
Interlayer Structures and Dynamics of Arsenate and Arsenite Intercalated Layered Double Hydroxides: A First Principles Study


State Key Lab for Mineral Deposits Research, School of Earth Sciences and Engineering, Nanjing University, Nanjing 210046, China
*
Author to whom correspondence should be addressed.
Minerals 2017, 7(4), 53; https://doi.org/10.3390/min7040053
Submission received: 26 January 2017
/
Revised: 13 March 2017
/
Accepted: 23 March 2017
/
Published: 30 March 2017
(This article belongs to the Special Issue Computational Geochemistry)
Abstract
:In this study, by using first principles simulation techniques, we explored the basal spacings, interlayer structures, and dynamics of arsenite and arsenate intercalated Layered double hydroxides (LDHs). Our results confirm that the basal spacings of NO3−-LDHs increase with layer charge densities. It is found that Arsenic (As) species can enter the gallery spaces of LDHs with a Mg/Al ratio of 2:1 but they cannot enter those with lower charge densities. Interlayer species show layering distributions. All anions form a single layer distribution while water molecules form a single layer distribution at low layer charge density and a double layer distribution at high layer charge densities. H2AsO4− has two orientations in the interlayer regions (i.e., one with its three folds axis normal to the layer sheets and another with its two folds axis normal to the layer sheets), and only the latter is observed for HAsO42−. H2AsO3− orientates in a tilt-lying way. The mobility of water and NO3− increases with the layer charge densities while As species have very low mobility. Our simulations provide microscopic information of As intercalated LDHs, which can be used for further understanding of the structures of oxy-anion intercalated LDHs.
1. Introduction
As a common pollutant in water bodies, Arsenic (As) has been threatening water supplies all over the world for a long time [1]. The concentration of As in fresh water is often lower than 1 μg/L [2], while high concentrations up to 50 μg/L and even 500 μg/L can be found in groundwater, especially those in Bangladesh, Vietnam, Hungary, Argentina, Mexico, and some regions in China [2,3]. As holds several oxidation states (−3, 0, 3, 5) [2] and can exist in both organic and inorganic forms [4], while only As(III) and As(V) are of importance in water contamination [3,4]. The distribution of As(III) and As(V) is governed by redox potential [3]. Oxidizing waters contain more As(V) species [3]. High level concentrations of arsenite have been found in rivers close to As(III)-dominated industrial effluent [5] and in underground water systems, for example, groundwater from Bangladesh [6] and Inner Mongolia [7]. The pH conditions of water also has an impact on the speciation of As. The predominant species of As(V) and As(III) are H2AsO4−, HAsO42−, and H2AsO3, respectively, in natural waters where the pH ranges from 6 to 9, and H2AsO3− becomes the predominant species of As(III) with a pH over 9.2 [3,8].
Numerous studies have been carried out to investigate As adsorption on various minerals, such as clay minerals [9,10,11,12,13,14,15,16,17] and (hydr)oxides [18,19,20,21,22,23,24,25,26,27]. Adsorption of As onto clay can occur directly on edge surfaces and indirectly through cation bridging on basal surfaces [17], which has been shown to be highly dependent on pH [9,10,12,13]. As can form inner sphere complexes on (hydr)oxides [18,19,22], which are confirmed by density functional theory (DFT) calculations [20,22,23,27].
Layered double hydroxides (LDHs) have the potential to remove oxyanion water contaminants such as arsenate [8,28,29,30,31], chromate [32,33,34], and phosphate [3,35,36,37,38,39] due to its large specific surface areas and high anion exchange capacity. LDHs, also known as hydrotalcite-like compounds or anionic clays, are a class of layered minerals with exchangeable anions in the gallery spaces [40]. The basic structure of LDH sheets is brucite [Mg(OH)2], with some divalent cations replaced by trivalent cations. The isomorphic substitutions lead to net positive charges, which are compensated by the interlayer anions. The composition of LDHs can be expressed by the general formula [M1-xIIMxIII(OH)2]x+(An−)x/n·mH2O, where MII and MIII represent the divalent and trivalent cations respectively, and An− stands for the anions in the gallery space such as CO32−, SO42−, Cl−, and NO3−.
Many experiments have been conducted to study the adsorption of As onto LDHs. Wang et al. [8] have studied the influence of layer charge densities on the adsorption capacity, and suggested that arsenate adsorbed predominantly on the external surface of Mg/Al = 4 LDHs while adsorption occurs on both external and interlayer surfaces at higher layer charge densities. Caporale et al. [29] compared the adsorption of arsenate onto Mg/Fe and Mg/Al LDHs, and their results show that Mg/Fe LDHs can absorb more As than Mg/Al LDHs. Yang et al. [41] found that the adsorption rate of arsenate onto LDHs increases with decreasing LDH particle size, while the adsorption capacity is independent of the particle size. Violante et al. [30] demonstrated that phosphate has a greater affinity to LDHs than arsenate. These experiments have focused on the influence on the adsorptions of As from layer charge density, different LDHs, particles sizes, and competitive adsorption from other anions. However, no molecular level insights were provided into the interaction between As species and LDHs.
All of the above experimental studies focus on macroscopic properties, such as adsorption capacity and adsorption kinetics of As adsorption onto LDHs. However, only a few studies have reported on the interlayer structures of oxy-anion intercalated LDHs due to the limitations of experimental techniques. For example, Martina et al. found that fatty anions can form monolayers and bilayers in the interlayer based on XRD data [42], and Vera et al. proposed that sulfate could graft to adjacent LDH layers after heating the sample above 250 °C [43]. Although macroscopic properties of As adsorption onto LDHs have been previously reported, further information is needed on the molecular structures of arsenate and arsenite intercalated LDHs. Previous studies have confirmed that the basal spacings of NO3−-LDHs increase with layer charge densities [44,45,46,47], but the underlying mechanism is still being discussed. Xu and Zeng [45] and Gago et al. [44] argued that the stack behavior of interlayer anions causes this change while Wang et al. [8,47] and Hui et al. [46] thought this change is triggered by the orientations of interlayer anions.
Molecular simulations are very useful for exploring microscopic interlayer properties of layered materials [48]. Several molecular dynamics (MD) simulations of LDHs have been reported [46,49,50,51,52,53]. For example, Kai et al. [51] and Kumar et al. [49] focused on the structures and energetics of LDHs. Chen et al. [52] studied the diffusion properties of interlayer species of Cl−-LDHs. In MD simulations, the accuracy of the force field underpins the reliability of the simulation results. However, to our knowledge, a force field suitable for arsenic-LDHs is still lacking and there is no simulation study on these systems up to now.
As a theoretical tool, first principles molecular dynamics (FPMD) combining electronic structures calculation with molecular dynamics sampling can overcome some drawbacks in experiments, for example, studying the interlayer structures of layered materials without the influences from outer surfaces. Recently, precise information of the speciation, structures, and acidities of As in aqueous solutions has been obtained by using FPMD [54], indicating that FPMD can provide precise information in As-bearing systems. The efforts to explore the structural and thermodynamic properties of layered minerals such as gibbsite [55] and clay minerals [56,57,58,59] also obtained results consistent with experiments. In this study, we employ first principles simulation techniques to investigate basal spacings and interlayer properties of arsenate and arsenite intercalated LDHs. Basal spacings are obtained by using cell optimization and compared with the available experimental data. The structures and dynamics of interlayer species are derived using FPMD. The distributions, orientations, and diffusion coefficients of interlayer species are characterized in detail. The atomic level information contributes to the understanding of the interactions between As and LDHs.
2. Methods
2.1. Models
The LDH structures used in this study were based on the powder X-ray diffraction data of hydrotalcite after refinement using the Rietveld method [60]. Part of the magnesium was replaced by aluminum to reach Mg/Al ratios of 2:1, 3:1, and 4:1. The isomorphic substitutions of Al for Mg in the octahedral sheets followed Loewenstein’s rule [51,61] at relatively low layer charge densities (with a Mg/Al ratio of 3:1 or 4:1), that is, no substitution sites were adjacent, while at a Mg/Al ratio of 2:1, there were two neighboring substitution sites because the periodic boundary conditions did not allow all the trivalent cations to satisfy the cation avoidance rule, and some studies [50] have proven that a few adjacent substitution sites will not affect the overall structures and dynamics properties of LDHs. Considering the limits of computational power, we modified the original model with three octahedral sheets to two sheets, which consists of 4 × 4 × 2 M-OH octahedra. Models with a Mg/Al ratio of 2:1, 3:1, and 4:1 contain 5, 4, and 3 substitution sites per sheet, respectively. The net positive charges of octahedral sheets were compensated by the interlayer anions H2AsO4−, HAsO42−, H2AsO3−, and NO3− (Figure 1). The number of As and water molecules was obtained from previous studies [8,46]. The basal spacings of our initial models were increased artificially to incorporate arsenic species. Oxy-anions were separated from each other by water molecules. The compositions of simulated systems are listed in Table 1. Periodic boundary conditions were imposed on the three directions (i.e., XYZ) for all calculations.
2.2. Simulation Details
All calculations were performed using the CP2K/QUICKSTEP package [62,63]. Electronic structures were calculated with DFT [64,65]. In the QUICKSTEP package, DFT is implemented with the hybrid Gaussian plane wave (GPW) approach [66], which uses a Gaussian basis set for the pseudo-wave functions and Kohn-Sham matrixes while a plane wave auxiliary basis is used to represent the electron density. The Perdew-Burke-Ernzerhof (PBE) functional was applied for the exchange-correlation [67] and Goedecker-Teter-Hutter (GTH) type pseudopotentials were used for the core configurations [68]. Intra- and inter-molecular dispersion effects were computed using the DFT-D3 functional [69]. The cutoff for the electronic density was set to 440 Ry.
The structures were firstly optimized through cell optimization using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm to obtain reasonable structures at different layer charge densities for H2AsO4−, HAsO42−, H2AsO3−, and NO3−-LDHs. With cell optimization, variable cell structure relaxation is performed. The optimized cell parameters are then used to derive the basal spacing of LDHs. After cell optimizations, arsenic containing systems at a Mg/Al ratio of 2:1 and NO3−-LDHs at different layer charge densities were selected (details in Section 3.2.1) to perform Born-Oppenheimer molecular dynamics (BOMD) calculations. BOMD calculates the wave function and electronic density of the whole system at every MD step. Both the cell optimization calculations and the BOMD simulations were carried out with a wave function optimization tolerance of 1.0 × 10−6. BOMD simulations were performed at a time step of 0.5 fs (femtosecond, 1 fs = 1.0 × 10−15 s) with the Nose-Hoover chain thermostat to control the temperature which was set to be 298 K. The simulation time of each BOMD (i.e., H2AsO4−, HAsO42−, and H2AsO3−-LDHs at 2:1 and NO3−-LDHs at 2:1, 3:1, and 4:1) was over 50 ps (picosecond, 1 ps = 1.0 × 10−12 s).
2.3. Simulation Analyses
The basal spacing of LDHs is obtained from the optimization run,
The spatial distributions of interlayer species are described by their number densities along the Z direction, which can be expressed as,
where <N (z − Δz⁄2, z + Δz⁄2)> indicates the average occurrence number with the Z coordinate in the range of (z − Δz⁄2, z + Δz⁄2) and S stands for the area of the basal surfaces.
The mobility of the interlayer species is characterized by the diffusion coefficient D, which is calculated according to the Einstein relation [70],
where the left hand side represents the mean squared displacement, N is the number of atoms of interest, is the position of the ith atom at time t, and d is the diffusion dimension.
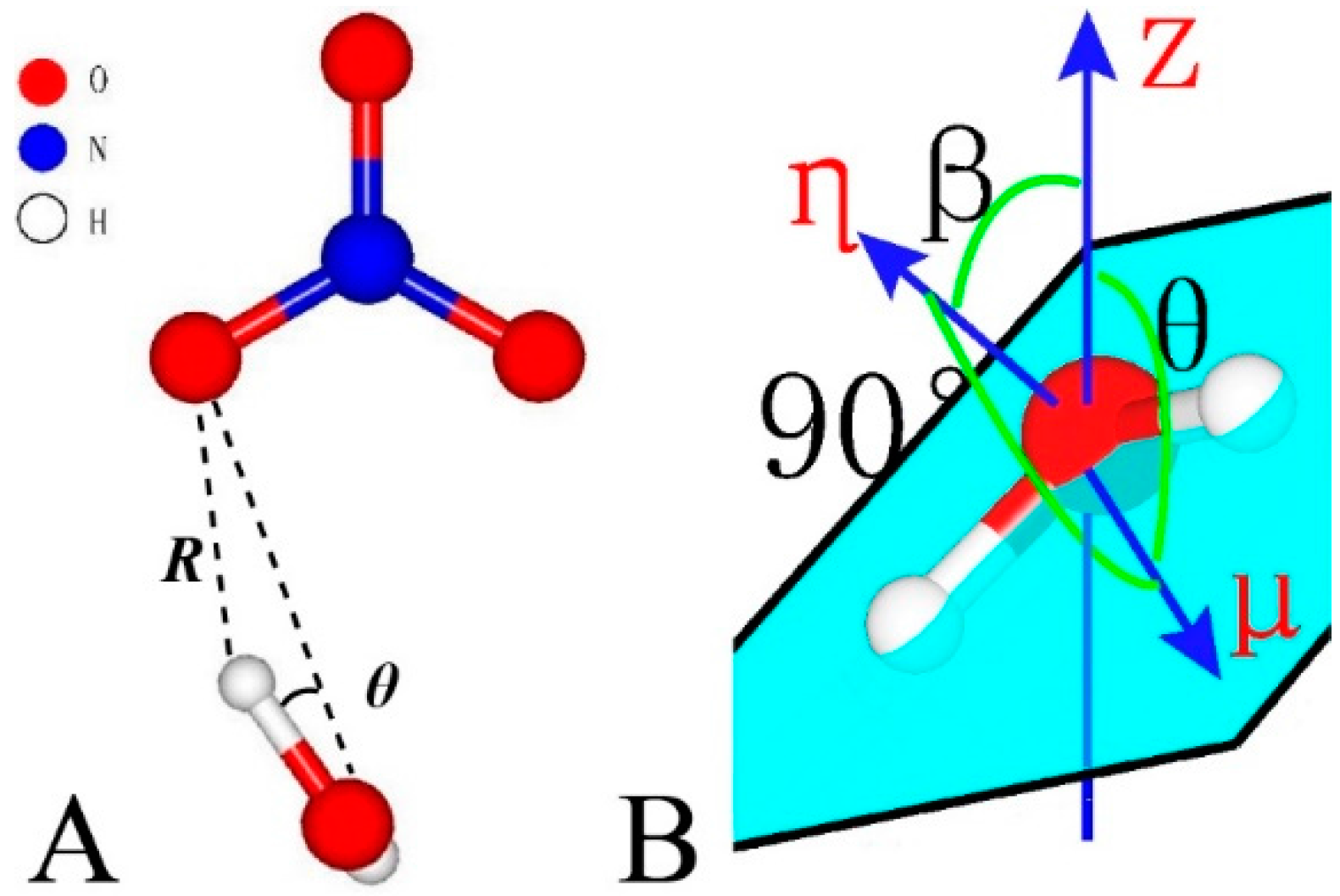
The criteria for hydrogen bonding adopted in this study stay the same with those commonly used in bulk water systems, that is, θ ≤ 30° and R ≤ 2.4 Å (Figure 2A). The orientations of water and NO3− are specified by θ and β [71]. As shown in Figure 2B, μ and η denote the directions of the dipole moment and the normal of the water molecule plane, respectively. Z stands for the vector (001). β is the angle between η and Z and θ is the angle between μ and Z.
3. Results and Discussion
3.1. Basal Spacing
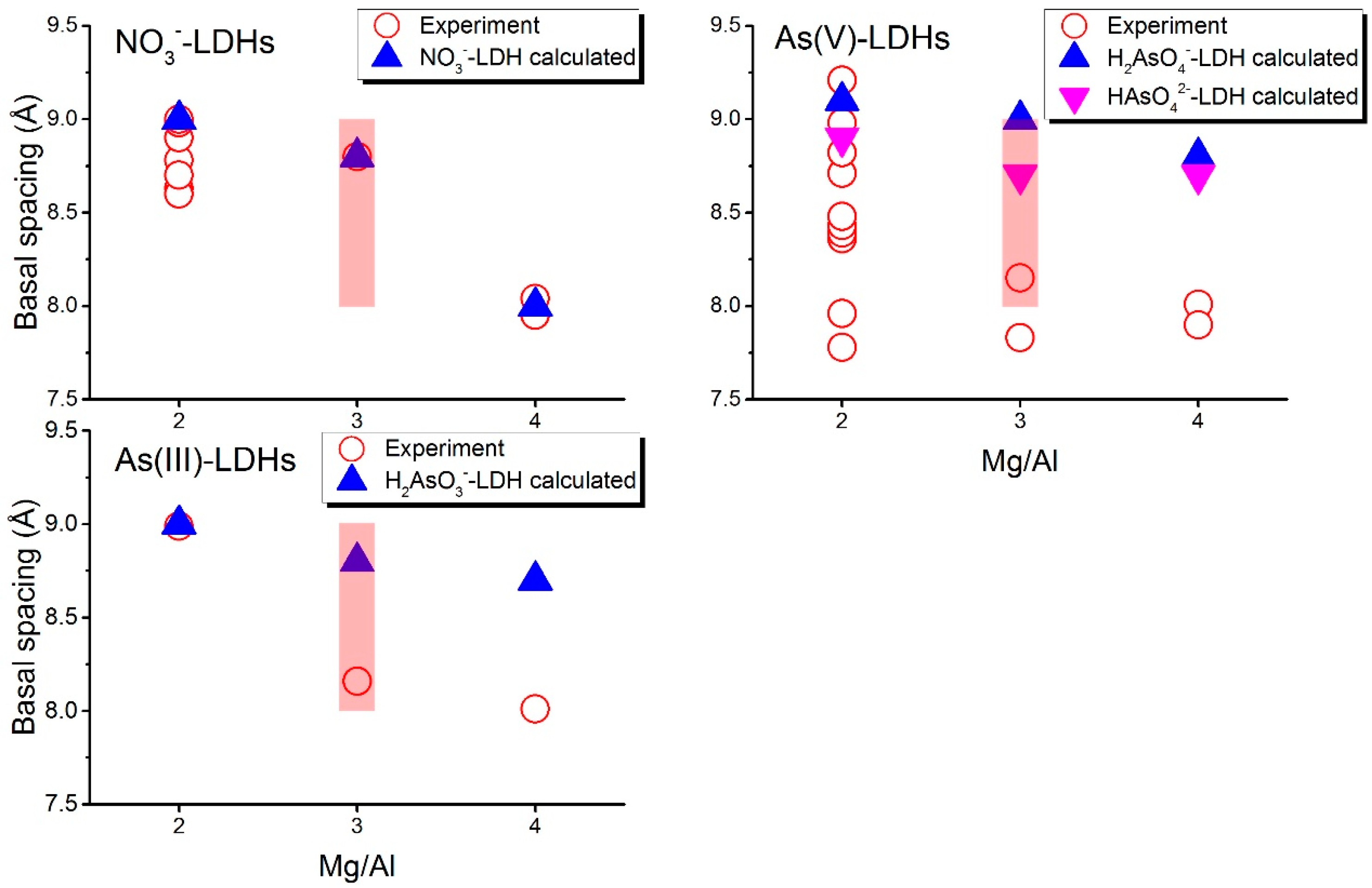
Figure 3 plots the d(003) values of the LDH systems. The basal spacings of NO3−-LDHs have been characterized by many experimental studies, which give close results [8,47,72,73,74,75,76,77,78]; thus the NO3−-LDHs can be used to validate our simulation settings. Here we did not include the experimental results obtained under very high pH conditions because the adsorptions of arsenic species are highly inhibited by OH− under high pH [79]. Experimental basal spacings for NO3−-LDHs with a Mg/Al ratio of 2:1 ranges from 8.63 Å to 8.98 Å (with an average value of 8.77 Å) and the calculated basal spacing is 9.0 Å. Our calculated basal spacing for NO3−-LDHs with a Mg/Al ratio of 3:1 and 4:1 equal 8.80 Å and 7.95 Å, respectively, which agree well with those from the experiments, 8.80 Å and 8.00 Å for 3:1 and 4:1, respectively. For 3:1, however, Wang et al. suggested that a wide range from 8.04 Å to 9.0 Å could be observed in the experiments [47]. This agreement validates our simulation set up. Our simulation further confirmed that the changes in the orientations of NO3− should be responsible for the changes in the basal spacing. We explain this point in Section 3.2.2.
H2AsO4− and HAsO42− co-exist at near-neutral pH conditions and they both occurred in the experimental systems [31], so the basal spacings for As(V) are presented together in Figure 3. The calculated basal spacings (i.e., 9.10 Å and 8.90 Å for H2AsO4− and HAsO42−, respectively) are consistent with some of the reported experiments [31,80] that are conducted under initial pH around 7 and 8 (range from 8.71 Å to 9.21 Å, Figure 3) at a Mg/Al ratio of 2:1, while large differences are found between our simulations and the other experiments [8,76] conducted under pH of 5 or higher than 9 (<8.48 Å, Figure 3). At lower layer charge densities (Mg/Al = 3:1 and 4:1), basal spacings from experiments are around 8.00 Å (8.15 Å and 7.83 Å at 3:1, 8.01 Å and 7.90 Å at 4:1 [8,80]), about 0.9 Å smaller than our calculated value (9.00 Å and 8.70 Å for 3:1, 8.80 Å and 7.90 Å for 4:1). For the H2AsO3− containing system at 2:1, the calculated basal spacing (i.e., 9.00 Å) is consistent with experiments (i.e., 8.99 Å [81]). At Mg/Al ratios of 3:1 and 4:1, our calculated basal spacings are 8.80 Å and 8.70 Å, respectively, while experiments gave 8.16 Å and 8.01 Å [81] for 3:1 and 4:1, respectively.
The basal spacing values of As LDHs with low layer charge densities from experiments are significantly smaller than those from our simulations, but they coincide with those of NO3−-LDHs (~8.0 Å at 4:1 and 8.04 Å–9.0 Å at 3:1 [47]). This implies that these species may not enter the gallery spaces of LDHs in some experiments, that is, the systems measured by those experiments are in fact still NO3−-LDHs. This supports the conclusion in a previous study where the authors believed that the adsorption of arsenate mainly occurred at external surfaces of low charge density LDHs [8]. Some experiments [8,76] at 2:1 gave much smaller values (<8.48 Å, Figure 3) than our calculations (9.10 Å and 8.90 Å for H2AsO4− and HAsO42−, respectively) which may be caused by the same reason. Based on these results, LDHs with a high layer charge density (Mg/Al = 2:1) should be more efficient for As removal.
3.2. Interlayer Structures
3.2.1. Distributions of Interlayer Species
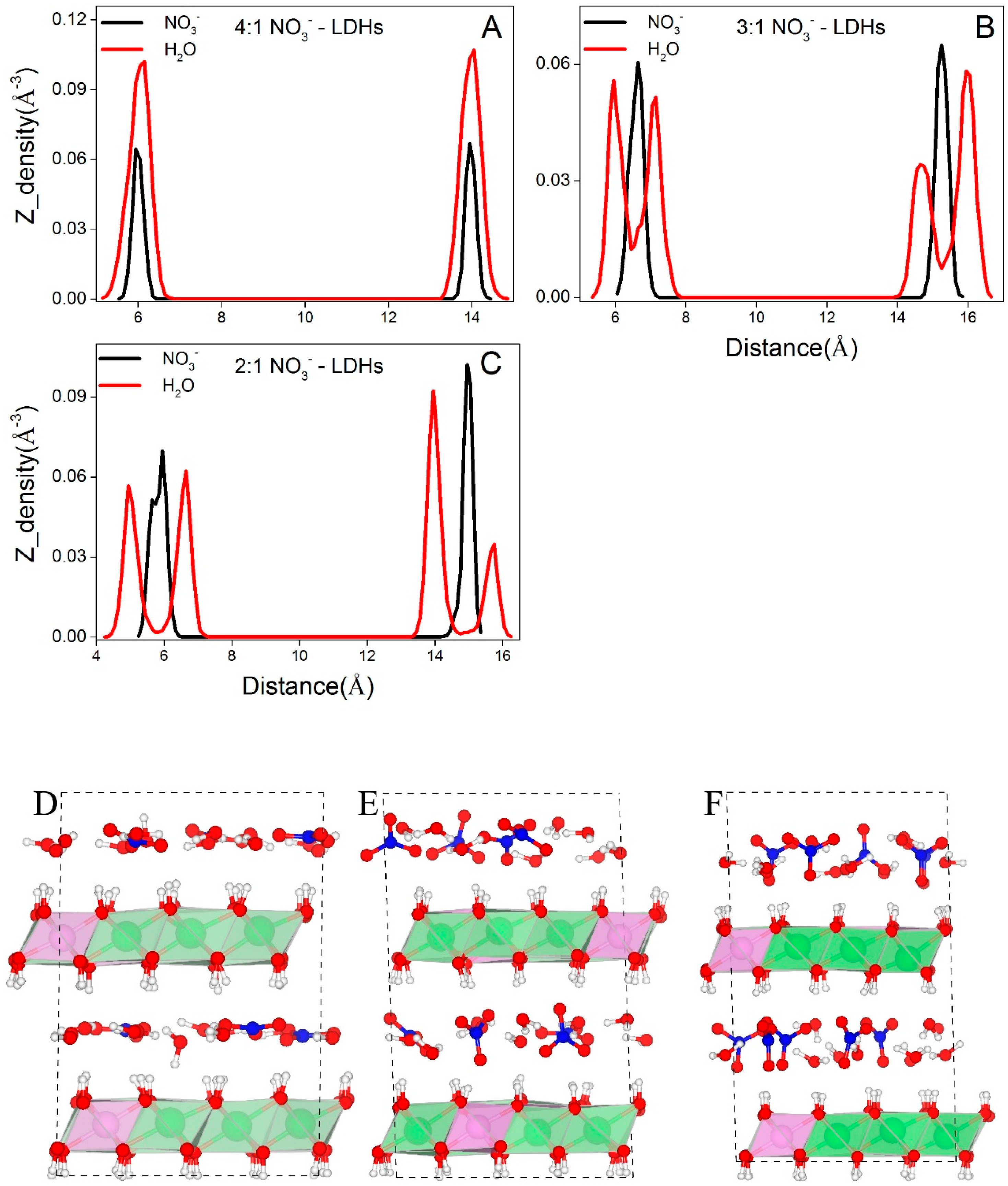
Figure 4 shows the distributions of water molecules and NO3− in NO3−-LDHs systems. At low layer charge density (Mg/Al = 4:1, Figure 4A,D), both water and NO3− exhibit a single layer distribution. At higher layer charge densities (Mg/Al = 3:1 and 2:1), water molecules form double layer distributions and NO3− retains the single layer distribution (Figure 4B,C,E,F).
The different layering distributions of water molecules at different layer charge densities are the result of the changes in the basal spacing. At 4:1, the calculated basal spacing of NO3−-LDHs is 7.95 Å and the thickness of the octahedral sheet is about 3.9 Å; thus the interlayer region is about 4 Å along the Z direction. Interlayer species can form hydrogen bonds with both the upper and the lower sheets. Therefore, the small interlayer space restricts interlayer species to a single layer distribution through hydrogen bonding. At higher layer charge densities, the calculated basal spacings are significantly larger, that is, 8.80 Å and 9.0 Å for 3:1 and 2:1, respectively; thus the interlayer regions are about 4.9 Å and 5.1 Å, respectively. According to the hydrogen bonding criteria presented in Figure 2, water molecules cannot form hydrogen bonds with both sheets even considering only the distance along the Z direction between water oxygen, and surface hydroxyls (marked as LDHs-OHs). As shown in Section 3.2.2, water molecules form hydrogen bonds with either the upper or the lower sheets and lead to the double layer distribution.
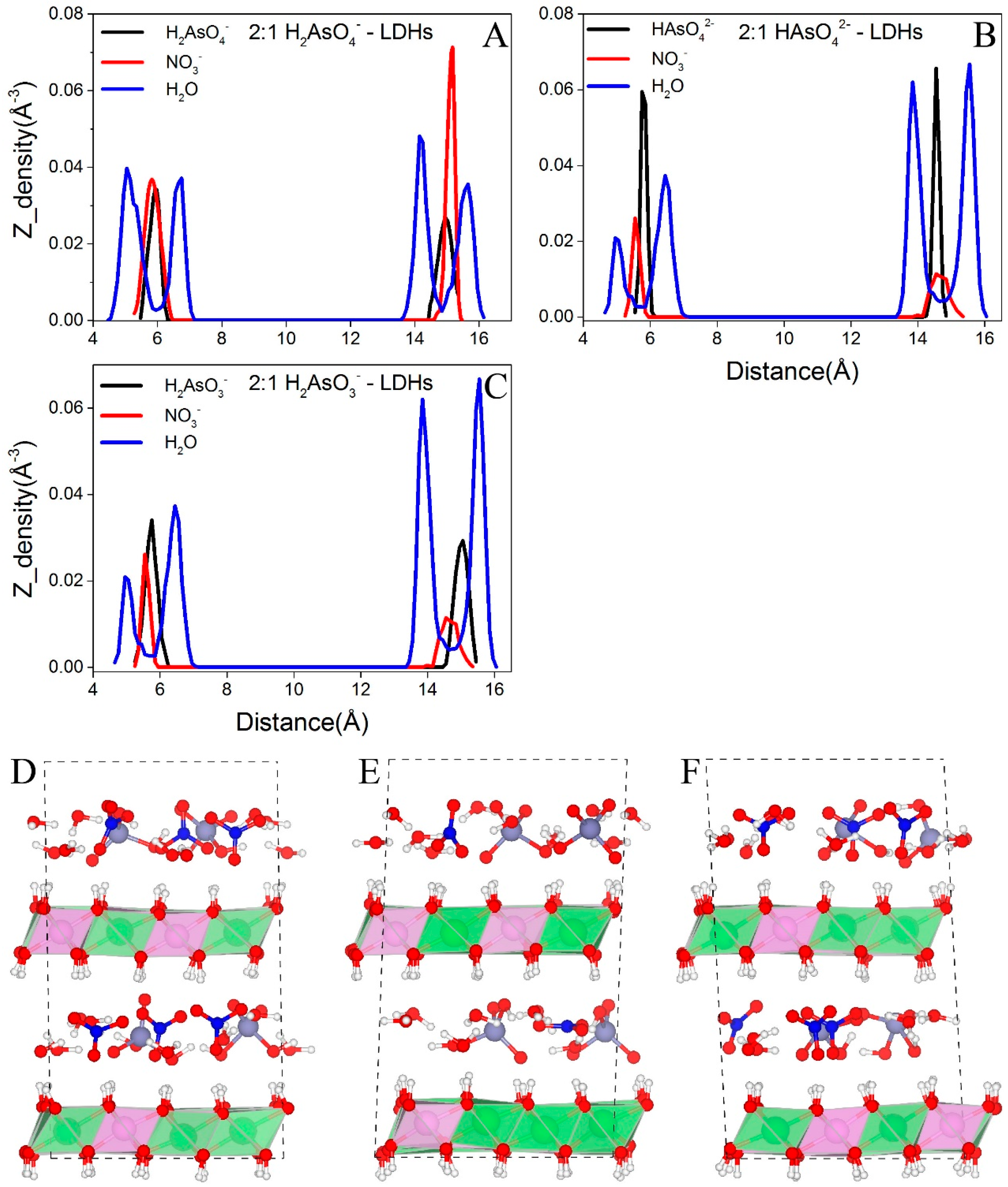
Figure 5 shows the distributions of interlayer species of H2AsO4−, HAsO42−, and H2AsO3−-LDHs. Only As containing systems with high layer charge density (i.e., Mg/Al = 2:1) are selected to perform BOMD simulations because in the experiments, arsenic species did not enter the gallery space at low layer charge density as discussed above. Water molecules in these systems form hydrogen bonds with either upper or lower LDHs-OHs and form a double layer distribution in the gallery space, which is similar to those water molecules in NO3−-LDHs at high layer charge densities. H2AsO4−, HAsO42−, and H2AsO3− locate around the central plane between two octahedral sheets and show a single layer distribution (Figure 5A–F, respectively).
3.2.2. Orientations of Interlayer Species
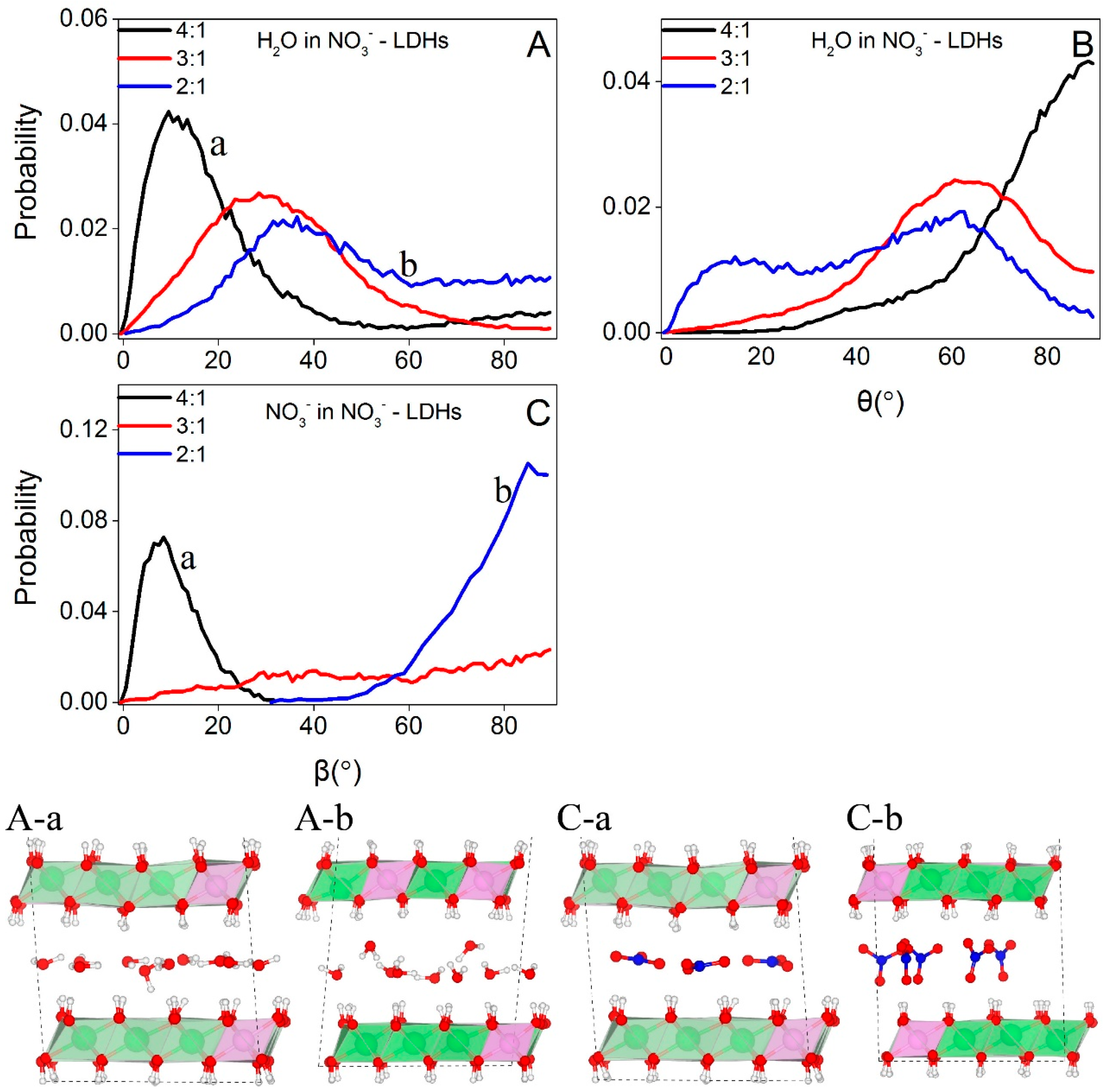
Figure 6 depicts the orientations of NO3− and water molecules at different layer charge densities. As shown in Figure 6, water molecules prefer an orientation parallel to the octahedral sheets at low charge density with the angle between the normal of the molecule plane and Z that peaked at about 10° (Figure 6A-a). This angle increases with the layer charge density and reaches about 35° at 2:1 (Figure 6A-b). As shown in Figure 6B, the probability distributions of the angles between water dipole moments and Z are centered at about 90° at low layer charge density, corresponding to the parallel orientation of the water plane (Figure 6A-a). With increasing layer charge densities, water dipole moments lose the parallel orientations and no preferred orientations are observed at Mg/Al = 2:1 (Figure 6A-b).
The probability distribution of the angle between the NO3− plane and the octahedral sheets peaks at about 10° (Figure 6C-a) and about 85° (Figure 6C-b) for Mg/Al = 4:1 and 2:1, respectively, indicating that NO3− prefers a parallel orientation at low layer charge density and a perpendicular orientation at 2:1. NO3− can switch between the two configurations at Mg/Al = 3:1 and thus no preferred orientations are observed (Figure 6C).
Combining the results that the angle between the NO3− plane and octahedral sheets increases with the layer charge density (Figure 6C and snapshots herein) and that the basal spacings increase with the layer charge density, our simulations suggest that it is the changes in the orientation of NO3− that trigger the increments in basal spacings, consistent with the previous studies [8,47].
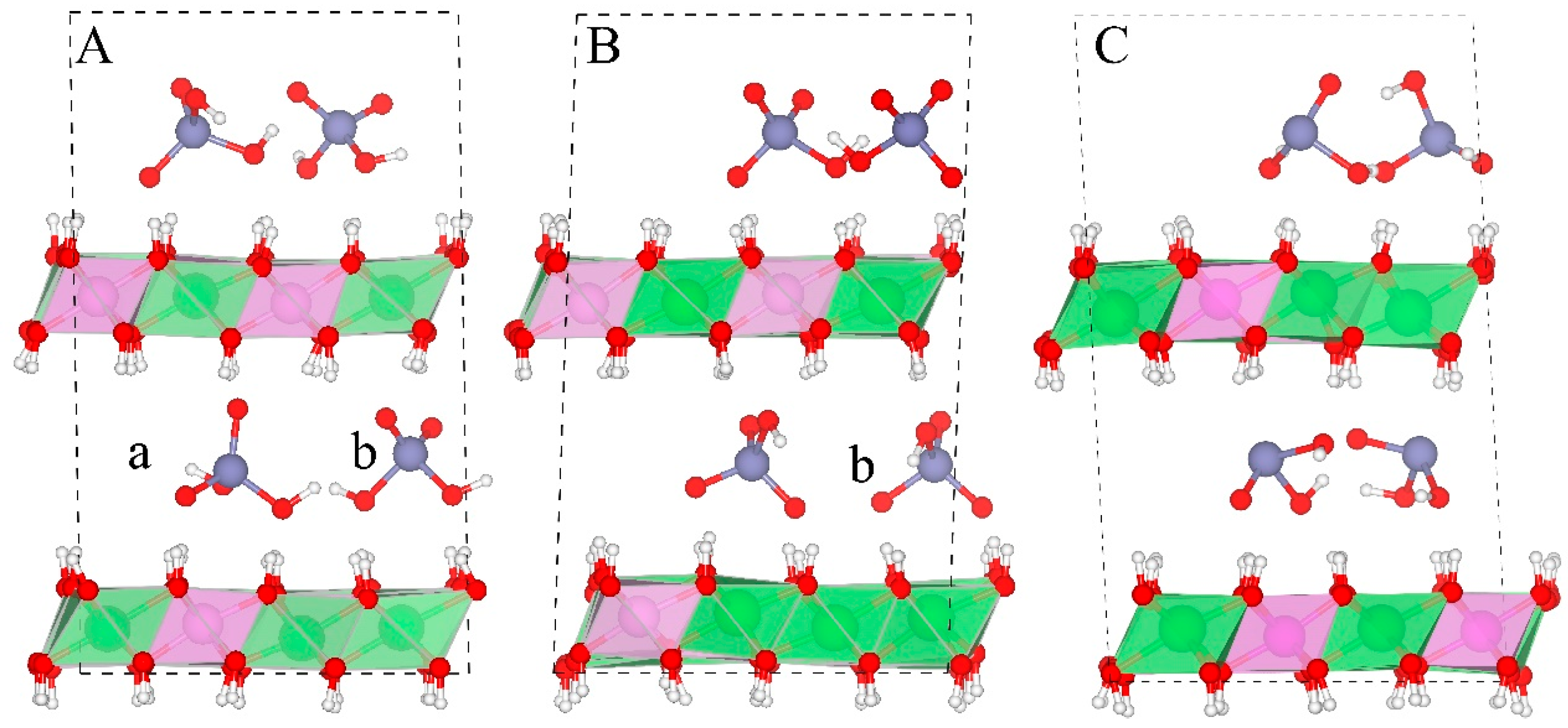
Figure 7 shows the orientations of As species in the gallery space. Two orientations are observed during the simulation for H2AsO4− (Figure 7A). Orientation a (Figure 7A-a) has one 3-fold symmetry axis of the As-O pyramid perpendicular to the layer sheets. Orientation b (Figure 7A-b) has one 2-fold symmetry axis of the As-O pyramid perpendicular to the layer sheets. These two orientations can switch between each other while b is the major orientation during the simulation (e.g., three out of four H2AsO4− anions have this orientation as shown in Figure 7A).The spatial range of these two orientations is equal to the height and the edge distance of the As-O tetrahedron, and (d is the distance between As and O, about 1.71 Å in our systems), respectively. In the HAsO42− containing system, only orientation b is observed during the whole simulation (Figure 7B). Based on the above observation, one can deduce that other 4-fold oxy-anions (e.g., PO43−, MoO42−, CrO42−, and SeO42−) should also have these two orientations.
3.3. Hydrogen Bonding of As Species
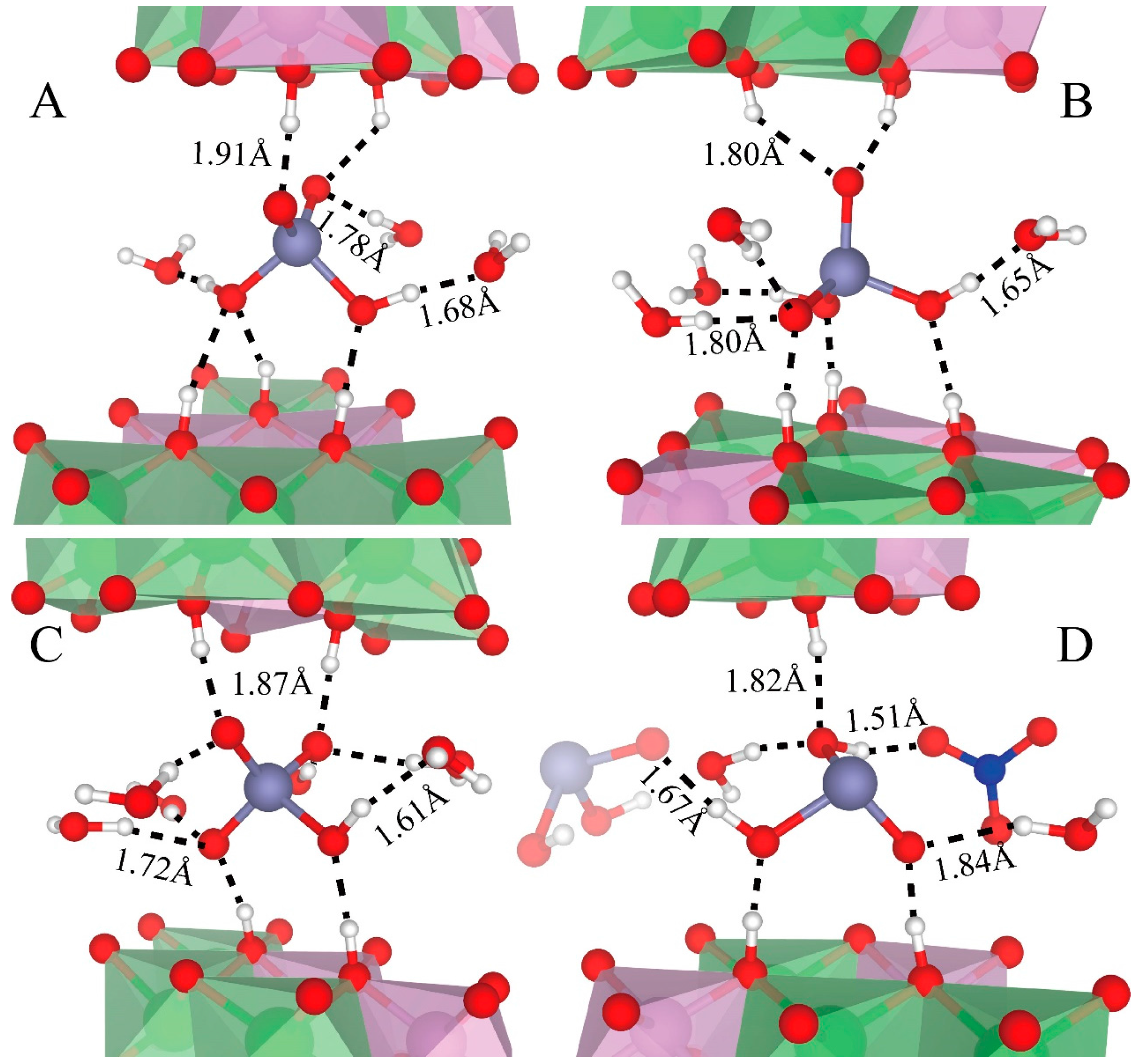
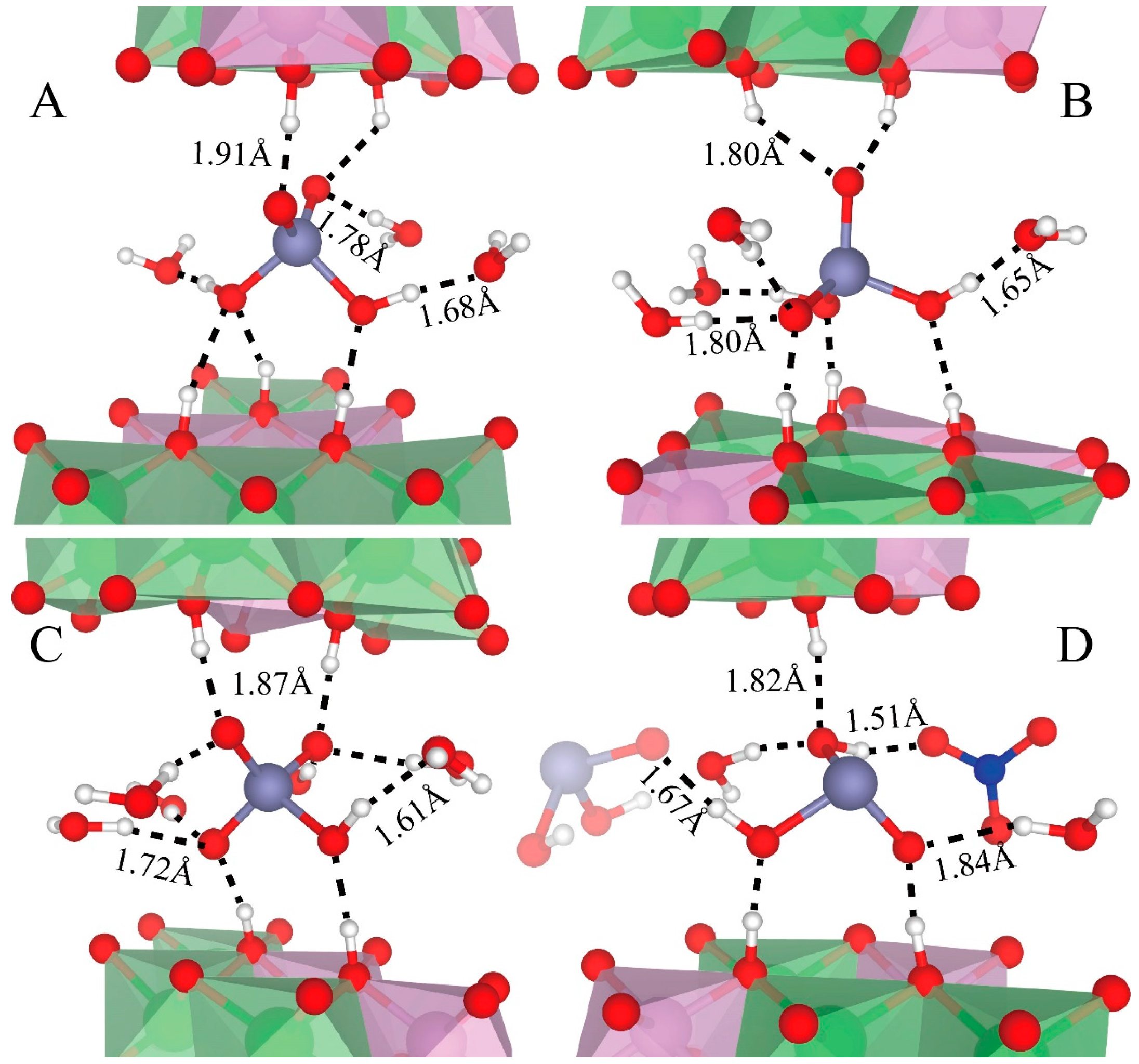
Figure 8 depicts the hydrogen bonds of H2AsO4− (Figure 8A,B), HAsO42− (Figure 8C), and H2AsO3− (Figure 8D). The oxygen of these species can accept two or three hydrogen bonds from water and LDHs-OHs (Figure 8). Their OHs donate one hydrogen bond to water molecules (Figure 8A–C) or NO3− (Figure 8D), but not to LDHs-OHs.
The average H-bond lengths are labeled in Figure 8. The hydrogen bonds donated by As species have smaller distances than those donated by water and LDHs-OHs, with an average distance of 1.68 Å for H2AsO4−, 1.61 Å for HAsO42−, and 1.67 Å for H2AsO3−. Hydrogen bonds accepted by As species from water have a distance ranging from 1.7 Å to 1.8 Å, with an average value of 1.78 Å for H2AsO4−, 1.72 Å for HAsO42−, and 1.84 Å for H2AsO3−. The hydrogen bonds accepted by the As species from LDHs-OHs are slightly longer, with an average distance of 1.91 Å for H2AsO4−, 1.87 Å for HAsO42−, and 1.82 Å for H2AsO3−.
3.4. Mobility of Interlayer Species
Table 2 lists the diffusion coefficients of the interlayer species of the modeled systems. The diffusion coefficients of water are of the order 10−13–10−12 m2/s, similar to those from a previous molecular dynamics study of Cl−-LDHs [52]. They are much lower than those of water in montmorillonite, i.e., 10−10 m2/s [82,83,84,85]. Diffusion coefficients of water and NO3− increase significantly with layer charge density from 4:1 to 3:1 and stay almost the same from 3:1 to 2:1. The possible reasons for the changes of mobility might be the electrostatic forces and basal spacings which are also functions of the layer charge densities. The electrostatic forces between anions and the octahedral sheets increase with the layer charge densities (i.e., 2:1 and 3:1), diminishing the mobility of the anions. This is, however, contradictory to our findings. On the other hand, as shown above, the basal spacing increases significantly from 4:1 to 3:1 (i.e., 8.0 Å to 8.8 Å) and therefore, interlayer species move more freely in 3:1 LDHs, leading to significant increments in the diffusion coefficients. From 3:1 to 2:1, the change in basal spacing is quite small (i.e., from 8.8 Å to 9.0 Å) and therefore, no significant change in the diffusion coefficients has been observed.
The calculated diffusion coefficients of As species are about one order of magnitude lower than those of water and NO3− at 2:1. This could also be attributed to the space effect, that is, the relatively larger sizes of these arsenic species leave them little space to move. The low mobility of arsenic species suggests that LDHs can be the potential materials for As disposal.
4. Conclusions
By using first principles simulation techniques, we studied the arsenite and arsenate intercalated LDHs. The basal spacings, interlayer structures, and diffusion coefficients of interlayer species were derived and analyzed in detail. The following conclusions have been made.
- Comparisons between computed and experimental basal spacings indicate that arsenite and arsenate can enter the gallery spaces of LDHs with a Mg/Al ratio of 2:1, but may not enter the gallery spaces of LDHs with Mg/Al of 3:1 or 4:1.
- All interlayer species show layering distributions. As species and NO3− form single layer distributions while water molecules form single layer distributions at low layer charge densities and double layer distributions at high layer charge densities.
- H2AsO4− has two orientations in the interlayer regions, i.e., one with its three fold axis normal to the layer sheets and another with its two fold axis normal to the layer sheets, while only the latter one is observed for HAsO42−. H2AsO3− orientates in a tilt-lying way.
- It is found that the mobility of water and NO3− increases with the layer charge densities. All As species show very low mobility (of the order 10−13 m2/s) in LDHs.
Acknowledgments
We acknowledge the National Science Foundation of China (Nos. 41222015, 41273074, and 41572027), Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase), the Foundation for the Author of National Excellent Doctoral Dissertation of China (No. 201228), Newton International Fellowship Program, and the financial support from the State Key Laboratory at Nanjing University. We are grateful to the High Performance Computing Center of Nanjing University for allowing us to use the IBM Blade cluster system.
Author Contributions
Xiandong Liu and Yingchun Zhang conceived and designed the simulations; Yingchun Zhang performed the simulations; Yingchun Zhang, Xiandong Liu, and Chi Zhang analyzed the data; All authors contributed to the preparation and writing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mandal, B.K.; Suzuki, K.T. Arsenic round the world: A review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2001, 17, 517–568. [Google Scholar] [CrossRef]
- Frankenberger, W.T., Jr. Environmental Chemistry of Arsenic; CRC Press: Baco Raton, FL, USA, 2001. [Google Scholar]
- Andreae, M.O. Distribution and speciation of arsenic in natural waters and some marine algae. Deep Sea Res. Part II Top. Stud. Oceanogr. 1978, 25, 391–402. [Google Scholar] [CrossRef]
- Andreae, M.O.; Andreae, T.W. Dissolved arsenic species in the schelde estuary and watershed, Belgium. Estuar. Coast. Shelf Sci. 1989, 29, 421–433. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G.; Huq, I.; Luo, Z.; Nicolli, H.B. International perspective on naturally occurring arsenic problems in groundwater. In Arsenic Exposure and Health Effects IV; Elsevier: Amsterdam, The Netherlands, 2001; pp. 9–25. [Google Scholar]
- Smedley, P.L.; Zhang, M.; Zhang, G.; Luo, Z. Arsenic and Other Redox-Sensitive Elements in Groundwater from the Huhhot Basin, Inner Mongolia; A A Balkema Publishers: Leiden, IL, USA, 2001; pp. 581–584. [Google Scholar]
- Wang, S.L.; Cheng, H.L.; Ming, K.W.; Chuang, Y.H.; Chiang, P.N. Arsenate adsorption by Mg/Al–NO3 layered double hydroxides with varying the Mg/Al ratio. Appl. Clay Sci. 2009, 43, 79–85. [Google Scholar] [CrossRef]
- Goldberg, S. Competitive adsorption of arsenate and arsenite on oxides and clay minerals. Soil Sci. Soc. Am. J. 2002, 66, 413–421. [Google Scholar] [CrossRef]
- Violante, A.; Pigna, M. Competitive sorption of arsenate and phosphate on different clay minerals and soils. Soil Sci. Soc. Am. J. 2002, 66, 1788–1796. [Google Scholar] [CrossRef]
- Dousova, B.; Lhotka, M.; Grygar, T.; Machovic, V.; Herzogova, L. In situ co-adsorption of arsenic and iron/manganese ions on raw clays. Appl. Clay Sci. 2011, 54, 166–171. [Google Scholar] [CrossRef]
- Ghorbanzadeh, N.; Jung, W.; Halajnia, A.; Lakzian, A.; Kabra, A.N.; Jeon, B.H. Removal of arsenate and arsenite from aqueous solution by adsorption on clay minerals. Geosyst. Eng. 2015, 18, 302–311. [Google Scholar] [CrossRef]
- Na, P.; Jia, X.M.; Bin, Y.; Li, Y.; Na, J.; Chen, Y.C.; Wang, L.S. Arsenic adsorption on Ti-pillared montmorillonite. J. Chem. Technol. Biotechnol. 2010, 85, 708–714. [Google Scholar] [CrossRef]
- Lin, Z.; Puls, R.W. Adsorption, desorption and oxidation of arsenic affected by clay minerals and aging process. Environ. Geol. 2000, 39, 753–759. [Google Scholar] [CrossRef]
- Saada, A.; Breeze, D.; Crouzet, C.; Cornu, S.; Baranger, P. Adsorption of arsenic (V) on kaolinite and on kaolinite-humic acid complexes—Role of humic acid nitrogen groups. Chemosphere 2003, 51, 757–763. [Google Scholar] [CrossRef]
- Bhowmick, S.; Chakraborty, S.; Mondal, P.; van Renterghem, W.; van den Berghe, S.; Roman-Ross, G.; Chatterjee, D.; Iglesias, M. Montmorillonite-supported nanoscale zero-valent iron for removal of arsenic from aqueous solution: Kinetics and mechanism. Chem. Eng. J. 2014, 243, 14–23. [Google Scholar] [CrossRef]
- Wainipee, W.; Cuadros, J.; Sephton, M.A.; Unsworth, C.; Gill, M.G.; Strekopytov, S.; Weiss, D.J. The effects of oil on As(V) adsorption on illite, kaolinite, montmorillonite and chlorite. Geochim. Cosmochim. Acta 2013, 121, 487–502. [Google Scholar] [CrossRef]
- Manning, B.; Fendorf, S.; Goldberg, S. Surface structures and stability of arsenic(III) on goethite: Spectroscopic evidence for inner-sphere complexes. Environ. Sci. Technol. 1998, 32, 2383–2388. [Google Scholar] [CrossRef]
- Farquhar, M.L.; Charnock, J.M.; Livens, F.R.; Vaughan, D.J. Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: An X-ray absorption spectroscopy study. Environ. Sci. Technol. 2002, 36, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.M.; Randall, S.R. Surface complexation of arsenic(V) to iron(III) (hydr)oxides: Structural mechanism from Ab initio molecular geometries and exafs spectroscopy. Geochim. Cosmochim. Acta 2003, 67, 4223–4230. [Google Scholar] [CrossRef]
- Tu, Y.J.; You, C.F.; Chang, C.K.; Wang, S.L. Xanes evidence of arsenate removal from water with magnetic ferrite. J. Environ. Manag. 2013, 120, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Root, R.A.; Farrell, J.; Ela, W.; Chorover, J. Effect of silicic acid on arsenate and arsenite retention mechanisms on 6-l ferrihydrite: A spectroscopic and batch adsorption approach. Appl. Geochem. 2013, 38, 110. [Google Scholar] [CrossRef] [PubMed]
- Ladeira, A.C.Q.; Ciminelli, V.S.T.; Duarte, H.A.; Alves, M.C.M.; Ramos, A.Y. Mechanism of anion retention from exafs and density functional calculations: Arsenic (V) adsorbed on gibbsite. Geochim. Cosmochim. Acta 2001, 65, 1211–1217. [Google Scholar] [CrossRef]
- Onanguema, G.; Morin, G.; Wang, Y.; Foster, A.L.; Juillot, F.; Calas, G.; Brown, G.E. Xanes evidence for rapid arsenic(III) oxidation at magnetite and ferrihydrite surfaces by dissolved O2 via Fe2+-mediated reactions. Environ. Sci. Technol. 2010, 44, 5416–5422. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.Q.; Paul, K.W.; Kubicki, J.D.; Sparks, D.L. Quantum chemical study of arsenic (III, V) adsorption on mn-oxides: Implications for arsenic(III) oxidation. Environ. Sci. Technol. 2009, 43, 6655. [Google Scholar] [CrossRef] [PubMed]
- Watts, H.D.; Tribe, L.; Kubicki, J.D. Arsenic adsorption onto minerals: Connecting experimental observations with density functional theory calculations. Minerals 2014, 4, 208–240. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Kwon, A.K.D.; Paul, A.K.W.; Sparks, D.L. Surface complex structures modelled with quantum chemical calculations: Carbonate, phosphate, sulphate, arsenate and arsenite. Eur. J. Soil Sci. 2007, 58, 932–944. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, J.H. Characterization of adsorbed arsenate on amorphous and nano crystalline mgfe-layered double hydroxides. J. Nanopharm. Res. 2011, 13, 887–894. [Google Scholar] [CrossRef]
- Caporale, A.G.; Pigna, M.; Dynes, J.J.; Cozzolino, V.; Zhu, J.; Violante, A. Effect of inorganic and organic ligands on the sorption/desorption of arsenate on/from Al–Mg and Fe–Mg layered double hydroxides. J. Hazard. Mater. 2011, 198, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Violante, A.; Pucci, M.; Cozzolino, V.; Zhu, J.; Pigna, M. Sorption/desorption of arsenate on/from Mg–Al layered double hydroxides: Influence of phosphate. J. Colloid Interface Sci. 2009, 333, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.V.; Kamath, P.V. Synthesis and characterization of arsenate-intercalated layered double hydroxides (ldhs): Prospects for arsenic mineralization. J. Colloid Interface Sci. 2009, 331, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.V.; Kamath, P.V. Chromate uptake characteristics of the pristine layered double hydroxides of mg with al. Solid State Sci. 2008, 10, 260–266. [Google Scholar] [CrossRef]
- Prasanna, S.V.; Rao, R.A.; Kamath, P.V. Layered double hydroxides as potential chromate scavengers. J. Colloid Interface Sci. 2007, 304, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.V.; Kamath, P.V.; Shivakumara, C. Synthesis and characterization of layered double hydroxides (ldhs) with intercalated chromate ions. Mater. Res. Bull. 2007, 42, 1028–1039. [Google Scholar] [CrossRef]
- Das, J.; Patra, B.S.; Baliarsingh, N.; Parida, K.M. Adsorption of phosphate by layered double hydroxides in aqueous solutions. Appl. Clay Sci. 2006, 32, 252–260. [Google Scholar] [CrossRef]
- Peng, C.; Hong, Z.; Chong, W.; Ma, H.; Hu, J.; Pu, Y.; Peng, L. Competitive adsorption characteristics of fluoride and phosphate on calcined Mg–Al–Co 3 layered double hydroxides. J. Hazard. Mater. 2012, 213–214, 100–108. [Google Scholar]
- Xiang, C.; Huang, X.; Wang, X.; Zhao, B.; Chen, A.; Sun, D. Phosphate adsorption from sewage sludge filtrate using zinc–aluminum layered double hydroxides. J. Hazard. Mater. 2009, 169, 958–964. [Google Scholar]
- Wang, S.L.; Cheng, C.Y.; Tzou, Y.M.; Liaw, R.B.; Chang, T.W.; Chen, J.H. Phosphate removal from water using lithium intercalated gibbsite. J. Hazard. Mater. 2007, 147, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Seida, Y.; Nakano, Y. Removal of phosphate by layered double hydroxides containing iron. Water Res. 2002, 36, 1306–1312. [Google Scholar] [CrossRef]
- He, J.; Wei, M.; Li, B.; Kang, Y.; Evans, D.G.; Duan, X. Preparation of layered double hydroxides. In Layered Double Hydroxides; Springer: Berlin/Heidelberg, Germany, 2006; pp. 89–119. [Google Scholar]
- Yang, L.; Dadwhal, M.; Shahrivari, Z.; Ostwal, M.; Liu, P.K.T.; Sahimi, M.; Tsotsis, T.T. Adsorption of arsenic on layered double hydroxides: Effect of the particle size. Ind. Eng. Chem. Res. 2006, 45, 4742–4751. [Google Scholar] [CrossRef]
- Meyn, M.; Beneke, K.; Lagaly, G. Anion-exchange reactions of layered double hydroxides. Inorg. Chem. 1990, 29, 5201–5207. [Google Scholar] [CrossRef]
- Constantino, V.R.; Pinnavaia, T.J. Basic properties of Mg2+1−xAl3+x layered double hydroxides intercalated by carbonate, hydroxide, chloride, and sulfate anions. Inorg. Chem. 1995, 34, 883–892. [Google Scholar] [CrossRef]
- Gago, S.; Pillinger, M.; Valente, A.A.; Santos, T.M.; João Rocha, A.; Gonçalves, I.S. Immobilization of oxomolybdenum species in a layered double hydroxide pillared by 2,2′-bipyridine-5,5′-dicarboxylate anions. Inorg. Chem. 2004, 43, 5422–5431. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Zeng, H.C. Abrupt structural transformation in hydrotalcite-like compounds Mg1−xAlx(OH)2(NO3)x·nH2O as a continuous function of nitrate anions. J. Phys. Chem. B 2001, 105, 1743–1749. [Google Scholar] [CrossRef]
- Li, H.; Jing, M.; Evans, D.G.; Zhou, T.; Li, F.; Xue, D. Molecular dynamics modeling of the structures and binding energies of α-nickel hydroxides and nickel−aluminum layered double hydroxides containing various interlayer guest anions. Chem. Mater. 2006, 18, 4405–4414. [Google Scholar] [CrossRef]
- Wang, S.L.; Wang, P.C. In situ xrd and atr-ftir study on the molecular orientation of interlayer nitrate in Mg/Al-layered double hydroxides in water. Colloids Surf. A Physicochem. Eng. Asp. 2007, 292, 131–138. [Google Scholar] [CrossRef]
- Cygan, R.T.; Greathouse, J.A.; Heinz, H.; Kalinichev, A.G. Molecular models and simulations of layered materials. J. Mater. Chem. 2009, 19, 2470–2481. [Google Scholar] [CrossRef]
- Kumar, P.P.; Kalinichev, A.G.; Kirkpatrick, R.J. Molecular dynamics simulation of the energetics and structure of layered double hydroxides intercalated with carboxylic acids. J. Phys. Chem. C 2007, 111, 13517–13523. [Google Scholar] [CrossRef]
- Pisson, J.; Morel, J.P.; Moreldesrosiers, N.; Taviotguého, C.; Malfreyt, P. Molecular modeling of the structure and dynamics of the interlayer species of znalcl layered double hydroxide. J. Phys. Chem. B 2008, 112, 7856–7864. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; Kang, H.; Zhang, H.; Yuan, S. Molecular simulation studies for intercalation of photoactive dyes into layered double hydroxide. Colloids Surf. A Physicochem. Eng. Asp. 2012, 402, 108–116. [Google Scholar] [CrossRef]
- Chen, M.; Shen, W.; Lu, X.; Zhu, R.; He, H.; Zhu, J. Jumping diffusion of water intercalated in layered double hydroxides. J. Phys. Chem. C 2016, 120, 12924–12931. [Google Scholar] [CrossRef]
- Newman, S.P.; Williams, S.J.; Coveney, P.V.; Jones, W. Interlayer arrangement of hydrated MgAl layered double hydroxides containing guest terephthalate anions: Comparison of simulation and measurement. J. Phys. Chem. B 1998, 102, 6710–6719. [Google Scholar] [CrossRef]
- Liu, X.; He, M.; Lu, X.; Wang, R. Structures and acidity constants of arsenite and thioarsenite species in hydrothermal solutions. Chem. Geol. 2015, 411, 192–199. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.; Sprik, M.; Lu, X.; Wang, R. Understanding surface acidity of gibbsite with first principles molecular dynamics simulations. Geochim. Cosmochim. Acta 2013, 120, 487–495. [Google Scholar] [CrossRef]
- Liu, X.; Lu, X.; Meijer, E.J.; Wang, R.; Zhou, H. Atomic-scale structures of interfaces between phyllosilicate edges and water. Geochim. Cosmochim. Acta 2012, 81, 56–68. [Google Scholar] [CrossRef]
- Liu, X.; Lu, X.; Sprik, M.; Cheng, J.; Meijer, E.J.; Wang, R. Acidity of edge surface sites of montmorillonite and kaolinite. Geochim. Cosmochim. Acta 2013, 117, 180–190. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.; Sprik, M.; Lu, X.; Wang, R. Surface acidity of 2:1-type dioctahedral clay minerals from first principles molecular dynamics simulations. Geochim. Cosmochim. Acta 2014, 140, 410–417. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Lu, X.; He, M.; Meijer, E.J.; Wang, R. Surface complexation of heavy metal cations on clay edges: Insights from first principles molecular dynamics simulation of Ni (II). Geochim. Cosmochim. Acta 2017, 203, 54–68. [Google Scholar] [CrossRef]
- Bellotto, M.; Rebours, B.; Olivier Clause, A.; Lynch, J.; Bazin, D.; Elkaïm, E. A reexamination of hydrotalcite crystal chemistry. J. Phys. Chem. B 1996, 100, 8527–8534. [Google Scholar] [CrossRef]
- Loewenstein, W. The distribution of aluminum in the tetrahedra of silicates and aluminates. Am. Mineral. 1954, 39, 92–96. [Google Scholar]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Lippert, B.G.; Hutter, J.; Parrinello, M. A hybrid Gaussian and plane wave density functional scheme. Mol. Phys. Int. J. Interface Chem. Phys. 1997, 92, 477–487. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B Condens. Matter 1995, 54, 1703–1710. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Marry, V.; Rotenberg, B.; Turq, P. Structure and dynamics of water at a clay surface from molecular dynamics simulation. Phys. Chem. Chem. Phys. 2008, 10, 4802–4813. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.-H.; Kwak, S.-Y.; Park, J.-S.; Jeong, Y.-J. Cellular uptake behavior of [γ-32p] labeled ATP–LDH. J. Mater. Chem. 2001, 11, 1671–1674. [Google Scholar] [CrossRef]
- Newman, S.P.; Jones, W. Comparative study of some layered hydroxide salts containing exchangeable interlayer anions. J. Solid State Chem. 1999, 148, 26–40. [Google Scholar] [CrossRef]
- Han, J.B.; Lu, J.; Wei, M.; Wang, Z.L.; Duan, X. Heterogeneous ultrathin films fabricated by alternate assembly of exfoliated layered double hydroxides and polyanion. Chem. Commun. 2008, 41, 5188–5190. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.H.; Jung, J.S.; Oh, J.M.; Man, P.; Jeong, J.; Kang, Y.K.; Han, O.J. Layered double hydroxide as an efficient drug reservoir for folate derivatives. Biomaterials 2004, 25, 3059–3064. [Google Scholar] [CrossRef] [PubMed]
- Bujdosó, T.; Patzkó, Á.; Galbács, Z.; Dékány, I. Structural characterization of arsenate ion exchanged mgal-layered double hydroxide. Appl. Clay Sci. 2009, 44, 75–82. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, J.; Zhang, Z.; Jin, R.; Zhang, X.; Yan, X.; Umar, A.; Guo, Z.; Wang, Q. Synthesis of polypropylene/Mg3Al–X (X = CO32−, NO3−, Cl−, SO42−) LDH nanocomposites using a solvent mixing method: Thermal and melt rheological properties. J. Mater. Chem. A 2013, 1, 9928–9934. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, R.; Osada, M.; Iyi, N.; Ebina, Y.; Takada, K.; Sasaki, T. Synthesis, anion exchange, and delamination of Co-Al layered double hydroxide: Assembly of the exfoliated nanosheet/polyanion composite films and magneto-optical studies. J. Am. Chem. Soc. 2006, 128, 4872–4880. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.-H.; Lim, T.-T.; Dong, Z. Application of layered double hydroxides for removal of oxyanions: A review. Water Res. 2008, 42, 1343–1368. [Google Scholar] [CrossRef] [PubMed]
- Grover, K.; Komarneni, S.; Katsuki, H. Synthetic hydrotalcite-type and hydrocalumite-type layered double hydroxides for arsenate uptake. Appl. Clay Sci. 2010, 48, 631–637. [Google Scholar] [CrossRef]
- Grover, K.; Komarneni, S.; Katsuki, H. Uptake of arsenite by synthetic layered double hydroxides. Water Res. 2009, 43, 3884–3890. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, L.H.; Zheng, Z.; Kang, M.L.; Li, C.; Liu, C.L. Molecular dynamics simulation of the diffusion of uranium species in clay pores. J. Hazard. Mater. 2013, 244–245, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yi, H.; Zhao, Y.; Min, F.; Song, S. Study on the differences of Na- and Ca-montmorillonites in crystalline swelling regime through molecular dynamics simulation. Adv. Powder Technol. 2016, 27, 779–785. [Google Scholar] [CrossRef]
- Myshakin, E.M.; Saidi, W.A.; Romanov, V.N.; Cygan, R.T.; Jordan, K.D. Molecular dynamics simulations of carbon dioxide intercalation in hydrated Na-montmorillonite. J. Phys. Chem. C 2013, 117, 141–155. [Google Scholar] [CrossRef]
- Boek, E.S. Molecular dynamics simulations of interlayer structure and mobility in hydrated Li-, Na- and K-montmorillonite clays. Mol. Phys. 2014, 112, 1472–1483. [Google Scholar] [CrossRef]
Figure 1.
Structure of H2AsO4−-Layered double hydroxides (LDHs) with a Mg/Al ratio of 2:1, water molecules are not presented here for clarity. H = white, N = blue, O = red, As = purple, Mg-OH octahedron = green, Al-OH octahedron = pink.
Figure 1.
Structure of H2AsO4−-Layered double hydroxides (LDHs) with a Mg/Al ratio of 2:1, water molecules are not presented here for clarity. H = white, N = blue, O = red, As = purple, Mg-OH octahedron = green, Al-OH octahedron = pink.
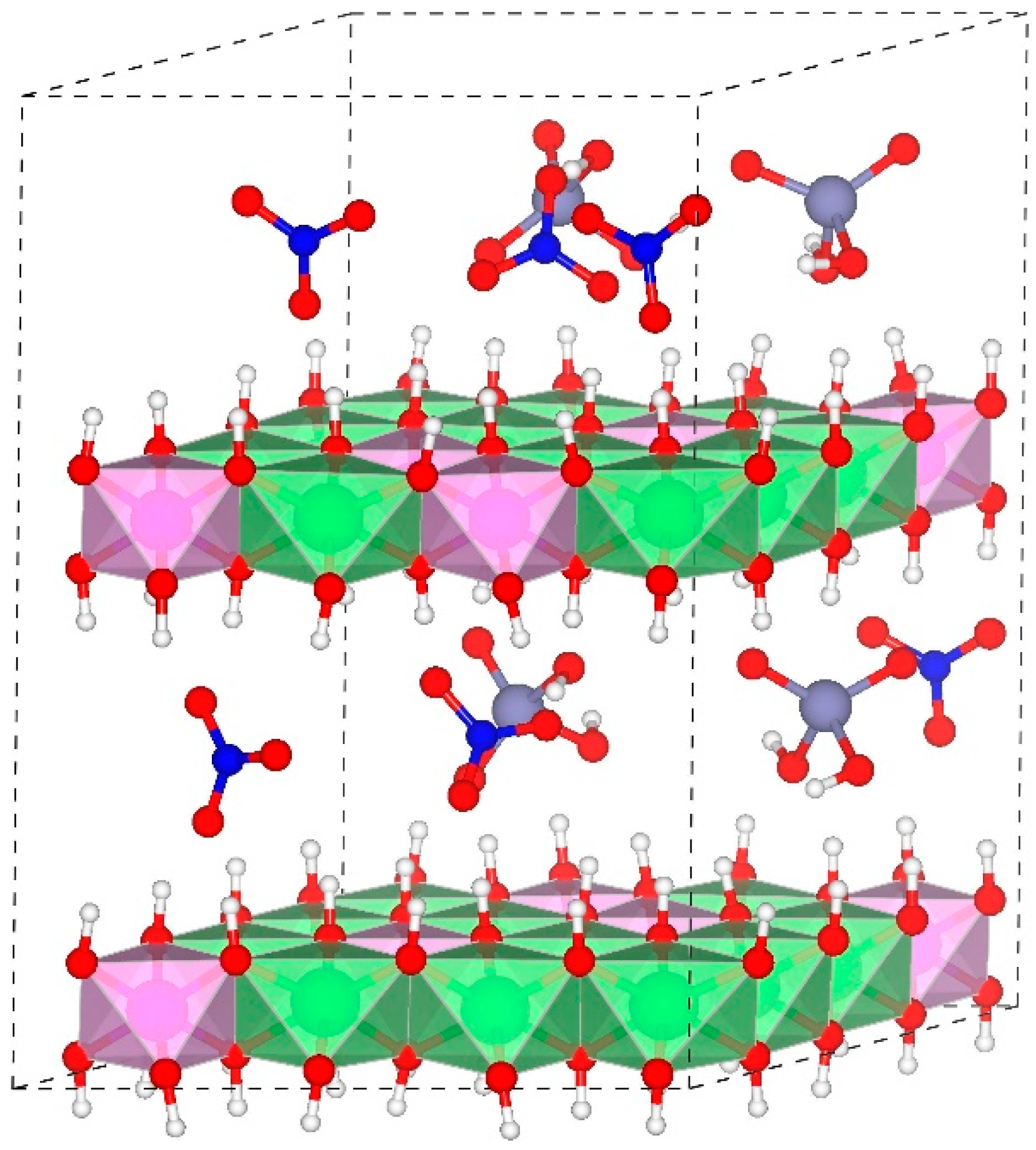
Figure 2.
Geometry parameters for hydrogen bonding (A) and molecule orientation (B).
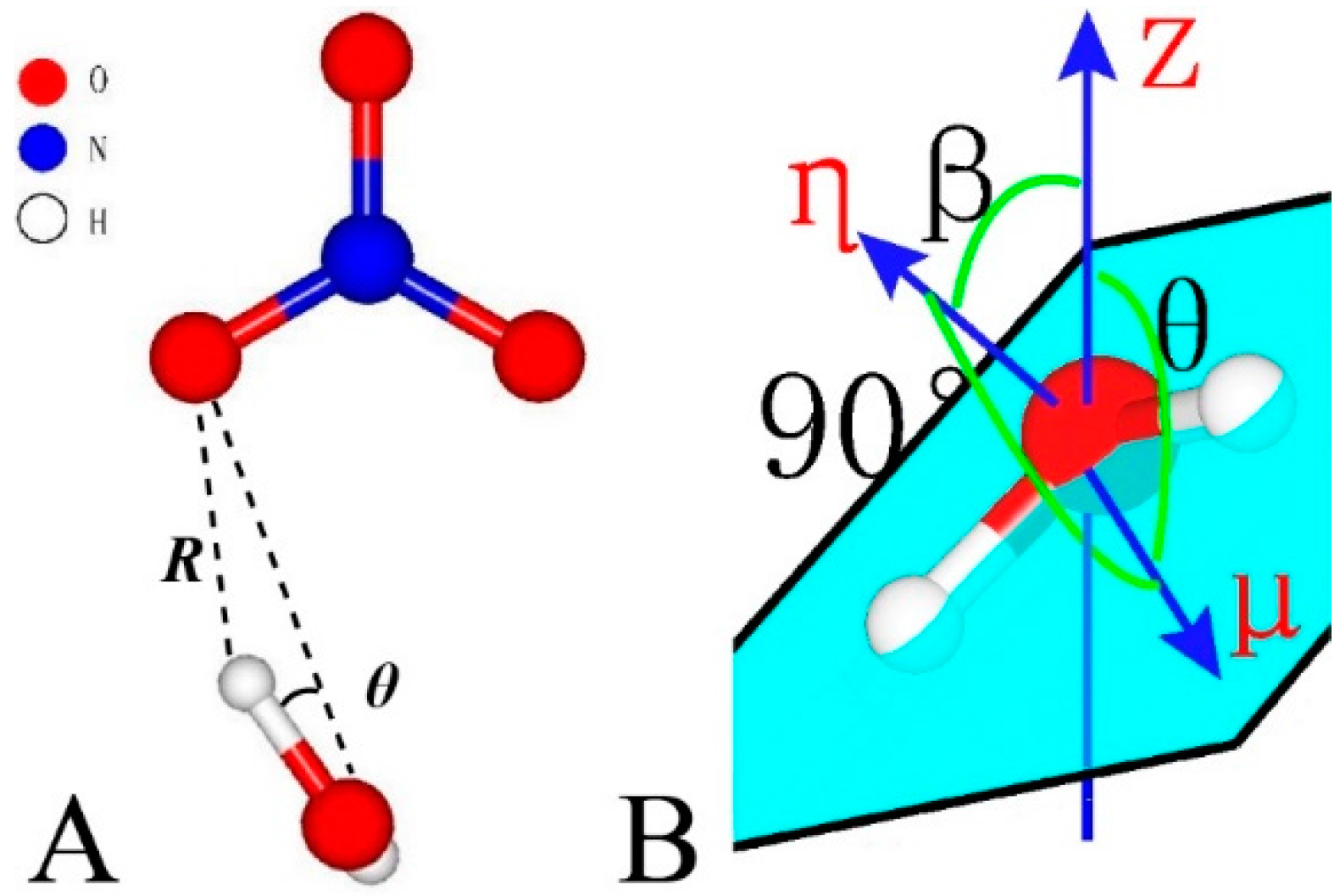
Figure 3.
Comparison of basal spacings between experiments (open symbols) and simulations (solid symbols). Experiments results are from references [8,47,72,73,74,75,76,78] for NO3− 2:1, [8,47,77] for NO3− 3:1, [8,47] for NO3− 4:1, [8,31,76,80] for As(V) 2:1, [8,80] for As(V) 3:1, and 4:1 and [81] for As(III). Shaded region presents the range of basal spacing for NO3−-LDHs at Mg/Al 3:1 suggested by Wang et al. [47].
Figure 3.
Comparison of basal spacings between experiments (open symbols) and simulations (solid symbols). Experiments results are from references [8,47,72,73,74,75,76,78] for NO3− 2:1, [8,47,77] for NO3− 3:1, [8,47] for NO3− 4:1, [8,31,76,80] for As(V) 2:1, [8,80] for As(V) 3:1, and 4:1 and [81] for As(III). Shaded region presents the range of basal spacing for NO3−-LDHs at Mg/Al 3:1 suggested by Wang et al. [47].
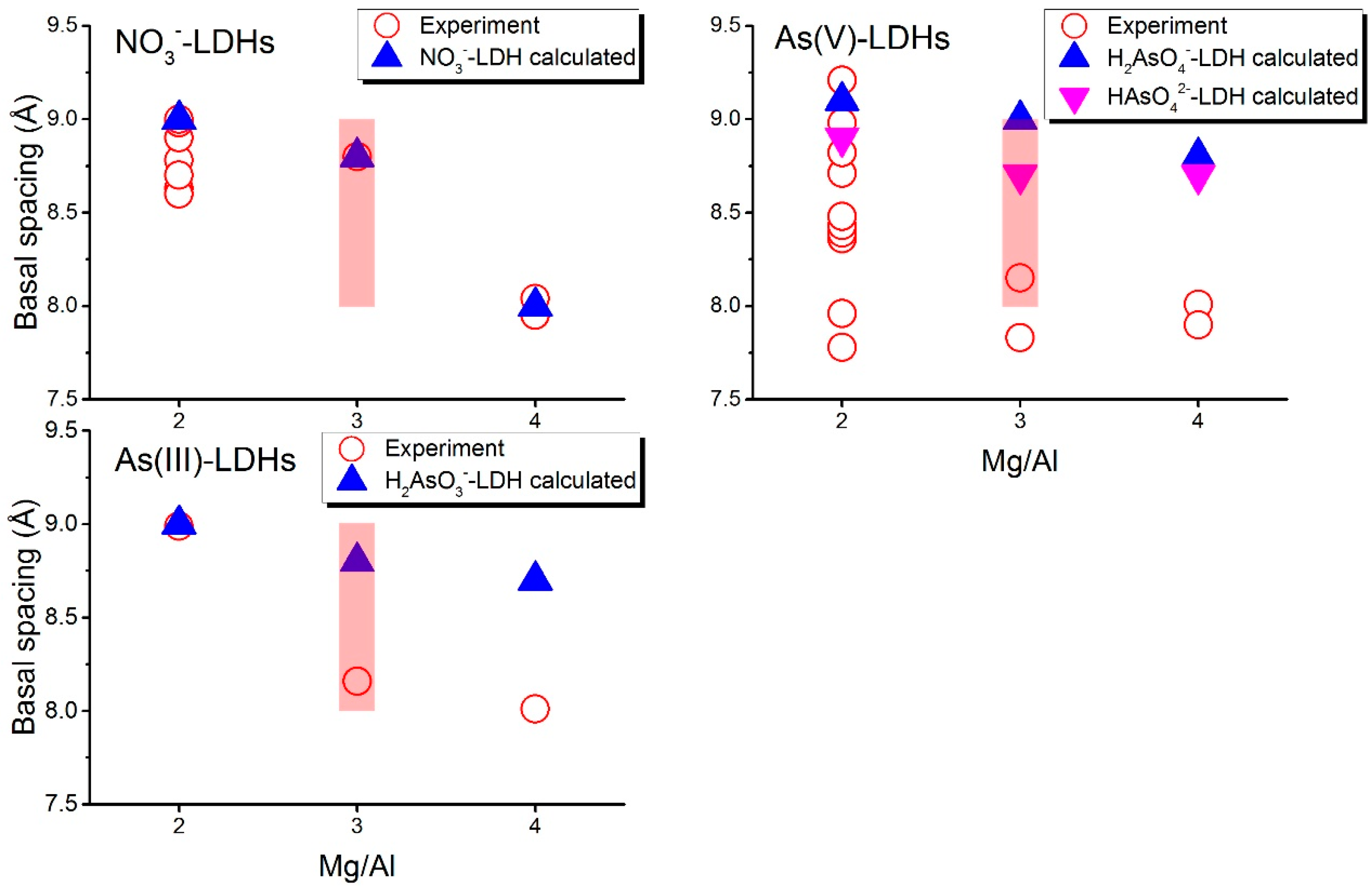
Figure 4.
Distributions of interlayer species of NO3−-LDHs at different layer charge densities. O in H2O and N in NO3− are selected to represent the corresponding molecules. (A–C) show the distributions of interlayer species along the z axis for NO3−-LDHs at Mg/Al ratios of 4:1, 3:1, and 2:1, respectively. (D–F) are the snapshots for NO3−-LDHs at 4:1, 3:1, and 2:1, respectively. Color profiles stay the same with Figure 1.
Figure 4.
Distributions of interlayer species of NO3−-LDHs at different layer charge densities. O in H2O and N in NO3− are selected to represent the corresponding molecules. (A–C) show the distributions of interlayer species along the z axis for NO3−-LDHs at Mg/Al ratios of 4:1, 3:1, and 2:1, respectively. (D–F) are the snapshots for NO3−-LDHs at 4:1, 3:1, and 2:1, respectively. Color profiles stay the same with Figure 1.

Figure 5.
Distributions of interlayer species in arsenic systems. O, N, and As are selected to represent H2O, NO3−, and arsenic oxy-anions, respectively. (A–C) exhibit the distributions of the interlayer species in H2AsO4−, HAsO42−, and H2AsO3−-LDHs and (D–F) present the snapshots for these system, respectively. Color profiles are the same as in Figure 1.
Figure 5.
Distributions of interlayer species in arsenic systems. O, N, and As are selected to represent H2O, NO3−, and arsenic oxy-anions, respectively. (A–C) exhibit the distributions of the interlayer species in H2AsO4−, HAsO42−, and H2AsO3−-LDHs and (D–F) present the snapshots for these system, respectively. Color profiles are the same as in Figure 1.
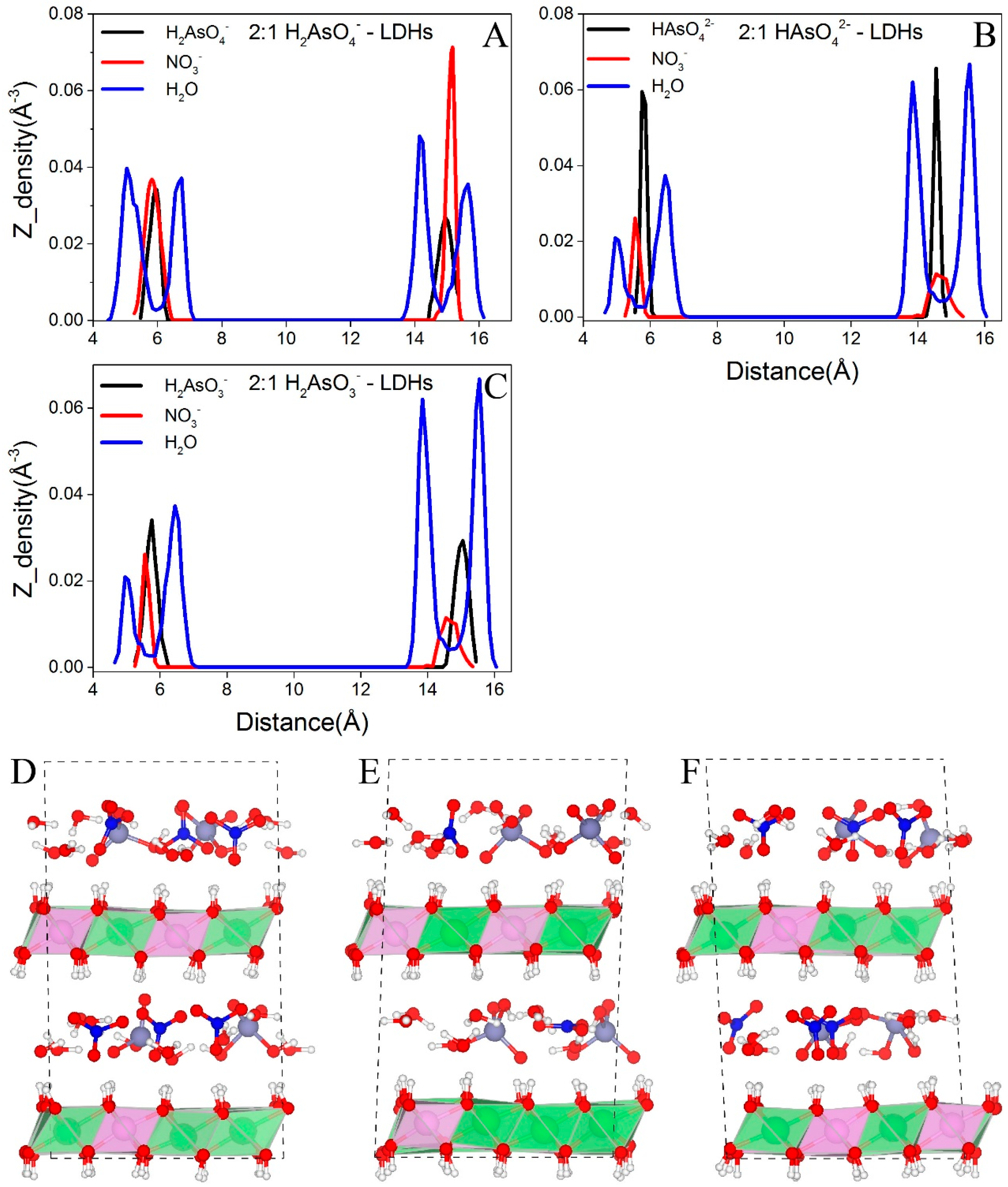
Figure 6.
Orientations of water and NO3− in NO3−-LDHs at different charge densities. (A,C) show the probability distributions of the angle between Z and the vector normal to the molecule plane while (a,b) correspond to Mg/Al ratios of 4:1 and 2:1, respectively. (B) shows the probability distributions of the angle between Z and the dipole moment of water. Color profiles stay the same with Figure 1.
Figure 6.
Orientations of water and NO3− in NO3−-LDHs at different charge densities. (A,C) show the probability distributions of the angle between Z and the vector normal to the molecule plane while (a,b) correspond to Mg/Al ratios of 4:1 and 2:1, respectively. (B) shows the probability distributions of the angle between Z and the dipole moment of water. Color profiles stay the same with Figure 1.
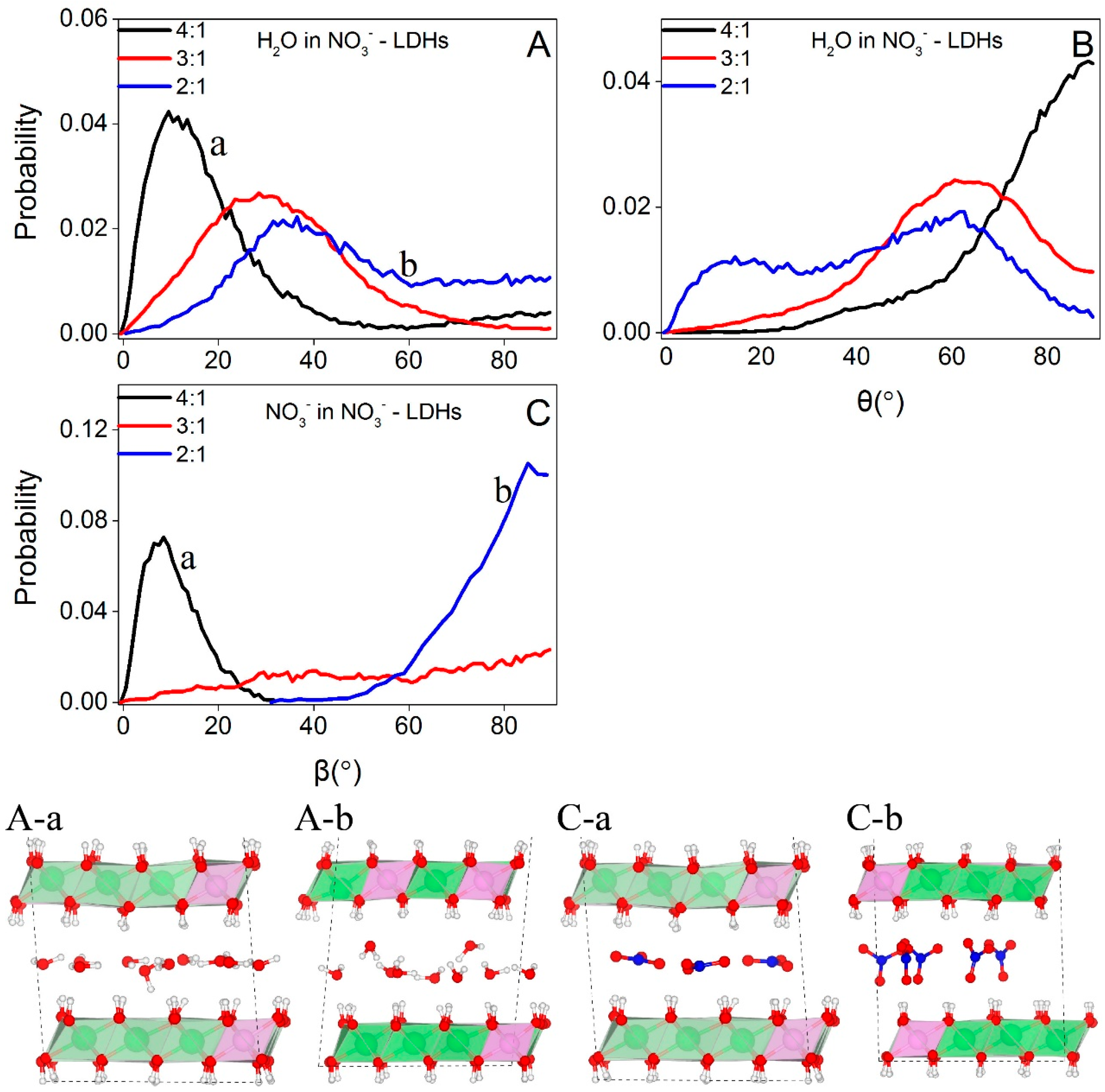
Figure 7.
Structures of arsenic oxy-anions in the gallery space. Water molecules and NO3− are removed for clarity. (A–C) show the interlayer structures of H2AsO4−, HAsO42−, and H2AsO3−-LDHs, respectively. (a,b) denote two orientations of the As(V) species. Color profiles stay the same as in Figure 1.
Figure 7.
Structures of arsenic oxy-anions in the gallery space. Water molecules and NO3− are removed for clarity. (A–C) show the interlayer structures of H2AsO4−, HAsO42−, and H2AsO3−-LDHs, respectively. (a,b) denote two orientations of the As(V) species. Color profiles stay the same as in Figure 1.
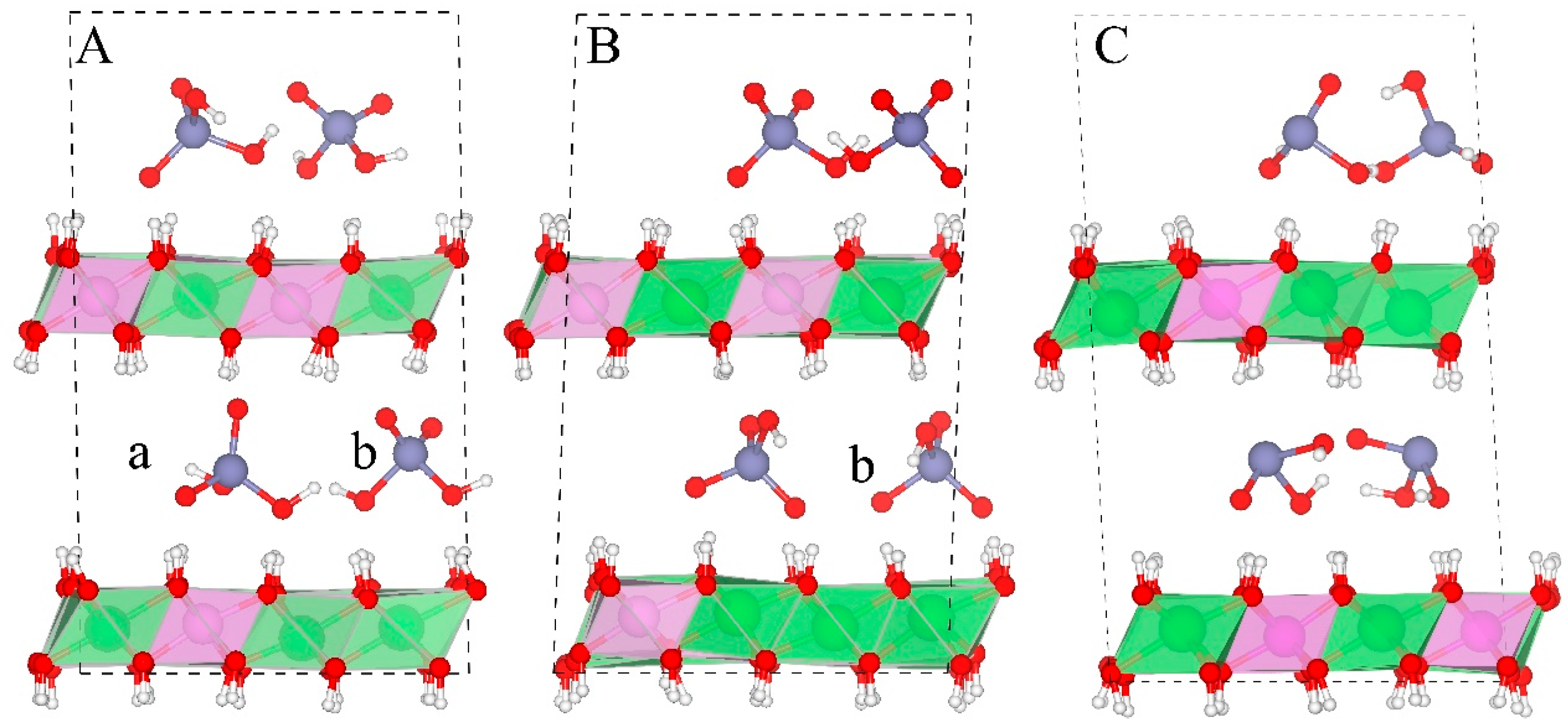
Figure 8.
Snapshots of hydrogen bonds formed between As species and other interlayer species and LDHs-OHs from octahedra surfaces. Only those water, NO3−, and LDHs-OHs are presented here for clarity. (A,B) depict the hydrogen bonding of H2AsO4− with different orientations as described in the main text. (C,D) show the hydrogen bonding of HAsO42− and H2AsO3−, respectively. The average distances between H and the acceptor are presented. Color profiles stay the same as in Figure 1.
Figure 8.
Snapshots of hydrogen bonds formed between As species and other interlayer species and LDHs-OHs from octahedra surfaces. Only those water, NO3−, and LDHs-OHs are presented here for clarity. (A,B) depict the hydrogen bonding of H2AsO4− with different orientations as described in the main text. (C,D) show the hydrogen bonding of HAsO42− and H2AsO3−, respectively. The average distances between H and the acceptor are presented. Color profiles stay the same as in Figure 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chemical compositions of modeled systems.
| Mg/Al | Systems | Formulae |
|---|---|---|
| 2:1 | H2AsO4− | Mg11Al5(OH)32(NO3)3(H2AsO4)2·(H2O)6 |
| HAsO42− | Mg11Al5(OH)32(NO3)(HAsO4)2·(H2O)8 | |
| H2AsO3− | Mg11Al5(OH)32(NO3)3(H2AsO3)2·(H2O)6 | |
| NO3− | Mg11Al5(OH)32(NO3)5·(H2O)8 | |
| 3:1 | H2AsO4− | Mg12Al4(OH)32(NO3)3(H2AsO4)·(H2O)7 |
| HAsO42− | Mg12Al4(OH)32(NO3)2(HAsO4)·(H2O)8 | |
| H2AsO3− | Mg12Al4(OH)32(NO3)3(H2AsO3)·(H2O)7 | |
| NO3− | Mg12Al4(OH)32(NO3)4·(H2O)8 | |
| 4:1 | H2AsO4− | Mg38Al10(OH)96(NO3)9(H2AsO4)·(H2O)25 |
| HAsO42− | Mg38Al10(OH)96(NO3)8(HAsO4)·(H2O)26 | |
| H2AsO3− | Mg38Al10(OH)96(NO3)9(H2AsO3)·(H2O)25 | |
| NO3− | Mg13Al3(OH)32(NO3)3·(H2O)9 |
Table 2.
Diffusion coefficients of interlayer species (in 1.0 × 10−13 m2/s).
| Mg/Al | H2O | NO3− | H2AsO4− | HAsO42− | H2AsO3− |
|---|---|---|---|---|---|
| 2:1 | 12.1 | 13.6 | 0.8 | 1.7 | 3.9 |
| 3:1 | 15.4 | 10.8 | - | - | - |
| 4:1 | 3.4 | 1.4 | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, Y.; Liu, X.; Zhang, C.; He, M.; Lu, X. Interlayer Structures and Dynamics of Arsenate and Arsenite Intercalated Layered Double Hydroxides: A First Principles Study. Minerals 2017, 7, 53. https://doi.org/10.3390/min7040053
AMA Style
Zhang Y, Liu X, Zhang C, He M, Lu X. Interlayer Structures and Dynamics of Arsenate and Arsenite Intercalated Layered Double Hydroxides: A First Principles Study. Minerals. 2017; 7(4):53. https://doi.org/10.3390/min7040053
Chicago/Turabian StyleZhang, Yingchun, Xiandong Liu, Chi Zhang, Mengjia He, and Xiancai Lu. 2017. "Interlayer Structures and Dynamics of Arsenate and Arsenite Intercalated Layered Double Hydroxides: A First Principles Study" Minerals 7, no. 4: 53. https://doi.org/10.3390/min7040053
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.