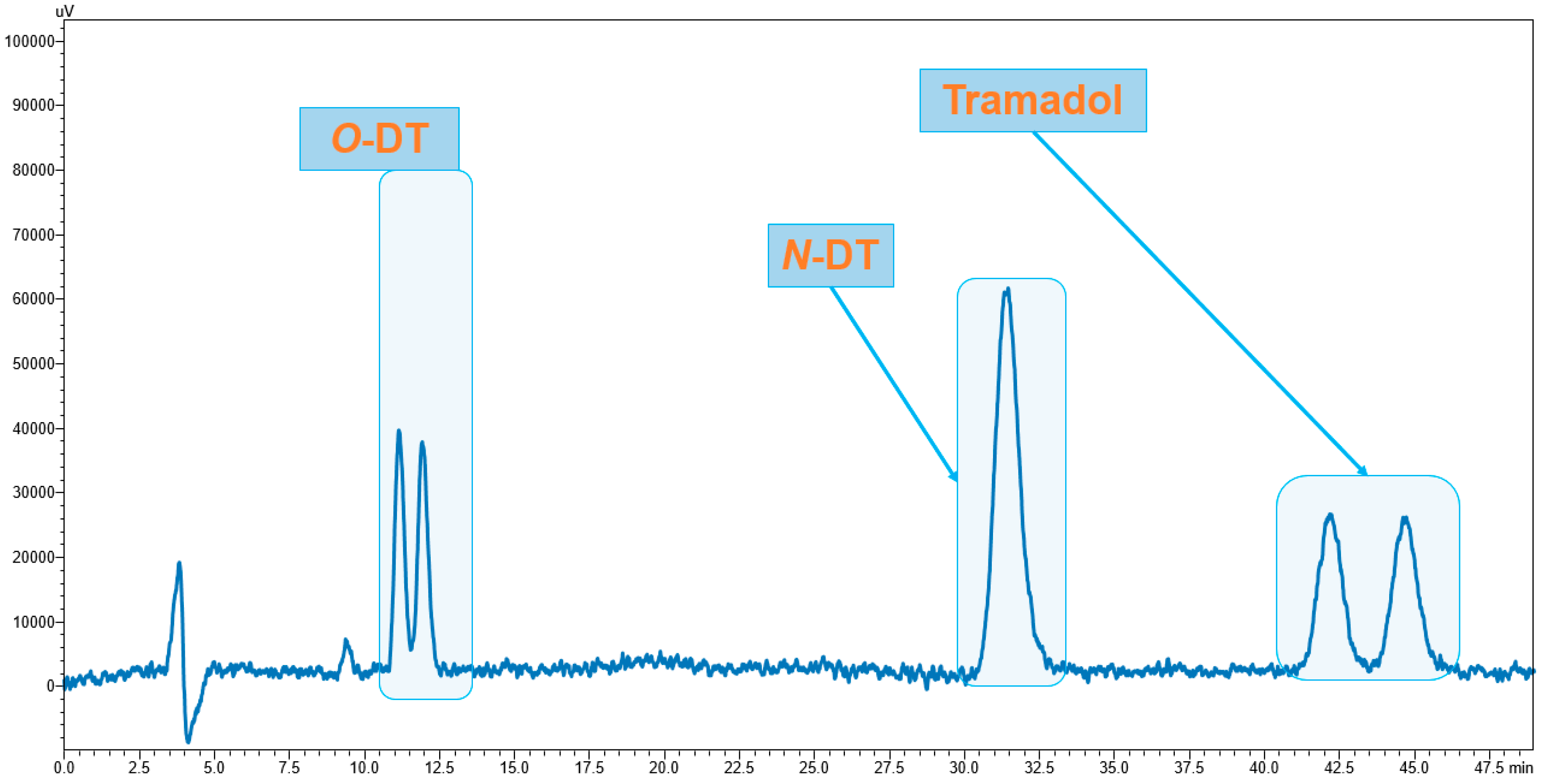
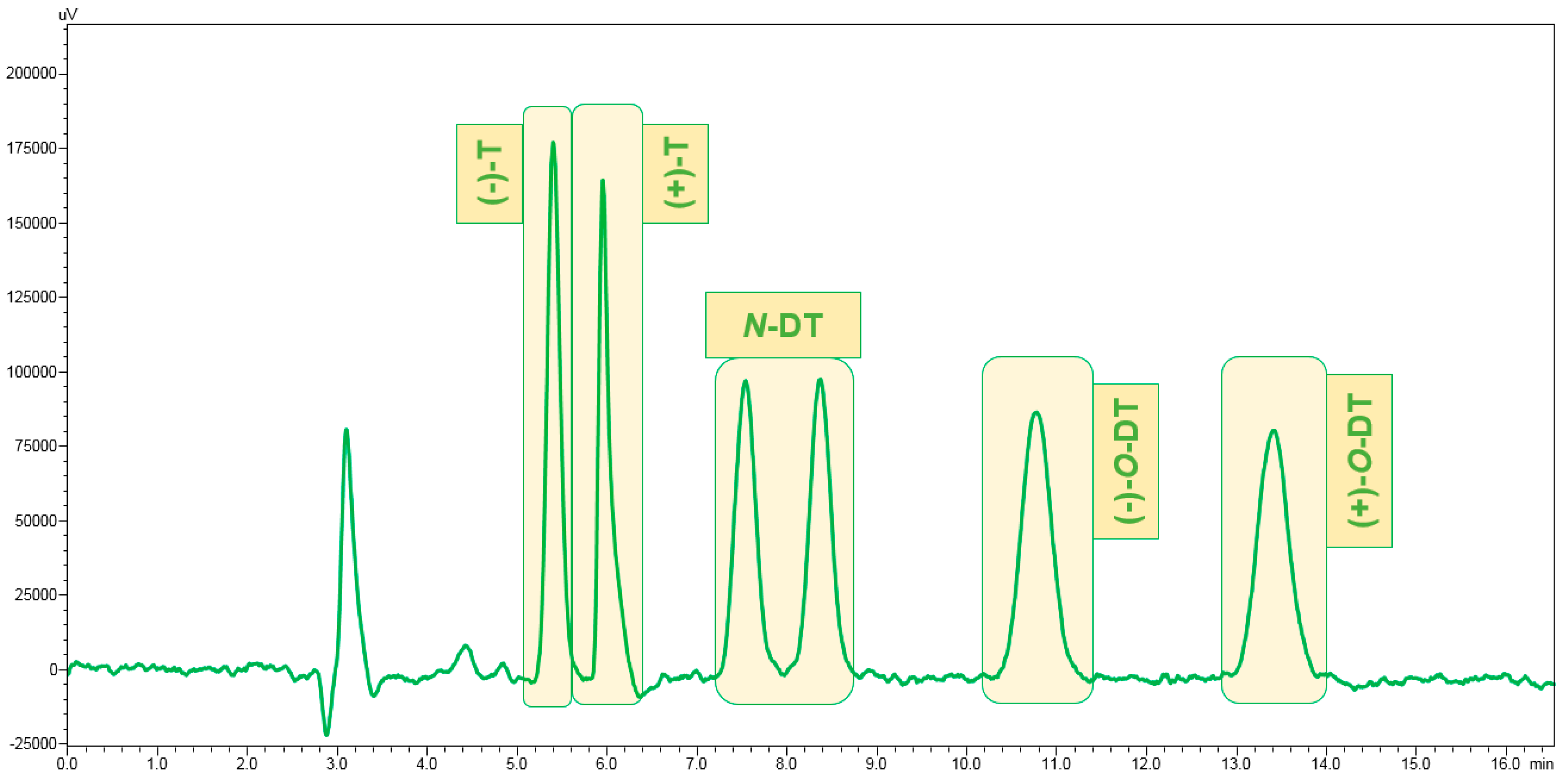
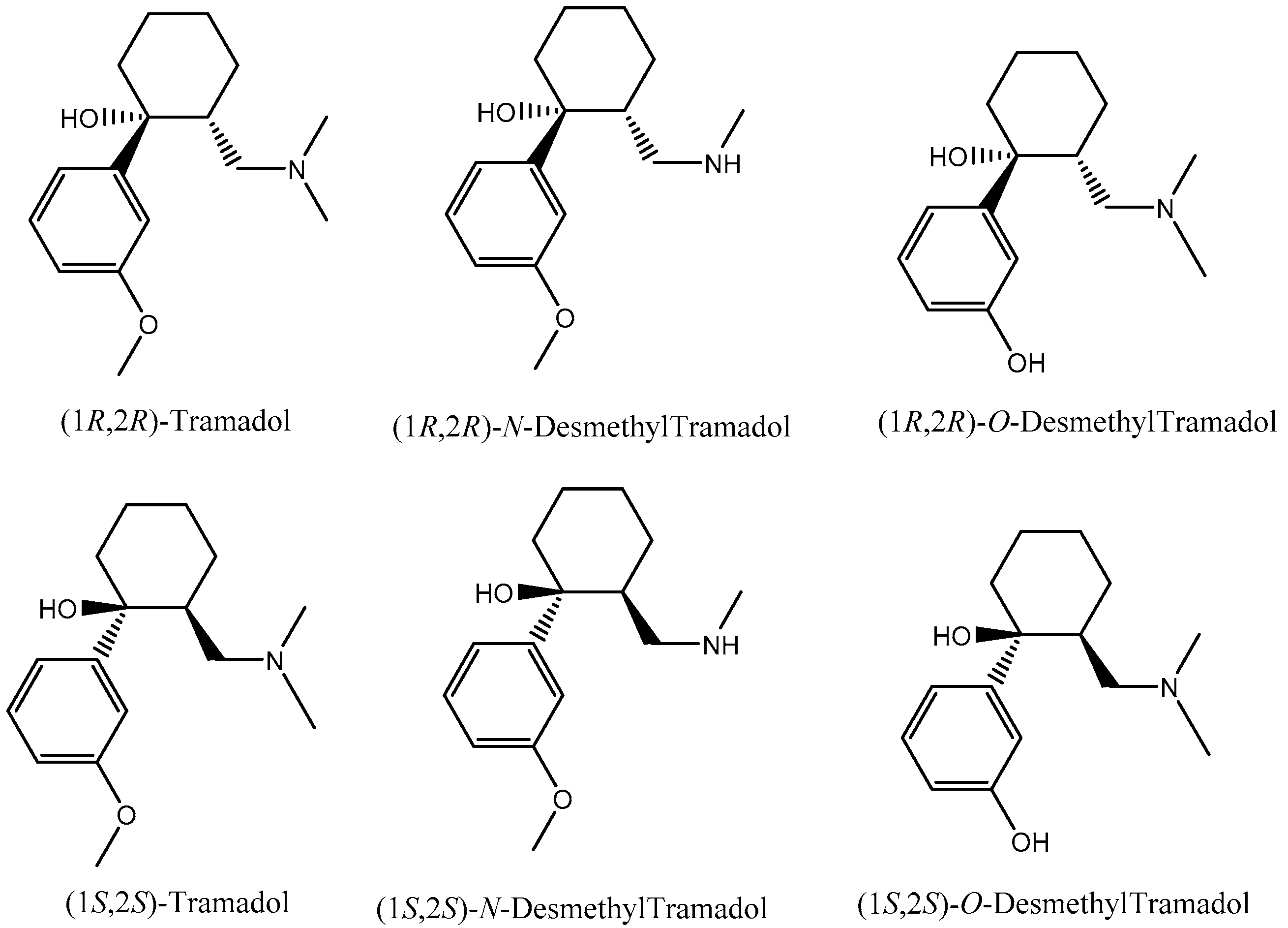
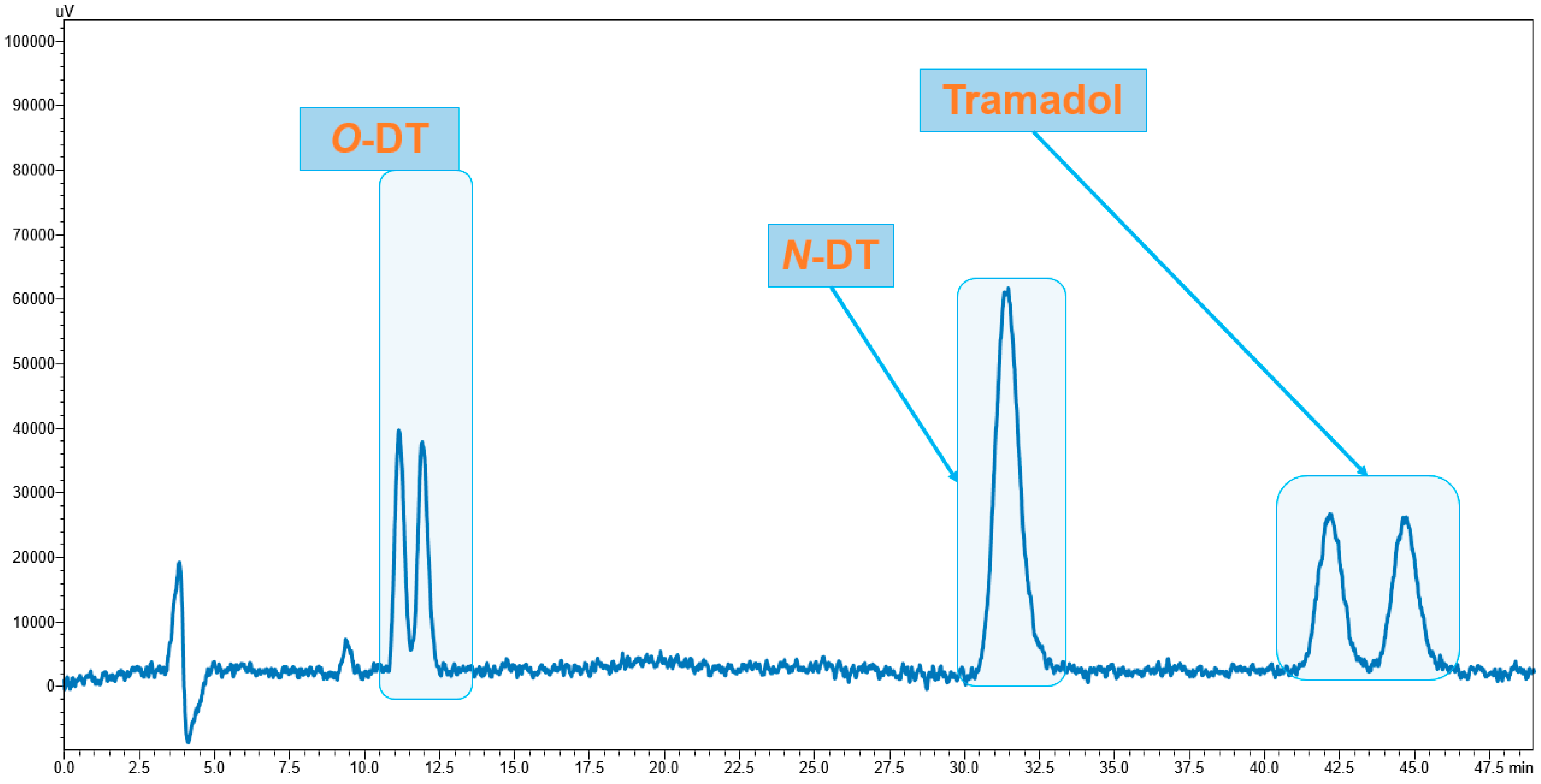
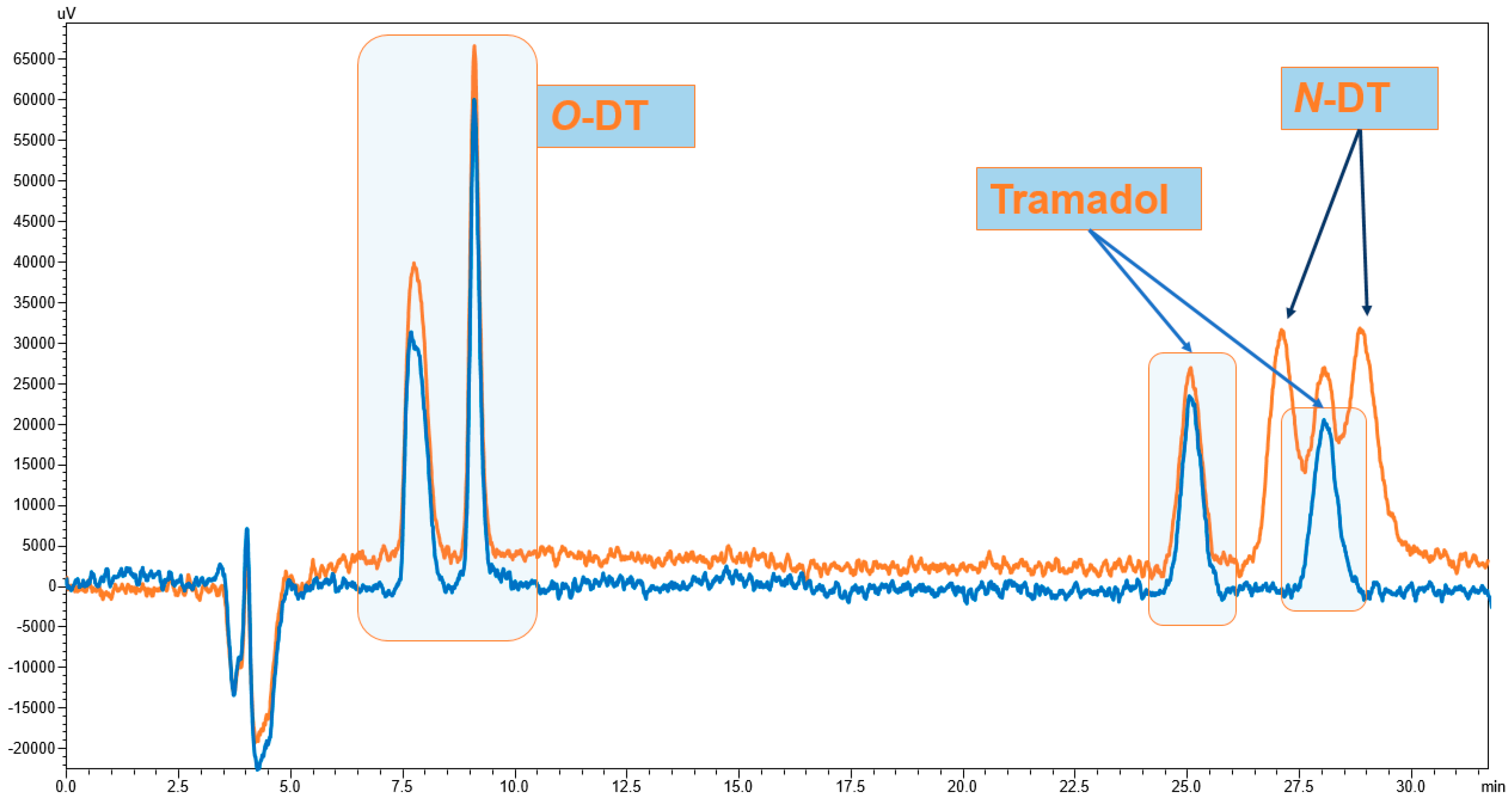
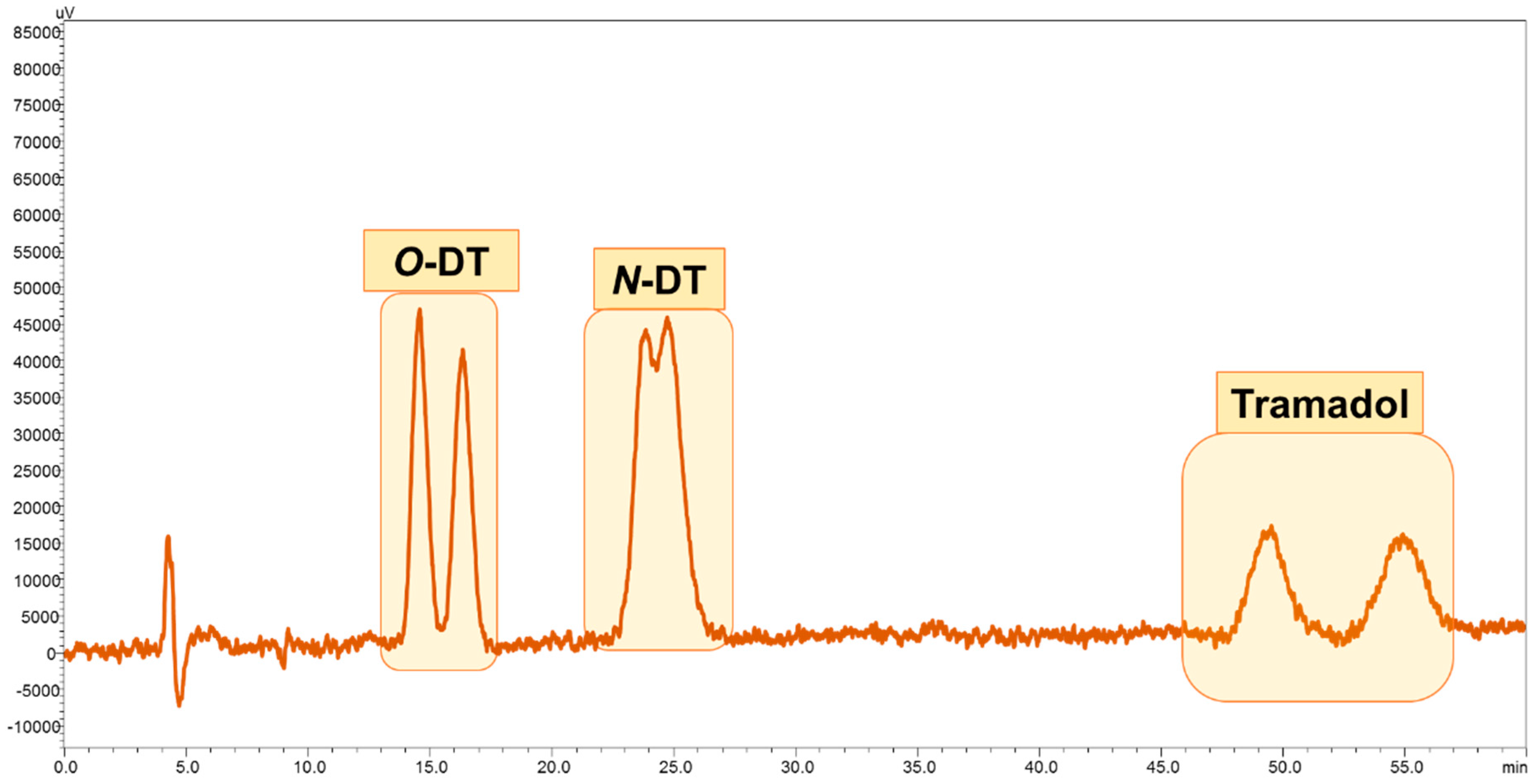
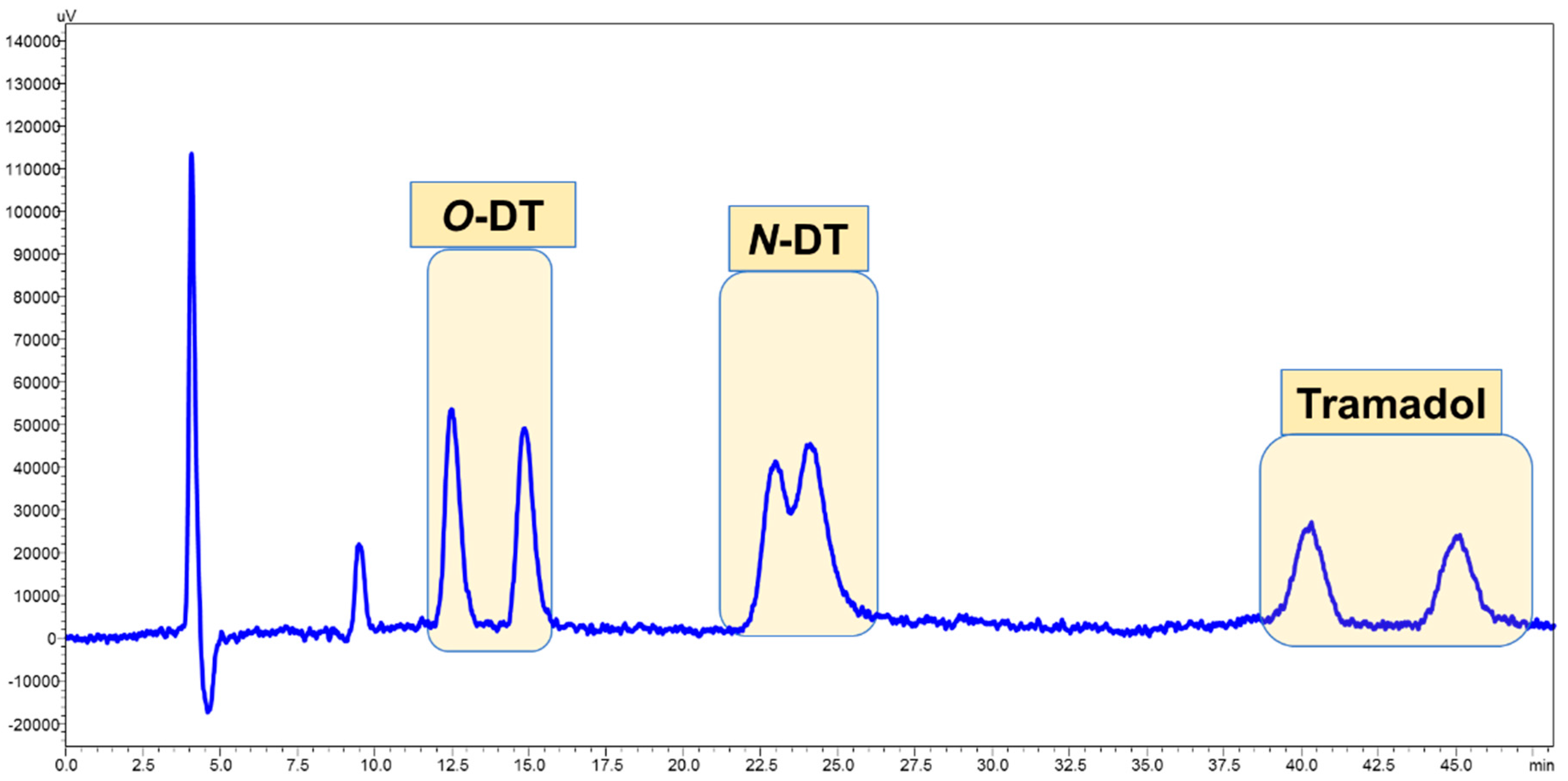
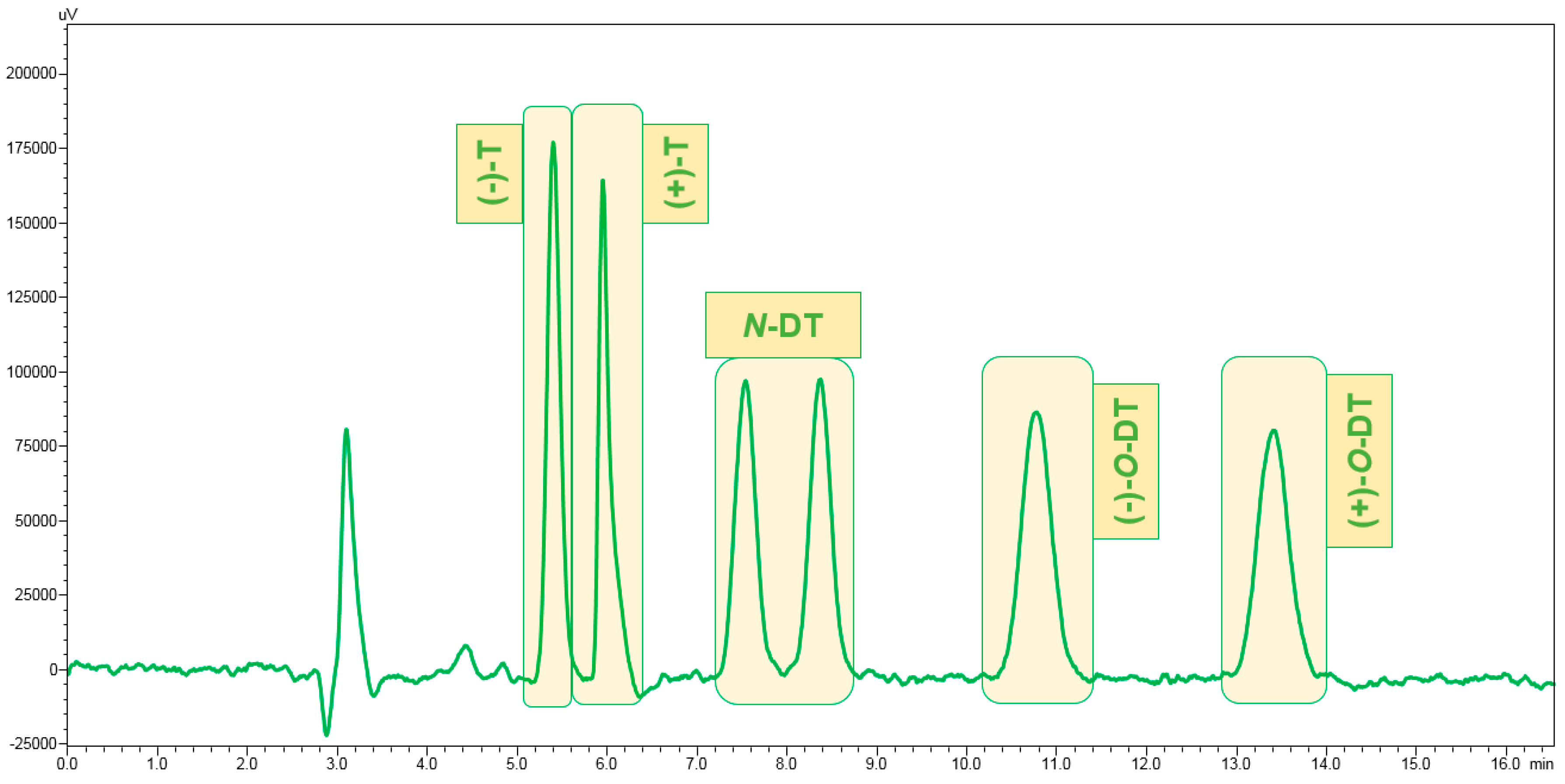
3.1. Enantiomeric Separation
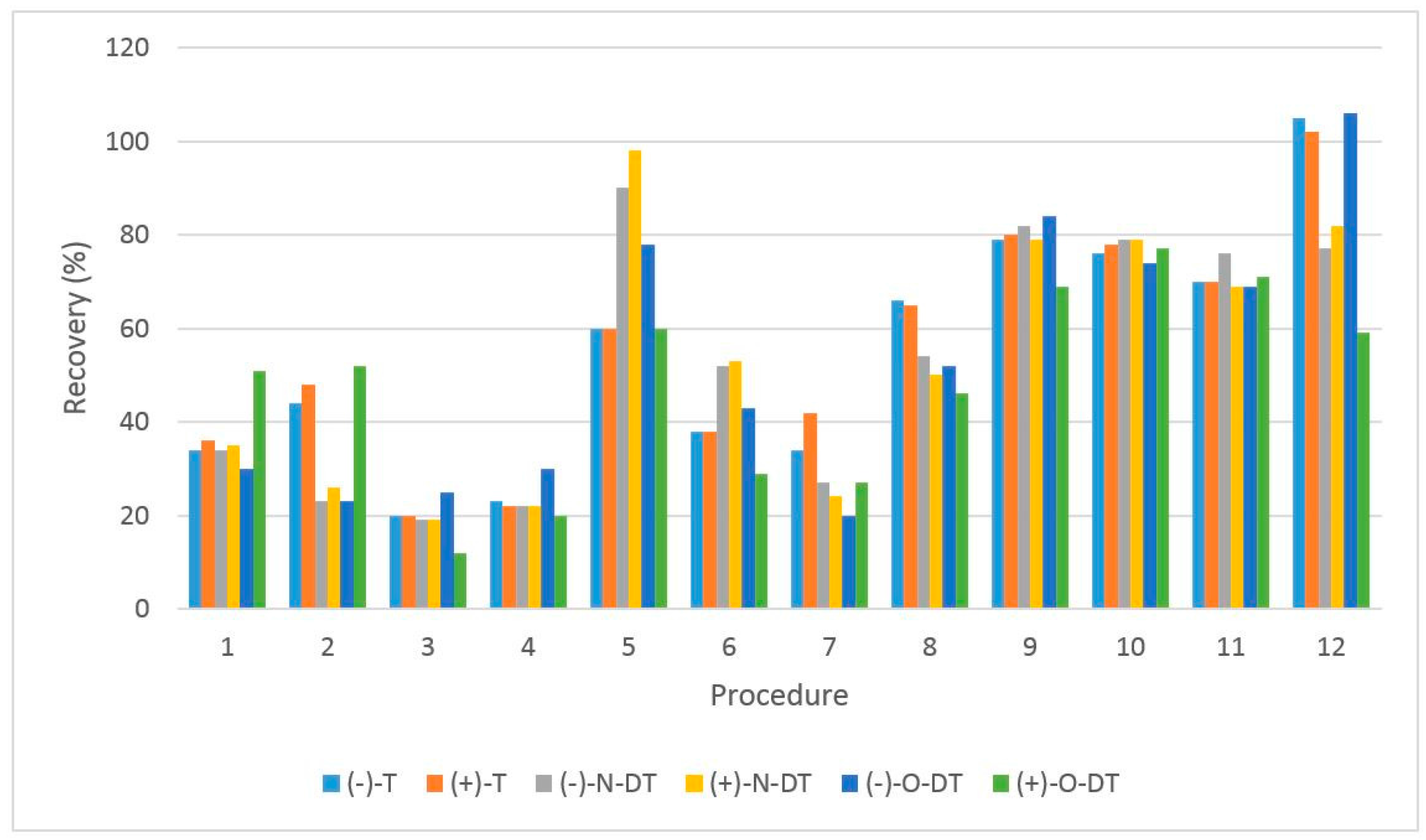
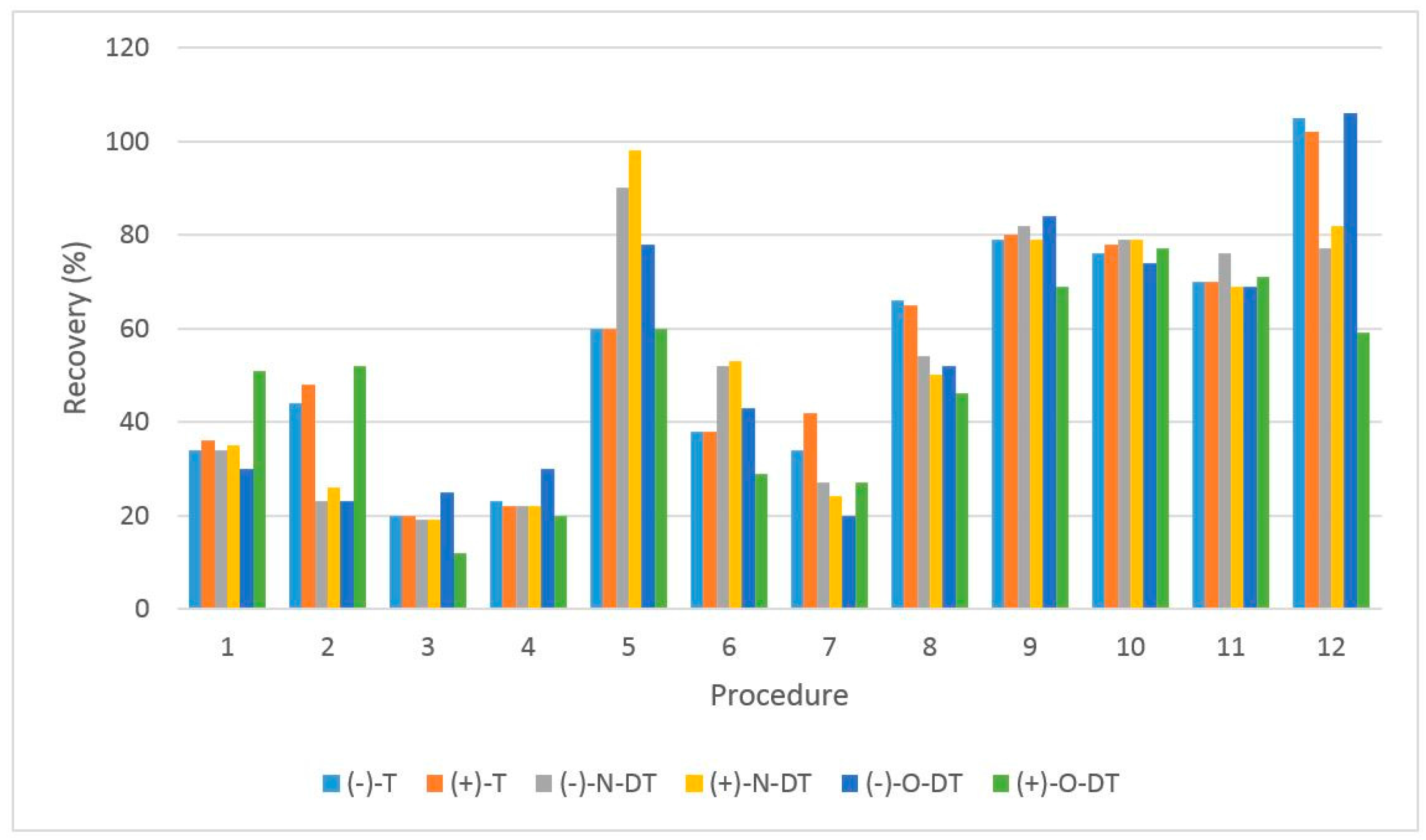
3.2. Optimization of the Sample Preparation Procedure
3.3. Method Validation
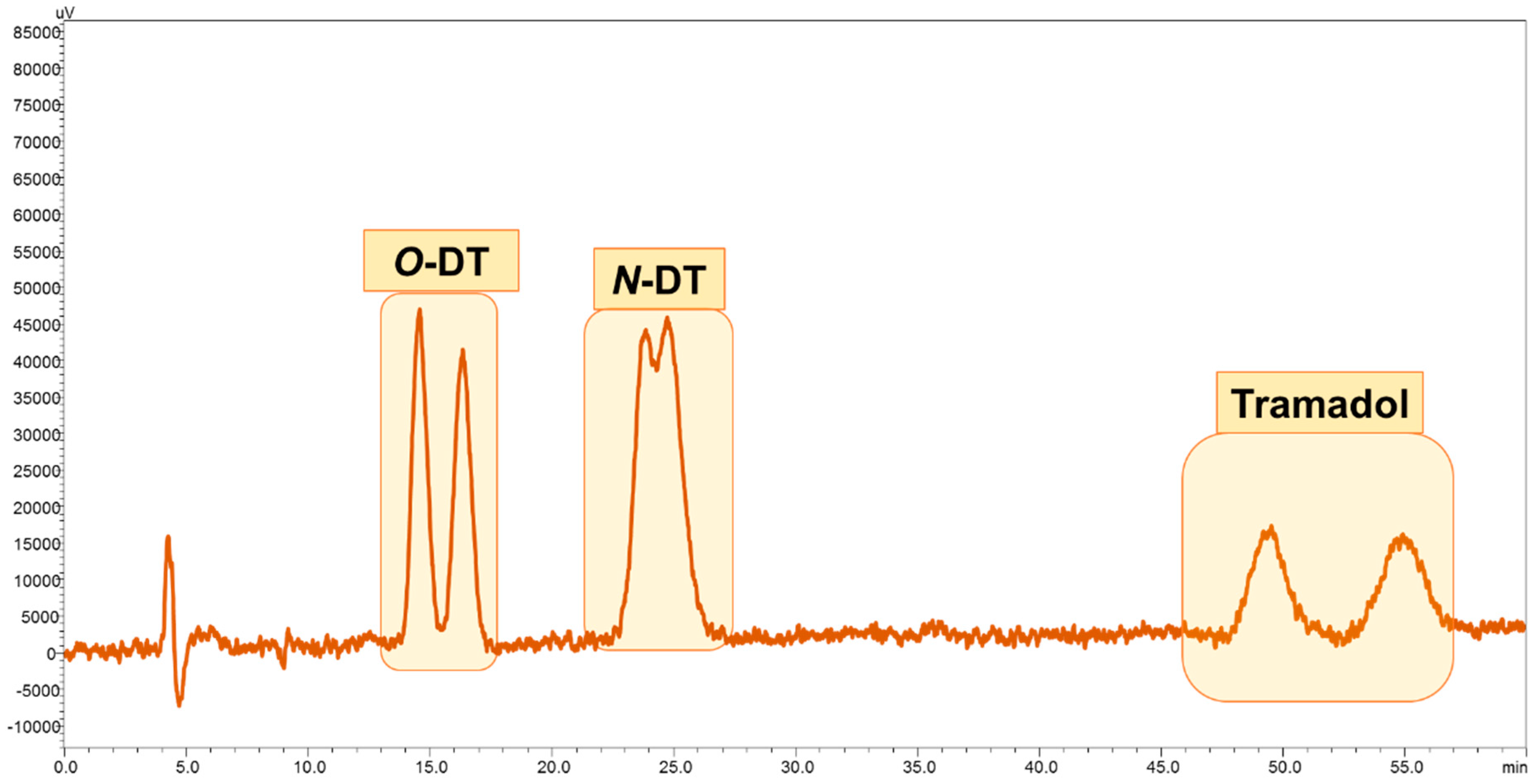
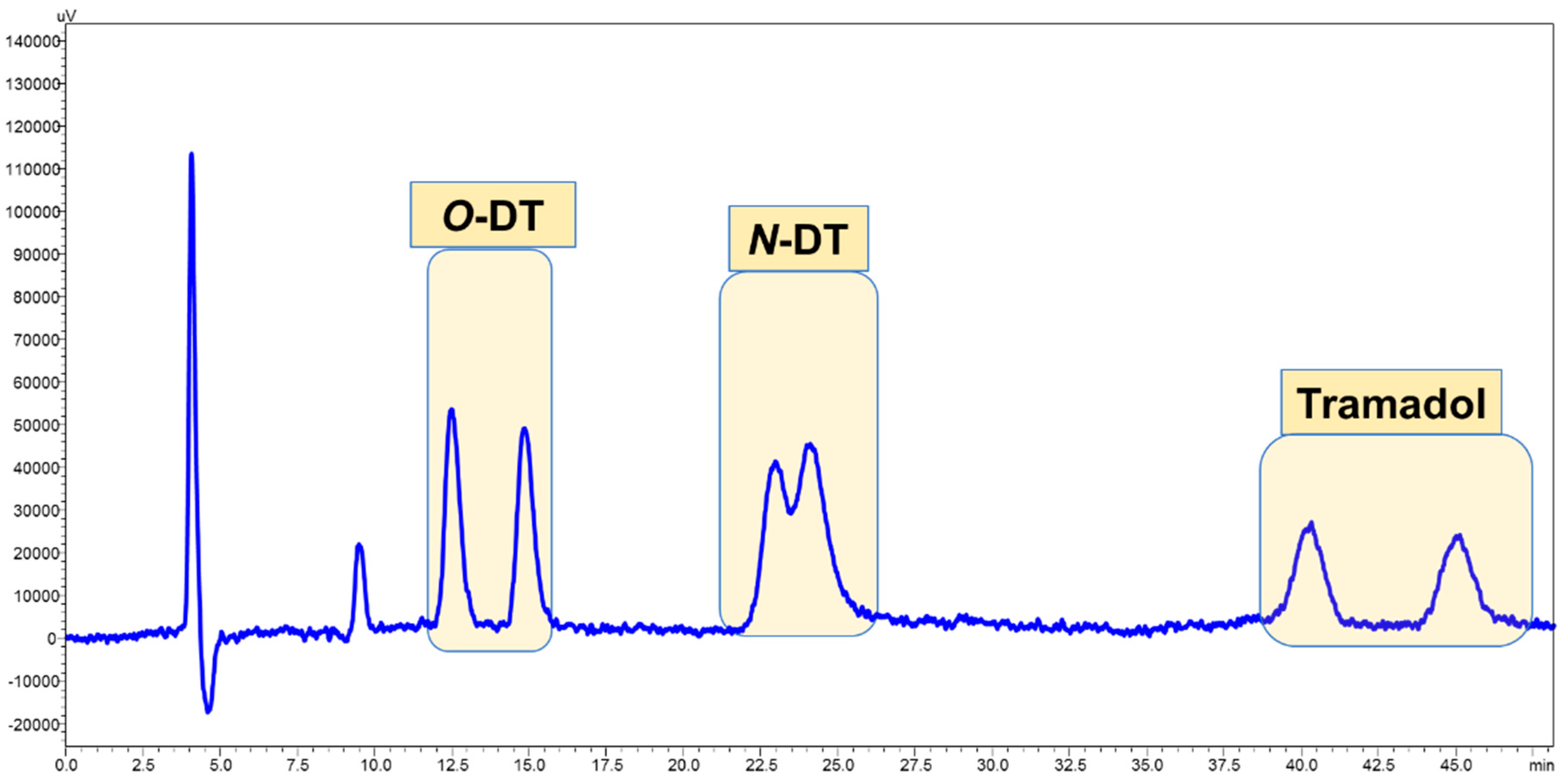
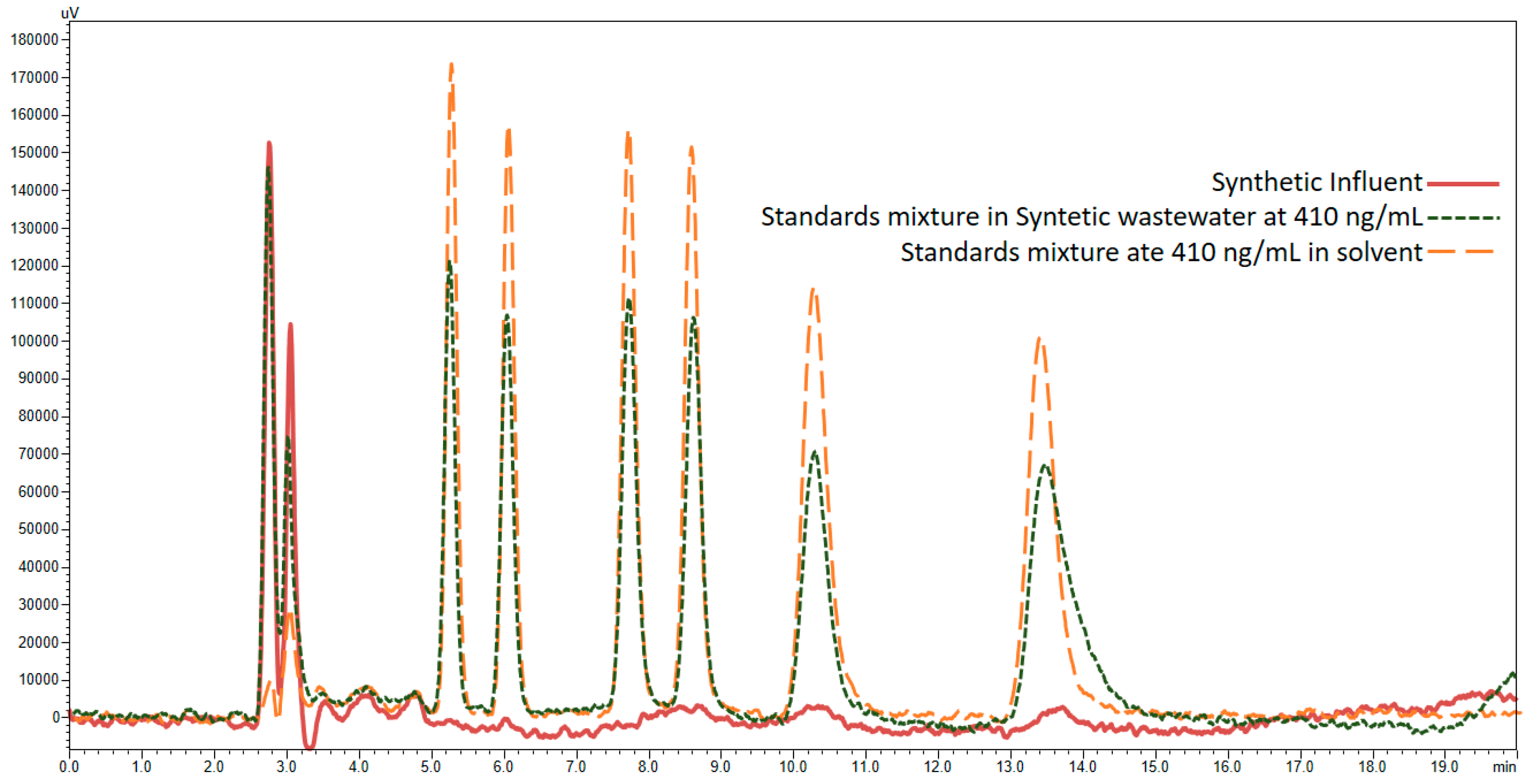
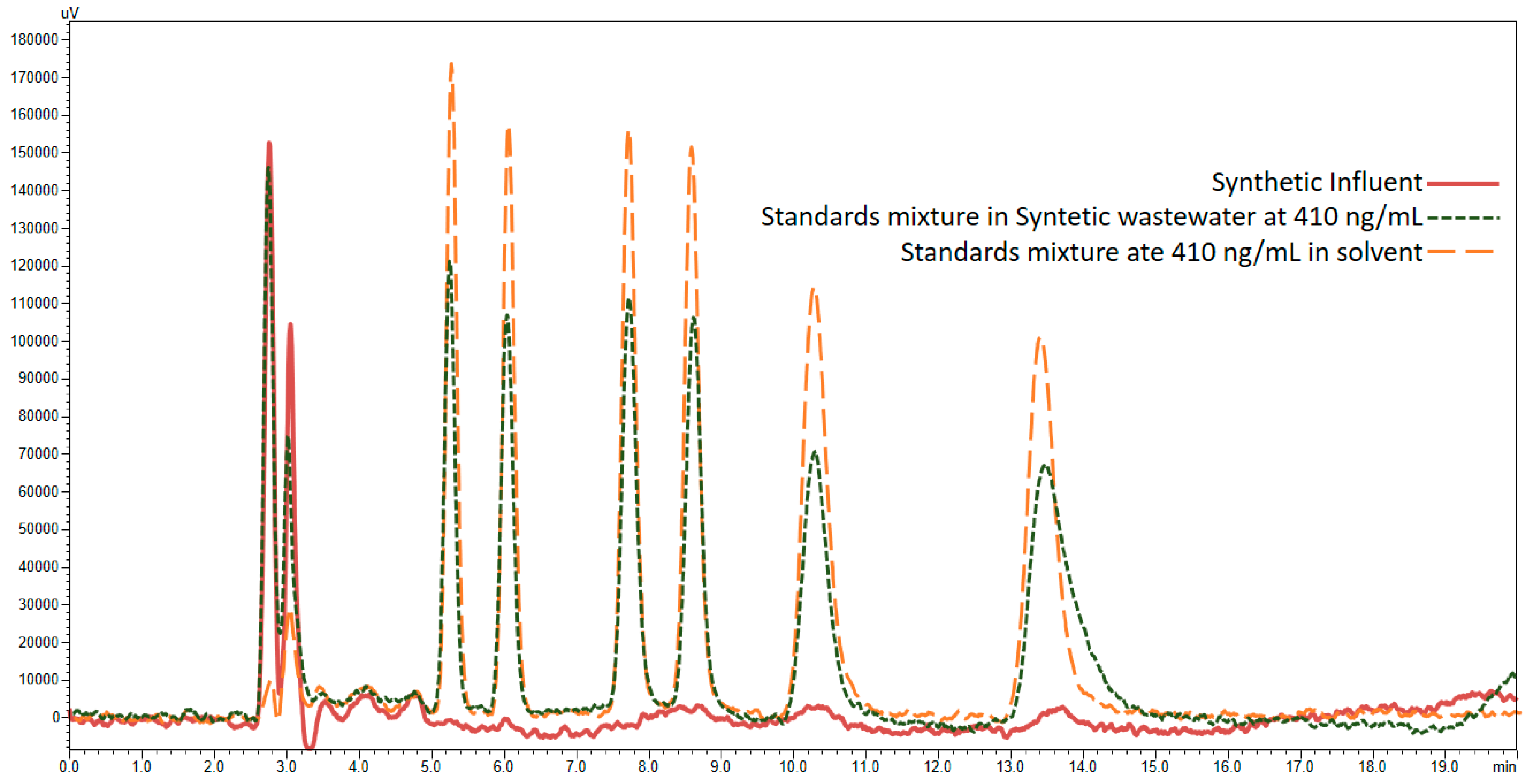
3.4. Application of Developed LC-FD Method in WWTP Samples
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Herrmann, M.; Olsson, O.; Fiehn, R.; Herrel, M.; Kümmerer, K. The significance of different health institutions and their respective contributions of active pharmaceutical ingredients to wastewater. Environ. Int. 2015, 85, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Cizmas, L.; Sharma, V.; Gray, C.; McDonald, T. Pharmaceuticals and personal care products in waters: Occurrence, toxicity, and risk. Environ. Chem. Lett. 2015, 13, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview. J. Chromatogr. B 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.; Castro, P.L.; Tiritan, M. Environmental fate of chiral pharmaceuticals: Determination, degradation and toxicity. In Environmental Chemistry for a Sustainable World; Lichtfouse, E., Schwarzbauer, J., Robert, D., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 3–45. [Google Scholar]
- Ribeiro, A.R.; Castro, P.M.L.; Tiritan, M.E. Chiral pharmaceuticals in the environment. Environ. Chem. Lett. 2012, 10, 239–253. [Google Scholar] [CrossRef]
- Stanley, J.K.; Ramirez, A.J.; Chambliss, C.K.; Brooks, B.W. Enantiospecific sublethal effects of the antidepressant fluoxetine to a model aquatic vertebrate and invertebrate. Chemosphere 2007, 69, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.K.; Ramirez, A.J.; Mottaleb, M.; Chambliss, C.K.; Brooks, B.W. Enantiospecific toxicity of the beta-blocker propranolol to daphnia magna and pimephales promelas. Environ. Toxicol. Chem. 2006, 25, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Rukhlenko, I.D.; Tepliakov, N.V.; Baimuratov, A.S.; Andronaki, S.A.; Gun’ko, Y.K.; Baranov, A.V.; Fedorov, A.V. Completely chiral optical force for enantioseparation. Sci. Rep. 2016, 6, 36884. [Google Scholar] [CrossRef] [PubMed]
- Kupai, J.; Rojik, E.; Huszthy, P.; Szekely, G. Role of chirality and macroring in imprinted polymers with enantiodiscriminative power. ACS Appl. Mater. Interfaces 2015, 7, 9516–9525. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Oliveira, A.; Costa, I.; Gouveia, C.A.; Carvalho, F.; Moreira, R.F.; Dinis-Oliveira, R.J. Simultaneous quantification of tramadol and O-desmethyltramadol in hair samples by gas chromatography-electron impact/mass spectrometry. Biomed. Chromatogr. 2013, 27, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Castrignano, E.; Lubben, A.; Kasprzyk-Hordern, B. Enantiomeric profiling of chiral drug biomarkers in wastewater with the usage of chiral liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. A 2016, 1438, 84–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, S.; Taschwer, M.; Schmid, M.G. Chiral separation of cathinone derivatives used as recreational drugs by HPLC-UV using a CHIRALPAK® AS-H column as stationary phase. Chirality 2012, 24, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.R.; Maia, A.S.; Moreira, I.S.; Afonso, C.M.; Castro, P.M.; Tiritan, M.E. Enantioselective quantification of fluoxetine and norfluoxetine by HPLC in wastewater effluents. Chemosphere 2014, 95, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; He, L.; Sun, W.; Cheng, Y.; Liu, J.; Ren, Z. Chiral liquid membrane for enantioselective separation of racemic ibuprofen by l-tartaric acid derivatives. RSC Adv. 2015, 5, 41729–41735. [Google Scholar] [CrossRef]
- Musshoff, F.; Madea, B. Fatality due to ingestion of tramadol alone. Forensic Sci. Int. 2001, 116, 197–199. [Google Scholar] [CrossRef]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef] [PubMed]
- Kusari, S.; Tatsimo, S.J.; Zuhlke, S.; Spiteller, M. Synthetic origin of tramadol in the environment. Angew. Chem. Int. Ed. 2016, 55, 240–243. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.A.; Mohamed, K.M.; Nasser, A.Y.; Button, J.; Holt, D.W. Simultaneous determination of tramadol, O-desmethyltramadol and n-desmethyltramadol in human urine by gas chromatography-mass spectrometry. J. Chromatogr. B 2013, 926, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.N.; Sharma, N.; Sanyal, M.; Shrivastav, P.S. An accurate, rapid and sensitive determination of tramadol and its active metabolite O-desmethyltramadol in human plasma by LC–MS/MS. J. Pharm. Biomed. Anal. 2009, 49, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; El-Abhar, H.; Saleh, S. The effect of nigella sativa oil against the liver damage induced by schistosoma mansoni infection in mice. J. Ethnopharmacol. 2002, 79, 1–11. [Google Scholar] [CrossRef]
- Awadalla, E.A.; Salah-Eldin, A. Histopathological and molecular studies on tramadol mediated hepato-renal toxicity in rats. IOSR J. Pharm. Biol. Sci. 2015, 10, 90–102. [Google Scholar]
- Tao, Q.; Stone, D.J., Jr.; Borenstein, M.R.; Jean-Bart, V.; Codd, E.E.; Coogan, T.P.; Desai-Krieger, D.; Liao, S.; Raffa, R.B. Gas chromatographic method using nitrogen–phosphorus detection for the measurement of tramadol and its O-desmethyl metabolite in plasma and brain tissue of mice and rats. J. Chromatogr. B Biomed. Sci. Appl. 2001, 763, 165–171. [Google Scholar] [CrossRef]
- Ardakani, Y.H.; Rouini, M.-R. Improved liquid chromatographic method for the simultaneous determination of tramadol and its three main metabolites in human plasma, urine and saliva. J. Pharm. Biomed. Anal. 2007, 44, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, R. Simultaneous analysis of tramadol, metoprolol and their metabolites in human plasma and urine by high performance liquid chromatography. Chin. Med. J. 2006, 119, 2013–2017. [Google Scholar] [PubMed]
- Zhao, L.M.; Chen, X.Y.; Cui, J.J.; Sunita, M.; Zhong, D.F. Determination of tramadol and its active metabolite O-desmethyltramadol in plasma and amniotic fluid using LC/MS/MS. Acta Pharm. Sin. 2004, 39, 458–462. [Google Scholar]
- Ceccato, A.; Chiap, P.; Hubert, P.; Crommen, J. Automated determination of tramadol enantiomers in human plasma using solid-phase extraction in combination with chiral liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1997, 698, 161–170. [Google Scholar] [CrossRef]
- Valle, M.; Pavón, J.M.; Calvo, R.; Campanero, M.A.; Trocóniz, I.F. Simultaneous determination of tramadol and its major active metabolite O-demethyltramadol by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl. 1999, 724, 83–89. [Google Scholar] [CrossRef]
- Ceccato, A.; Vanderbist, F.; Pabst, J.Y.; Streel, B. Enantiomeric determination of tramadol and its main metabolite O-desmethyltramadol in human plasma by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2000, 748, 65–76. [Google Scholar] [CrossRef]
- Ruda, S.; Cherkaoui, S.; Dayer, P.; Fanali, S.; Veuthey, J.L. Simultaneous stereoselective analysis of tramadol and its main phase I metabolites by on-line capillary zone electrophoresis-electrospray ionization mass spectrometry. J. Chromatogr. A 2000, 868, 295–303. [Google Scholar] [CrossRef]
- Juzwin, S.J.; Wang, D.C.; Anderson, N.J.; Wong, F.A. The determination of RWJ-38705 (tramadol N-oxide) and its metabolites in preclinical pharmacokinetic studies using LC–MS/MS. J. Pharm. Biomed. Anal. 2000, 22, 469–480. [Google Scholar] [CrossRef]
- Gan, S.H.; Ismail, R. Validation of a high-performance liquid chromatography method for tramadol and O-desmethyltramadol in human plasma using solid-phase extraction. J. Chromatogr. B Biomed. Sci. Appl. 2001, 759, 325–335. [Google Scholar] [CrossRef]
- Nobilis, M.; Kopecky, J.; Kvetina, J.; Chladek, J.; Svoboda, Z.; Vorisek, V.; Perlik, F.; Pour, M.; Kunes, J. High-performance liquid chromatographic determination of tramadol and its O-desmethylated metabolite in blood plasma: Application to a bioequivalence study in humans. J. Chromatogr. A 2002, 949, 11–22. [Google Scholar] [CrossRef]
- Campanero, M.A.; García-Quetglas, E.; Sádaba, B.; Azanza, J.R. Simultaneous stereoselective analysis of tramadol and its primary phase I metabolites in plasma by liquid chromatography: Application to a pharmacokinetic study in humans. J. Chromatogr. A 2004, 1031, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Ardakani, Y.H.; Mehvar, R.; Foroumadi, A.; Rouini, M.-R. Enantioselective determination of tramadol and its main phase I metabolites in human plasma by high-performance liquid chromatography. J. Chromatogr. B 2008, 864, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Fawcett, J.P. Improved HPLC method for the simultaneous determination of tramadol and O-desmethyltramadol in human plasma. J. Chromatogr. B 2005, 821, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Rouini, M.-R.; Ardakani, Y.H.; Soltani, F.; Aboul-Enein, H.Y.; Foroumadi, A. Development and validation of a rapid HPLC method for simultaneous determination of tramadol, and its two main metabolites in human plasma. J. Chromatogr. B 2006, 830, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Mehvar, R.; Elliott, K.; Parasrampuria, R.; Eradiri, O. Stereospecific high-performance liquid chromatographic analysis of tramadol and its O-demethylated (M1) and N,O-demethylated (M5) metabolites in human plasma. J. Chromatogr. B 2007, 852, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Curticapean, A.; Muntean, D.; Curticapean, M.; Dogaru, M.; Vari, C. Optimized HPLC method for tramadol and O-desmethyl tramadol determination in human plasma. J. Biochem. Biophys. Methods 2008, 70, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Vlase, L.; Leucuta, S.E.; Imre, S. Determination of tramadol and O-desmethyltramadol in human plasma by high-performance liquid chromatography with mass spectrometry detection. Talanta 2008, 75, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.E.; Davies, P.; Lubben, A.; Kasprzyk-Hordern, B. Determination of chiral pharmaceuticals and illicit drugs in wastewater and sludge using microwave assisted extraction, solid-phase extraction and chiral liquid chromatography coupled with tandem mass spectrometry. Anal. Chim. Acta 2015, 882, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH Steering Committee. ICH Q2B Validation of Analytical Procedures: Methodology; (CPMP/ICH/281/95); European Agency for the Evaluation of Medicinal Products, International Commission on Harmonisation: London, UK, 1996. [Google Scholar]
- Lämmerhofer, M. Chiral recognition by enantioselective liquid chromatography: Mechanisms and modern chiral stationary phases. J. Chromatogr. A 2010, 1217, 814–856. [Google Scholar] [CrossRef] [PubMed]
- De Campos Lourenço, T.; Cassiano, N.M.; Cass, Q.B. Fases estacionárias quirais para cromatografia líquida de alta eficiência. Quim. Nova 2010, 33, 2155–2164. [Google Scholar] [CrossRef]
- Ward, T.J.; Ward, K.D. Chiral separations: Fundamental review 2010. Anal. Chem. 2010, 82, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Kalíková, K.; Riesová, M.; Tesařová, E. Recentchiral selectors for separation in HPLC and CE. Cent. Eur. J. Chem. 2012, 10, 450–471. [Google Scholar] [CrossRef]
- Scriba, G.K.E. Chiral recognition mechanisms in analytical separation sciences. Chromatographia 2012, 75, 815–838. [Google Scholar] [CrossRef]
- Fernandes, C.; Tiritan, M.; Pinto, M. Small molecules as chromatographic tools for HPLC enantiomeric resolution: Pirkle-type chiral stationary phases evolution. Chromatographia 2013, 76, 871–897. [Google Scholar] [CrossRef]
- Haginaka, J. Recent progresses in protein-based chiral stationary phases for enantioseparations in liquid chromatography. J. Chromatogr. B 2008, 875, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.; Vu, V.A.; Matthijs, N.; Maftouh, M.; Massart, D.L.; Vander Heyden, Y. Screening approach for chiral separation of pharmaceuticals: Part I. Normal-phase liquid chromatography. J. Chromatogr. A 2002, 947, 69–83. [Google Scholar] [CrossRef]
- Andersson, M.E.; Aslan, D.; Clarke, A.; Roeraade, J.; Hagman, G. Evaluation of generic chiral liquid chromatography screens for pharmaceutical analysis. J. Chromatogr. A 2003, 1005, 83–101. [Google Scholar] [CrossRef]
- Cass, Q.B.; Batigalhia, F. Enantiomeric resolution of a series of chiral sulfoxides by high-performance liquid chromatography on polysaccharide-based columns with multimodal elution. J. Chromatogr. A 2003, 987, 445–452. [Google Scholar] [CrossRef]
- Fernandes, C.; Palmeira, A.; Santos, A.; Tiritan, M.E.; Afonso, C.; Pinto, M.M. Enantioresolution of chiral derivatives of xanthones on (S,S)-whelk-O1 and l-phenylglycine stationary phases and chiral recognition mechanism by docking approach for (S,S)-whelk-O1. Chirality 2013, 25, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Chytil, L.; Matouskova, O.; Cerna, O.; Pokorna, P.; Vobruba, V.; Perlik, F.; Slanar, O. Enantiomeric determination of tramadol and O-desmethyltramadol in human plasma by fast liquid chromatographic technique coupled with mass spectrometric detection. J. Chromatogr. B 2010, 878, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Musshoff, F.; Madea, B.; Stuber, F.; Stamer, U.M. Enantiomeric determination of tramadol and O-desmethyltramadol by liquid chromatography-mass spectrometry and application to postoperative patients receiving tramadol. J. Anal. Toxicol. 2006, 30, 463–467. [Google Scholar] [CrossRef] [PubMed]
- ICH, M. Q2b—Validation of analytical procedures: Methodology. In Proceedings of the International Conference on Harmonization Expert Working Group, Geneva, Switzerland, 6 Novermber 1996. [Google Scholar]
- Stamer, U.M.; Musshoff, F.; Kobilay, M.; Madea, B.; Hoeft, A.; Stuber, F. Concentrations of tramadol and O-desmethyltramadol enantiomers in different CYP2D6 genotypes. Clin. Pharmacol. Ther. 2007, 82, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.A.; Roelofse, J.A.; Shipton, E.A. Pharmacokinetics of oral tramadol drops for postoperative pain relief in children aged 4 to 7 years—A pilot study. Anesth. Prog. 2002, 49, 109–112. [Google Scholar] [PubMed]
- Paar, W.D.; Poche, S.; Gerloff, J.; Dengler, H.J. Polymorphic CYP2D6 mediates O-demethylation of the opioid analgesic tramadol. Eur. J. Clin. Pharmacol. 1997, 53, 235–239. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Procedure | Cartridge | Conditioning | Washing | Elution (1st Step) | Elution (2nd Step) |
|---|---|---|---|---|---|
| 1 | MCX | 8 mL MeOH 8 mL H2O | 8 mL H2O 8 mL MeOH | 8 mL of 5% NH4OH solved in ACN/MeOH 60:40 | - |
| 2 | MCX | 8 mL MeOH 8 mL H2O | 8 mL 2% Formic Acid | 8 mL MeOH | 8 mL of 5% NH4OH solved in MeOH |
| 3 | MCX | 8 mL MeOH 8 mL H2O | 8 mL 2% Formic Acid | 12 mL of 10% NH4OH solved in MeOH | - |
| 4 | MCX | 8 mL EtOH 8 mL H2O | 8 mL 2% Formic Acid | 8 mL EtOH | 8 mL of 5% NH4OH solved in EtOH |
| 5 | MCX | 8 mL EtOH 8 mL Formic acid 2% | 8 mL 2% Formic Acid | 8 mL of 0.6% Formic Acid solved in EtOH | 8 mL of 5% NH4OH solved in EtOH |
| 6 | MCX | 8 mL EtOH 8 mL H2O (pH = 2 adjusted with HCl) | 8 mL 2% Formic Acid | 8 mL EtOH | 8 mL of 5% NH4OH solved in EtOH |
| 7 | HLB | 10 mL MetOH 10 mL H2O | 10 mL H2O | 10 mL MeOH | - |
| 8 | MCX | 8 mL MeOH 8 mL H2O | 8 mL 2% Formic Acid | 12 mL of 10% NH4OH solved in MeOH | - |
| 9 | MCX | 8 mL EtOH 8 mL H2O | 8 mL 2% Formic Acid | 8 mL EtOH | 8 mL of 5% NH4OH solved in EtOH |
| 10 | MCX | 8 mL EtOH 8 mL 2% Formic Acid | 8 mL 2% Formic Acid | 8 mL of 0.6% Formic Acid solved in EtOH | 12 mL of 5% NH4OH solved in EtOH |
| 11 | MCX | 8 mL EtOH 8 mL 2% Formic Acid | 8 mL 2% Formic Acid | 12 mL of 5% NH4OH solved in EtOH | - |
| 12 | MCX | 8 mL EtOH 8 mL 2% Formic Acid | 8 mL 2% Formic Acid | 8 mL of 0.6% Formic Acid solved in EtOH | 12 mL of 5% NH4OH solved in EtOH |
| Column | Elution Mode | Mobile Phase: Proportion (v/v) | Tramadol | N-DT | O-DT | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K1 | K2 | α | Rs | K1 | K2 | α | Rs | K1 | K2 | α | Rs | |||
| Lux Cellulose-2 | Normal | Hex/IPA/DEA: 90:10:0.1 ** | 0.73 | 0.89 | 1.22 | 1.00 | 1.13 | 1.62 | 1.44 | 3.02 | 1.55 | 3.04 | 1.96 | 5.53 |
| Hex/IPA/DEA: 90:10:0.05 ** | 4.31 | 4.66 | 1.08 | 0.92 | 4.53 | 5.57 | 1.23 | 2.75 | 9.86 | 14.7 | 1.49 | 4.10 | ||
| Hex/EtOH/DEA: 96:4: 0.1 ** | 0.98 | 1.07 | 1.11 | 1.12 | 1.91 | - | 1.00 | - | 3.56 | 4.16 | 1.17 | 2.42 | ||
| Reversed | ACN (5 mM ammonium formate/0.1% DEA)/H2O: 35:65 * | 5.75 | 6.13 | 1.07 | 1.49 | 4.29 | - | 1.00 | - | 1.29 | 1.42 | 1.11 | 0.949 | |
| ACN (5 mM ammonium formate/0.1% DEA)/H2O: 30:70 * | 9.98 | 10.6 | 1.07 | 1.89 | 7.19 | - | 1.00 | - | 1.91 | 2.11 | 1.11 | 1.31 | ||
| Lux Cellulose-4 | Reversed | ACN (5 mM ammonium formate/0.1% DEA)/H2O: 35:65 ** | 6.32 | 7.21 | 1.14 | 2.79 | 10.7 | 11.4 | 1.07 | 1.26 | 1.25 | 1.66 | 1.33 | 2.31 |
| MeOH (5 mM ammonium formate/0.05% DEA)/H2O: 55:45 ** | 10.7 | 11.9 | 1.12 | 2.18 | 4.62 | 4.83 | 1.05 | 0.236 | 2.43 | 2.85 | 1.17 | 1.43 | ||
| ACN:EtOH (10 mM ammonium formate/0.1% DEA) /H2O: 17.5:17.5:65 ** | 8.91 | 10.1 | 1.13 | 2.69 | 4.65 | 4.92 | 1.06 | 0.36 | 2.07 | 2.65 | 1.28 | 2.62 | ||
| Normal | Hex/EtOH/DEA—96:4:0.1 *** | 0.74 | 0.920 | 1.24 | 2.16 | 1.43 | 1.70 | 1.19 | 1.85 | 2.48 | 3.33 | 1.34 | 4.16 | |
| Analyte | E | Nominal Conc. Range (ng L−1) | Calibration Curve Equation | r2 | IDL (ng L−1) | IQL (ng L−1) | MDL (ng L−1) | MQL (ng L−1) |
|---|---|---|---|---|---|---|---|---|
| Tramadol | (−)-T | 28–168 | y = 6944.4x − 93525 | 0.9902 | 10 | 35 | 8 | 28 |
| (+)-T | y = 7421.2x − 113599 | 0.9901 | 10 | 35 | 8 | 28 | ||
| N-DT | (−)-N-DT | 28–168 | y = 6128.8x + 4247 | 0.9958 | 10 | 35 | 8 | 28 |
| (+)-N-DT | y = 5973.8x + 33396 | 0.9939 | 10 | 35 | 8 | 28 | ||
| O-DT | (−)-O-DT | 56–196 | y = 30420x − 2 × 106 | 0.9954 | 25 | 70 | 20 | 56 |
| (+)-O-DT | y = 44269x − 2 × 106 | 0.9948 | 25 | 70 | 20 | 56 |
| Analyte | Nominal Concentration (ng mL−1) | E | 1st Day | 2nd Day | 3rd Day | Inter-Day RSD (%) | Recovery (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | RSD (%) | Accuracy (%) | RSD (%) | Accuracy (%) | RSD (%) | |||||
| Tramadol | 75 | (−)-T | 89.6 | 7.7 | 88.5 | 4.9 | 94.3 | 3.1 | 6.5 | 87.1 |
| (+)-T | 90.6 | 8.3 | 91.4 | 3.7 | 93.3 | 5.9 | 6.4 | 88.9 | ||
| 150 | (−)-T | 80.1 | 4.6 | 80.6 | 1.6 | 82.1 | 6.5 | 4.9 | 78.4 | |
| (+)-T | 81.0 | 6.7 | 80.4 | 0.3 | 82.9 | 5.8 | 5.4 | 78.3 | ||
| 205 | (−)-T | 105.6 | 1.0 | 82.2 | 8.0 | 97.9 | 8.5 | 12.5 | 97.8 | |
| (+)-T | 70.3 | 1.1 | 84.0 | 3.8 | 90.0 | 8.4 | 11.7 | 83.2 | ||
| N-DT | 75 | (−)-N-DT | 99.1 | 8.2 | 82.0 | 8.8 | 105.7 | 3.5 | 11.7 | 99.2 |
| (+)-N-DT | 114.1 | 8.8 | 91.2 | 3.9 | 100.0 | 8.9 | 11.7 | 99.4 | ||
| 150 | (−)-N-DT | 98.3 | 1.7 | 91.9 | 6.2 | 97.5 | 6.1 | 5.8 | 97.6 | |
| (+)-N-DT | 87.8 | 5.6 | 81.3 | 1.5 | 90.6 | 6.7 | 6.8 | 88.2 | ||
| 205 | (−)-N-DT | 77.0 | 6.3 | 85.6 | 4.0 | 105.4 | 7.9 | 16.7 | 85.9 | |
| (+)-N-DT | 79.4 | 4.9 | 89.6 | 3.7 | 108.7 | 7.4 | 16.3 | 89.8 | ||
| O-DT | 75 | (−)-O-DT | 94.9 | 5.0 | 92.5 | 8.2 | 96.9 | 4.5 | 14.8 | 72.2 |
| (+)-O-DT | 96.0 | 6.8 | 97.1 | 8.9 | 97.5 | 9.2 | 9.5 | 83.6 | ||
| 150 | (−)-O-DT | 97.0 | 9.9 | 93.2 | 5.0 | 96.3 | 7.8 | 10.6 | 87.4 | |
| (+)-O-DT | 99.9 | 4.6 | 98.5 | 7.2 | 99.4 | 3.6 | 5.9 | 98.1 | ||
| 205 | (−)-O-DT | 81.7 | 7.0 | 90.0 | 4.5 | 97.9 | 3.8 | 14.9 | 82.0 | |
| (+)-O-DT | 90.2 | 5.9 | 95.6 | 6.3 | 106.3 | 8.9 | 14.5 | 92.2 | ||
| Sample nº | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| Effluent | Influent | Effluent | Influent | |||
| Concentration (ng L−1) | Tramadol | (−)-T | 235.8 | 357.9 | 325.1 | 350.0 |
| (+)-T | 118.7 | 233.6 | 314.9 | 233.8 | ||
| N-DT | (−)-N-DT | <QL | <QL | <QL | <QL | |
| (+)-N-DT | 43.7 | 63.9 | 62.1 | 72.7 | ||
| O-DT | (−)-O-DT | 60.8 | 69.7 | 71.6 | 69.5 | |
| (+)-O-DT | 57.7 | 86.7 | 95.4 | 106.7 | ||
| Enantiomeric Fraction | Tramadol | EF1 | 0.67 | 0.61 | 0.51 | 0.60 |
| N-DT | EF1 | ≈0 | ≈0 | ≈0 | ≈0 | |
| O-DT | EF1 | 0.51 | 0.45 | 0.43 | 0.39 | |
| DRE (%) | Tramadol | (−)-T | 65.9 | 92.9 | ||
| (+)-T | 50.8 | 134.7 * | ||||
| N-DT | (−)-N-DT | NC | NC | |||
| (+)-N-DT | 68.4 | 85.4 | ||||
| O-DT | (−)-O-DT | 87.2 | 103.0 * | |||
| (+)-O-DT | 66.6 | 89.4 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, C.; Ribeiro, C.; Maia, A.S.; Gonçalves, V.; Tiritan, M.E.; Afonso, C. Enantiomeric Separation of Tramadol and Its Metabolites: Method Validation and Application to Environmental Samples. Symmetry 2017, 9, 170. https://doi.org/10.3390/sym9090170
Silva C, Ribeiro C, Maia AS, Gonçalves V, Tiritan ME, Afonso C. Enantiomeric Separation of Tramadol and Its Metabolites: Method Validation and Application to Environmental Samples. Symmetry. 2017; 9(9):170. https://doi.org/10.3390/sym9090170
Chicago/Turabian StyleSilva, Cátia, Cláudia Ribeiro, Alexandra S. Maia, Virgínia Gonçalves, Maria Elizabeth Tiritan, and Carlos Afonso. 2017. "Enantiomeric Separation of Tramadol and Its Metabolites: Method Validation and Application to Environmental Samples" Symmetry 9, no. 9: 170. https://doi.org/10.3390/sym9090170