1. Introduction
Arsenic is one of the most toxic inorganic pollutants in aquatic systems [
1]. Its source is mainly geogenic, entering the environment through volcanic emissions, hydrothermal systems [
2], mineral erosion, or by reductive dissolution of iron hydroxides, such as in the case of Bangladesh [
3]. Although the input from anthropogenic sources (mining, fossil fuels) is lower than natural sources, these can have a strong impact, generating local pollution episodes [
2]. There are mainly three mechanisms that explain arsenic liberation into natural environments [
4]: (1) oxidation of arsenic sulphides, (2) competitive desorption, and (3) reductive dissolution of iron oxides. Although each has been extensively investigated under several experimental conditions, there is still a lack of knowledge as to how these processes take place in specific environments.
Some climatic models predict that, by the end of the century, one of the consequences of global warming will be a sea level rise of up to 1.1 m above the current level [
5]. This sea level change will establish a new geochemical context in the oxidation zone of mine tailings and acid sulphate soils near the shoreline, where the stability of sorbent minerals under seawater flood would be uncertain. In this paper, the effects of seawater intrusion on arsenic adsorbed to or co-precipitated with major sorbent minerals, present in acid soils or mine tailings, are simulated. Synthesized jarosite, schwertmannite, goethite, and ferrihydrite were used to represent different arsenic uptake scenarios. The aim of these experiments is to establish the stability and transport of this metalloid in these coastal environments under changing environmental conditions.
Iron oxides behave as excellent sorbents for a variety of contaminants in almost every environment. They have been widely used in the extraction of heavy metals from natural [
6] and industrial sources [
7], which has generated a great number of studies about mechanisms of surface interactions [
8,
9,
10,
11,
12]. These minerals are common in environments with high availability of metal sulphides exposed to oxidizing conditions, as in acid sulphate soils, in the oxidation zone of mine tailings, or ore deposits [
13,
14]. In these systems, oxidation of the primary sulphide (pyrite) is capable of releasing large amounts of protons, sulphate, Fe(II), and trace metals (Equation (1)) [
15]. Once Fe(III) is produced by oxidation of Fe(II), which may be strongly accelerated by microbial activity under low pH conditions, Fe(III) becomes the primary oxidant of pyrite [
16].
At circumneutral pH, ferrihydrite (5Fe
2O
3∙9(H
2O)) is commonly the first mineral phase to precipitate from hydrolysis of ferric solutions [
17]. Ferrihydrite is able to transform into crystalline phases of higher thermodynamic stability as goethite (α-FeOOH) or hematite (Fe
2O
3) [
18]. When the pH of the system is acidic enough (pH 2–4) and there is a high sulphate concentration, jarosite (Na, K, H
3O)∙[Fe
3(SO
4)
2(OH)
6]) (pH ~ 2) and schwertmannite (Fe
16O
16(OH)
12∙(SO
4)
2) constitute the main phases [
13]. Between pH 2.5 and 4, schwertmannite is, perhaps, the principal secondary mineral that forms from acid drainage [
19,
20].
In natural environments, redox processes commonly control the solubility of iron oxides. This occurs when interaction occurs between dissolved species, such as H
+, OH
− or other metal ions, with the hydroxyl groups present on the oxides surfaces [
21,
22]. Adsorption and formation of surface complexes with reducing species is a reaction that generates an electron transfer, reducing Fe(III) to Fe(II) [
21]. Weaker Fe(II) bonds enhance reductive dissolution and release of species from oxides surfaces. For example, in Alberta (Canada) microbial activity in acid sulphate soils released significant amounts of As when the Eh was below +100 mV [
23]. Mine tailings at the former Delnite gold mine in Northern Ontario showed that As(V) reduction and mobilization occurred due to reductive dissolution of goethite, influenced by a biosolid-cover [
24]. Reductive dissolution also takes place in remediated marine shore tailings deposits [
14]. Iron oxides under reductive conditions formed a Fe-Mn plume, which developed toward the shoreline where oxidizing and higher pH conditions triggered Fe(III) oxide-hydroxide precipitation.
When arsenic is present during hydrolysis of Fe(III) it can co-precipitate. In some minerals it can form part of the structure, for instance in jarosite at the
TO
4 site [
25,
26], or it can be adsorbed as surface complexes [
27]. In relation to iron oxides, arsenate tends to be adsorbed as outer-sphere complexes [
28], while arsenite can be adsorbed either as inner or outer-sphere complexes [
28,
29].
Different studies using Extended X-ray Absorption Fine Structure (EXAFS) and Fourier transform infrared spectroscopy (FTIR) have attempted to establish the possible bond between arsenic and iron oxide surfaces without reaching consensus to date. Waychunas
et al. [
30] stated that, due to thermodynamic constraints, there is only a low chance for the formation of mononuclear monodentate and bidentate complexes. Some authors established that bidentate binuclear complexes are most likely to form due to their higher thermodynamic stability [
31,
32,
33]. MICRO-EXAFS spectra of individual Fe(III) oxy-hydroxide grains point to inner-sphere bidentate-binuclear forms as the predominant As(V) complex and the existence of a second sphere corresponding mainly to bidentate-mononuclear [
34]. Furthermore, other authors postulate that As complexes at the surface of goethite would be exclusively monodentate complexes [
35] and when the As load increases significantly or pH level is greater than 6, then, only bidentate binuclear complexes are formed [
30,
33,
36].
Some species can compete with As for available vacancies restricting the metalloid adsorption. Studies on ferrihydrite indicate that the extent of adsorption can be affected by ionic competition, mainly by PO
4 >> organic ligands > SO
4 > Cl [
37,
38]. When As is part of the structure of iron oxides, the long term release is controlled by media dissolution, pH-dependent sorption/desorption, ion exchange, and transformation processes [
27,
39].
Tailings disposal in bays and beaches has been a widely used practice in the past and is still performed in places like Papua New Guinea and Indonesia [
40]. Lihir gold mine on Niolam Island in Papua New Guinea annually produces over 35 Mt of waste rock which are dumped into nearshore deepwater valleys, and about 100,000 ML of post-processing tailings slurry deposited at depth from a sub-surface pipeline [
41]. By representing a potential risk to marine ecosystems, one of the main challenges in this field is to determine the geochemical stability and pollution potential of tailings in flooded environments [
42].
In Chañaral (Chile) about 220 Mt of tailings fill the homonymous bay covering an area of about 4 km
2. Dold [
43] indicated the presence of an oxidation zone (>1 m) with significant amounts of arsenic associated with secondary Fe(III) oxide-hydroxides and jarosite. The instability of the sorbent phases and the intrusion of seawater, a product of variations within the coastal cycle, promoted the release of dissolved As, Mo, and colloidal adsorbed Cu and Zn into the ocean, generating an impact over the meiofaunal assemblages through increasing the bioavailability of heavy metals [
44] and there has been no sign of recovery 35 years after ceasing disposal [
45]. During the summer, high rates of evaporation promote capillary transport of Cu and Zn, which precipitate as efflorescent salts on the tailings surface, increasing the risk of metal exposure for the local community [
43,
46]. Remediation experiences of similar systems have been successfully implemented in environments where the hydrological characteristics allowed it (e.g., Bahia de Ite, Peru [
14]).
Concentrations of arsenic in open ocean seawater usually do not exceed 2 µg/L [
47]. It is found mainly as arsenate (As (V)), although, concentration of arsenite (As(III)) in coastal waters affected by anthropogenic activity can be as high as 19% of As
Total [
48]. Arsenic in oxidizing environments can be mainly found as protonated species of AsO
43−. Biological activity plays an important role in marine arsenic speciation, reducing arsenate to arsenite, given the low thermodynamic stability of arsenite in oxidizing environments (As(III)/As(V) ≈ 10
−26) [
49]. It also plays a role in the formation of monomethylarsenic acid and dimethylarsenic acid; however, the concentration of this arsenic species is much smaller than the inorganic forms.
Low concentrations of organic matter present in seawater (1–3 mg/L CH
3O [
50]) do not allow reductive dissolution to be an effective mechanism for As release, though in coastal environments organic matter is usually more abundant than that found in the open ocean, which could be an important factor in speciation and solubility of iron [
51].
Although As(III) species are mainly limited by biological processes, As(III) can be found in large quantities in polluted mining areas, especially in reducing groundwater environments [
48]; however, it was not considered for the purpose of this study.
2. Materials and Methods
Sorbent minerals used for this experiment were synthesized according to several procedures [
13,
17,
52,
53,
54,
55,
56,
57]. No sample of schwertmannite with co-precipitated arsenic was synthesized, instead, a natural sample form the acid mine drainage of Monte Romero mine (Iberian Pyrite Belt, Spain) was used [
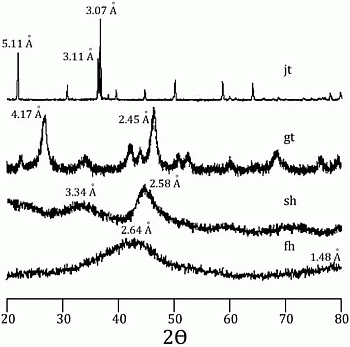
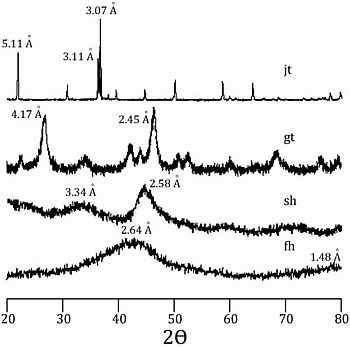
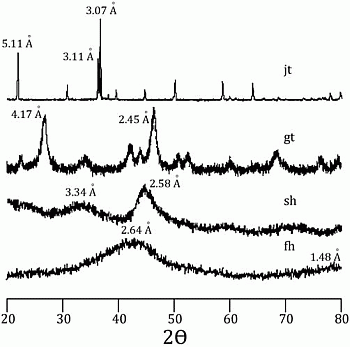
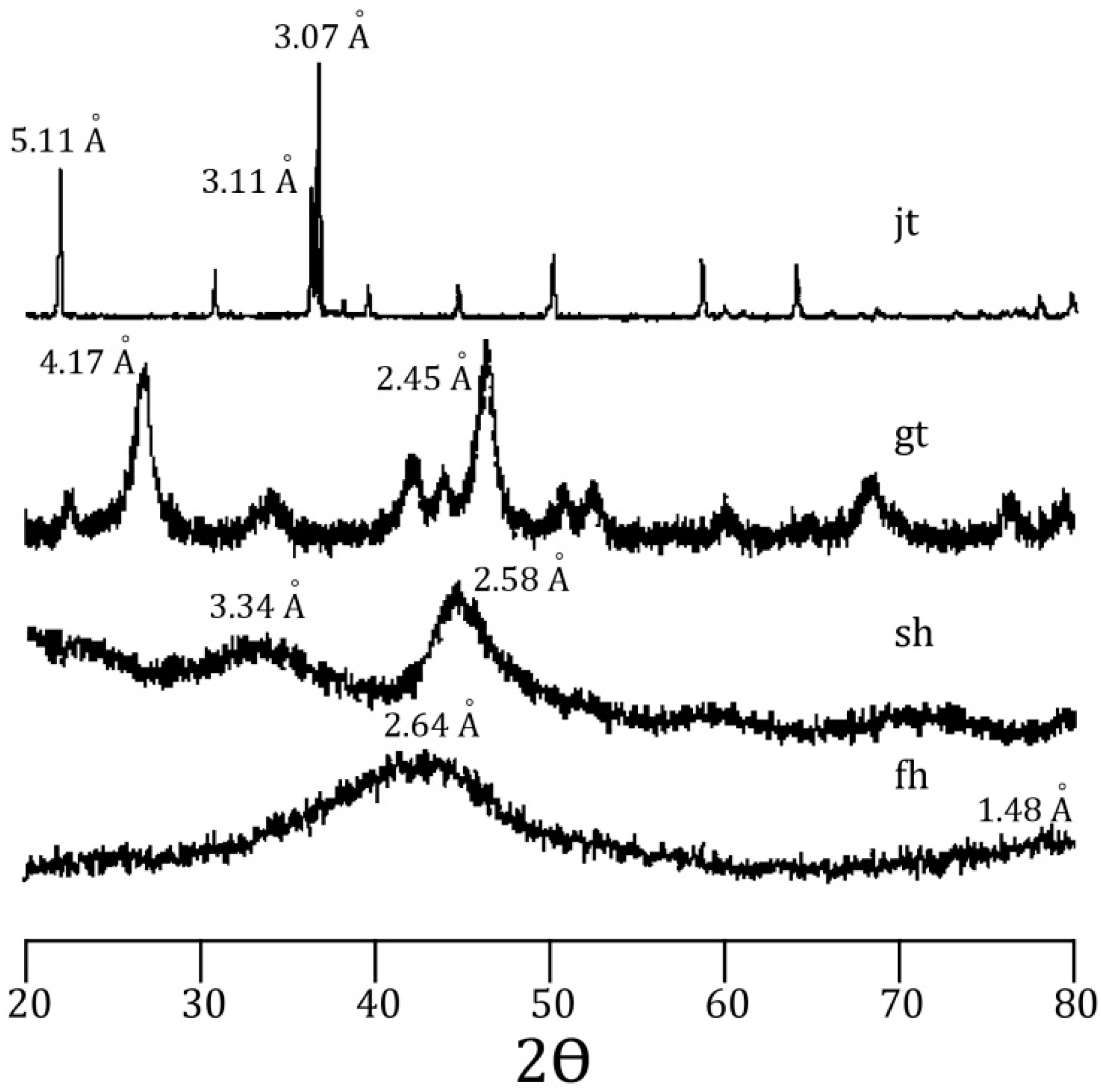
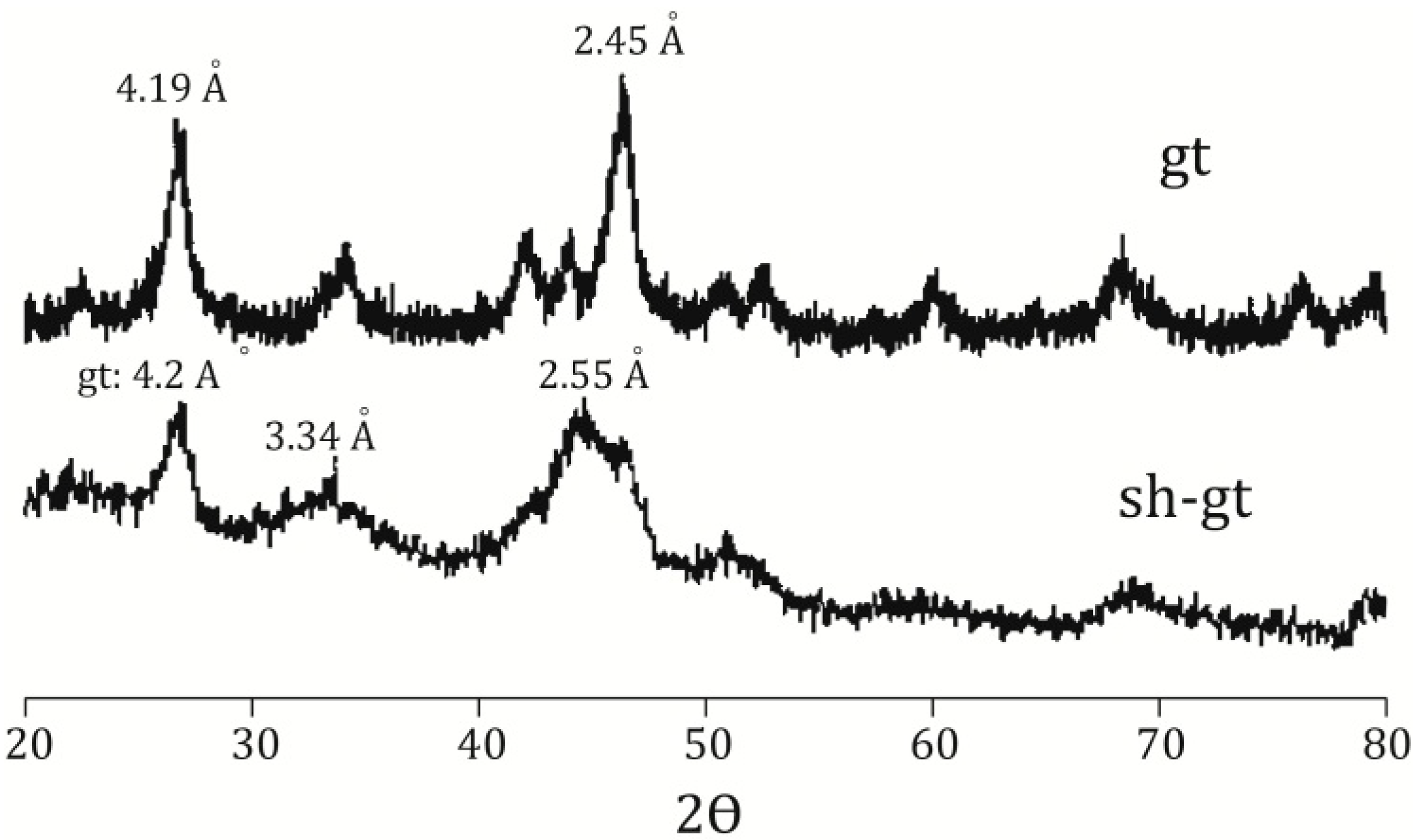
58]. Incorporation of arsenic was considered to be by surface adsorption and co-precipitation. Every mineral was characterized using a Rigaku RADII-C X-ray diffractometer (XRD) (35 kV, 15 mA) from 20° to 100° 2θ using a range of 0.05° 2θ and a counting time of 10 s per step. X-ray diffraction patterns confirmed the correct synthesis for the procedures used and are presented in
Figure 1 and
Figure 2. Further information about the experimental procedure can be found in the supplementary information section. In order to simulate As release from Fe(III) oxide-hydroxides and jarosite during seawater intrusion, the synthesized minerals were contacted with seawater during a 25-day period.
Figure 1.
X-ray diffraction pattern for jarosite (jt), goethite (gt), schwertmanntite (sh), and ferrihydrite (fh).
Figure 1.
X-ray diffraction pattern for jarosite (jt), goethite (gt), schwertmanntite (sh), and ferrihydrite (fh).
Figure 2.
X-ray diffraction pattern for goethite (gt) and schwertmannite (sh-gt) with adsorbed As.
Figure 2.
X-ray diffraction pattern for goethite (gt) and schwertmannite (sh-gt) with adsorbed As.
Twenty-five liters of seawater were taken 10 km offshore Concepción, Chile, by a research vessel. The chemical analysis of the seawater was carried out using titration methods with AgNO
3 for Cl
− and turbidimetry for SO
42−. As and Fe were both measured using an atomic absorption spectrometer (AAS), model Hitachi Z-8100 Polarized Zeeman (Hitachi, Tokyo, Japan). For the Fe analysis, Chelex-100 resin was used to pre-concentrate the sample with HNO
3, which was then flushed to 10 mL. Arsenic hydrides were generated in the presence of H
2SO
4, HNO
3, and HClO
4 which were later used for readings (
Table 1 and
Table 2). The respective detection limits were 5 µg/L for Fe and 1 µg/L for As.
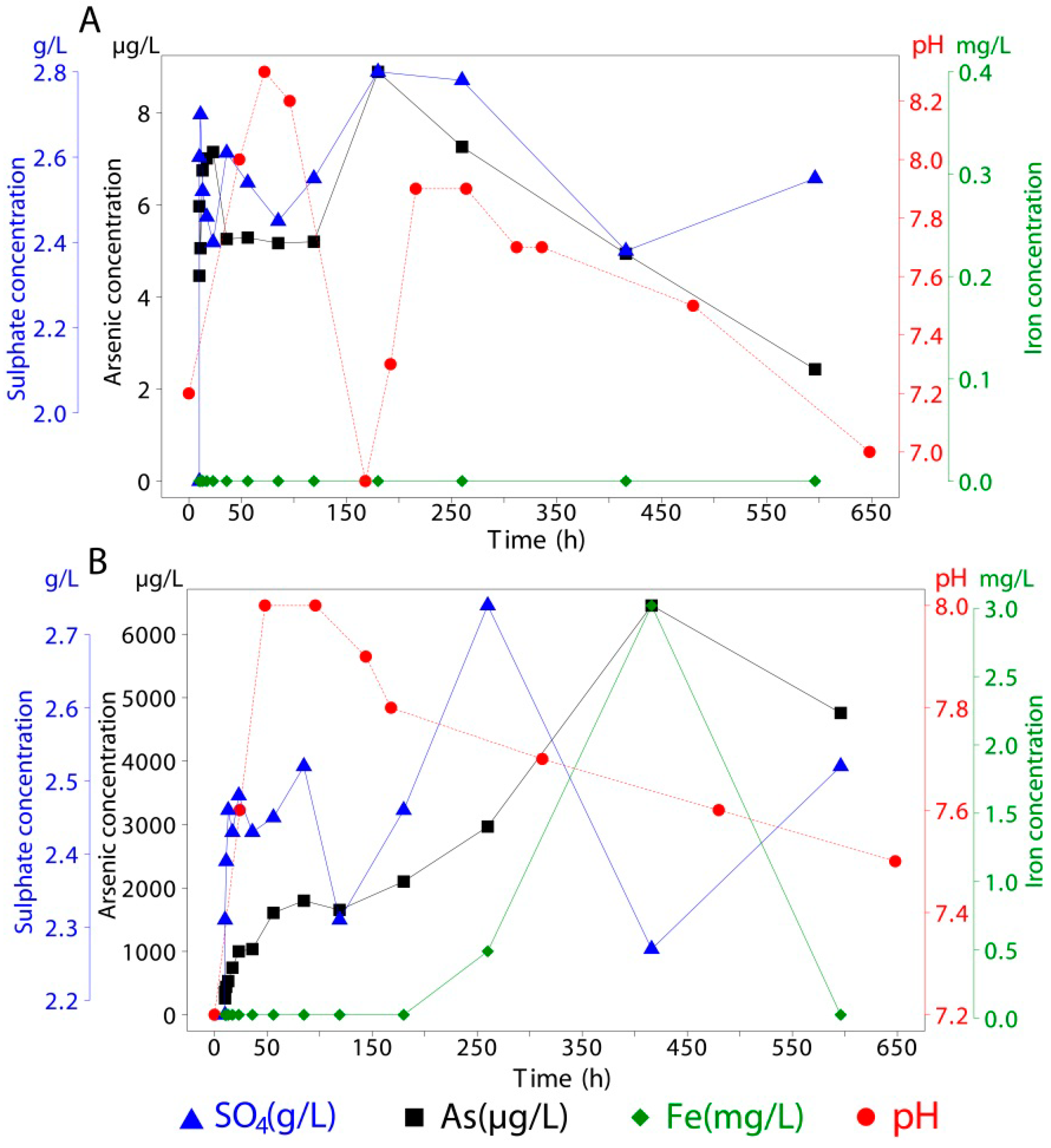
Adsorbed and co-precipitated arsenic, associated with mineral samples, was measured using the same instrumental techniques as for sea water but with different acids for liberating the arsenic. Arsenic was detected in all samples as concentrations were above the detection limit. For every mineral used in this experiment, 1.5 g of the synthesized material (natural schwertmannite for co-precipitated As) was brought into contact with 400 mL of seawater in sealed vessels for 596 h (~25 days). During this period, continuous stirring was applied (
Figure 3). Over the duration of the experiments, 14 extractions (15 mL each) were taken, of which about 40% took place within the first 24 h. This was decided in order to track the period with the greatest release kinetics [
59]. Every extraction was filtered using a 0.45 µm membrane filter, acidified with 50 µL of HNO
3 and stored at 4 °C until analysis. Concentrations of the released species are available in
Table 3,
Table 4,
Table 5 and
Table 6. One of the limitations of this study is that pH and extractions were made at different times so there is some uncertainty regarding the correlation of these variables. However, long term trends, indicative of their relative behavior, can be identified in the figures and
Table 7. All analyses were conducted in GEA Facilities (Instituto de Geología Económica Aplicada, Universidad de Concepción, Chile). In this work, computations involving arsenic and surface speciation were performed using PhreePlot [
60]. Thermodynamic constants were taken from the wateq4f database. For the modeling of surface speciation, a charge distribution multisite ion complexation (CDMUSIC) model was chosen.
Table 1.
Composition of sea water used in this study and comparison with literature values.
Table 1.
Composition of sea water used in this study and comparison with literature values.
| Ions/Metals | This work | Nordstrom [61] | Turekian [62] |
|---|
| Cl (g/L) | 19.5 | 19.35 | 19.4 |
| SO4 (g/L) | 2.6 | 2.71 | 2.58 |
| As (µg/L) | 1 | - | 2.6 |
| Fe (mg/L) | <0.008 | 0.002 | 0.0034 |
Table 2.
Arsenic load in minerals used for this study.
Table 2.
Arsenic load in minerals used for this study.
| Mineral | As (wt %) | As(mol/g) | As (mg/kg) |
|---|
| Schwertmannite | Co-precipitated | 0.56 | 7.47 × 10−5 | 21 |
| Adsorbed | 0.81 | 1.08 × 10−4 | 30.375 |
| Ferrihydrite | Co-precipitated | 2.97 | 3.96 × 10−4 | 111.375 |
| Adsorbed | 2.96 | 3.95 × 10−4 | 111 |
| Jarosite | Co-precipitated | 0.16 | 2.14 × 10−5 | 6 |
| Adsorbed | 0.38 | 5.07 × 10−5 | 14.25 |
| Goethite | Co-precipitated | 0.71 | 9.48 × 10−5 | 26.625 |
| Adsorbed | 3.34 | 4.46 × 10−4 | 125.25 |
Figure 3.
Magnetic stirrer with vessels containing schwertmannite samples.
Figure 3.
Magnetic stirrer with vessels containing schwertmannite samples.
Table 3.
As release from ferrihydrite.
Table 3.
As release from ferrihydrite.
| Experiment type | Without As | Co-precipitated As | Adsorbed As |
|---|
| Hours | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As |
|---|
| (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) |
|---|
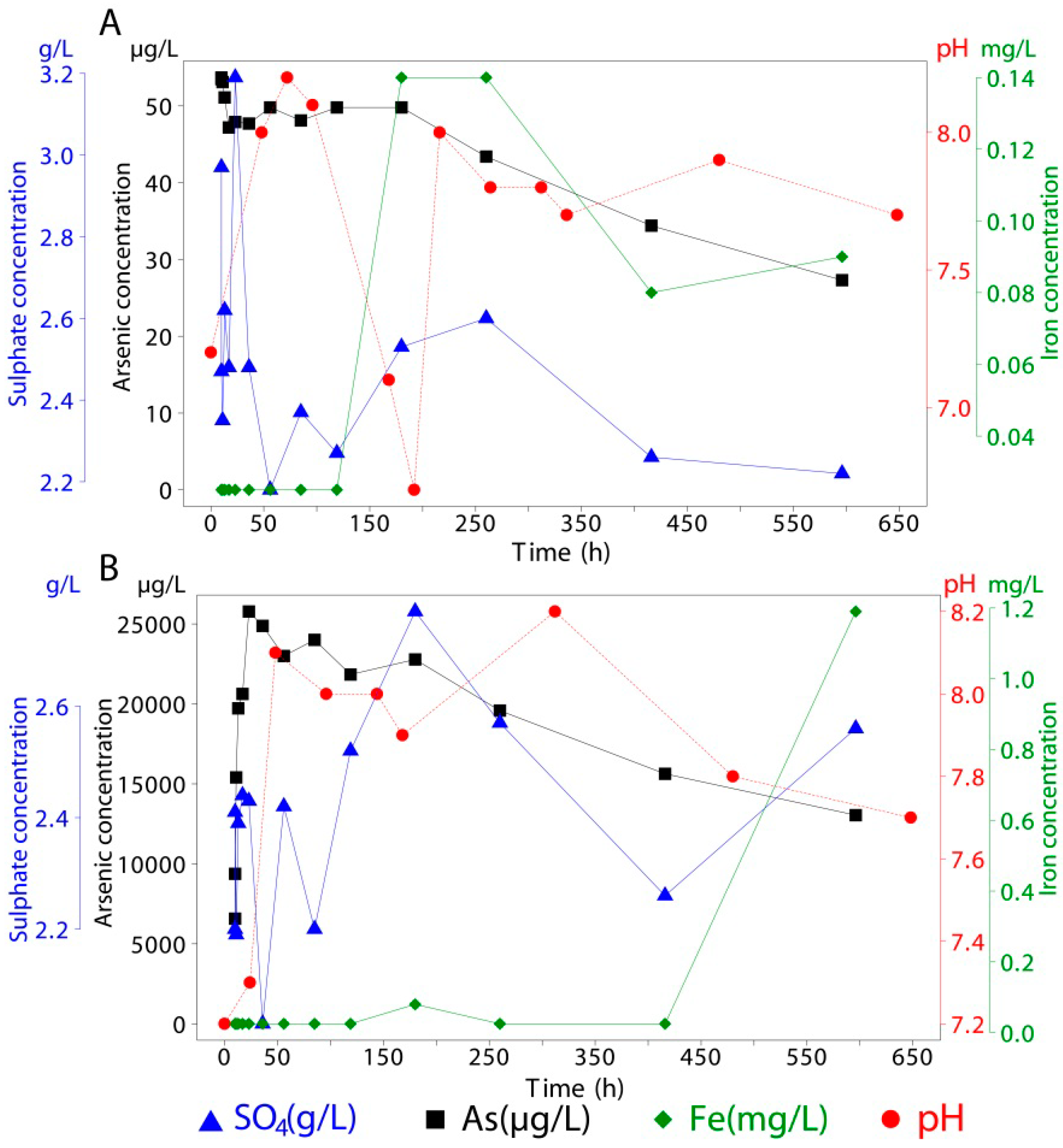
| 10 | 19.78 | 2.46 | <0.05 | <1 | 19.92 | 2.37 | <0.05 | 253 | 19.62 | 1.53 | <0.05 | 1227 |
| 10 | 19.45 | 2.33 | <0.05 | <1 | 19.43 | 2.49 | <0.05 | 260 | 19.36 | 2.48 | <0.05 | 1556 |
| 11 | 19.73 | 2.25 | <0.05 | <1 | 19.55 | 2.36 | <0.05 | 260 | 19.72 | 2.29 | <0.05 | 1533 |
| 13 | 19.55 | 2.35 | <0.05 | <1 | 19.94 | 2.36 | <0.05 | 258 | 19.73 | 2.30 | <0.05 | 1366 |
| 17 | 19.41 | 2.45 | <0.05 | <1 | 19.65 | 2.24 | <0.05 | 263 | 19.80 | 2.25 | <0.05 | 1254 |
| 23 | 21.74 | 2.39 | <0.05 | 5 | 19.37 | 2.47 | <0.05 | 224 | 19.63 | 2.47 | <0.05 | 1088 |
| 36 | 20.65 | 2.35 | <0.05 | 2 | 19.08 | 2.31 | <0.05 | 226 | 19.44 | 2.26 | <0.05 | 950 |
| 56 | 19.56 | 2.22 | <0.05 | 2 | 19.09 | 2.06 | <0.05 | 217 | 19.58 | 2.28 | <0.05 | 849 |
| 85 | 19.45 | 2.63 | <0.05 | 8 | 19.63 | 2.57 | <0.05 | 189 | 18.93 | 2.51 | <0.05 | 836 |
| 119 | 19.46 | 2.32 | <0.05 | - | 19.12 | 2.17 | <0.05 | 188 | 19.39 | 2.46 | <0.05 | 692 |
| 180 | 19.50 | 2.24 | <0.05 | <1 | 19.50 | 2.63 | <0.05 | 173 | 19.50 | 2.44 | <0.05 | 693 |
| 260 | 19.50 | 2.39 | <0.05 | <1 | 19.50 | 2.34 | <0.05 | 178 | 19.50 | 2.55 | <0.05 | 682 |
| 416 | 19.50 | 2.14 | <0.05 | <1 | 19.50 | 2.75 | <0.05 | 118 | 19.50 | 2.11 | <0.05 | 308 |
| 596 | 19.50 | 2.43 | <0.05 | <1 | 19.50 | 2.72 | <0.05 | 76 | 19.50 | 2.34 | <0.05 | 273 |
Table 4.
As release from schwertmannite.
Table 4.
As release from schwertmannite.
| Experiment type | Without As | Co-precipitated As | Adsorbed As |
|---|
| Hours | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As |
|---|
| (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) |
|---|
| 10 | 19.73 | 2.31 | <0.05 | <1 | 21.29 | 2.50 | <0.05 | <1 | 19.64 | 2.65 | <0.05 | <1 |
| 10 | 19.51 | 2.76 | <0.05 | <1 | 20.00 | 2.40 | <0.05 | <1 | 20.28 | 2.42 | <0.05 | <1 |
| 11 | 19.73 | 2.62 | <0.05 | <1 | 19.68 | 2.27 | <0.05 | 2 | 19.94 | 2.52 | <0.05 | <1 |
| 13 | 19.92 | 2.38 | <0.05 | <1 | 18.93 | 2.43 | <0.05 | <1 | 19.12 | 2.48 | <0.05 | <1 |
| 17 | 19.35 | 2.52 | <0.05 | <1 | 19.21 | 2.38 | <0.05 | <1 | 18.96 | 2.40 | <0.05 | 3 |
| 23 | 19.09 | 2.70 | <0.05 | <1 | 19.05 | 2.64 | 0.42 | 9 | 19.16 | 2.43 | <0.05 | 2 |
| 36 | 19.15 | 2.56 | <0.05 | <1 | 18.96 | 2.45 | 0.14 | 2 | 20.30 | 2.50 | 0.96 | 7 |
| 56 | 19.23 | 2.55 | 0.16 | <1 | 19.13 | 2.49 | 0.17 | <1 | 19.10 | 2.77 | <0.05 | 1 |
| 85 | 19.33 | 2.54 | 0.24 | <1 | 19.15 | 2.69 | 0.18 | 1 | 19.22 | 2.21 | <0.05 | 2 |
| 119 | 19.99 | 2.24 | 0.20 | 1 | 19.36 | 2.51 | 0.27 | <1 | 19.03 | 2.69 | <0.05 | 2 |
| 180 | 19.50 | 2.64 | 0.20 | <1 | 19.50 | 2.93 | 0.26 | 1 | 19.50 | 2.76 | <0.05 | 3 |
| 260 | 19.50 | 2.68 | 0.26 | 7 | 19.50 | 2.75 | 0.29 | 2 | 19.50 | 2.70 | <0.05 | 2 |
| 416 | 19.50 | 2.49 | 0.20 | 2 | 19.50 | 2.29 | 0.33 | 3 | 19.50 | 2.20 | <0.05 | 2 |
| 596 | 19.50 | 2.35 | 0.10 | 2 | 19.50 | 2.39 | 0.42 | 2 | 19.50 | 2.32 | <0.05 | 1 |
Table 5.
As release from goethite.
Table 5.
As release from goethite.
| Experiment type | Without As | Co-precipitated As | Adsorbed As |
|---|
| Hours | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As |
|---|
| (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) |
|---|
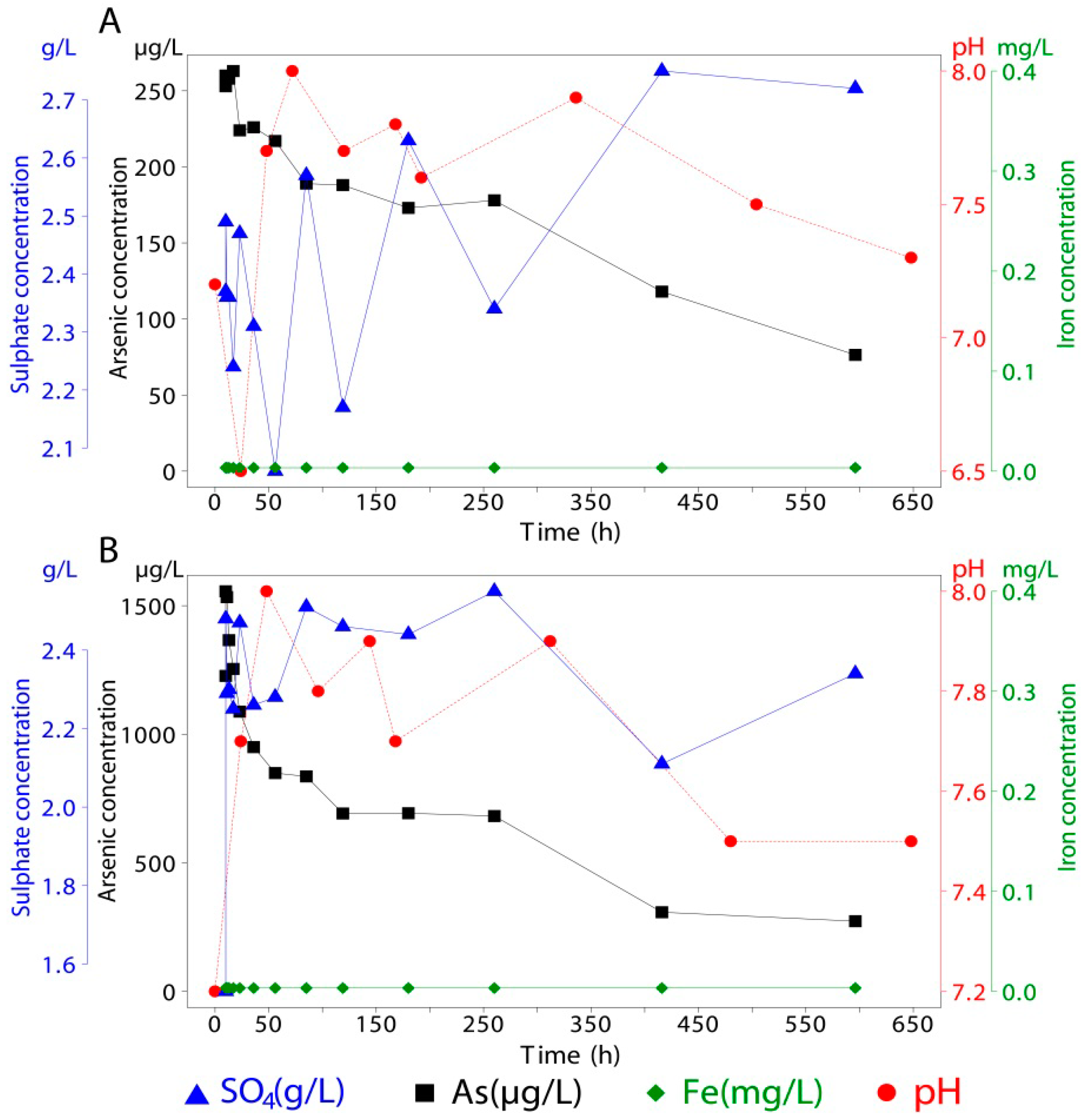
| 10 | 19.54 | 2.38 | <0.05 | <1 | 18.97 | 2.97 | <0.05 | 54 | 19.63 | 2.20 | <0.05 | 6,572 |
| 10 | 19.44 | 2.37 | <0.05 | <1 | 19.59 | 2.47 | <0.05 | 54 | 19.87 | 2.41 | <0.05 | 9,378 |
| 11 | 19.44 | 2.14 | <0.05 | <1 | 19.54 | 2.35 | <0.05 | 53 | 19.60 | 2.19 | <0.05 | 15,404 |
| 13 | 19.30 | 2.35 | <0.05 | <1 | 19.44 | 2.62 | <0.05 | 51 | 19.25 | 2.39 | <0.05 | 19,721 |
| 17 | 20.28 | 2.20 | <0.05 | <1 | 19.44 | 2.48 | <0.05 | 47 | 19.28 | 2.44 | <0.05 | 20,620 |
| 23 | 19.27 | 2.20 | <0.05 | <1 | 19.39 | 3.19 | <0.05 | 48 | 19.25 | 2.43 | <0.05 | 25,776 |
| 36 | 19.35 | 2.14 | <0.05 | <1 | 19.97 | 2.48 | <0.05 | 48 | 19.53 | 2.03 | <0.05 | 24,873 |
| 56 | 19.33 | 2.19 | <0.05 | <1 | 19.71 | 2.18 | <0.05 | 50 | 19.99 | 2.42 | <0.05 | 22,994 |
| 85 | 19.33 | 2.29 | <0.05 | <1 | 19.65 | 2.37 | <0.05 | 48 | 19.19 | 2.20 | <0.05 | 24,000 |
| 119 | 19.71 | 2.51 | <0.05 | 1 | 19.09 | 2.27 | <0.05 | 50 | 19.40 | 2.52 | <0.05 | 21,834 |
| 180 | 19.50 | 2.69 | <0.05 | 3 | 19.50 | 2.53 | 0.14 | 50 | 19.50 | 2.77 | 0.08 | 22,776 |
| 260 | 19.50 | 2.62 | <0.05 | 6 | 19.50 | 2.60 | 0.14 | 43 | 19.50 | 2.57 | <0.05 | 19,578 |
| 416 | 19.50 | 2.07 | 0.08 | 1 | 19.50 | 2.26 | 0.08 | 34 | 19.50 | 2.26 | <0.05 | 15,627 |
| 596 | 19.50 | 2.34 | 0.20 | 2 | 19.50 | 2.22 | 0.09 | 27 | 19.50 | 2.56 | 1.19 | 13,043 |
Table 6.
As release from jarosite.
Table 6.
As release from jarosite.
| Experiment type | Without As | Co-precipitated As | Adsorbed As |
|---|
| Hours | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As | Cl | SO4 | Fe | As |
|---|
| (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) | (g/L) | (g/L) | (mg/L) | (µg/L) |
|---|
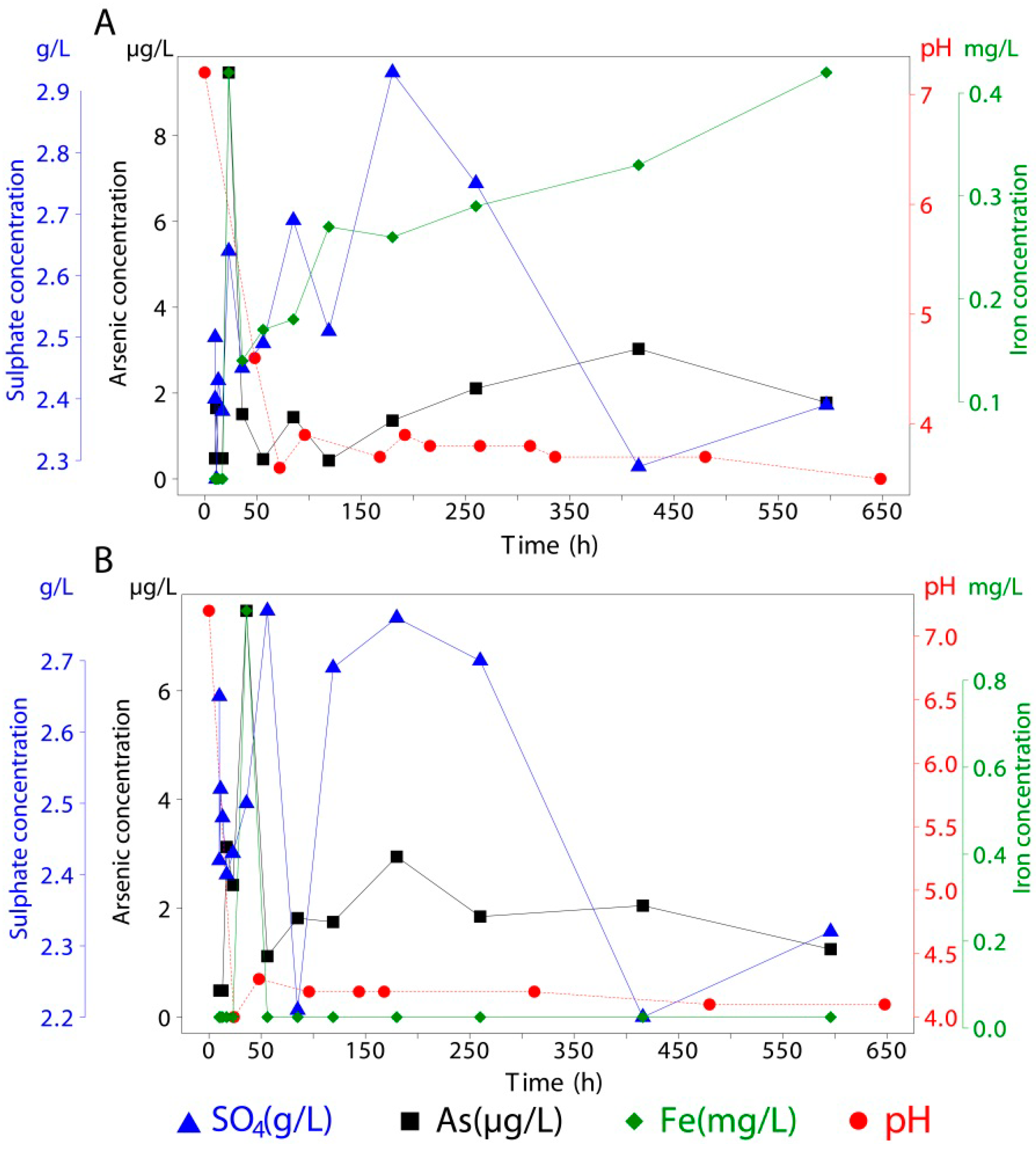
| 10 | 19.63 | 2.68 | <0.05 | <1 | 19.80 | 1.84 | <0.05 | 4 | 19.93 | 2.18 | <0.05 | 261 |
| 10 | 19.36 | 2.44 | <0.05 | <1 | 19.77 | 2.60 | <0.05 | 6 | 19.64 | 2.31 | <0.05 | 362 |
| 11 | 19.34 | 2.19 | <0.05 | <1 | 19.66 | 2.70 | <0.05 | 5 | 19.23 | 2.39 | <0.05 | 441 |
| 13 | 19.43 | 2.27 | <0.05 | <1 | 19.68 | 2.52 | <0.05 | 7 | 19.42 | 2.46 | <0.05 | 532 |
| 17 | 19.69 | 2.40 | <0.05 | <1 | 19.45 | 2.46 | <0.05 | 7 | 19.68 | 2.43 | <0.05 | 744 |
| 23 | 19.67 | 2.56 | <0.05 | <1 | 19.34 | 2.40 | <0.05 | 7 | 19.79 | 2.48 | <0.05 | 1000 |
| 36 | 19.35 | 2.39 | <0.05 | <1 | 19.24 | 2.61 | <0.05 | 5 | 19.45 | 2.43 | <0.05 | 1037 |
| 56 | 19.35 | 2.37 | <0.05 | 5 | 19.29 | 2.54 | <0.05 | 5 | 19.34 | 2.45 | <0.05 | 1607 |
| 85 | 19.58 | 2.33 | <0.05 | <1 | 19.26 | 2.45 | <0.05 | 5 | 19.21 | 2.52 | <0.05 | 1799 |
| 119 | 19.68 | 2.72 | <0.05 | <1 | 19.36 | 2.55 | <0.05 | 5 | 19.28 | 2.31 | <0.05 | 1654 |
| 180 | 19.50 | 2.56 | <0.05 | 2 | 19.50 | 2.80 | <0.05 | 9 | 19.50 | 2.46 | <0.05 | 2101 |
| 260 | 19.50 | 2.70 | <0.05 | <1 | 19.50 | 2.78 | <0.05 | 7 | 19.50 | 2.74 | 0.49 | 2968 |
| 416 | 19.50 | 2.17 | <0.05 | <1 | 19.50 | 2.38 | <0.05 | 5 | 19.50 | 2.27 | 3.02 | 6452 |
| 596 | 19.50 | 2.32 | <0.05 | 1 | 19.50 | 2.55 | <0.05 | 2 | 19.50 | 2.52 | <0.05 | 4758 |
Table 7.
pH during seawater saturation.
Table 7.
pH during seawater saturation.
| Ferrihydrite |
| Without As | Hours | 0 | 24 | 48 | 72 | 120 | 168 | 192 | 336 | 504 | 648 | | | |
| pH | 7.2 | 6.4 | 7.7 | 8.0 | 7.7 | 7.7 | 7.3 | 7.9 | 7.3 | 7.4 | | | |
| Co-precipitated As | Hours | 0 | 24 | 48 | 72 | 120 | 168 | 192 | 336 | 504 | 648 | | | |
| pH | 7.2 | 6.5 | 7.7 | 8.0 | 7.7 | 7.8 | 7.6 | 7.9 | 7.5 | 7.3 | | | |
| Adsorbed As | Hours | 0 | 24 | 48 | 96 | 144 | 168 | 312 | 480 | 648 | | | | |
| pH | 7.2 | 7.7 | 8.0 | 7.8 | 7.9 | 7.7 | 7.9 | 7.5 | 7.5 | | | | |
| Schwertmannite |
| Without As | Hours | 0 | 24 | 72 | 96 | 120 | 192 | 216 | 240 | 288 | 336 | 360 | 504 | 672 |
| pH | 7.2 | 5.2 | 3.6 | 3.3 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.5 | 3.5 | 3.6 |
| Co-precipitated As | Hours | 0 | 48 | 72 | 96 | 168 | 192 | 216 | 264 | 312 | 336 | 480 | 648 | |
| pH | 7.2 | 4.6 | 3.6 | 3.9 | 3.7 | 3.9 | 3.8 | 3.8 | 3.8 | 3.7 | 3.7 | 3.5 | |
| Adsorbed As | Hours | 0 | 24 | 48 | 96 | 144 | 168 | 312 | 480 | 648 | | | | |
| pH | 7.2 | 4.0 | 4.3 | 4.2 | 4.2 | 4.2 | 4.2 | 4.1 | 4.1 | | | | |
| Jarosite |
| Without As | Hours | 0 | 24 | 72 | 96 | 120 | 192 | 216 | 240 | 288 | 336 | 360 | 504 | 672 |
| pH | 7.2 | 7.9 | 6.9 | 8.0 | 8.0 | 6.8 | 7.6 | 7.9 | 7.8 | 7.6 | 7.4 | 5.4 | 5.9 |
| Co-precipitated As | Hours | 0 | 48 | 72 | 96 | 168 | 192 | 216 | 264 | 312 | 336 | 480 | 648 | |
| pH | 7.2 | 8.0 | 8.3 | 8.2 | 6.9 | 7.3 | 7.9 | 7.9 | 7.7 | 7.7 | 7.5 | 7.0 | |
| Adsorbed As | Hours | 0 | 24 | 48 | 96 | 144 | 168 | 312 | 480 | 648 | | | | |
| pH | 7.2 | 7.6 | 8.0 | 8.0 | 7.9 | 7.8 | 7.7 | 7.6 | 7.5 | | | | |
| Goethite |
| Without As | Hours | 0 | 24 | 72 | 96 | 120 | 192 | 216 | 240 | 288 | 336 | 360 | 504 | 672 |
| pH | 7.2 | 6.8 | 5.7 | 8.2 | 8.1 | 7.2 | 6.8 | 8.0 | 7.7 | 7.7 | 7.7 | 7.8 | 7.3 |
| Co-precipitated As | Hours | 0 | 48 | 72 | 96 | 168 | 192 | 216 | 264 | 312 | 336 | 480 | 648 | |
| pH | 7.2 | 8.0 | 8.2 | 8.1 | 7.1 | 6.7 | 8.0 | 7.8 | 7.8 | 7.7 | 7.9 | 7.7 | |
| Adsorbed As | Hours | 0 | 24 | 48 | 96 | 144 | 168 | 312 | 480 | 648 | | | | |
| pH | 7.2 | 7.3 | 8.1 | 8.0 | 8.0 | 7.9 | 8.2 | 7.8 | 7.7 | | | | |
4. Conclusions
During this laboratory study, some common Fe(III) hydroxides and oxyhydroxy-sulphates, including ferrihydrite, goethite, schwertmannite, and jarosite, present in the oxidation zone of mine tailings and in acid soils, showed efficacy as arsenic sorbents under several experimental conditions. Goethite and ferrihydrite were able to adsorb large loads of arsenic at their surfaces (~3 wt %) while schwertmannite and jarosite were able to incorporate less than 0.8 and 0.3 wt % of arsenic, respectively. Once in contact with seawater, each mineral showed different sorbent capacities, depending on the type of arsenic load and mineral stability.
Our results demonstrated that during seawater intrusion in coastal tailings, arsenic release can be attributed mainly to ion exchange and dissolution processes. Ferrihydrite and schwertmannite, two meta-stable minerals with large reactive surface areas, were less likely to release arsenic due to a higher ZPC and schwertmannite’s buffering capacity that acidified the pH of contact seawater. Moreover, highly stable goethite and jarosite showed the greatest release among minerals with adsorbed arsenic.
In general terms, synthesized minerals with co-precipitated arsenic were less inclined to liberation in comparison with syntheses with adsorbed arsenic, where ion exchange was a key parameter for liberation of the metalloid.
Schwertmannite and ferrihydrite presented the highest retention; however in the first case it mostly depended on the pH buffering capacity. It can be hypothesized that, under a real scale flooding scenario, schwertmannite would not be able to acidify the seawater in the same way it did in this experiment and that sea water geochemistry would dominate. In this case, a lower retention would be expected. In the medium term, transformation processes can release significant amounts of As that would not be fully retained by new sorbents. In the case of schwertmannite, exchange between sulphate and several species can affect stability and increase transformation rates to goethite [
70]. The increase in dissolved iron and sulphate from schwertmannite towards the end of the experiment may indicate low stability or the beginning of transformation/dissolution processes. Ferrihydrite demonstrated its importance in coastal environments by representing a sink for arsenic at alkaline pH. However, the meta-stable nature for this mineral implies that in the long term dissolution and transformation processes can release arsenic regardless.
Goethite, another common stable mineral in the coastal environment, was not able to retain adsorbed arsenic after contact with seawater. This was due to the lower stability that surfaces complexes presented under seawater conditions. Although some authors have proposed bidentate complexes as the main linkage for arsenic in goethite’s surface, in this case the release of 20% TLC would be more in accordance with less stable monodentate complexes as proposed by Loring [
35].
In coastal environments, stability of sorbent minerals should be considered as a whole, taking into account interaction between sorbents, fate and transport of toxic elements, surface complexation, interaction with seawater species, transformation into stable minerals, and dissolution processes.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




