About the Genetic Mechanisms of Apatites: A Survey on the Methodological Approaches


Dipartimento di Scienze della Terra, Università degli Studi di Torino, Via Valperga Caluso 35, I-10125 Torino, Italy
*
Author to whom correspondence should be addressed.
Minerals 2017, 7(8), 139; https://doi.org/10.3390/min7080139
Submission received: 15 June 2017
/
Revised: 21 July 2017
/
Accepted: 1 August 2017
/
Published: 5 August 2017
(This article belongs to the Special Issue Nucleation of Minerals: Precursors, Intermediates and Their Use in Materials Chemistry)
Abstract
:Apatites are properly considered as a strategic material owing to the broad range of their practical uses, primarily biomedical but chemical, pharmaceutical, environmental and geological as well. The apatite group of minerals has been the subject of a huge number of papers, mainly devoted to the mass crystallization of nanosized hydroxyapatite (or carboapatite) as a scaffold for osteoinduction purposes. Many wet and dry methods of synthesis have been proposed. The products have been characterized using various techniques, from the transmission electron microscopy to many spectroscopic methods like IR and Raman. The experimental approach usually found in literature allows getting tailor made micro- and nano- crystals ready to be used in a wide variety of fields. Despite the wide interest in synthesis and characterization, little attention has been paid to the relationships between bulk structure and corresponding surfaces and to the role plaid by surfaces on the mechanisms involved during the early stages of growth of apatites. In order to improve the understanding of their structure and chemical variability, close attention will be focused on the structural complexity of hydroxyapatite (HAp), on the richness of its surfaces and their role in the interaction with the precursor phases, and in growth kinetics and morphology.
1. Introduction
As described in two excellent review papers [1,2] the term “Apatite” represents a group of minerals of a very wide interest. As a matter of fact, extraction of phosphor from apatite ores provides, to this day, fertilizers to feed the Earth population; apatites are used as well in geological dating techniques and studies of rare earth element variation in rocks. In the applications related to the Materials Science, apatite is the basic raw material used in the fluorescent lighting industry, while its unique crystal-chemical properties make it fundamental in the production of laser devices with controllable properties. The industrial process named PIMS (phosphate-induced-metal-stabilization) intervenes in the environmental remediation and the crystal structure properties of the used apatites favour the storage of radiactive waste as substituents in the Ca-sites. Concerning the research in the health field, it is worth outlining that two important members of apatites (Hydroxyapatite, HAp, and Carbonated apatite, CAp) represent both the inorganic pillar building hard tissues such as bones, theet, antlers of mammals and their pathological calcified tissues (i.e., those generated by various diseases). Having considered that apatites are studied since more that two centuries and according to their wide field of interest, it is not surprising that the total amount of currently available publications on the “Apatite” subject exceeds 40,000 with the annual increase for, at least, 2000 papers.
In this review, we do not claim to open a wide-range discussion on this subject, but we will confine our attention to the genetic mechanisms of apatite crystals, with a peculiar care for the relations between the bulk structure of the crystal and its corresponding face-by-face surface structures, in order to improve the knowledge of the interfaces that form when the crystal interacts with its growth medium. This way of methodological thinking represents the unavoidable consequence ofthe scale-change which affects the crystals when going from geological (cm, mm) to industrial (mm-μm) to biomaterial (μm-nm) size. In fact, surface kinetics becomes more and more important as much as crystal size decreases, and the features of the crystal surfaces dominate the kinetics of both nucleation and crystal growth in its early stages, since interfacial tension, adsorption and/or absorption, and epitaxy (if any) play a fundamental role in determining the anisotropy of the growth kinetics and then the final growth morphology [3,4,5].
Our reasoning will develop according to the following steps:
- (1)
- A sketch of the apatite bulk structure, drawn according the most realistic related symmetry space groups, focusing mainly on Hydroxyapatite (HAp) and, secondarily, on its structurally derivated, the Carbonated apatite (CAp).
- (2)
- A comparison between the stable surface profiles of the main crystallographic {hkl} forms, i.e., the hexagonal prism and pinacoids, and the monoclinic pinacoids.
- (3)
- The ambiguities arising in interpreting the growth morphology, when the hexagonal symmetry of apatite is assumed “a priori” and the twinning occurrence is not considered as a “habit modifier”.
- (4)
- The role of the epitaxy when another phosphate works as a precursor of the apatite (e.g., the case of the Monetite assisted growth of micrometric HAp crystals).
- (5)
- The growth of nanosized vs. micrometric HAp crystals.
2. Crystal-Chemistry and Bulk Structure of Apatites: Hydroxy-Apatite as a “Case Study”
According to Dorozhkin [1], apatites belong to the wide family of Calcium ortho-phosphates, characterized by a structure based on the tetrahedral ion . From the crystal-chemical point of view, all apatites are defined by a Ca/P molar ratio (Ca/P, mr) ranging between 1.5 and 1.67, as illustrated in Table 1. Ca/P molar ratio can slightly exceed 1.67, when ions enter the crystal lattice replacing and/or ions in CAp [6].
All other Ca orthophosphates have 0.5 < (Ca/P, mr) < 1.5, while the amorphous calcium phosphates, ACPs, [CaxHy(PO4)z·nH2O, n= 3–4.5; 15%–20% H2O] have 1.2 < (Ca/P, mr) < 2.2 and the Tetracalcium phosphate (TetCP), mineral hilgenstockite, Ca4(PO4)2O, has (Ca/P, mr) = 2.0. Roughly speaking, from Table 2 it follows that the symmetry of apatites schematically belongs either to the hexagonal or to monoclinic system, HAp belonging to both, according to the crystallization temperature and chemical purity of the sample. This is the reason why we will focus our attention on HAp polymorphism, which is not yet fully understood but represents a fruitful case study for equilibrium and growth morphology of apatites.
2.1. The Hexagonal Setting
(i) The P63/m setting. Itwas first assigned to a synthetic single crystal (by X-ray diffraction) without determining the position of H atoms [7]; later on, it was confirmed by neutron diffraction on a natural crystal containing ~0.3% of F replacing the OH groups [8]. Furthermore, this space group has been experimentally determined in crystal obtained by hydrothermal synthesis [9,10,11] and in natural samples [12] where the OH dipoles are partially replaced by simple cations such as F and Cl. As concerns the theoretical investigation on the surface properties, the P63/m setting has been preferentially adopted to predict the theoretical growth morphology [13], to determine the surface relaxation [14], to simulate the molecular adsorption on the most important HAp surfaces [15,16,17,18] and to evaluate the formation energies of Na and K replacing the Ca ions of HAp [19].
(ii) The P63 setting. The symmetry mirror m is not consistent with an ordered alignment of the OH dipoles, since the alternating O and H occupying special positions should be equidistant. The symmetry reduction P63/m→ P63 brings all OH dipoles with the same orientation in every structure channel (ferroelectric ordering); moreover, it is energetically favored and then has been systematically chosen, in the ab initio quantum-mechanical calculations, to simulate either the specific surface energies or the interaction of biomolecules with highly occurring {hkl} crystal forms [26,27,28].
2.2. The Monoclinic Setting
In the monoclinic HAp structure, the OH dipoles lie on the screw 21 axes which run along the structure channels. Owing to the action of the glide plane, the OH groups point upward and downward in alternate nearest neighbor channels (antiparallel orientation). The number of investigations about the monoclinic HAp polymorph is small compared with that on the hexagonal one. This is surely due to the difficulty of obtaining the pure monoclinic phase, the relative percentage between the two polymorphs varying from a synthesis run to another [29,30] and to the high nucleation frequency of monoclinic twins [31].
It has been theoretically shown that the monoclinic polymorph is energetically favored at low temperature [15,32,33,34,35], with respect to the hexagonal one, the reversible transition between them occurring at 480.5 K in heating and at 477.5 K in the cooling process.
In Figure 1 the structures assumed by HAp are drawn, projected along the direction of the channels. In Figure 1a the P63/m setting shows the perfect hexagonality of the [001] zone. In Figure 1b the setting P21/c outlines the alternating OH dipoles in contiguous channels. Even if the two structures look very similar each other (the transition enthalpy between them being quite low: 130 J/mol [36,37]), a deep difference arises when comparing the homologous faces in the two settings.
2.3. The Complexity of Carbonated Apatites
Special care has to be deserved to the carbonated apatites, CAp, since much discussion and controversy still exist about how carbonate ions are incorporated into the apatite lattice and hence how its symmetry is affected. Carbonate in bones reaches approximately 7 wt %, while in tooth enamel is less than 3.5 wt %. When carbonate ions enter the structure channels, in the OH sites, the A-type substitution is obtained, while B-type is generated when carbonate ions enter in the sites. As concerns the existence domains, it seems that A-type can be produced only through solid-state reactions (at 1000 °C), whereas the B-type can be found in biological apatite and obtained from solutions in the temperature range of 50–100 °C. Owing to the structural disorder intrinsic to the partial substitutions in both A- and B-types, and to the difficulty in preparing sufficient large crystals for an X-ray structural analysis, the precise configuration of the carbonate ions in the CAp structure has not been determined yet [6].
Thus, it should not be surprising if different space groups (belonging to the hexagonal or trigonal systems) have been hypothesized for CAp. As an example, three structural locations of type B were found for the carbonate ion in a complex, disordered type A-B carbonate apatite, with space group P63/m belonging to a crystal synthesized at 3 GPa and at 1400 °C [39].
Another CAp crystal, of type A, investigated by the same authors, was found domain-disordered having symmetry [40].
A third space group () was foundon a single crystal, grown by a CaCO3 flux method, having chemical formula Ca9.75[(PO4)5.5(CO3)0.5]CO3. In this crystal all the A-sites, corresponding to OH-sitesin HAp were substituted by CO3 ions [41].
In any case, suggestions are lacking to explain the growth mechanisms of CAp, i.e., to identify those crystal faces where the carbonated species present in the growth medium (as CO2 or , or ) preferentially react, in order to be absorbed in the growing crystal bulk. With the aim at giving a contribution to these aspects, in Section 5 we will describe how Ca-carbonates can affect both structure and morphology of growing CAp single crystals.
3. Solved and Unsolved Problems about the Relationship between Monoclinic and Hexagonal HAp Growth Behavior
The relationships between the “hypothesized” HAp hexagonal space groups (P63/m, P63, ) and the monoclinic one (P21/b) remains an open debate, to date. A non-exhaustive list of solved and unsolved problems are illustrated in the following:
(i) “HAp: hexagonal or monoclinic?” [42] and “Crystallographic structure of human tooth enamel by electron microscopy and X-ray diffraction: hexagonal or monoclinic?” [43] are representative titles of recent works concerning apatites growing in a bio-environment (bio-apatites).
(ii) Human single HAp nano-crystals (enamel and dentine) investigated by convergent beam electron- diffraction (CBED) and automated electron-diffraction tomography (ADT), recently showed [44] that P63 works instead of P63/m S.G. and that this experimental evidence is of prime importance for understanding the influence of electric fields on the morphogenesis process of calcified tissues at the nano-scale [45].
(iii) An intriguing question has been recently asked about the “symmetry violation” or “symmetry breaking” by Tao et al. [46]: “Why does apatite, which has a nominal hexagonal symmetry, form platelets with the c axis in the plane of the platelets, in violation of the underlying HAp crystallographic symmetry, and why do the platelets grow with their axes parallel to the fibril axis?”. A more precise problem [47] stands in line with this way of thinking: “Focusing on nano-HAp morphology, needlelike [48,49,50,51] and platelike [52,53,54] nano-particles can be prepared, while in bone tissue only the second type seems to be present [55,56,57]. For both morphologies the prevailing surface terminations are of the {010} type, which in the case of needlelike nano-HAp are the lateral facets of the hexagonal nano-particles elongated along the crystallographic c-axis, while, for the platelike nano-particles they are the basal facets of nano-particles preferentially grown along both the c- and a- (or b-) axis [53,54,58] breaking the crystal symmetry through a mechanism still matter of investigation [52,58].
(iv) Determining the outmost surface profiles of the most frequently observed {hkl} forms can be a risky attempt.
(a) From experiments: direct information from experiments are rare and doubtful. High resolution electron microscopy (HRTEM) was used to characterize the atomic structure of the “hexagonally shaped region” limited by the {01.0} faces [59]. The investigation on the crystalline-amorphous interfaces formed by electron-beam damage and on the grain boundaries parallel to the hexagonal prism, suggested that the HAp structure is terminated at the plane connecting the OH columns. However, the Authors “assumed” the crystal structure according to the hexagonal symmetry, on the ground that the differences between the monoclinic and the hexagonal phase are negligible in this kind of study. We will see, later on, that the deep differences between the surface profiles of hexagonal an monoclinic phases cannot be neglected, if one aims at understanding the complexity of growth kinetics (→growth morphology) in pure growth medium and/or in the presence of habit modifiers. More recently, Ospina et al. [60] used the HRTEM technique to analyze the surface profiles of HAp nanoparticles (supposed to be structurally hexagonal…) precipitated from aqueous solution at 37 °C with preferential growth along the [001] direction and large (01.0) faces. The simulated nanocrystal was used to investigate the two different (01.0) faces observed in a single nanoparticle of 50 nm length and 8.49 nm width. This analysis permitted the characterization of the non-stoichiometric HAp surface terminations [59,61,62]. Starting from the HRTEM results, periodic density functional theory (DFT) was used to model and refine hydrated (01.0) HAp surfaces with the two different terminations in order to obtain information about the structural modification and chemical environment around the Ca-sites.
(b) From a theoretical point of view:
- The first paper in which the surface profiles of HAp were systematically examined and growth morphology was predicted and compared with the observed one, appeared on 1986. Terpstra et al. [13] applied the “connected net” method, based on the morphological theory by Hartman-Perdok [63] to the hexagonal (P63/m) HAp structure. Starting from a qualitative evaluation of the Ca–PO4 and Ca–OH bonds, and assuming that the ordering of the OH dipoles has any influence on the Ca–OH bonds, the Authors argued that one can expect that “…monoclinic HAp should have the same morphological appearance of the hexagonal HAp”; accordingly, “…the lowering of the apatite symmetry (P63/m → P21/b) due to the ordering of OH is not likely to be the cause of the occurrence of the plate-like apatitic crystals in calcified tissues”.
- Surface relaxation on two opposite (100) free surfaces of a P63/m HAp crystal, was invoked to explain the “… symmetry breaking effect on growth morphologies, producing equilibrium platelet morphologies even when these are inconsistent with the symmetry of the crystal” [14,64]. However, the Authors are conscious that platelet growth morphology does not occur when HAp is precipitated in vitro. Hence they speculated that “…it may be possible that in vivo growth of apatite the body may manipulate the growth conditions (carbonate impurities, organic molecules found in bones) so as to allow the platelet growth mechanism to work”.
- An interesting attempt was performed by De Leeuw and Rabone [17], who used molecular dynamics simulations to evaluate the interaction of citric acid with the hexagonal (P63/m) prismatic (01.0) and pinacoidal (00.1) surfaces in an aqueous environment. The prismatic surfaces were assumed as terminated by PO4 groups (nothing being revealed about the termination of the pinacoid). Surface energies were calculated for dehydrated (in vacuum) surfaces, followed by those calculated in aqueous environment and, finally, with citric acid in an aqueous environment. The large difference in the adsorption energies for these two kind of surfaces indicates that “…citric acid would be a much better growth inhibitor of the {01.0} prismatic form, slowing down its growth rate by binding to surface growth sites”. As a consequence, the crystal shape of HAp grown in the presence of citric acid would become along [001], the direction of the OH channels, with expression of the prismatic form, as compared to the crystal morphology in the absence of citric acid. It is worth noting that this work, published on 2007, allowed outlining, for the first time, the influence of the different surface structure of two important HAp forms, even if the molecular adsorption is assumed within the “large constraints” of the Molecular Dynamic Simulation.
- Astala and Stott [61] carried out a first-principles study about water adsorption on the {01.0}, {00.1} and {10.1} forms of hexagonal (P63/m) HAp. In particular, they determined the surface energy in vacuum of the pinacoid {00.1} having considered the approach to modeling the surface profiles proposed by Rulis et al. [62]. In this model, the sole constraint imposed was that “… surface must be constructed so that the corresponding slabs were charge neutral”. Moreover, they considered that, in HAp, the orientation of the OH group along the [001] channels (c axis) reduces the symmetry such that the top and bottom of the 001 slab surfaces are no longer symmetric. Accordingly, these Authors introduced the term “symmetry breaking” limited to the form {00.1}. As concerns the other important form, the hexagonal {01.0} prism, the problem of the “symmetry breaking” does not longer exist since the OH dipoles lie parallel to the {01.0} surfaces. However, this form can terminate with three different profiles, according to the geometric 01.0 cut applied to the bulk of the crystal. One of them corresponds to a stoichiometric slab (Ca/P ratio being 1.67); the two out of three are non-stoichiometric: a Ca-rich slab (Ca/P ratio = 1.75) and a PO4 rich slab (Ca/P ratio = 1.5). This is surely interesting, but something was wrong, not only from a crystallographic point of view but also because the growth mechanism of the flat faces (F-faces, in the sense of Hartman-Perdok [63]) was not respected. In fact, the space group (P63/m) implies that the slices of thickness d002 and d010 should be centrosymmetric and hence their total dipole moment should vanish (owing to the electrical stability of the slice). Hence, the symmetry breaking attributed to the steps growing on both sides of the {00.1} form is not self-consistent.
- A sensible step forward was taken by the Ugliengo et al. [19], who carried out a periodic B3LYP study of hexagonal P63 HAp (00.1) surface modelled by thin layer slabs [19]. The (00.1) surface coming out from P63 group is intrinsically polar, owing to the OH dipoles, all iso-oriented perpendicularly to the 00.1 planes. Nevertheless, the convergence of , the specific surface energy value for the hexagonal (00.1) form, has been ascertained for a slab thickness varying from a minimum of one layer (~7Å) to a maximum of 9 layers (~60Å). For the homologous monoclinic (non-polar) P21/b HAp (00.1) surface the convergence is, obviously, more rapidly obtained. For the sake of comparison, the two asymptotic values are: = 1107 and = 1337 erg cm−2. In a successive paper [26], it has been explained why the right choice of the P63 group has been made: as a matter of fact, “… the quantum-mechanical simulation of the hexagonal HAp cannot be performed within P63/m space group because of the unphysical duplication of each OH group by the m mirror plane”. In the same paper, the surface profiles of hexagonal prismatic {01.0} and pyramidal {10.1} forms were investigated as well. On the ground of Astala and Stott [61] and according to the Authors way of thinking, the {01.0} non-polar surfaces can be imagined as a stacking of electro-neutral layers …-ABA-ABA-…where the A-type has the composition Ca3(PO4)2 while the B-type corresponds to the composition Ca4(PO4)2(OH)2. The specific surface energy related to these “stoichiometric” surfaces is = 1709 erg cm−2. On the ground of recent findings [59,61], two other “non-stoichiometric” {01.0} surfaces were also considered: [B-AA-B-AA-B], Ca-rich, being Ca/P = 1.71 and [AA-B-AA-B-AA], P-rich, being Ca/P = 1.62. Regrettably, the corresponding values were not calculated, since thesurface energy cannot be evaluated using the standard formula, adopted by the Authors, that is valid for stoichiometric slab only.
Notwithstanding, all these surfaces (both stoichiometric and non-stoichiometric) were usefully adopted to calculate the interaction energy of adsorbed single molecules such as: water, glycine, glutamic acid, lysine and carbon oxide [26,27].
- It is worth recollecting as well a remarkable paper recently publishedby Putnis et al. [65] about the growth kinetics measured by AFM on a (010) prismatic face of a synthetic HAp crystal growing from aqueous solution [65]. It was observed that HAp crystallization occurred by either classical spiral growth or non-classical particle-attachment from various supersaturated solutions at near-physiological conditions, suggesting these mechanisms do not need to be mutually exclusive. Moreover, this work represents “… the first evidence of time-resolved morphology evolution during precursor-particle attachment processes, ranging from primary spheroidal particles of different sizes to triangular and hexagonal solids formed by kinetically accessible organized assembly and aggregation”.
When summarizing, the critical points illustrated in the present Section show that the largest majority of the researches has been done on the hexagonal HAp polymorphs, especially those concerning the interfaces crystal/growth medium (both in vacuum and in the presence of a condensed mother phase, with and without specific impurities). Nevertheless, any satisfactory explanation has not been done, till now, about the “symmetry breaking”. In other words, if the space groups is P63/m or P63, how can a hexagonal prism (built by six equivalent faces) degenerate in a set of six faces where two out of them dominate the growth morphology, so determining the final appearance of platelets [001] elongated, instead of the expected hexagonal rods?
A few simple questions arise. Are we sure that HAp crystals are always hexagonal, especially when nucleated at low temperatures from aqueous solution? Moreover, what should be the action of the very frequent twinning on the final morphology of the monoclinic polymorph?
According to our experience about adsorption phenomena in crystal growth, selective adsorption/absorption occurs only when the adsorbing surfaces are crystallographically different [66,67]: hence, something should be wrong when neglecting that monoclinic HAp structure could be “necessarily” more rich than the hexagonal one, at least for the variety of the surface profiles exposed to the mother phase.
In the following Section we will deal with the method of investigation we propose to outline the marked difference between the surfaces generated by monoclinic and hexagonal HAp polymorphs, along with the related consequences on their equilibrium and growth morphologies. To do this, it would be more fruitful to begin with the more complex monoclinic phase.
4. Approaching the Morphology of the Monoclinic HAp through the Hartman-Perdok Method
Recently [38], we determined the ab initio theoretical equilibrium shape (ES) of the monoclinic HAp, in vacuum and at 0K; neither vibrational nor configurational entropy were taken into account. Stable surface profiles of the different {hkl} forms were obtained by applying the constraints intrinsic to the Hartman-Perdok method [63] (HP-method, hereinafter). To do this: (i) the character (flat-F, stepped-S, kinked-K) of the {hkl} forms was identified, through the search of the most important periodic bond chains (PBC) building up the crystal structure; (ii) the corresponding crystal slices of thickness dhkl were considered, respecting the space group constraints; (iii) all the possible surface profiles related to every dhkl slice were drawn, respecting the conditions of both electro-neutrality and vanishing of the dipole moment component perpendicular to the hkl plane; (iv) the specific surface energy (γhkl) was calculated, for each hkl surface profile, to draw the ES of the single crystal, by applying the selective rules required by the Gibbs-Wulff’s theorem [3].
It is worth outlining as well that the steps (i) → (iv) were needed to investigate both crystallography and genetic aspects of the HAp twins, to find their original composition planes (OCP) and to evaluate the energy values associated to their formation [68]. Moreover, steps (i) → (iii) were also needed as the preliminary requirement to interpret the epitaxial interactions between HAp and monetite (CaHPO4), used as a precursor phase favoring the 2D-heterogeneous nucleation that allows 3D HAp to appear beyond its usual growth conditions [69].
Finally, we really must spend a few words in favor of applying the HP method: it is somewhat laborious, since it cannot be successfully automated; nevertheless, its results obtained for a long time are reassuring, especially when dealing with low symmetry and complex crystal structures.
Computational details are described in reference [38]. The parameters (in Å) of the adopted optimized cell were: a0 = 9.3253; b0 = 6.9503; c0 = 18.6436; β = 119.972°, referred to the space group P21/c and to 4 unit formulas [i.e., Ca20(OH)4(PO4)12]. The setting P21/c has been preferred to P21/b because the diad traditionally coincides with the y-axis, in the monoclinic system.
4.1. The Surface Profiles of the Faces in the [010] Zone, i.e., Parallel to the HAp Channels
According to the HP analysis, three flat forms can be found in the [010] zone: {100}, {} and {001}, related to the thicknesses , and , respectively. The thickness of these slices fulfills the systematic extinction rules coming out from the space group P21/c. In Figure 2 the monoclinic HAp structure is projected along the [010] direction and the surface profiles of the three forms are drawn, as comes out from choosing one of the possible [010] PBC, i.e., the PBC [010]A. At first sight, one could say that the surface profiles of the three forms are practically indistinguishable: then, one could expect that both their surface and attachment energies should also have similar values. Consequently, it is not surprising that the ensemble built by the three monoclinic pinacoids {100}, {} and {001} might be easily confused with the “really hexagonal” prism related to the hexagonal HAp polymorph. However, from a deeper examination of Figure 2 it follows that:
- Neither PO4 tetrahedra nor Ca ions lie on the ideal separation surface between two adjacent slices and hence both ions are not shared between adjacent slices. Further, the , and slices do not show dipole moments perpendicular to their surfaces. Accordingly, the surface profiles of the three pinacoids {100}, {} and {001} do not have to be reconstructed.
- The OH ions in the channels are screened from the mother phase by the outmost layers of each slice populated by PO4 and Ca ions not directly bound to the OH in the channels.
- The and slices are center-symmetric, while the slice contains only the 21 screw axes parallel to the [010] channels: this implies that a growing HAp monoclinic crystal shows two types of slices, according to whether the OH dipoles lying in the middle of the slice are oriented along the positive or negative sense of the y axis. On the contrary, and slices do not show polarity parallel to the slice itself, which could unavoidably affect the difference of both surface and attachment energies of {100} and {} with respect to the {001} form.
There are two other ways of building a PBC running along the channel axis [010]: they are both associated to a general PBC [010]B, one being center-symmetric while the other one runs along the axis 21 // [010]. Both these PBCs do not contain the OH dipoles in their centers, at variance with the [010]A PBC. Here we will confine our attention to the effect of this new choice on the terminations of the monoclinic HAp surfaces; for deeper details the reader is kindly invited to refer to the original paper [38]. Concerning the new alternative terminations:
- the outmost layers of the new surfaces are populated not only by the PO4 tetrahedra, but also by Ca ions and OH dipoles;
- the occupancies of the outmost layers must be reduced by 50%, as follows from the constraints (symmetry, charge, stoichiometry) imposed by the frontiers between adjacent , and slices. In fact, these frontiers pass through the centers of mass of PO4, Ca and OH ions.
Summing up, this is the way we have got over the problem of the surfaces exposed towards the vacuum: a few simple physical rules, expressed by HP method, were able to generate a lot of different (and self-consistent) surface profiles for each {hkl} form.
4.2. The Specific Surface Energies of the Monoclinic HAp and Its Equilibrium Shape (ES) Calculated at 0 K
In Table 3 both structural and energetic features of the {001} pinacoid are described. One can see that HP method allowed evaluation of the dispersion of the surface energy (γ001) values. Nine {001} surface profiles can be obtained: one out of them is terminated by the PO4 tetrahedra (see Figure 2a) and does not need to be reconstructed, while other eight different configurations of the outmost layer can be obtained when surface reconstruction is needed, owing to the structure of the two [010]B PBCs. In the latter case, the γ001 values are distributed in two sets, one half of them being lower and the other one higher than their mean value, 1692 erg cm−2, which is only 1.1% lower than the one corresponding to the unique profile obtained from the PBC [010]A.
The just mentioned investigation procedure was applied to the two other pinacoids {} and {100}, to the basal {010} pinacoid and to the three monoclinic prisms {012}, {110} and {} (see Figure 3b, for indexing). It followed that:
- The surfaces of the three pinacoids {001}, {} and {100}, parallel to the OH channels, can be terminated either by only PO4 or by the complete set composed by PO4, Ca and OH ions.
- The basal {010} pinacoid, which is perpendicular to the OH channels, can be terminated either by only Ca or by the set made by PO4, Ca and OH ions.
- The three monoclinic prisms {012}, {110} and {} can terminate either by Ca ions (as for the {012} prism) or by PO4 ions (as for the {110} and {} prisms). It follows that the terms like “Ca rich” and “P rich” along with the terms like “stoichiometric” and “non-stoichiometric” lose meaning. On the contrary, “reconstructed, or non-reconstructed, outmost layer”, “polar, or non-polar, dhkl slice”, “relative disposition of ions in a given reconstruction”, assume a precise crystallographic meaning, unambiguous in order to describe both equilibrium and growth crystal properties.
- The lowest values of the calculated specific surface energies, for every {hkl} form, was used to draw the ES of HAp at 0K. As shown in Figure 7 of reference [38], the monoclinic ES can be described by a pseudo-hexagonal “prism” truncated by the {010} pinacoid, which reaches the lowest γ value of the entire crystal when its surface is Ca-terminated ( = 1041 erg cm−2). The pseudo-hexagonality of the “prism” arises from the closeness of the lowest γ values of the three pinacoids: = 1546 erg cm−2; = 1525 erg cm−2; = 1515 erg cm−2, all these values being obtained when the outmost layers of these forms are reconstructed and exhibit an outmost population made by PO4, Ca and OH ions. The ES is completed by the presence of the small–sized quasi-equivalent prisms {012}, {110} and {}.
Finally, it is worth stressing once again that the {001} form is the most anisotropic in the [010] zone, since it is the only one (at variance with the {} and {100} forms) in which the OH dipoles alternate up/down within the successive d002 slices, irrespective of the termination of its outmost layer. This marked difference does not play an appreciable role at equilibrium (in the vacuum), as we just demonstrated. A different situation has to be envisaged when dealing with both foreign adsorption and growth kinetics, as we will discuss later on.
Summing up, we can assess that distinguishing the athermal ES of the monoclinic HAp from that of the hexagonal one should be quite difficult.
4.3. About the Growth Morphology of the Monoclinic HAp: A Comparison with the Hexagonal One
In Figure 3a we consider the shape of a micrometric HAp crystal we grew from pure aqueous solution under mild hydrothermal conditions. The sketch in Figure 3b represents a reasonable attempt at indexing its crystallographic forms: the pseudo-hexagonality could be misleading, but the different development of the {001} and {100} forms largely plays in favor of the monoclinic 2/m symmetry. A comparison with the theoretical shape of a crystal showing a 6/m symmetry is made in Figure 3c: all the six faces belonging to the hexagonal {01.0} prism must have the same extension, along with the hexagonal {10.2} bi-pyramid. This is to say that growth kinetics does not allow confusing the two HAp symmetry systems.
Let’s consider, as an example, a face (001) belonging to the form {001} and a contiguous face (100), belonging to the form {100}, as drawn in Figure 3a,b. It is theoretically known that they are flat-F faces and then can advance either by 2D-nucleation and/or by spiral growth; moreover, this kind of face kinetics was experimentally observed in the Putnis research team [65]. As a consequence, their growth rate will depend (a part the temperature, the supersaturation and the composition of the growth medium), from the structure of the steps running on their flat surfaces, because the thermodynamic parameter ruling layer-by-layer growth (2D or spiral) is the specific edge energy of the steps limiting either the 2D nuclei, or the spiral pattern, or both.
Concerning the form {001}, one can reasonably imagine that the shape of a 2D nucleus (and then of a spiral) developing on this face is a rectangle limited by the following kind of steps: two, different each other, parallel to the [010] direction and two, equivalent, parallel to the [100] axis. The surface structure of one out of the [010] steps is that of the {100} forms, while that of the other one is comes out from the {} form; the surface structure of the two symmetry equivalent steps parallel to the [100] axis originates from the {012} form. Remembering that the surface structure of the {100} and {} forms are practically the same, the 2D nuclei developing on the {001} form must have a quasi- mm symmetry.
This is no longer valid for nuclei (and/or spirals) which develop on the two {100} and {} forms adjacent to {001}. In fact, the steps running along the [010] direction borrow the {001} and {} structure, on the {100} form and the {001} and {100} structure, on the {} form, respectively. Summing up, the two pinacoids {100} and {} could have a very similar growth rate (due to the closeness of their surface patterns), while the pinacoid {001} could experience a different kinetic behavior.
As a consequence, the HAp growth shape (GS) viewed along the direction of the OH channels should be anisotropic with respect to the quasi-hexagonal ES: this means that, at constant temperature of crystallization and composition of the growth medium, the anisotropic aspect ratio will change with the supersaturation.
It is well known that F faces, crossed by screw dislocations outcrops, are characterized by a complex growth rate (RF/kT) dependence on the supersaturation (∆μ). In fact, the growth isotherms [(RF/kT) vs.∆μ] show: (i) a parabolic law, at low ∆μ values; (ii) an exponential law when the 2D nucleation occurs in between the spiral steps, at medium-high supersaturation; (iii) and finally a linear law at higher ∆μ values. Let’s call and the isotherms related to the form {001} and to the two other pinacoids {100} and {}; owing to the reasons just exposed, these curves cannot have the same trend with respect to the supersaturation and then can cross at a given critical supersaturation ∆μcr. This implies that they sharply differ at high ∆μ values, where , as we will show in a forthcoming paper: in this case the Growth Shape I can be obtained (Figure 4, center). In the supersaturation domains where ∆μ approaches ∆μcr, , the aspect ratio tends to the Growth shape II (Figure 4, right side). Finally, when the growth system tends to the equilibrium (∆μ →0), the crystal assumes a pseudo-hexagonal shape (Figure 4, left side).
Applying the same reasoning, we already demonstrated that both HAP rods and/or platelets should be strongly elongated along the direction of the OH channels [38].
A preliminary conclusion can be drawn: the formation of HAp platelets can be explained by the cooperation of two factors: (i) the asymmetrical behavior of the growth steps running on the form {001} with respect to those developing on the {100} and {} couple; (ii) the role exerted by the supersaturation of the growth system. Hence, “symmetry breaking” cannot be invoked, because the platelets are originated as a monoclinic HAp phase which enhances its asymmetric properties during growth, contrarily to the hexagonal one which could not change its aspect ratio with supersaturation.
5. The Growth of Nanosized vs. Micrometric HAp Crystals, in the Frame of the Experimental Methods
As briefly mentioned in the Introduction, the research about the structure and properties of apatites was strongly sector-based since the early studies. In 1951, referring to the crowd of scientists interested in the field, Jaffe [70] wrote: “Although all these people have had the same problems, they have not always been sufficiently acquainted with each other’s work”. Then, she stated: “Each group of scientists concerned with the apatite minerals has its own problem which it has attempted to solve in its own way” denouncing the failing communication inside the scientific community. As in many other fields of sciences, these statements are still as relevant today.
We are firmly convinced that biomedical and geo-mineralogical research, even if their experimental method and goals are different, could master as a common language both crystal growth and structural crystallography. The intrinsic complementarity of these disciplines could represent the most efficient and rapid way to overcome the remaining unsolved problems between structure and growth of apatite, both in nature and laboratory.
As already mentioned, the discussion about the symmetry of HAp involved a large number of papers, the most extensively studied being the hexagonal polymorph. Only a few papers deal with the monoclinic form, relegating its appearance to the biologically precipitated HAp [42] or the cases of high-temperature crystallization of HAp [20,29,30,31].
The continuous research of a rigorous routine of synthesis of apatites (along with all the possible chemical and structural variations) is the topic of a huge number of multidisciplinary papers published since the early years of the XX century. The interest in the synthesis of apatites is mainly related to the importance of the mineral in biomineralization and its biocompatibility.
During the years, many solid-state reactions, melt growth, wet chemical methods and hydrothermal methods have been proposed in order to obtain reproducible results in apatite crystallization. In most cases, authors obtained the mass crystallization of nano-sized and often poorly crystalline HAp. Then, a wide range of techniques was applied to answer the queries arising from the natural variability of composition and structure of HAp crystals. A thorough collection of the scientific production prior to 1951 was reported by Jaffe [70].
Only a few efforts resulted in the hydrothermal growth of large HAp crystals for diffraction and morphology investigations. In 1956, Perloff and Posner [71] hydrolized monetite (CaHPO4) working at low hydrothermal conditions (300 °C, water vapor pressure evaluated of 87 bar for 10 days). They modified a previous work by Schleede and coworkers [72] and obtained large HAp crystals about 300 µm long. In 1966, Jullman and Mosebach [73] obtained large hydroxyapatite crystals (2.35 mm long and 0.2 mm large) by direct precipitation from a solution of calcium nitrate, sodium hydroxyde, and potassium dihydrogen phosphate at 500 °C. In 1968, Kirn and Leidheiser [74] suggested a modification to the Perloff’s method, hydrolyzing monetite at 350 °C and about 600 bar in a diluted phosphoric acid solution. The final pH ranged around 2.5, and it was found that, at this point, the growth reached its end-point.
In their paper, Schleede and coworkers [72] did not describe the size and crystal quality of HAp crystals obtained hydrolizing brushite (CaHPO4·2H2O) under hydrothermal conditions but, recently, Ma and Liu [42] modified the original method by Schleede and demonstrated that monoclinic HAp could be obtained at low temperature without recourse to specific impurities.
5.1. The Role of the Epitaxy on the Growth of HAp from a Phosphate Precursor and the Control of Nucleation Frequency and Growth Rate by Supersaturation
Very recently [69], we described a procedure of growth of large HAp crystals with controlled size under mild hydrothermal conditions (210 °C and water vapor autogenic pressure evaluated of 20 bar). In this work, we followed the path suggested by Perloff and Kirn. As described in our paper, we obtained large, needle-like HAp crystals by the hydrolysis of monetite (CaHPO4) in water and in diluted phosphoric acid.
Free calcium and phosphate ions, conductivity and pH were monitored during the reaction. We observed large and well-finished HAp crystals generated working at pH lower or near the lower limit of the stability field of the HAp (calculated at pH ≈ 4.5). Thus, the growth of HAp crystals cannot be explainedin terms of 3D homogeneous nucleation. We demonstrated both experimentally and theoretically that monetite acts as a heterogeneous substrate for the nucleation of HAp, the two phases showing excellent epitaxial agreement [69]. This finding allows us to stress that, in the presence of a suitable substrate, the heterogeneous nucleation of HAp on monetite can occur, even if the system is unsaturated with respect to the nucleating phase.
On the other hand, if brushite substitutes monetite as a precursor of HAp at the same T, p conditions, the mass crystallization of nanosized HAp occurs, as described by many authors, although in the case of brushite excellent epitaxial relationships with HAp are fulfilled. In a forthcoming paper [75], we will demonstrate that the interplay between excellent epitaxial relationships and supersaturation rules the quality of the crystals obtained. The rate of reaction of brushite in water results to be markedly higher with respect to that of monetite, leading to a sudden increase in the supersaturation of the solution with respect to HAp and therefore, to a rise in the nucleation frequency.
In fact, the sudden formation of a huge number of nuclei is, of course, related to the supersaturation reached during the procedure. In turn, the supersaturation value is related to the concentration of ionic species into the reactor and to the pH that could be varied in order to move the system nearly out of stability and, accordingly, to lower the related supersaturation. For that reason, at the end of our paper [69] we stressed the importance of the control over the pH value to improve both bulk and surface crystal quality. High supersaturation values lead to mass crystallization of small nuclei owing to the relationship between the supersaturation and the nucleation frequency of the phase. In light of that, the issue of the determination of trustworthy values of solubility becomes crucial to guarantee the effective control over the operational supersaturation. It is worth recollecting here that we adopted, as the thermodynamic supersaturation, the difference between the chemical potential of a growth unit in the mother phase and in the crystal: ∆μ = (kT) ln β, where β = (1+ σv) = [(a−aeq)/aeq] represents the ratio between the activities in the volume of the mother phase and at equilibrium. Concerning J3D (number of 3D nuclei per unit volume and time), i.e., thenucleation frequency of a 3D phase, we remember that J3D ≈ exp (−∆G3D/(kT)) where ∆G3D indicates the activation energy for 3D nucleation: ∆G3D = (f × Ω2 × γ3)/(∆μ)2. Here, f is a factor form depending on the shape of the nucleus, Ω is the volume of the growth unit in the crystal and γ is the averaged value of the specific surface energy of the crystallizing nucleus.
5.2. The Issue of the Solubility and Its Consequences on the Nucleation Frequency
The need for a reassessment of the solubility of calcium phosphates has been stressed by many authors [71] and we already pointed out [69] the attention to the lack of reliable data on the solubility of HAp from the literature. These data are of primary importance when growing crystals because of the relationship between the solubility of a solid phase, the supersaturation of the solution and, accordingly, the nucleation frequency. Many papers dealing with the solubility of HAp have been published during the years, being the HAp stability of crucial importance mainly for biomedical applications. The values of the solubility product vary over a range of 1011, according to Van Wazer [76] and many other authors [77,78,79,80]. A unique value of solubility is controversial. In 1953 and successively in 1955, Neuman [81,82] and his collaborators declared the principle of solubility product non-enforceable in the case of HAp, due to the chemical variability of the solid calcium phosphate system. Other reports claim the system does not obey to the principle of the solubility product constant based on the stoichiometry of the salt. Some models entail the formation of surface complexes [83] or dependence from the pH of the surface stoichiometry [84,85] and surface reactions [86,87,88]. Following these models, the solid/solution equilibrium is achieved only by the external layers of the crystalline HAp, having a slightly different composition with respect to the bulk. This composition varies along with the chemical composition of the solution and all the possible chemical reactions at the surface. On the contrary, according to some authors, the effect of the surface on the HAp solubility has been disregarded [89].
Moreover, the preparation of pure HAp for solubility measurements is often not free from contamination. For example, small amounts of carbonate in solution modify a lot the value of the solubility product according to the papers by Greenwald [90,91,92]. Additionally, the precipitation of acid byproducts like OCP or Ca8H2(PO4)5·5H2O is a recurring event, and their presence will modify the solubility product value.
Furthermore, the matter of the incongruent dissolution of calcium phosphates is widely debated by many authors [86,87,88,93]. In this case, the chemical composition of the solid and the solute may differ, leading to a misinterpretation of the solubility measurements. On the other hand, some reports claimed the existence of a single solubility product, as Clark did in his papers. In fact, in 1955, Clark and Peech [94] presented a solubility diagram for HAp in soils. At the same time, Clark [77] proposed a further paper, demonstrating that, within the variability due to analytical errors, HAp possesses a unique solubility product over a wide range of experimental conditions.
Besides the issue of the solubility, also the application ofthe right growth conditions knowing the stability field of HAp plays a major role in HAp synthesis. Many papers were dedicated to the subject. In 1974, Skinner [95] determined the equilibrium phase diagrams for the system CaO-P2O5-H2O growing HAp at temperature ranging from 300 to 600 °C in hydrothermal conditions. In 1997, Andrade and coworkers [96,97] come through an extensive thermodynamic analysis of the system Ca-P-H2O. They developed the Eh-pH and PaCa-pH diagrams varying the temperature and the activities of calcium and phosphorous in the system.
As one can infer from the charts, the HAp dominates at pH > 4 at any temperature, with a strong dependence on the activity of calcium in solution and on the Eh.
Summarizing, when referring to the issue of solubility, the traditional methods of solubility determination by the addition of large amounts of solid do not work properly in the calcium phosphate system. This may be related to many problems: (i) the incongruent dissolution of Ca-phosphates in water; (ii) the uncommonly high number of phases and equilibria involved in speciation of Ca-phosphates in solution, many of those discarded from calculations for simplification; (iii) the retrograde HAp solubility with respect to temperature, as reported by McDowell [98]; (iv) the Ca/P ratio. If non-stoichiometric HAp is used for solubility determination, the solubility value obtained will change with this ratio; (v) the chemical contamination by foreign ions; (vi) the surface effects on HAp solubility.
When obtaining HAp from direct precipitation by mixing of calcium and phosphate solutions, as reported by most of the papers about HAp precipitation, authors are compelled to work at high concentration of the former solutions. Moreover, in order to avoid the precipitation of the acid phases of calcium phosphate, authors usually force the system toward basic conditions (pH ranging between 8 and 10), moving the strongly concentrated solution into the field of HAp stability. A careful control of the solution supersaturationis hindered by the absence of reliable data about HAp solubility. The lack of monitoring of the supersaturation leads to the conventional mass crystallization of nano-sized HAp.
The growth of HAp by hydrolysis of a calcium phosphate (mainly monetite or brushite as in the papers by Perloff [71] and Schleede [72]) under hydrothermal conditions discourage, to some degree, the authors to work at very high supersaturation. In fact, the hydrolysis obtained at high temperature slows down the achievement of the supersaturation both due to the kinetics of dissolution of the precursor and to the retrograde behavior of HAp solubility with respect to temperature.
Summarizing, in our just quoted paper [69], we demonstrated the effectiveness of the low supersaturation growth obtained by the hydrolysis of monetite at low hydrothermal conditions. As stated in the previous paragraph, reducing the concentration of calcium and phosphate in solution and moving the system toward low pH values, near the low-pH boundary of the stability field of HAp lowers the supersaturation of the system with respect to HAp. This, in turn, lowers the HAp nucleation frequency allowing the crystallization of large individuals, which are needed for both structural and morphological investigations. Both in the case of monetite and brushite, we invoked the significance of excellent parametric coincidences between host and guest phases in order to promote the 2D heterogeneous nucleation and epitaxial growth (2D heterogeneous nucleation) of the latter, lowering the energy required for the nucleation. The amazing differences between the results are mainly due to the kinetics of dissolution and supersaturation.
Moreover, this approach let us demonstrate that, also during hydrolysis, at high dissolution rates, the first generation of crystals has a “tape-like” or “ribbon-like” [20] behavior typically ascribed to biological apatites. Therefore, the flat, undeniably monoclinic habit of small HAp crystals can be obtained when working at high supersaturation, which favors nucleation instead of crystal growth and without claiming the effect of either specific impurities or high crystallization temperature.
5.3. The Effect of the Presence of Carbonate Ions in Solution
The observation of the morphological effect of carbonated species present in the growth medium provides information about the crystal faces involved in the adsorption/absorption of those impurities.
It is experimentally demonstrated that the presence of carbonated species affects both the morphology and structure of apatites depending on their concentration and on the pH of the solution [75]. In fact, the pH strongly affects speciation of calcium carbonate in solution and, consequently, the surfaces that possibly interact with those surface specific impurities.
In the presence of a carbon dioxide saturated solution, the pH ranges in the stability field of the CO2 and HCO3− carbon species. In such conditions, the growth rates of the {100} and {} forms of monoclinic HAp differentiate with respect to the {001}. The interaction between apatite and impurities in solution results in a growth morphology strongly modified, from the typical pseudo–prismatic extensively described in a previous work [69] (Figure 5a) to a tabular one (Figure 5b). The tabular morphology is dominated by extended faces ascribable to the {001} pinacoid, the only one among the three kinetically strongly differentiated (see Figure 4).
Moving toward high pH values, in the stability field of the CO3=, the morphology of the apatite crystals (Figure 5c) is one more time strongly modified, with hexagonal prisms frequently twinned and terminated by flat pinacoids. In this case, carbonate enters the crystal modifying the crystal structure as well, as demonstrated by IR and Raman spectroscopic analisys [75].
We suggest explaining the whole morphological and structural modification in the light of the cooperative effect [67,69,99] between Ca-carbonates in solution and Ca-apatite surfaces, considering both the modification of growth morphology and the stabilization of carbonate-containing apatites as the consequence of the epitaxy as the key mechanism during crystal growth.
6. Conclusions
Experimental and theoretical investigations developed over the last sixty years on the complex relationships between chemical composition, crystal structure and crystal morphology of apatites, showed that many unsolved problems arise owing to the structural anisotropy of the ions (, ) filling the channels of apatites. Hydroxyapatite (HAp) represents the case study in which the anisotropy, due to the presence of the dipoles in the structure channels, reduces the structure symmetry to the lowest one, both for hexagonal (P63) and monoclinic (P21/c) crystals. In this review, we focused our attention on the monoclinic HAp polymorph both because its structure does not show ambiguity [20,21] and because it should be more fruitful considering the lowest instead of the highest symmetry, when one has to interpret the wide morphological variations that HAp undergoes during growth.
The approach we adopted to predict the surface morphology of monoclinic HAp, both for equilibrium and growth, is that by Hartman-Perdok [63], that represents a self-consistent way not only to find the character of a growing face but also to obtain all the outmost surface profiles of a given {hkl} crystal form, either when the surface reconstruction is not necessary or when different reconstructed surface profiles are needed to render the surface compatible with the constraints of the electrical stability.
This allowed us to prove that the calculated equilibrium shape (ES) of a monoclinic HAp single crystal is markedly pseudo-hexagonal; as a consequence, this can be misleading when the growth shapes (GS) of both single and twinned grown crystals are considered [38,68]. As a matter of fact, careful examination of experimental growth shapes observed either in nature (both in geological and bio-mineralized samples) or in laboratory grown crystals from pure aqueous solutions, allows to say that the HAp hexagonality very often is nothing else but a manner of speaking. Moreover, the HAp platelets which grew elongated along the crystal channels, in the early stages of laboratory crystals and in mineralized biological tissues (tooth enamel, bones), are not due to the breaking of the hexagonal symmetry but to different growth rates of the different monoclinic pinacoids in zone with the crystal channels, according to the varying supersaturation values experienced by the growth solution in a closed system.
Furthermore, investigation about the surface profiles, carried out through the Hartman-Perdok method applied to both HAp and monetite (CaHPO4), was also proved to be necessary as a preliminary treatment to determine which kind of interaction occurs between a pre-existingmonetite and the 3D growth of HAp. In fact, it has been found (both experimentally and theoretically) that monetite can be reliably used as a precursor phase that favors the 2D epitaxial nucleation of HAp that allows the hydroxyapatite to form beyond its usual growth conditions [69]. This finding represents a useful pathway to interpret other relationships between a precursor and the HAp crystal, when the precursor is not an amorphous phase.
Acknowledgments
Special thanks are given to two anonymous reviewers for their useful suggestions and comments.
Author Contributions
All the authors conceived the work; Linda Pastero and Dino Aquilano wrote the paper; Marco Bruno made the figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M. The many facets of apatite. Am. Mineral. 2015, 100, 1033–1039. [Google Scholar] [CrossRef]
- Kern, R. The equilibrium form of a crystal. In Morphology of Crystals: Part A: Fundamentals; Sunagawa, I., Ed.; Terra Scientific Publishing Company/Tokyo; D. Reidel Publishing Company: Tokyo, Japan, 1987; pp. 77–206. [Google Scholar]
- Kern, R.; Métois, J.J.; Le Lay, G. Basic mechanisms in the early stages of epitaxy. In Current Topics in Materials Science; Kaldis, E., Ed.; North-Holland Publishing Company: Amsterdam, The Netherland, 1979; Volume 3, pp. 131–419. [Google Scholar]
- Kern, R. Adsorption, absorption, versus crystal growth. Cryst. Res. Technol. 2013, 48, 727–782. [Google Scholar] [CrossRef]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng. C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- Posner, A.S.; Perloff, A.; Diorio, A.F. Refinement of the hydroxyapatite structure. Acta Crystallogr. 1958, 11, 308–309. [Google Scholar] [CrossRef]
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal Structure of Hydroxyapatite. Nature 1964, 204, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Eysel, W.; Roy, D.M. Hydrothermal flux growth of hydroxyapatites by temperature oscillation. J. Cryst. Growth 1973, 20, 245–250. [Google Scholar] [CrossRef]
- Arends, J.; Schuthof, J.; van der Lindwen, W.H.; Bennema, P.; van der Berg, P.J. Preparation of pure hydroxyapatite single crystals by hydrothermal recrystallization. J. Cryst. Growth 1979, 46, 213–220. [Google Scholar] [CrossRef]
- Mengeot, M.; Harvill, M.L.; Gilliam, O.R. Hydrothermal growth of calcium hydroxyapatite single crystals. J. Cryst. Growth 1973, 19, 199–203. [Google Scholar] [CrossRef]
- Hughes, J.M.; Cameron, M.; Crowley, K.D. Structural variations in natural F, OH, and Cl apatites. Am. Mineral. 1989, 74, 870–876. [Google Scholar]
- Terpstra, R.A.; Bennema, P.; Hartman, P.; Woensdregt, C.F.; Perdok, W.G.; Senechal, M.L. F faces of apatite and its morphology: Theory and observation. J. Cryst. Growth 1986, 78, 468–478. [Google Scholar] [CrossRef]
- Lee, W.T.; Dove, M.T.; Salje, E.K.H. Surface relaxations in hydroxyapatite. J. Phys. Condens. Matter 2000, 12, 9829–9841. [Google Scholar] [CrossRef]
- De Leeuw, N.H. Molecular Dynamics Simulations of the Growth Inhibiting Effect of Fe2+, Mg2+, Cd2+, and Sr2+ on Calcite Crystal Growth. J. Phys. Chem. B 2002, 106, 5241–5249. [Google Scholar] [CrossRef]
- Filgueiras, M.R.T.; Mkhonto, D.; de Leeuw, N.H. Computer simulations of the adsorption of citric acid at hydroxyapatite surfaces. J. Cryst. Growth 2006, 294, 60–68. [Google Scholar] [CrossRef]
- De Leeuw, N.H.; Rabone, J.A.L. Molecular dynamics simulations of the interaction of citric acid with the hydroxyapatite (0001) and (011ˉ0) surfaces in an aqueous environment. CrystEngComm 2007, 9, 1178. [Google Scholar] [CrossRef]
- Matsunaga, K.; Murata, H. Formation Energies of Substitutional Sodium and Potassium in Hydroxyapatite. Mater. Trans. 2009, 50, 1041–1045. [Google Scholar] [CrossRef] [Green Version]
- Corno, M.; Orlando, R.; Civalleri, B.; Ugliengo, P. Periodic B3LYP study of hydroxyapatite (001) surface modelled by thin layer slabs. Eur. J. Mineral. 2007, 19, 757–767. [Google Scholar] [CrossRef]
- Elliott, J.C. Monoclinic Space Group of Hydroxyapatite. Nature 1971, 230, 72. [Google Scholar] [CrossRef]
- Elliott, J.C.; Young, R.A. Conversion of Single Crystals of Chlorapatite into Single Crystals of Hydroxyapatite. Nature 1967, 214, 904–906. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates. Biomatter 2011, 1, 121–164. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Dong, Z.L. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. Sect. B Struct. Sci. 2003, 59, 1–16. [Google Scholar] [CrossRef]
- Mathew, M.; Takagi, S. Structures of biological minerals in dental research. J. Res. Natl. Inst. Stand. Technol. 2001, 106, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Ortophosphates; Elsevier: Amsterdam, The Netherland, 1994; ISBN 9781483290317. [Google Scholar]
- Corno, M.; Rimola, A.; Bolis, V.; Ugliengo, P. Hydroxyapatite as a key biomaterial: Quantum-mechanical simulation of its surfaces in interaction with biomolecules. Phys. Chem. Chem. Phys. 2010, 12, 6309. [Google Scholar] [CrossRef] [PubMed]
- Bolis, V.; Busco, C.; Martra, G.; Bertinetti, L.; Sakhno, Y.; Ugliengo, P.; Chiatti, F.; Corno, M.; Roveri, N. Coordination chemistry of Ca sites at the surface of nanosized hydroxyapatite: Interaction with H2O and CO. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2012, 370, 1313–1336. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanan, K.; Young, R.A. Significant precision in crystal structural details. Holly Springs hydroxyapatite. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1969, 25, 1534–1543. [Google Scholar] [CrossRef]
- Elliott, J.C.; Mackie, P.E.; Young, R.A. Monoclinic Hydroxyapatite. Science 1973, 180, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Ikoma, T.; Yamazaki, A.; Nakamura, S.; Akao, M. Preparation and Structure Refinement of Monoclinic Hydroxyapatite. J. Solid State Chem. 1999, 144, 272–276. [Google Scholar] [CrossRef]
- Suetsugu, Y.; Tanaka, J. Crystal growth and structure analysis of twin-free monoclinic hydroxyapatite. J. Mater. Sci. Mater. Med. 2002, 13, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Treboux, G.; Layrolle, P.; Kanzaki, N.; Onuma, K.; Ito, A. Symmetry of posner’s cluster. J. Am. Chem. Soc. 2000, 122, 8323–8324. [Google Scholar] [CrossRef]
- Hochrein, O.; Kniep, R.; Zahn, D. Atomistic simulation study of the order/disorder (monoclinic to hexagonal) phase transition of hydroxyapatite. Chem. Mater. 2005, 17, 1978–1981. [Google Scholar] [CrossRef]
- Haverty, D.; Tofail, S.A.M.; Stanton, K.T.; McMonagle, J.B. Structure and stability of hydroxyapatite: Density functional calculation and Rietveld analysis. Phys. Rev. B 2005, 71, 94103. [Google Scholar] [CrossRef]
- Corno, M.; Busco, C.; Civalleri, B.; Ugliengo, P. Periodic ab initio study of structural and vibrational features of hexagonal hydroxyapatite Ca10(PO4)6(OH)2. Phys. Chem. Chem. Phys. 2006, 8, 2464. [Google Scholar] [CrossRef] [PubMed]
- Suda, H.; Yashima, M.; Kakihana, M.; Yoshimura, M. Monoclinic ↔ Hexagonal Phase Transition in Hydroxyapatite Studied by X-ray Powder Diffraction and Differential Scanning Calorimeter Techniques. J. Phys. Chem. 1995, 99, 6752–6754. [Google Scholar] [CrossRef]
- Ikoma, T.; Yamazaki, A.; Nakamura, S.; Masaru, A. Phase Transition of Monoclinic Hydroxyapatite. Netsu Sokutei 1998, 25, 141–149. [Google Scholar]
- Aquilano, D.; Bruno, M.; Rubbo, M.; Massaro, F.R.; Pastero, L. Low Symmetry Polymorph of Hydroxyapatite. Theoretical Equilibrium Morphology of the Monoclinic Ca5(OH)(PO4)3. Cryst. Growth Des. 2014, 14, 2846–2852. [Google Scholar] [CrossRef]
- Fleet, M.E.; Liu, X. Location of type B carbonate ion in type A–B carbonate apatite synthesized at high pressure. J. Solid State Chem. 2004, 177, 3174–3182. [Google Scholar] [CrossRef]
- Fleet, M.E.; Liu, X. Local structure of channel ions in carbonate apatite. Biomaterials 2005, 26, 7548–7554. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, Y.; Takahashi, Y.; Okamura, F.P.; Tanaka, J. Structure Analysis of A-Type Carbonate Apatite by a Single-Crystal X-Ray Diffraction Method. J. Solid State Chem. 2000, 155, 292–297. [Google Scholar] [CrossRef]
- Ma, G.; Liu, X.Y. Hydroxyapatite: Hexagonal or monoclinic? Cryst. Growth Des. 2009, 9, 2991–2994. [Google Scholar] [CrossRef]
- Reyes-Gasga, J.; Martinéz-Piñeiro, E.L.; Brès, É.F. Crystallographic structure of human tooth enamel by electron microscopy and X-ray diffraction: Hexagonal or monoclinic? J. Microsc. 2012, 248, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Mugnaioli, E.; Reyes-Gasga, J.; Kolb, U.; Hemmerlé, J.; Brès, E.F. Evidence of noncentrosymmetry of human tooth hydroxyapatite crystals. Chem. A Eur. J. 2014, 20, 6849–6852. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Dolhaine, H.; Du Chesne, A.; Heinz, S.; Hochrein, O.; Laeri, F.; Podebrad, O.; Vietze, U.; Weiland, T.; Kniep, R. Biomimetic Morphogenesis of Fluorapatite-Gelatin Composites: Fractal Growth, the Question of Intrinsic Electric Fields, Core/Shell Assemblies, Hollow Spheres and Reorganization of Denatured Collagen. Eur. J. Inorg. Chem. 1999, 1999, 1643–1653. [Google Scholar] [CrossRef]
- Tao, J.; Battle, K.C.; Pan, H.; Salter, E.A.; Chien, Y.-C.; Wierzbicki, A.; De Yoreo, J.J. Energetic basis for the molecular-scale organization of bone. Proc. Natl. Acad. Sci. USA 2015, 112, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Sakhno, Y.; Ivanchenko, P.; Iafisco, M.; Tampieri, A.; Martra, G. A step toward control of the surface structure of biomimetic hydroxyapatite nanoparticles: Effect of carboxylates on the {010} P-rich/Ca-rich facets ratio. J. Phys. Chem. C 2015, 119, 5928–5937. [Google Scholar] [CrossRef]
- Yao, X.; Yao, H.; Li, G.; Li, Y. Biomimetic synthesis of needle-like nano-hydroxyapatite templated by double-hydrophilic block copolymer. J. Mater. Sci. 2010, 45, 1930–1936. [Google Scholar] [CrossRef]
- Shuai, C.; Feng, P.; Nie, Y.; Hu, H.; Liu, J.; Peng, S. Nano-hydroxyapatite improves the properties of β-tricalcium phosphate bone scaffolds. Int. J. Appl. Ceram. Technol. 2013, 10, 1003–1013. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, H.; Zhang, L.; Li, Y.; Wei, S. In situ synthesis and in vitro biocompatibility of needle-like nano-hydroxyapatite in agar-gelatin co-hydrogel. Mater. Lett. 2013, 104, 8–12. [Google Scholar] [CrossRef]
- Ito, H.; Oaki, Y.; Imai, H. Selective synthesis of various nanoscale morphologies of hydroxyapatite via an intermediate phase. Cryst. Growth Des. 2008, 8, 1055–1059. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ono, S.; Hirakura, S.; Oaki, Y.; Imai, H. Morphological variation of hydroxyapatite grown in aqueous solution based on simulated body fluid. CrystEngComm 2012, 14, 1143–1149. [Google Scholar] [CrossRef]
- Bertinetti, L.; Tampieri, A.; Landi, E.; Ducati, C.; Midgley, P.A.; Coluccia, S.; Martra, G. Surface Structure, Hydration, and Cationic Sites of Nanohydroxyapatite: UHR-TEM, IR, and Microgravimetric Studies. J. Phys. Chem. C 2007, 111, 4027–4035. [Google Scholar] [CrossRef]
- Sakhno, Y.; Bertinetti, L.; Iafisco, M.; Tampieri, A.; Roveri, N.; Martra, G. Surface Hydration and Cationic Sites of Nanohydroxyapatites with Amorphous or Crystalline Surfaces: A Comparative Study. J. Phys. Chem. C 2010, 114, 16640–16648. [Google Scholar] [CrossRef]
- Eppell, S.J.; Tong, W.; Lawrence Katz, J.; Kuhn, L.; Glimcher, M.J. Shape and size of isolated bone mineralites measured using atomic force microscopy. J. Orthop. Res. 2001, 19, 1027–1034. [Google Scholar] [CrossRef]
- Fratzl, P.; Gupta, H.S.; Paschalis, E.P.; Roschger, P. Structure and mechanical quality of the collagen–mineral nano-composite in bone. J. Mater. Chem. 2004, 14, 2115–2123. [Google Scholar] [CrossRef]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. R Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Delgado-López, J.M.; Frison, R.; Cervellino, A.; Gómez-Morales, J.; Guagliardi, A.; Masciocchi, N. Crystal Size, Morphology, and Growth Mechanism in Bio-Inspired Apatite Nanocrystals. Adv. Funct. Mater. 2014, 24, 1090–1099. [Google Scholar] [CrossRef]
- Sato, K.; Kogure, T.; Iwai, H.; Tanaka, J. Atomic-Scale {101ˉ0} Interfacial Structure in Hydroxyapatite Determined by High-Resolution Transmission Electron Microscopy. J. Am. Ceram. Soc. 2002, 85, 3054–3058. [Google Scholar] [CrossRef]
- Ospina, C.A.; Terra, J.; Ramirez, A.J.; Farina, M.; Ellis, D.E.; Rossi, A.M. Experimental evidence and structural modeling of nonstoichiometric (010) surfaces coexisting in hydroxyapatite nano-crystals. Colloids Surf. B Biointerfaces 2012, 89, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Astala, R.; Stott, M.J. First-principles study of hydroxyapatite surfaces and water adsorption. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 78, 75427. [Google Scholar] [CrossRef]
- Rulis, P.; Yao, H.; Ouyang, L.; Ching, W.Y. Electronic structure, bonding, charge distribution, and X-ray absorption spectra of the (001) surfaces of fluorapatite and hydroxyapatite from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 245410. [Google Scholar] [CrossRef]
- Hartman, P. Structure and morphology. In Crystal Growth: An Introduction; Hartman, P., Ed.; North-Holland Publishing Company: Amsterdam, The Netherland, 1973; pp. 367–402. [Google Scholar]
- Lee, W.T.; Salje, E.K.H.; Dove, M.T. Effect of surface relaxations on the equilibrium growth morphology of crystals: Platelet formation. J. Phys. Condens. Matter 1999, 11, 7385–7410. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Zhang, W.; Putnis, C.V.; Putnis, A. Direct Observation of Spiral Growth, Particle Attachment, and Morphology Evolution of Hydroxyapatite. Cryst. Growth Des. 2016, 16, 4509–4518. [Google Scholar] [CrossRef]
- Bruno, M. The reconstruction of dipolar surfaces: A preliminary step for adsorption modeling. Cryst. Res. Technol. 2013, 48, 811–818. [Google Scholar] [CrossRef]
- Aquilano, D.; Pastero, L. Anomalous mixed crystals: A peculiar case of adsorption/absorption. Cryst. Res. Technol. 2013, 48, 819–839. [Google Scholar] [CrossRef]
- Aquilano, D.; Bruno, M.; Rubbo, M.; Pastero, L.; Massaro, F.R. Twin Laws and Energy in Monoclinic Hydroxyapatite, Ca5(PO4)3(OH). Cryst. Growth Des. 2015, 15, 411–418. [Google Scholar] [CrossRef]
- Pastero, L.; Aquilano, D. Monetite-Assisted Growth of Micrometric Ca-Hydroxyapatite Crystals from Mild Hydrothermal Conditions. Cryst. Growth Des. 2016, 16, 852–860. [Google Scholar] [CrossRef]
- Jaffe, E.B. Abstracts of the Literature on Synthesis of Apatites and Some Related Phosphates; U.S. Geological Survey: Washington, DC, USA, 1951.
- Perloff, A.; Posner, A.S. Preparation of Pure Hydroxyapatite Crystals. Science 1956, 124, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Schleede, H.A.; Schmidt, W.; Kindt, H. Zur Kenntnis der Calciumphosphate und Apatite. Z. Elektrochem. 1932, 38, 633–641. [Google Scholar]
- Jullman, H.; Mosebach, R. Zur Synthese, Licht- und Doppelbrechung des Hydroxylapatits. Z. Naturf. B 1966, 21, 493–494. [Google Scholar] [CrossRef]
- Kirn, J.F.; Leidheiser, H. Progress in efforts to grow large single crystals of hydroxyapatite. J. Cryst. Growth 1968, 2, 111–112. [Google Scholar] [CrossRef]
- Pastero, L.; Bruno, M.; Rubbo, M.; Camara, F.; Aquilano, D. Growth of large Ca-Hydroxyapatite crystals from aqueous solution. In Proceedings of the IV Meeting of the Italian and Spanish Crystallographic Associations, Tenerife, Spain, 21–25 June 2016. [Google Scholar]
- Van Wazer, J.R. Phosphorus and its Compounds, Bd. 1: Chemistry. Angew. Chem. 1961, 73, 552. [Google Scholar] [CrossRef]
- Clark, J.S. Solubility criteria for the existence of hydroxyapatite. Can. J. Chem. 1955, 33, 1696–1700. [Google Scholar] [CrossRef]
- Larsen, S. Solubility of Hydroxyapatite. Nature 1966, 212, 605. [Google Scholar] [CrossRef]
- Chen, Z.F.; Darvell, B.W.; Leung, V.W.H. Hydroxyapatite solubility in simple inorganic solutions. Arch. Oral Biol. 2004, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.B.; Darvell, B.W. Calcium Phosphate Solubility: The Need for Re-Evaluation. Cryst. Growth Des. 2009, 9, 639–645. [Google Scholar] [CrossRef]
- Neuman, W.F.F.; Neuman, M.W.W. The Nature of the Mineral Phase of Bone. Chem. Rev. 1953, 53, 1–45. [Google Scholar] [CrossRef]
- Levinskas, G.; Neuman, W. The Solubility of Bone Mineral. I. Solubility Studies of Synthetic Hydroxylapatite. J. Phys. Chem. 1955, 59, 164–168. [Google Scholar] [CrossRef]
- Rootare, H.M.; Deitz, V.R.; Carpenter, F.G. Solubility product phenomena in hydroxyapatite-water systems. J. Colloid Sci. 1962, 17, 179–206. [Google Scholar] [CrossRef]
- Bell, L.C.; Mika, H. The pH dependence of the surface concentrations of calcium and phosphorous on hydroxyapatite in aqueous solutions. J. Soil Sci. 1979, 30, 247–258. [Google Scholar] [CrossRef]
- Mika, H.; Bell, L.C.; Kruger, B.J. The role of surface reactions in the dissolution of stoichiometric hydroxyapatite. Arch. Oral Biol. 1976, 21, 697–701. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Inorganic chemistry of the dissolution phenomenon: The dissolution mechanism of calcium apatites at the atomic (ionic) level. Comments Inorg. Chem. 1999, 20, 285–299. [Google Scholar] [CrossRef]
- Dorozhkin, S. V Surface Reactions of Apatite Dissolution. J. Colloid Interface Sci. 1997, 191, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. A review on the dissolution models of calcium apatites. Prog. Cryst. Growth Charact. Mater. 2002, 44, 45–61. [Google Scholar] [CrossRef]
- Chuong, R. Experimental Study of Surface and Lattice Effects on the Solubility of Hydroxyapatite. J. Dent. Res. 1973, 52, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, I. The solubility of calcium phosphate. I. The effect of pH and ofamount of solid phase. J. Biol. Chem. 1942, 143, 703–710. [Google Scholar]
- Greenwald, I. The solubility of calcium phosphate: II. The solubility product. J. Biol. Chem. 1942, 143, 711–714. [Google Scholar]
- Greenwald, I. The effect of phosphate on the solubility of calcium carbonate and of bicarbonate on the solubility of calcium and magnesium phosphates. J. Biol. Chem. 1945, 161, 697–704. [Google Scholar] [PubMed]
- Kaufman, H.W.; Kleinberg, I. Studies on the incongruent solubility of hydroxyapatite. Calcif. Tissue Int. 1979, 27, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.S.; Peech, M. Solubility Criteria for the Existence of Calcium and Aluminum Phosphates in Soils1. Soil Sci. Soc. Am. J. 1955, 19, 171. [Google Scholar] [CrossRef]
- Skinner, H.C.W. Studies in the basic mineralizing system, CaO-P2O5-H2O. Calcif. Tissue Res. 1974, 14, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.C.; Ogasawara, T.; Silva, F.T. Hydrothermal Crystallization of HAp. Proc 2nd Int. Symp. Apatite. 1997, 1, 4147. [Google Scholar]
- Byrappa, K.; Yoshimura, M. Hydrothermal growth of some selected crystals. In Handbook of Hydrothermal Technology; Noyes Publications/William Andrew Publishing, LLC: Norwich, NY, USA, 2001; pp. 287–299. ISBN 0-8155-1445-X. [Google Scholar]
- McDowell, H.; Gregory, T.M.; Brown, W.E. Solubility of Ca5(PO4)3OH in the system Ca(OH)2-H3PO4-H2O at 5,15,25 and 37 °C. J. Res. Natl. Bur. Stand. Chem. 1977, 81A, 273–281. [Google Scholar] [CrossRef]
- Pastero, L.; Aquilano, D. CaCO3 (Calcite)/Li2CO3 (zabuyelite) anomalous mixed crystals. Sector zoning and growth mechanisms. Cryst. Growth Des. 2008, 8, 3451–3460. [Google Scholar] [CrossRef]
Figure 1.
(a) The P63/m setting of a HAp crystal showing the perfect hexagonality of the [001] zone, being the axial parameters a0 = b0; (b) In the setting P21/c, the alternating up-down OH− dipoles in contiguous channels markedly outline the monoclinicity of the structure. The [010] direction coincides with the 21 axis and is perpendicular to the plane of the drawing, according to the convention adopted in [38].
Figure 1.
(a) The P63/m setting of a HAp crystal showing the perfect hexagonality of the [001] zone, being the axial parameters a0 = b0; (b) In the setting P21/c, the alternating up-down OH− dipoles in contiguous channels markedly outline the monoclinicity of the structure. The [010] direction coincides with the 21 axis and is perpendicular to the plane of the drawing, according to the convention adopted in [38].

Figure 2.
Projection along the structure channels of the HAp polymorph. The space group P21/c has been chosen, as in Figure 1b. Hence the channels run along the [010] direction parallel to the 21 diad axis. Slices are defined respecting the systematic extinction rules of the P21/c space group. (a) The structure of the slice of thickness , limiting the pinacoid {001}, shows that in two adjacent slices the OH dipoles are oriented in opposite sense. On the contrary, in (b,c) the structures of the slices and , limiting the pinacoids {} and {100}, respectively, show a strong similarity (they are not identical) since, within each slice, the OH dipoles alternate up-down.
Figure 2.
Projection along the structure channels of the HAp polymorph. The space group P21/c has been chosen, as in Figure 1b. Hence the channels run along the [010] direction parallel to the 21 diad axis. Slices are defined respecting the systematic extinction rules of the P21/c space group. (a) The structure of the slice of thickness , limiting the pinacoid {001}, shows that in two adjacent slices the OH dipoles are oriented in opposite sense. On the contrary, in (b,c) the structures of the slices and , limiting the pinacoids {} and {100}, respectively, show a strong similarity (they are not identical) since, within each slice, the OH dipoles alternate up-down.
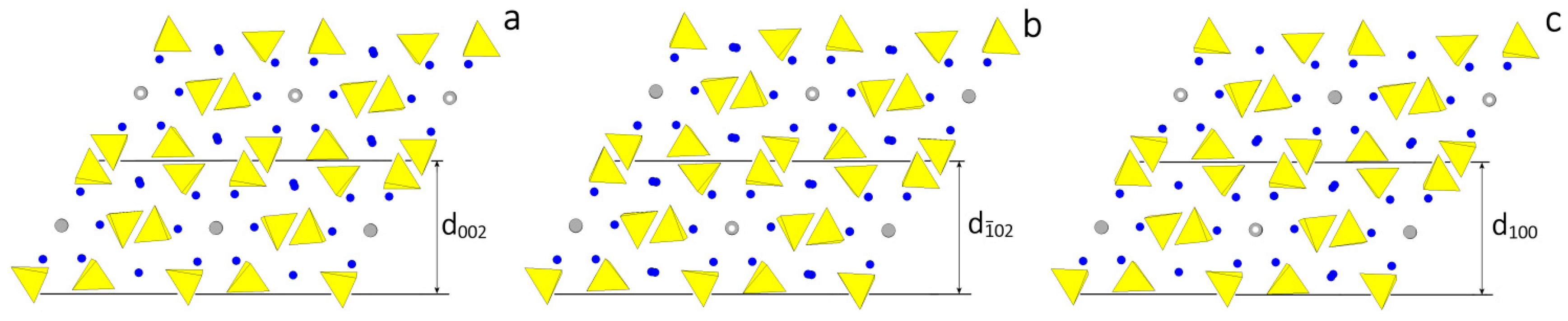
Figure 3.
(a) SEM picture of our HAp crystal grown under mild hydrothermal conditions. It belongs to the monoclinic polymorph, and shows three consecutive pinacoids: {001}, {100} and {}, all parallel to the [010] zone axis, along with the three corresponding prisms: {012}, {110}, and {}, as drawn in (b); where the theoretical (2/m) morphology is represented with different sizes and colors associated to different crystallographic forms. The theoretical (6/m) morphology is drawn in (c). It is related to the space group P63/m, with the [001] direction parallel to the 63 axis. The comparison between the three frames makes evident that the growth shape of the monoclinic polymorph could be easily confused with the hexagonal one.
Figure 3.
(a) SEM picture of our HAp crystal grown under mild hydrothermal conditions. It belongs to the monoclinic polymorph, and shows three consecutive pinacoids: {001}, {100} and {}, all parallel to the [010] zone axis, along with the three corresponding prisms: {012}, {110}, and {}, as drawn in (b); where the theoretical (2/m) morphology is represented with different sizes and colors associated to different crystallographic forms. The theoretical (6/m) morphology is drawn in (c). It is related to the space group P63/m, with the [001] direction parallel to the 63 axis. The comparison between the three frames makes evident that the growth shape of the monoclinic polymorph could be easily confused with the hexagonal one.
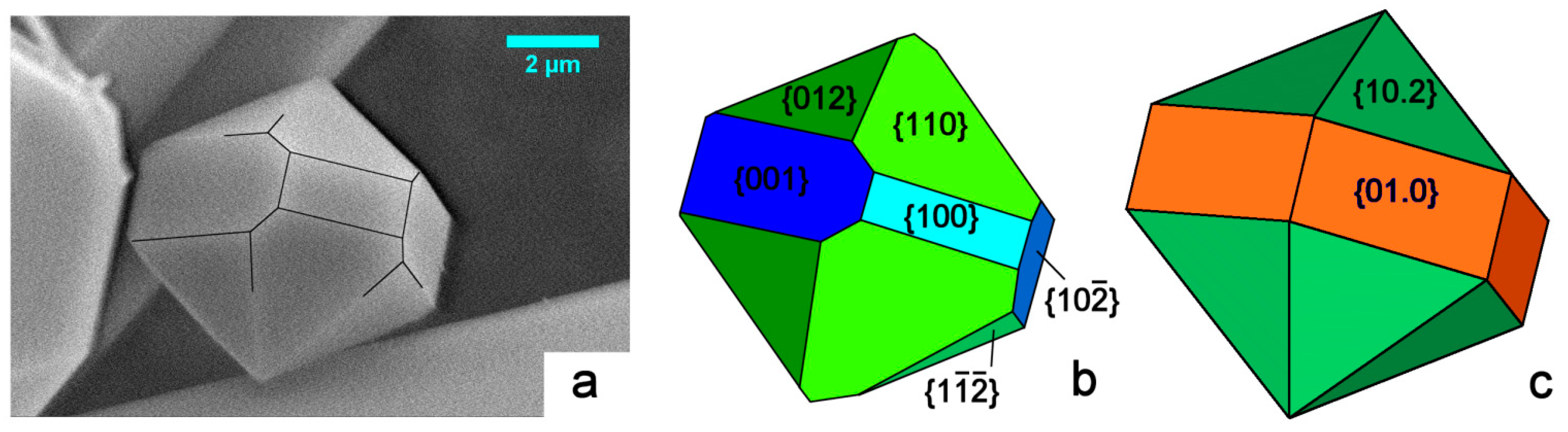
Figure 4.
Sketch of a monoclinic P21/c HAp crystal viewed along the OH channels direction. The equilibrium shape at 0 K is clearly pseudo-hexagonal, with the two {100} and {} forms showing a quasi-identical surface profile (left side). The growth shape I does occur when the supersaturation is high (early stages of growth in a closed system), owing to the anisotropy of the growth kinetics between the {001} pinacoid and the two kinetically close {100} and {} forms (center). The growth shape II, tending to the pseudo-hexagonality, is recovered when all the growth isotherms intersect, at a critical supersaturation value or when the system approaches the equilibrium (right side).
Figure 4.
Sketch of a monoclinic P21/c HAp crystal viewed along the OH channels direction. The equilibrium shape at 0 K is clearly pseudo-hexagonal, with the two {100} and {} forms showing a quasi-identical surface profile (left side). The growth shape I does occur when the supersaturation is high (early stages of growth in a closed system), owing to the anisotropy of the growth kinetics between the {001} pinacoid and the two kinetically close {100} and {} forms (center). The growth shape II, tending to the pseudo-hexagonality, is recovered when all the growth isotherms intersect, at a critical supersaturation value or when the system approaches the equilibrium (right side).
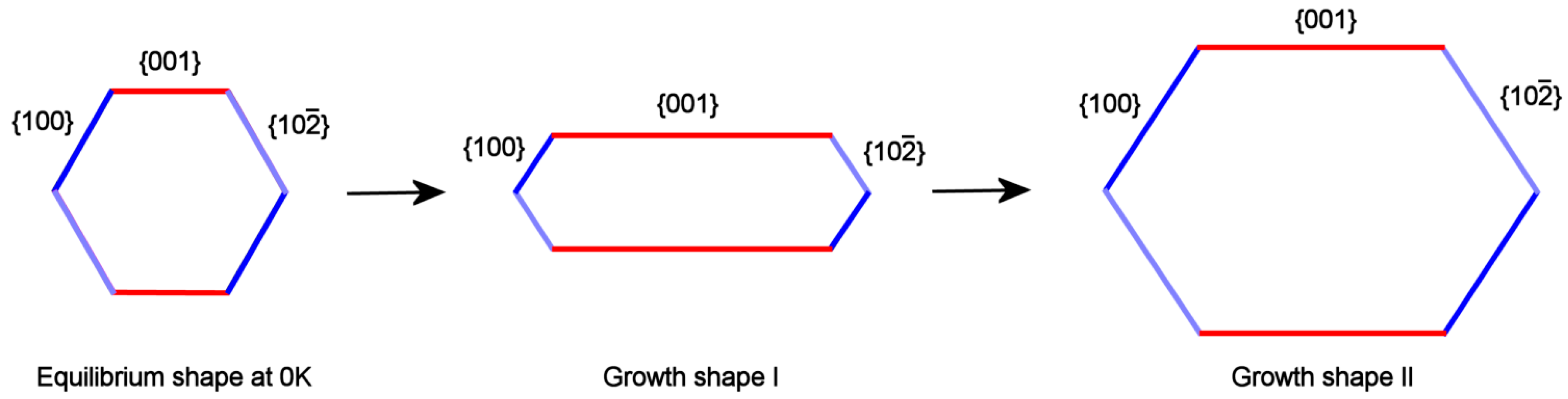
Figure 5.
HAp grown from CO2—free solution (a) Apatite grown from CO2 saturated solution (b) and CAp (c).
Figure 5.
HAp grown from CO2—free solution (a) Apatite grown from CO2 saturated solution (b) and CAp (c).


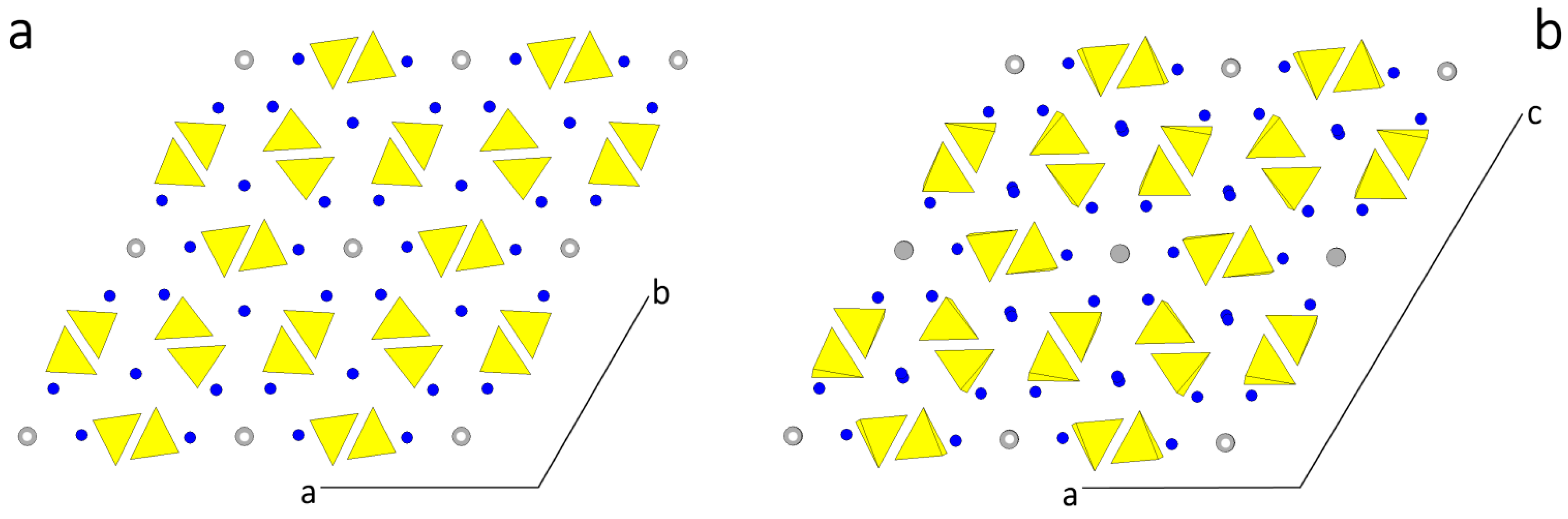{kind=link}
{kind=link}
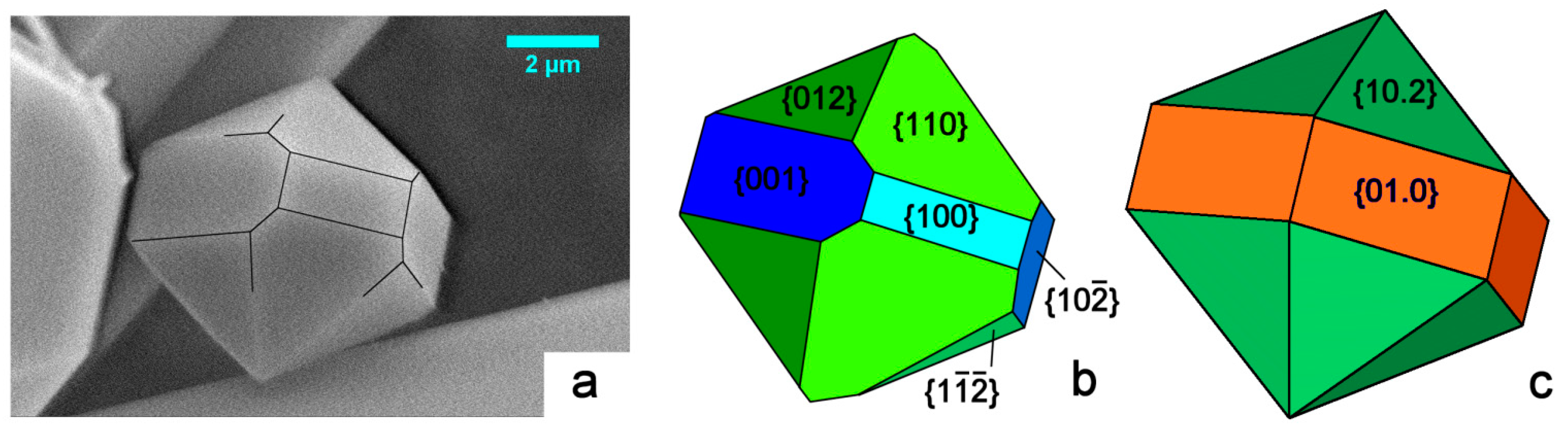{kind=link}
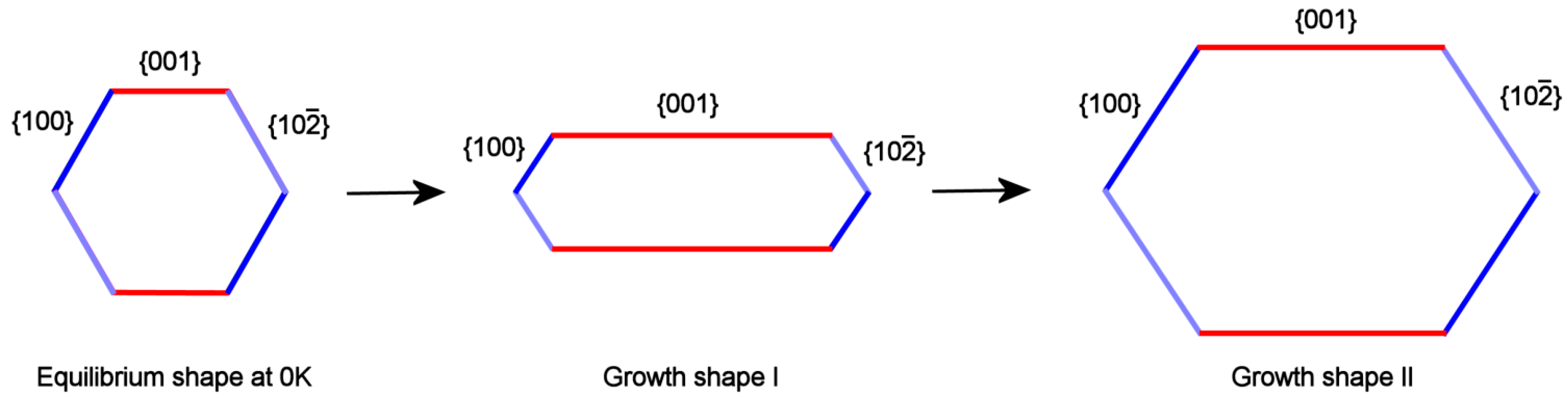{kind=link}
{kind=link}
Table 1.
The main minerals of the Apatite group, their Ca/P molar ratio and chemical composition.
| Ca/P Molar Ratio | Mineral Name | Chemical Composition |
|---|---|---|
| 1.5–1.67 | Calcium-deficient hydroxyapatite (Ca-def HAp) | Ca10−x(HPO4)x(PO4)6−x (OH)2−x(0 < x < 1) |
| 1.67 | Hydroxyapatite (HAp) | Ca10(PO4)6(OH)2 |
| 1.67 | End-member, A-type, carbonated apatite | Ca10(PO4)6CO3 |
| ≥1.67 | End-member, B-type, carbonated hydroxylapatite | Ca10−x[(PO4)6−2x (CO3)2x](OH)2(0 < x < 1) |
| ≥1.67 | Mixed A-type and B-type carbonated apatite | Ca10−x[(PO4)6−2x (CO3)2x]CO3(0 < x < 1) |
| 1.67 | Fluorapatite (FAp) | Ca10(PO4)6F2 |
| 1.67 | Oxyapatite (OAp), mineral voelckerite | Ca10(PO4)6O |
Table 2.
The main minerals of the Apatite group, along with their lattice parameters (in Å) and Space Groups, as it comes out from literature.
Table 2.
The main minerals of the Apatite group, along with their lattice parameters (in Å) and Space Groups, as it comes out from literature.
| Structure | Space Group | a0 | b0 | c0 | α | β | γ | Reference |
|---|---|---|---|---|---|---|---|---|
| HAp (Hydroxy-apatite) | ||||||||
| hexagonal | P63/m | 9.4302 | 9.4302 | 6.8911 | 90° | 90° | 120° | [7,8,9,10,11,12,13,14,15,16,17,18,19] |
| monoclinic | P21/b | 9.4214 | 2 × a0 | 6.881 | 90° | 90° | 120° | [20,21,22] |
| FAp (fluor-apatite) | ||||||||
| hexagonal | P63/m | 9.367 | 9.3973 | 6.8782 | 90° | 90° | 120° | [12,23,24,25] |
| OAp (oxy-apatite) | ||||||||
| hexagonal | P | 9.432 | 9.432 | 6.88 | 90.3° | 90° | 119.9° | [1,23,24,25] |
Table 3.
Surface features and corresponding specific surface energy values related to the {001} form*.
Table 3.
Surface features and corresponding specific surface energy values related to the {001} form*.
| Monoclinic HApForm | PBC of Reference | Surface Termination | Is the Outmost Layer Shared between Adjacent Slices? | Surface Reconstruction Needed | γ001(erg cm−1) |
|---|---|---|---|---|---|
| {001} | [010]B | PO4, Ca, OH | yes | yes | 1546, 1613, 1666,1691 |
| [010]A | PO4 | no | no | 1712 | |
| [010]B | PO4, Ca, OH | yes | yes | 1738,1741,1742,1793 |
*The unique γ001 value in the row referred to the PBC [010]A was obtained from the d002 slice terminated by PO4 ions, as drawn in Figure 2a. First and third row γ001 values refer to the eight profiles that can be obtained from the different disposition of PO4, Ca and OH ions when reconstructing the outmost layers of the alternative d002 slices, coming out from the two PBCs [010]B.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pastero, L.; Bruno, M.; Aquilano, D. About the Genetic Mechanisms of Apatites: A Survey on the Methodological Approaches. Minerals 2017, 7, 139. https://doi.org/10.3390/min7080139
AMA Style
Pastero L, Bruno M, Aquilano D. About the Genetic Mechanisms of Apatites: A Survey on the Methodological Approaches. Minerals. 2017; 7(8):139. https://doi.org/10.3390/min7080139
Chicago/Turabian StylePastero, Linda, Marco Bruno, and Dino Aquilano. 2017. "About the Genetic Mechanisms of Apatites: A Survey on the Methodological Approaches" Minerals 7, no. 8: 139. https://doi.org/10.3390/min7080139
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.