Structural and Electronic Properties of Different Terminations for Quartz (001) Surfaces as Well as Water Molecule Adsorption on It: A First-Principles Study



Abstract
:
1. Introduction
2. Computational Method and Model
2.1. Computational Method
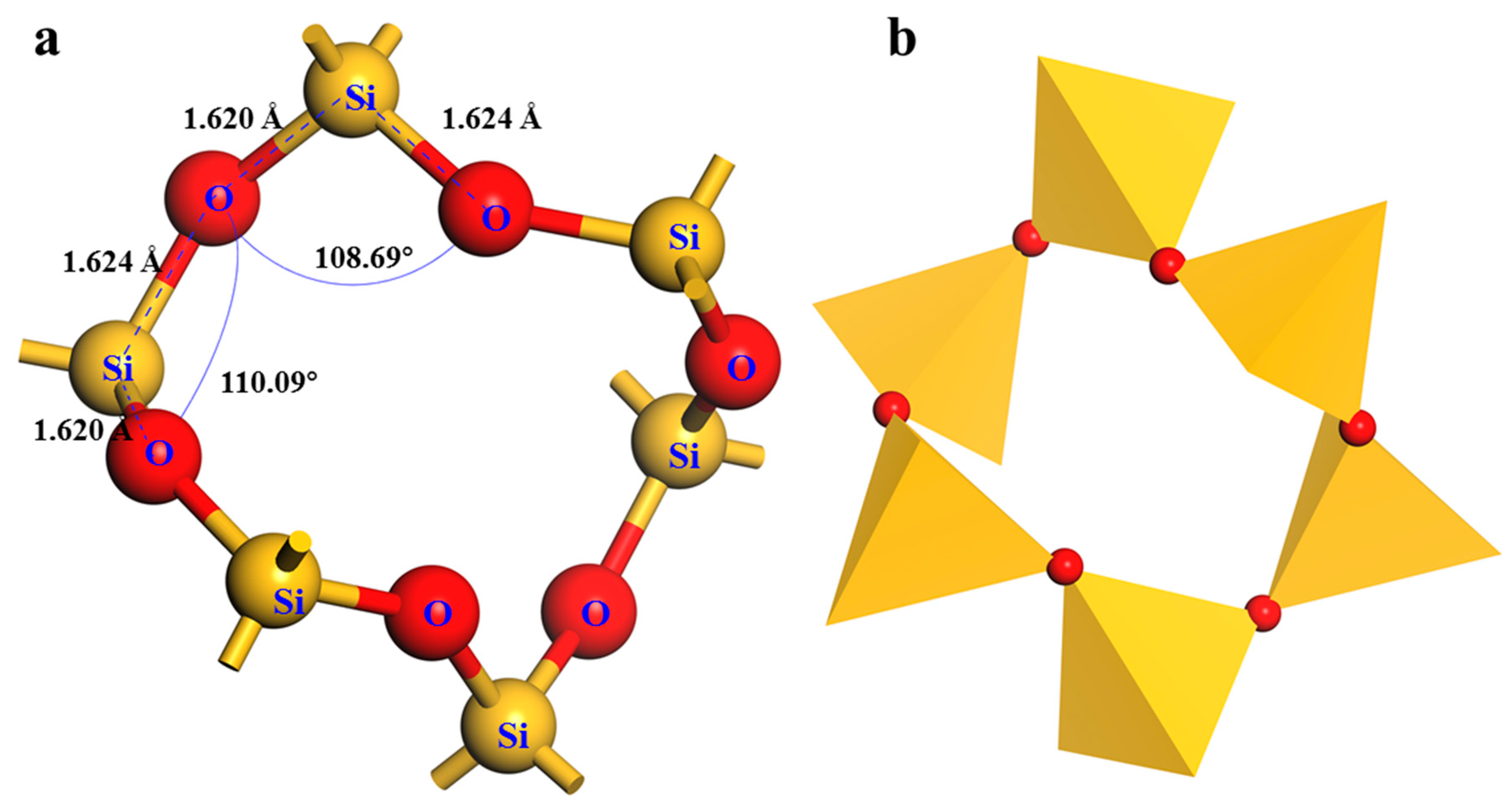
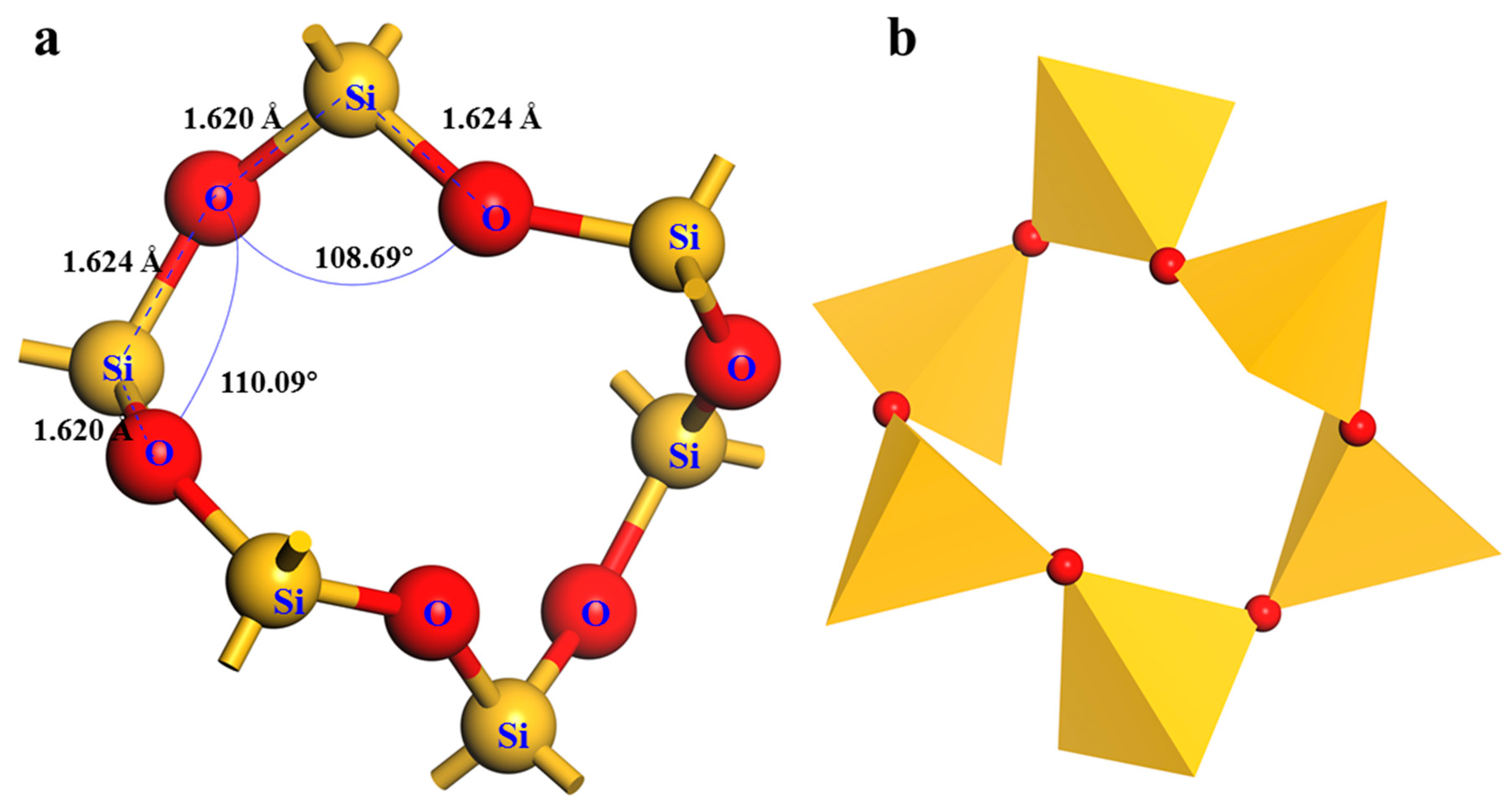
2.2 Quartz Crystal Cell Optimization
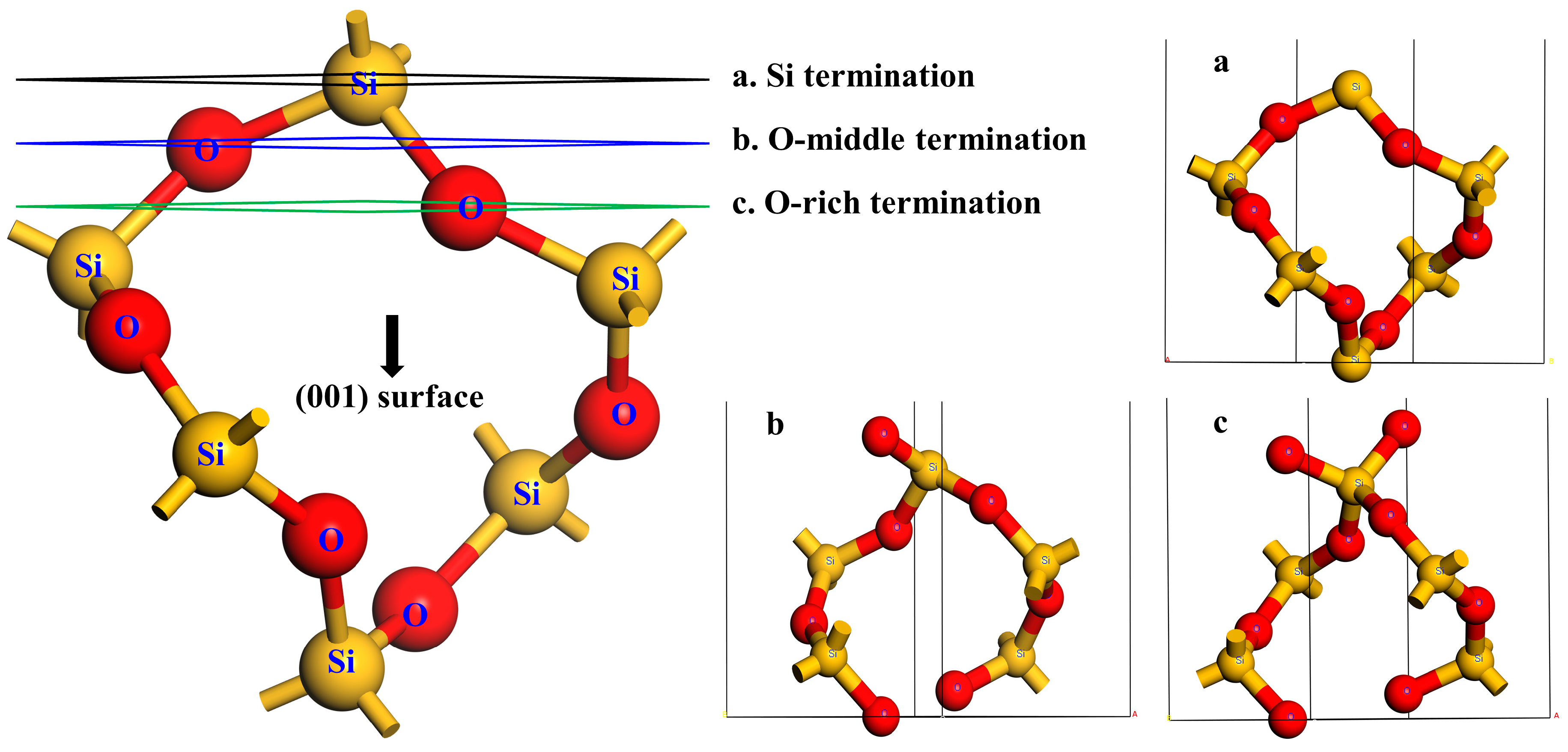
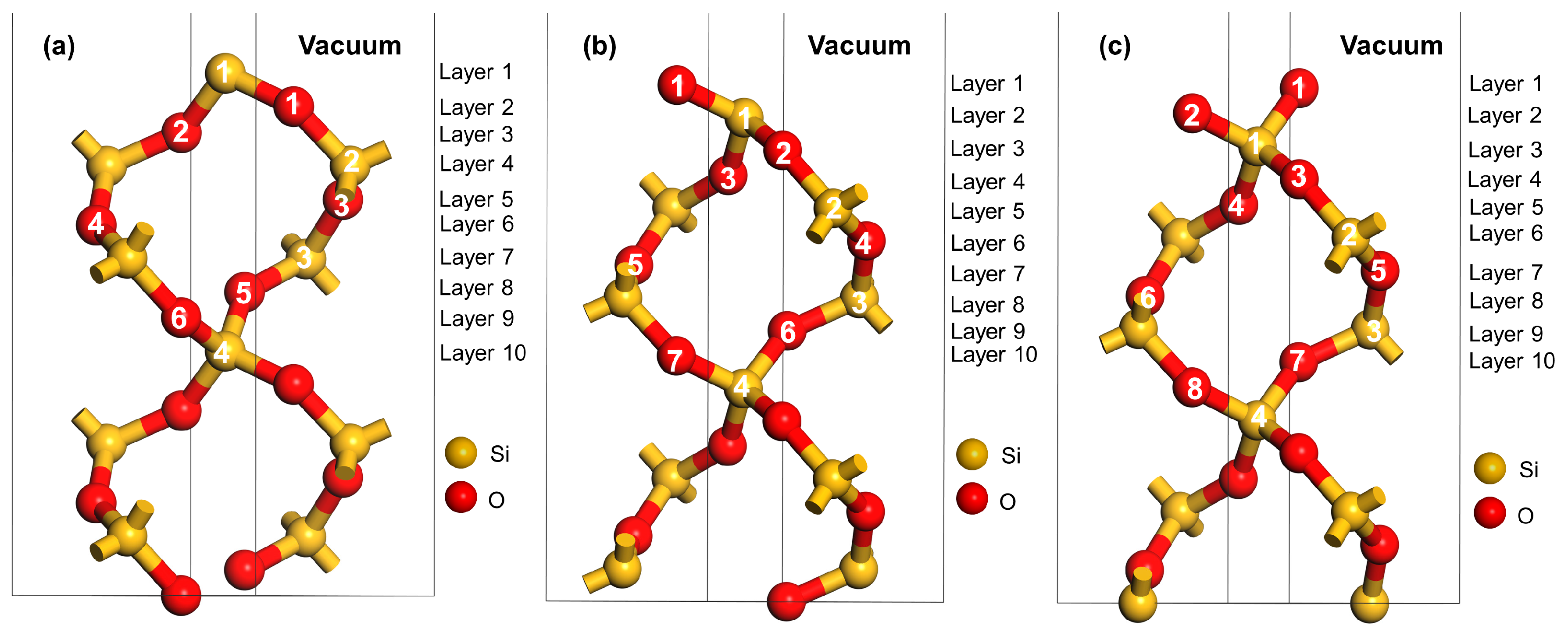
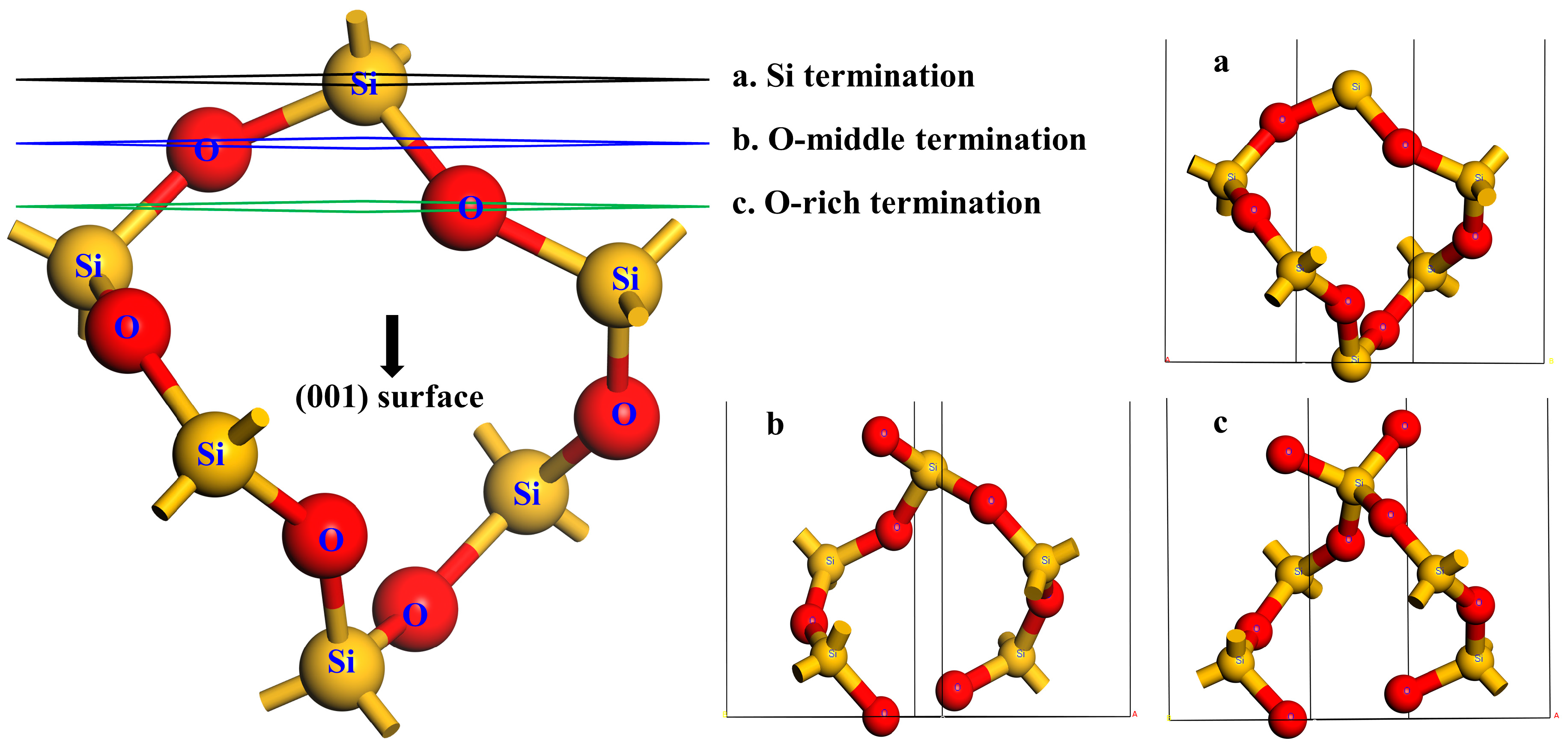
2.3. Surface Models and Calculation of Surface Energy
3. Results and Discussion
3.1. Surface Structural and Electronic Properties of Different Terminations for Quartz (001) Surface
3.1.1. Surface Energy
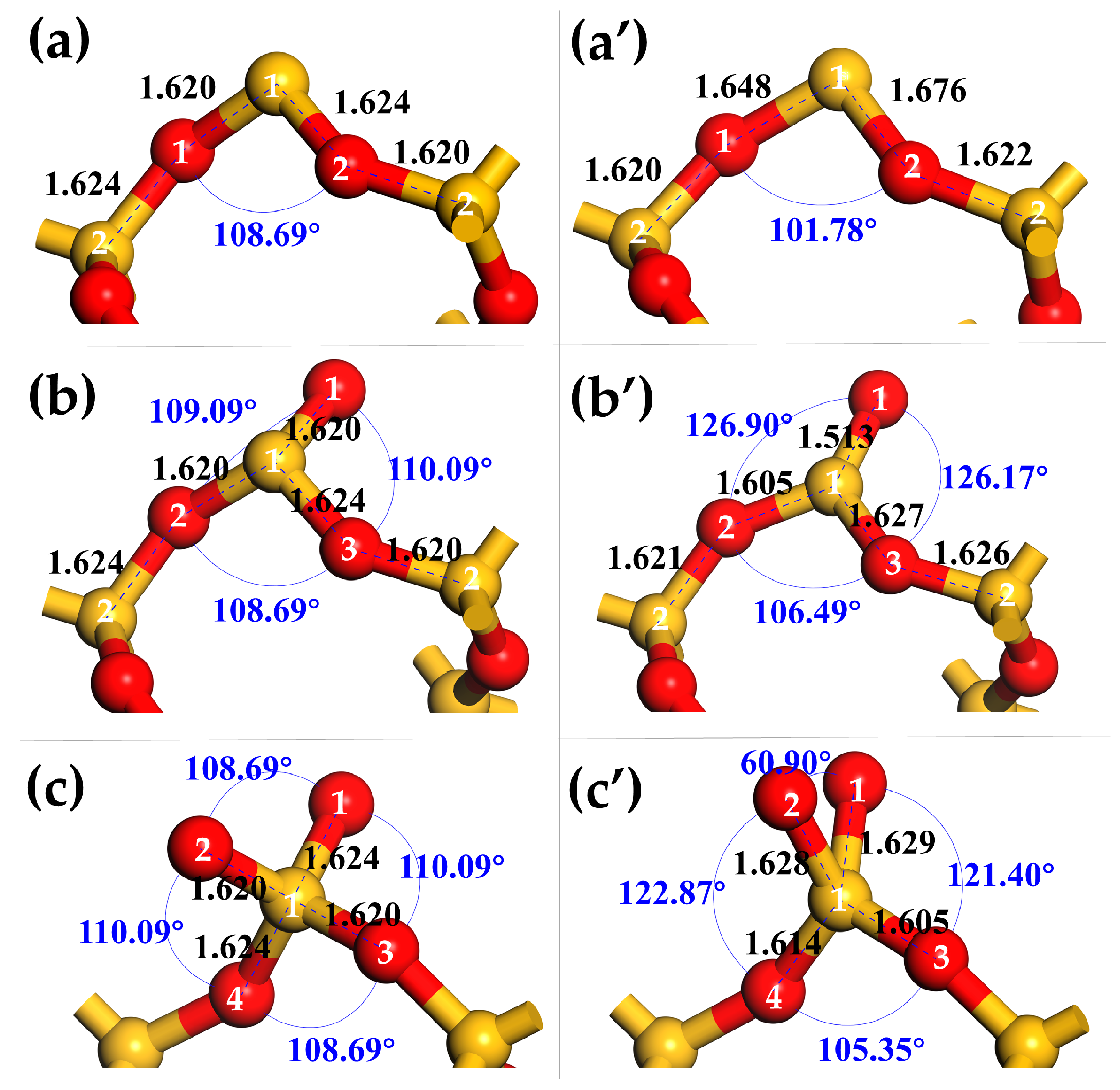
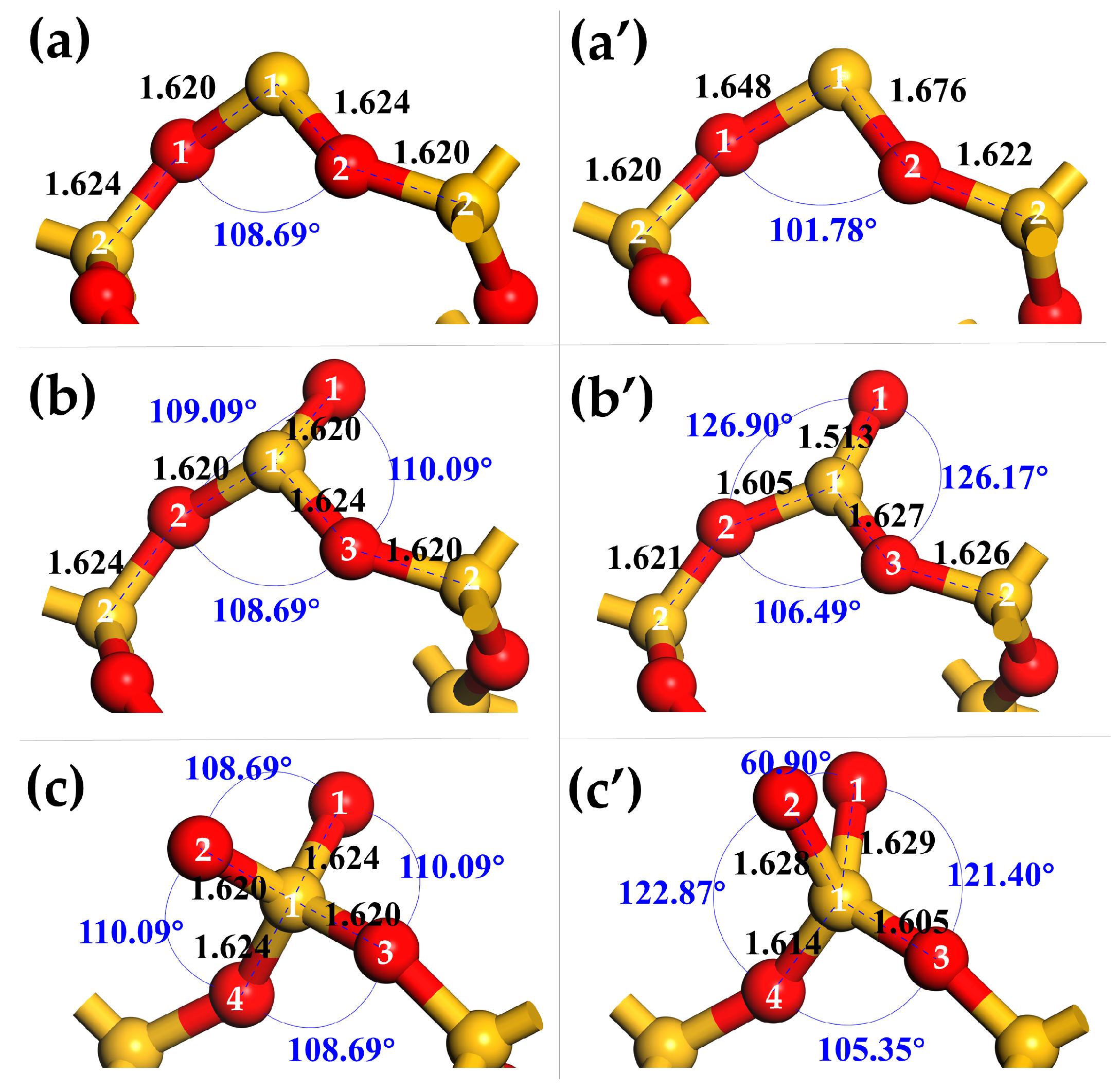
3.1.2. Surface Relaxation
3.1.3. Surface Atomic Mulliken Charge Distribution
3.1.4. Surface Atomic Mulliken Bond Population
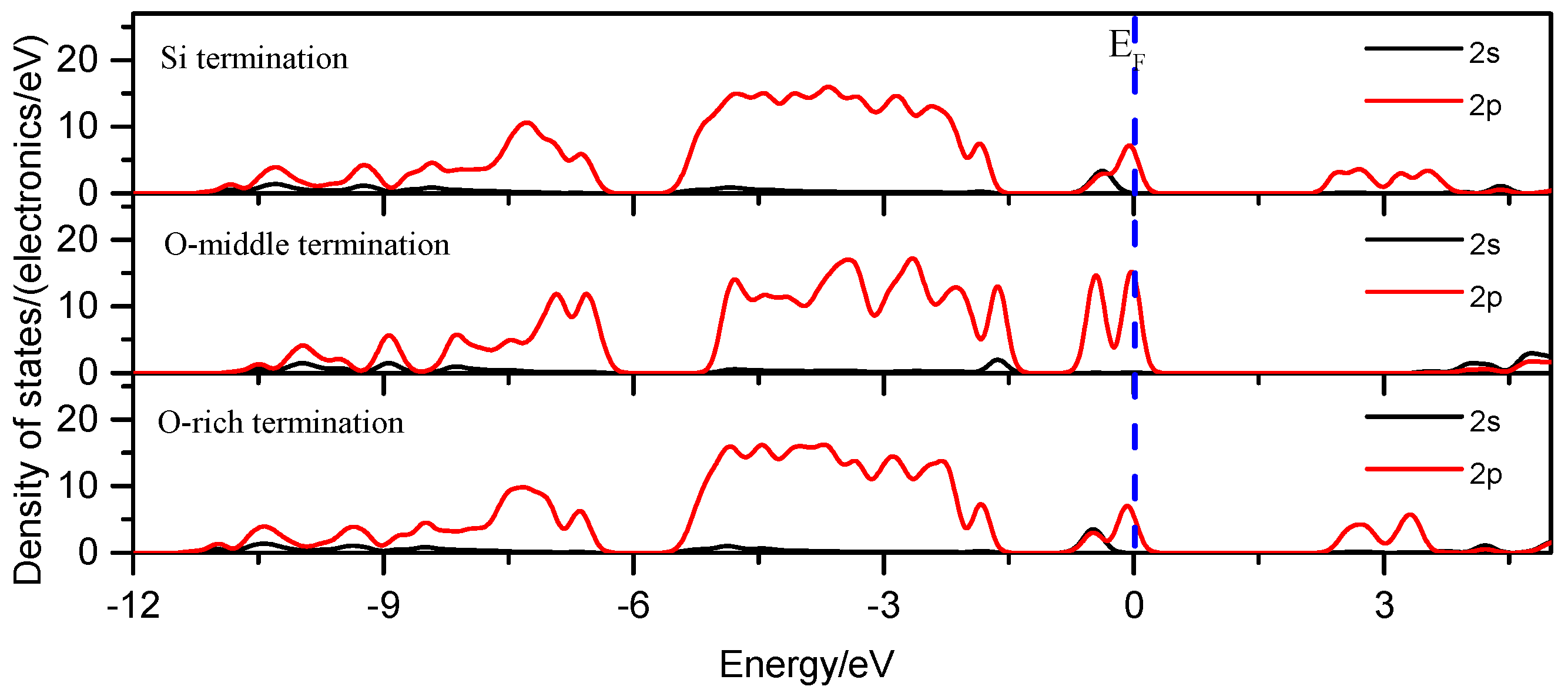
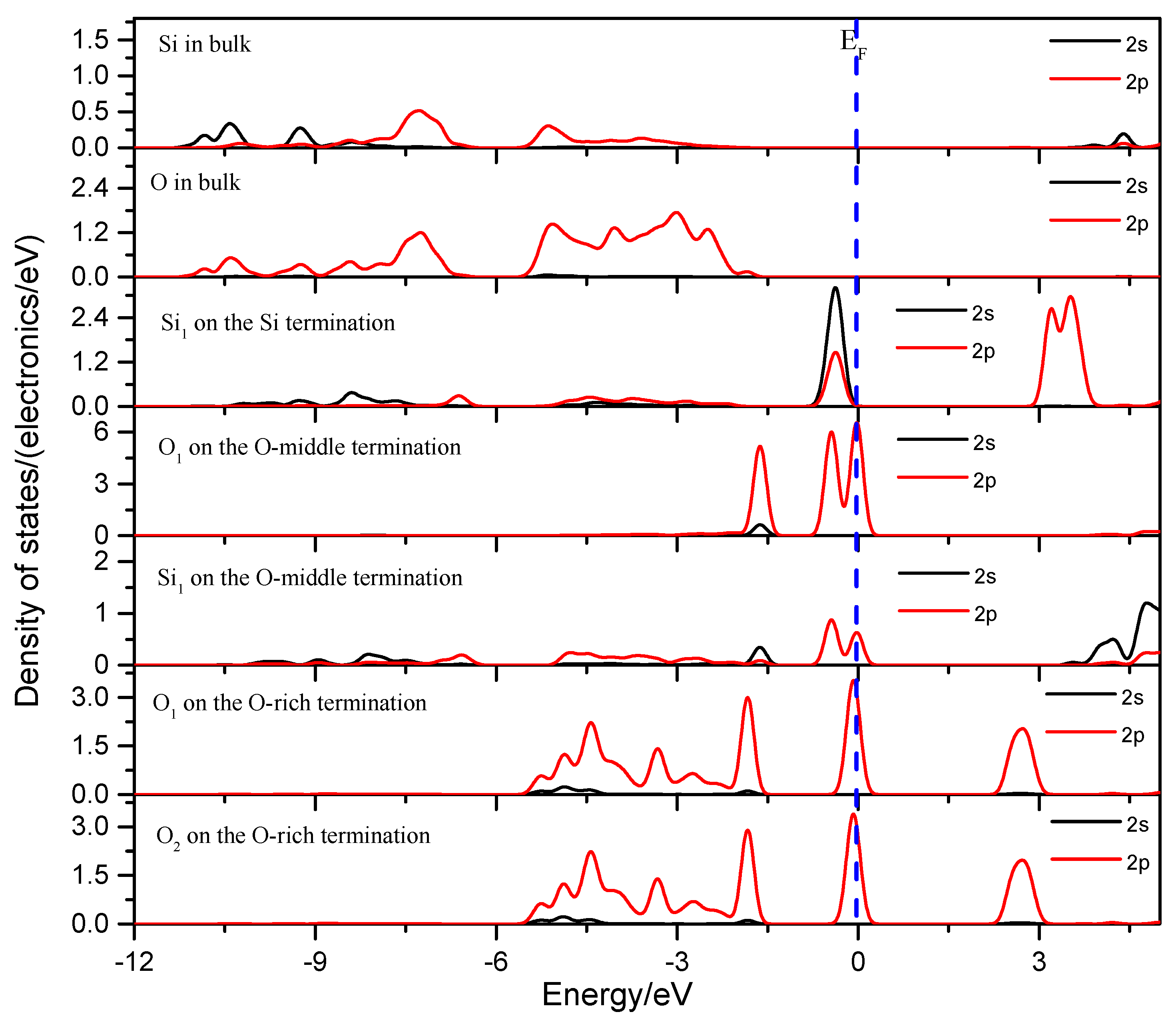
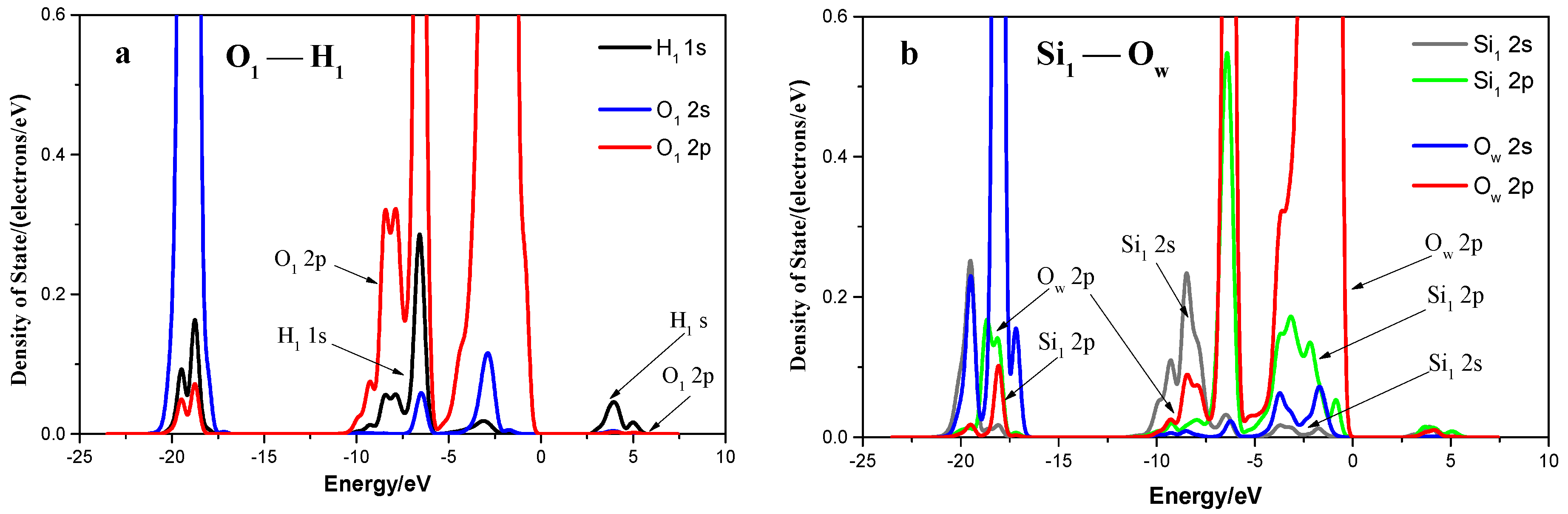
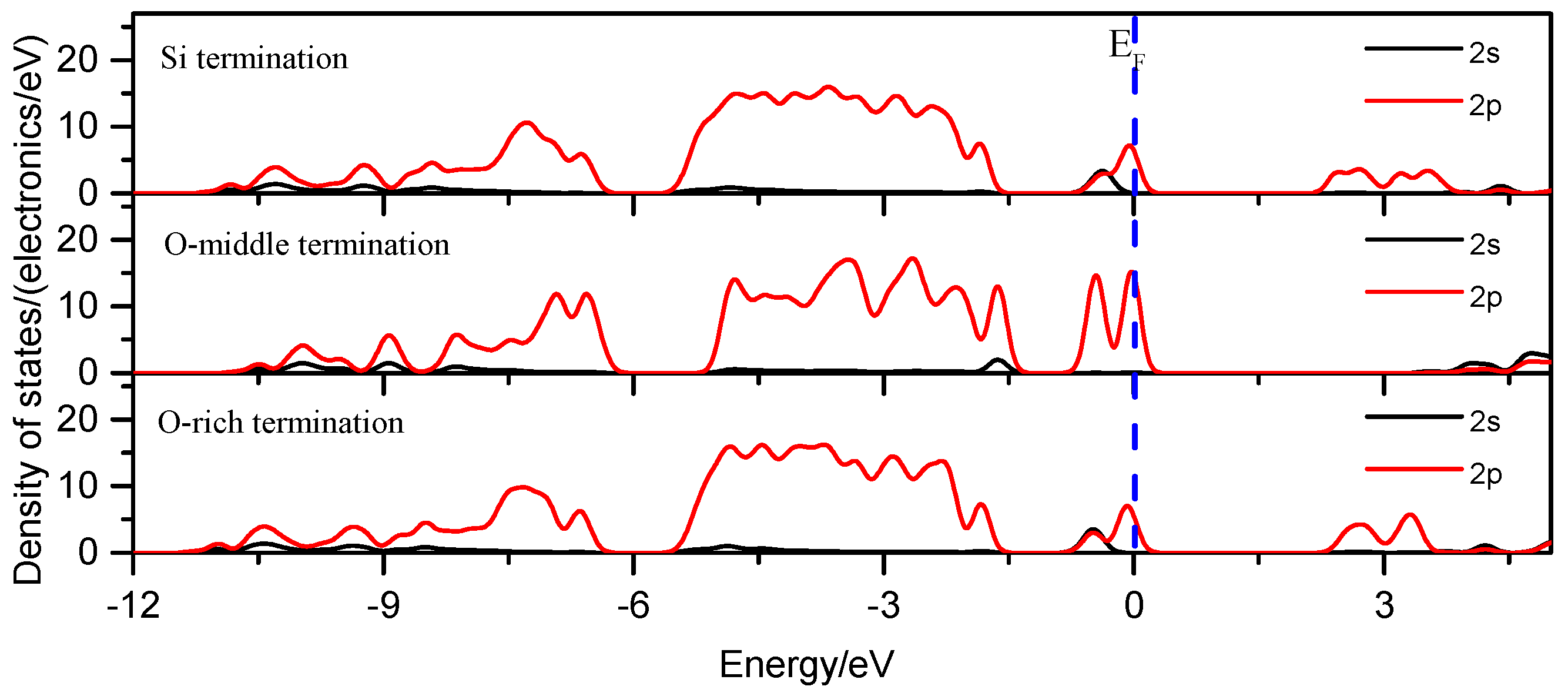
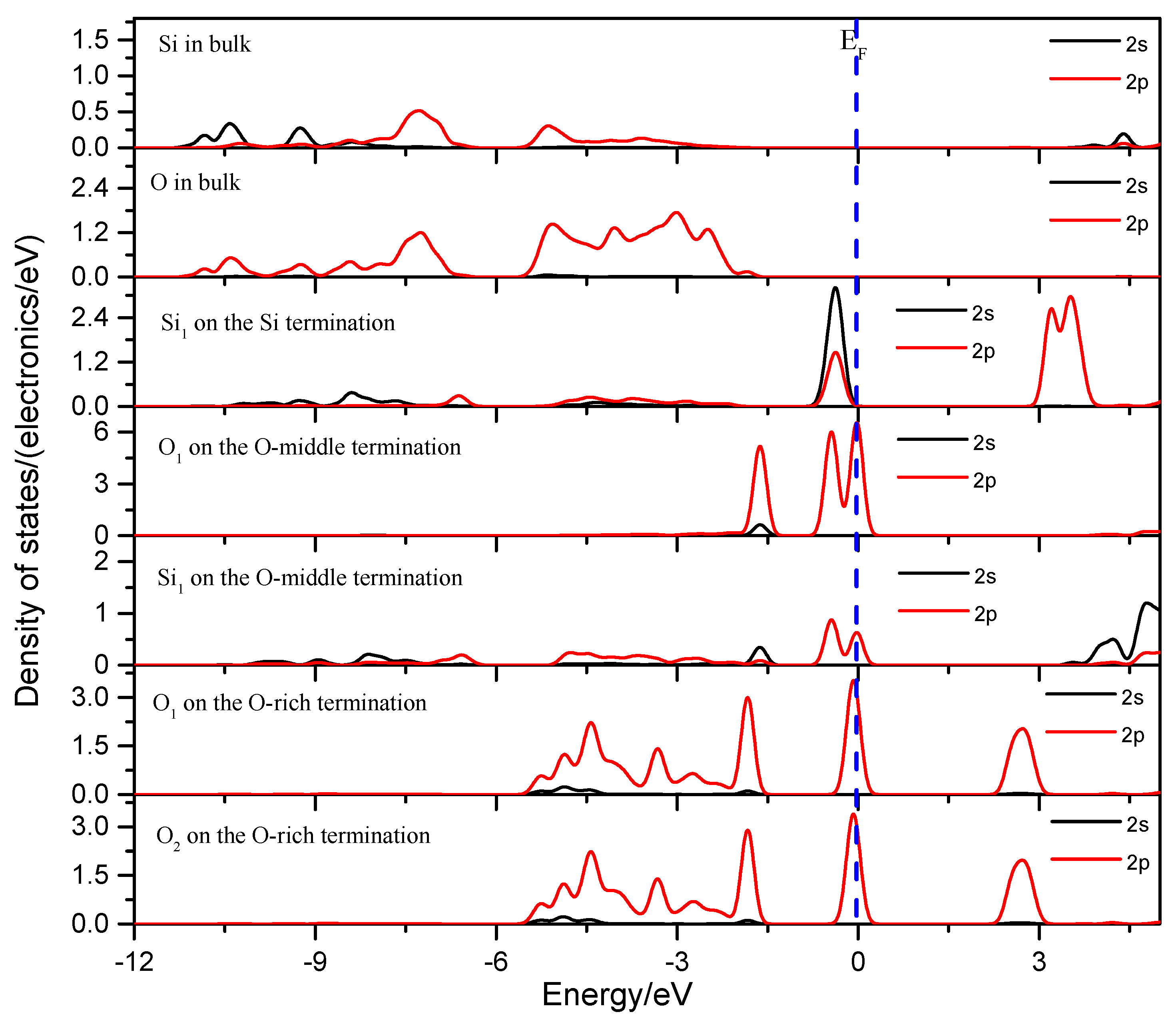
3.1.5. Density of States
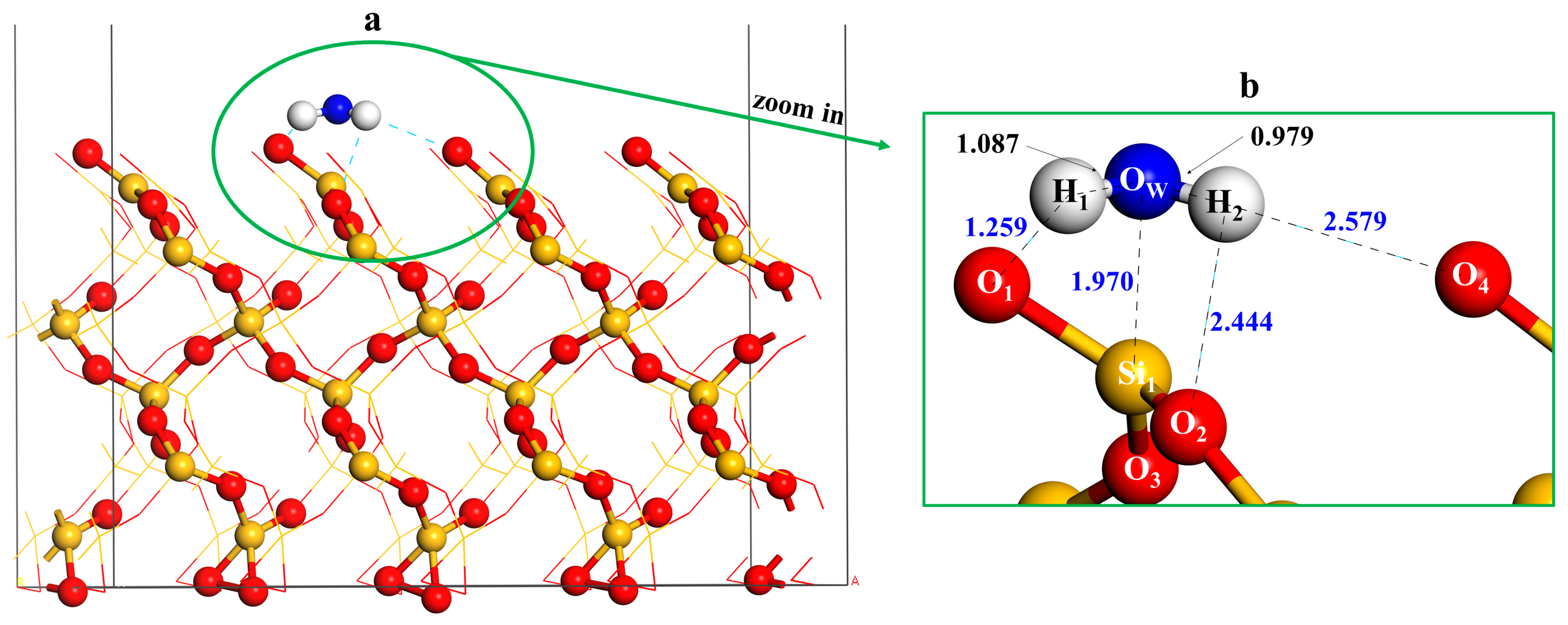
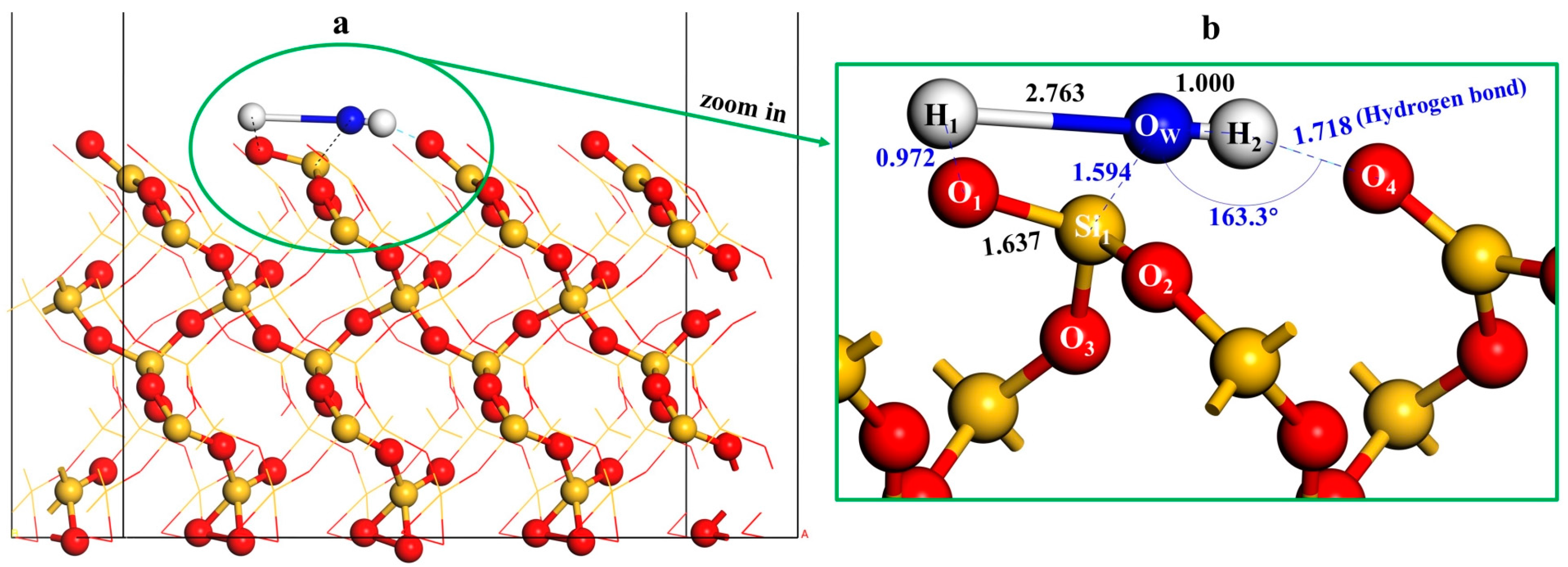
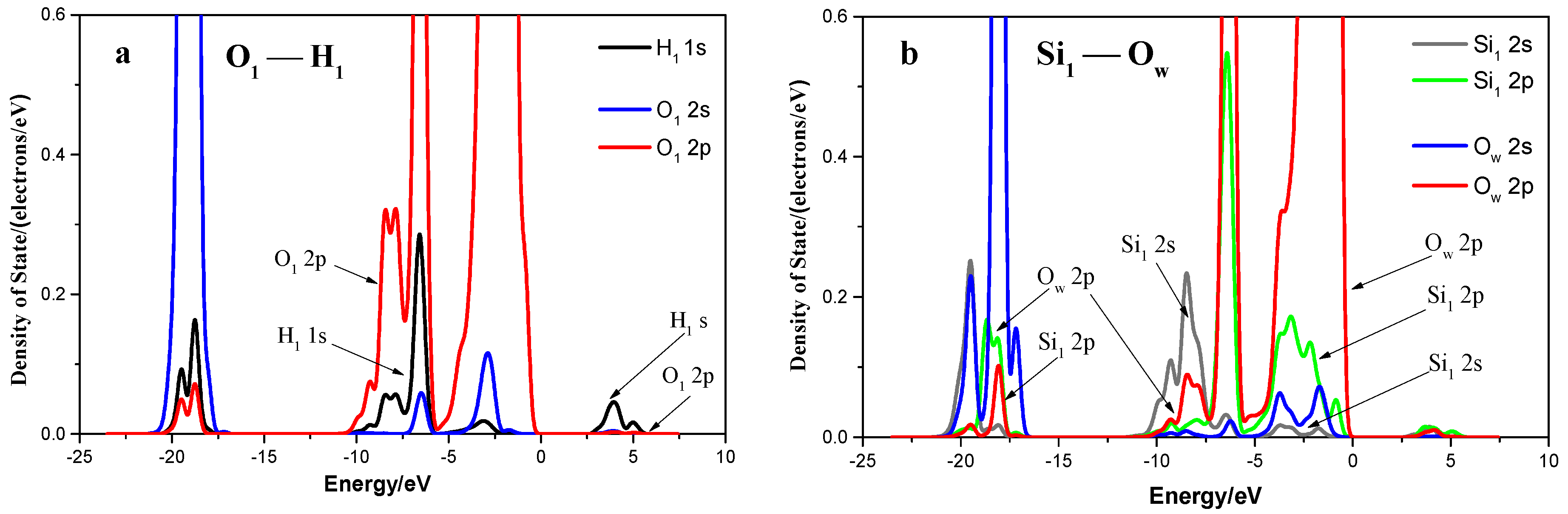
3.2. Adsorption of an Isolated Water Molecule on Quartz (001) Surface
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Su, J.W.; Esmaeilzadeh, H.; Zhang, F.; Yu, Q.; Cernigliaro, G.; Xu, J.; Sun, H.W. An ultrasensitive micropillar-based quartz crystal microbalance device for real-time measurement of protein immobilization and protein-protein interaction. Biosens. Bioelectron. 2018, 99, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Koenig, M.; Rodenhausen, K.B.; Eichhorn, K.; Oertel, U.; Schubert, M.; Stamm, M.; Uhlmann, P. Quartz crystal microbalance with coupled spectroscopic ellipsometry-study of temperature-responsive polymer brush systems. Appl. Surf. Sci. 2017, 421, 843–851. [Google Scholar] [CrossRef]
- Kim, B.; Jahng, J.; Khan, R.M.; Park, S.; Potma, E.O. Eigenmodes of a quartz tuning fork and their application to photoinduced force microscopy. Phys. Rev. B 2017, 95, 75440. [Google Scholar] [CrossRef]
- Meng, Q.Y.; Yuan, Z.T.; Feng, Q.M.; Ou, L.M. The effect of quartz on the flotation of fine wolframite with octyl hydroxamic acid. Minerals 2017, 7, 186. [Google Scholar] [CrossRef]
- Liu, A.; Fan, M.Q.; Fan, P.P. Interaction mechanism of miscible DDA–Kerosene and fine quartz and its effect on the reverse flotation of magnetic separation concentrate. Miner. Eng. 2014, 65, 41–50. [Google Scholar] [CrossRef]
- Chen, Y.W.; Cheng, H.P. Structure and stability of thin water films on quartz surfaces. Appl. Phys. Lett. 2010, 97, 161909. [Google Scholar] [CrossRef]
- Zhao, C.H.; Chen, J.H.; Li, Y.Q.; Huang, D.W.; Li, W.Z. DFT study of interactions between calcium hydroxyl ions and pyrite, marcasite, pyrrhotite surfaces. Appl. Surf. Sci. 2015, 355, 577–581. [Google Scholar] [CrossRef]
- Liu, A.; Fan, M.; Li, Z.; Fan, J. Non-polar oil assisted DDA flotation of quartz I: Interfacial interaction between dodecane oil drop and mineral particle. Int. J. Miner. Process. 2017, 168, 1–8. [Google Scholar] [CrossRef]
- Liu, A.; Fan, M.; Li, Z.; Fan, J. Non-polar oil assisted DDA flotation of quartz II: Effect of different polarity oil components on the flotation of quartz. Int. J. Miner. Process. 2017, 168, 25–34. [Google Scholar] [CrossRef]
- Li, X.B.; Zhang, Q.; Hou, B.; Ye, J.J.; Mao, S. Flotation separation of quartz from collophane using an amine collector and its adsorption mechanisms. Powder Technol. 2017, 318, 224–229. [Google Scholar] [CrossRef]
- Ulusoy, U.; Yekeler, M.; Hiçyılmaz, C. Determination of the shape, morphological and wettability properties of quartz and their correlations. Miner. Eng. 2003, 16, 951–964. [Google Scholar] [CrossRef]
- Luo, B.B.; Zhu, Y.M.; Sun, C.Y.; Li, Y.J.; Han, Y.X. Flotation and adsorption of a new collector α-Bromodecanoic acid on quartz surface. Miner. Eng. 2015, 77, 86–92. [Google Scholar] [CrossRef]
- Steurer, W.; Apfolter, A.; Koch, M.; Sarlat, T.; Søndergård, E.; Ernst, W.E.; Holst, B. The structure of the α-quartz (0001) surface investigated using helium atom scattering and atomic force microscopy. Surf. Sci. 2007, 601, 4407–4411. [Google Scholar] [CrossRef]
- Kawasaki, M.; Onuma, K.; Sunagawa, I. Morphological instabilities during growth of a rough interface: AFM observations of cobbles on the (0001) face of synthetic quartz crystals. J. Cryst. Growth 2003, 258, 188–196. [Google Scholar] [CrossRef]
- Geatches, D.L.; Clark, S.J.; Greenwell, H.C. Role of clay minerals in Oil-Forming reactions. J. Phys. Chem. A 2010, 114, 3569–3575. [Google Scholar] [CrossRef] [PubMed]
- Prigiobbe, V.; Negreira, A.S.; Lim, D.; Wilcox, J. Density functional theory calculations of the interaction of olivine with water. Energy Procedia 2013, 37, 5875–5883. [Google Scholar] [CrossRef]
- Escamilla-Roa, E.; Huertas, F.; Hernández-Laguna, A.; Sainz-Díaz, C. A DFT study of the adsorption of glycine in the interlayer space of montmorillonite. Phys. Chem. Chem. Phys. 2017, 19, 14961–14971. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Liguori, S.; Jew, A.D.; Brown, G.E.J.; Wilcox, J. Theoretical and experimental investigations of mercury adsorption on hematite surfaces. J. Air Waste Manag. Assoc. 2018, 68, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.H.; Guo, J. A DFT study on the effect of lattice impurities on the electronic structures and floatability of sphalerite. Miner. Eng. 2010, 23, 1120–1130. [Google Scholar] [CrossRef]
- Long, X.H.; Chen, J.H.; Chen, Y. Adsorption of ethyl xanthate on ZnS (110) surface in the presence of water molecules: A DFT study. Appl. Surf. Sci. 2016, 370, 11–18. [Google Scholar] [CrossRef]
- Qiu, Y.Q.; Cui, W.Y.; Li, L.J.; Ye, J.J.; Wang, J.; Zhang, Q. Structural, electronic properties with different terminations for fluorapatite (001) surface: A first-principles investigation. Comput. Mater. Sci. 2017, 126, 132–138. [Google Scholar] [CrossRef]
- Wang, F.; Li, K.; Zhou, N.G. Structural, electronic properties and stability of AlCMn3 (111) surfaces by first-principles calculations. Appl. Surf. Sci. 2014, 289, 351–357. [Google Scholar] [CrossRef]
- Chen, Y.W.; Cao, C.; Cheng, H.P. Finding stable α-quartz (0001) surface structures via simulations. Appl. Phys. Lett. 2008, 93, 181911. [Google Scholar] [CrossRef]
- Goumans, T.P.M.; Wander, A.; Brown, W.A.; Catlow, C.R.A. Structure and stability of the (001) α-quartz surface. Phys. Chem. Chem. Phys. 2007, 9, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Wang, E.G. Water adsorption on hydroxylated alpha-quartz (0001) surfaces: From monomer to flat bilayer. Phys. Rev. B 2006, 73, 035406. [Google Scholar] [CrossRef]
- Wander, M.C.F.; Clark, A.E. Structural and dielectric properties of quartz—Water interfaces. J. Phys. Chem. C 2008, 112, 19986–19994. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Qiu, Y.Q.; Li, L.J.; Ye, J.J.; Cui, W.Y. The first principles of the crystal structure and active sites of calcite. Chin. J. Eng. 2017, 39, 487–493. [Google Scholar]
- Deng, J.S.; Wen, S.M.; Liu, J.; Wu, D.D.; Feng, Q.C. Adsorption and activation of copper ions on chalcopyrite surfaces: A new viewpoint of self-activation. Trans. Nonferrous Met. Soc. China 2014, 24, 3955–3963. [Google Scholar] [CrossRef]
- Chen, J.H.; Lan, L.H.; Chen, Y. Computational simulation of adsorption and thermodynamic study of xanthate, dithiophosphate and dithiocarbamate on galena and pyrite surfaces. Miner. Eng. 2013, 46–47, 136–143. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.H. The first-principle study of the effect of lattice impurity on adsorption of CN on sphalerite surface. Miner. Eng. 2010, 23, 676–684. [Google Scholar] [CrossRef]
- Chen, J.H.; Long, X.H.; Chen, Y. Comparison of multilayer water adsorption on the hydrophobic galena (PbS) and hydrophilic pyrite (FeS2) surfaces: A DFT study. J. Phys. Chem. C 2014, 118, 11657–11665. [Google Scholar] [CrossRef]
- Chen, J.H.; Long, X.H.; Zhao, C.H.; Kang, D.; Guo, J. DFT calculation on relaxation and electronic structure of sulfide minerals surfaces in presence of H2O molecule. J. Cent. South Univ. 2014, 21, 3945–3954. [Google Scholar] [CrossRef]
- Long, X.H.; Chen, Y.; Chen, J.H.; Xu, Z.H.; Liu, Q.X.; Du, Z. The effect of water molecules on the thiol collector interaction on the galena (PbS) and sphalerite (ZnS) surfaces: A DFT study. Appl. Surf. Sci. 2016, 389, 103–111. [Google Scholar] [CrossRef]
- Rignanese, G.M.; Charlier, J.C.; Gonze, X. First-principles molecular-dynamics investigation of the hydration mechanisms of the (0001) α-quartz surface. Phys. Chem. Chem. Phys. 2004, 6, 1920–1925. [Google Scholar] [CrossRef]
- Du, Z.M.; de Leeuw, N.H. Molecular dynamics simulations of hydration, dissolution and nucleation processes at the α-quartz (0001) surface in liquid water. Dalton Trans. 2006, 22, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Gor, G.Y.; Bernstein, N. Adsorption-Induced surface stresses of the Water/Quartz interface: Ab initio molecular dynamics study. Langmuir 2016, 32, 5259–5266. [Google Scholar] [CrossRef] [PubMed]
- Notman, R.; Walsh, T.R. Molecular dynamics studies of the interactions of water and amino acid analogues with quartz surfaces. Langmuir 2009, 25, 1638–1644. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift für Kristallographie 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Kihara, K. An X-ray study of the temperature dependence of the quartz structure. Eur. J. Mineral. 1990, 2, 63–78. [Google Scholar] [CrossRef]
- Chen, J.H. The Solid Physics of Sulphide Minerals Flotation; Central South University Press: Changsha, China, 2015. [Google Scholar]
- Zhao, C.H.; Chen, J.H.; Long, X.H.; Guo, J. Study of H2O adsorption on sulfides surfaces and thermokinetic analysis. J. Ind. Eng. Chem. 2014, 20, 605–609. [Google Scholar] [CrossRef]
- Chen, J.; Ke, B.; Lan, L.; Li, Y. Influence of Ag, Sb, Bi and Zn impurities on electrochemical and flotation behaviour of galena. Miner. Eng. 2015, 72, 10–16. [Google Scholar] [CrossRef]
- Jiang, W.; Gao, Z.Y.; Sun, W.; Gao, J.; Hu, Y.H. A Density Functional Theory Study on the Effect of Lattice Impurities on the Electronic Structures and Reactivity of Fluorite. Minerals 2017, 7, 160. [Google Scholar] [CrossRef]
- Chen, Y.W.; Chu, I.H.; Wang, Y.; Cheng, H.P. Water thin film-silica interaction on α-quartz (0001) surfaces. Phys. Rev. B 2011, 84, 155444. [Google Scholar] [CrossRef]
- Pan, D.; Liu, L.M.; Tribello, G.A.; Slater, B.; Michaelides, A.; Wang, E. Surface energy and surface proton order of ice Ih. Phys. Rev. Lett. 2008, 101, 155703. [Google Scholar] [CrossRef] [PubMed]
- Feibelman, P.J. Partial dissociation of water. Science 2002, 295, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.J.; David, S.S. Enantiospecific adsorption of amino acids on hydroxylated quartz (1010). Phys. Chem. Chem. Phys. 2010, 12, 8016–8023. [Google Scholar]
- Van Erp, T.S.; Meijer, E.J. Hydration of methanol in water. A DFT-based molecular dynamics study. Chem. Phys. Lett. 2001, 333, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Xu, L.F.; Wang, E.G.; Gao, S. Vibrational recognition of Hydrogen-Bonded water networks on a metal surface. Phys. Rev. Lett. 2002, 89, 176104. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Meng, S.; Xu, L.F.; Wang, E.G. Ice tessellation on a hydroxylated silica surface. Phys. Rev. Lett. 2004, 92, 146102. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Chen, J.H.; Chen, Y.; Guo, J. Density functional theory study of influence of impurity on electronic properties and reactivity of pyrite. Trans. Nonferrous Met. Soc. 2011, 21, 1887–1895. [Google Scholar] [CrossRef]
- Zhao, C.H.; Chen, J.H.; Wu, B.Z.; Long, X.H. Density functional theory study on natural hydrophobicity of sulfide surfaces. Trans. Nonferrous Met. Soc. 2014, 24, 491–498. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atomic Layer Number | Surface Energy/(J·m−2) | ||
|---|---|---|---|
| Si Termination | O-Middle Termination | O-Rich Termination | |
| 9 | 2.928 | 1.999 | 2.920 |
| 12 | 3.124 | 2.000 | 2.915 |
| 15 | 2.898 | 1.997 | 2.907 |
| 18 | 2.896 | 1.995 | 2.896 |
| 21 | 2.896 | 1.995 | 2.893 |
| 24 | 2.897 | 1.999 | 2.899 |
| Vacuum Thickness/Å | Surface Energy/(J·m−2) | ||
|---|---|---|---|
| Si Termination | O-Middle Termination | O-Rich Termination | |
| 8 | 2.905 | 1.986 | 2.894 |
| 10 | 2.896 | 1.995 | 2.896 |
| 15 | 2.896 | 1.969 | 2.892 |
| 18 | 2.899 | 1.969 | 2.894 |
| 20 | 2.894 | 1.972 | 2.892 |
| Atoms | Si Termination | O-Middle Termination | O-Rich Termination | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C* | x | y | z | C* | x | y | z | C* | x | y | z | |
| O1 | 2 | 0.12 | 0.07 | −0.02 | 1 | 0.07 | 0.05 | 0.20 | 1 | −2.24 | 0.06 | −0.11 |
| O2 | 2 | −0.11 | −0.21 | −0.01 | 2 | 0.58 | 0.17 | 0.05 | 1 | 2.11 | 0.49 | 0.21 |
| O3 | 2 | 0.12 | −0.03 | 0.03 | 2 | −0.10 | −0.29 | −0.05 | 2 | 0.40 | 0.69 | −0.35 |
| O4 | 2 | 0.06 | 0.04 | 0.01 | 2 | 0.26 | −0.30 | −0.04 | 2 | −0.77 | 0.02 | −0.21 |
| O5 | 2 | 0.03 | −0.11 | 0.06 | 2 | −0.01 | 0.00 | 0.04 | 2 | −0.25 | −0.46 | −0.22 |
| O6 | 2 | 0.11 | 0.04 | 0.03 | 2 | −0.05 | −0.32 | 0.04 | 2 | −0.40 | −0.17 | −0.08 |
| O7 | 2 | 0.05 | 0.08 | 0.01 | 2 | 0.20 | 0.20 | −0.03 | 2 | 0.33 | −0.61 | −0.12 |
| O8 | 2 | 0.41 | 0.22 | 0.09 | 2 | −0.01 | 0.03 | −0.05 | 2 | 0.63 | −0.04 | 0.11 |
| Si1 | 2 | −2.76 | −0.29 | 0.05 | 4 | −1.81 | −0.77 | −0.18 | 4 | −0.04 | 0.23 | −0.31 |
| Si2 | 4 | 0.08 | −0.06 | 0.00 | 4 | 0.19 | 0.02 | 0.01 | 4 | −0.23 | 0.07 | −0.22 |
| Si3 | 4 | 0.10 | −0.02 | 0.24 | 4 | 0.18 | −0.02 | 0.01 | 4 | 0.08 | −0.29 | −0.09 |
| Si4 | 4 | 0.13 | 0.02 | 0.05 | 4 | 0.18 | 0.02 | −0.01 | 4 | 0.20 | −0.05 | 0.05 |
| Atoms | s | p | Total | Charge/e | |
|---|---|---|---|---|---|
| Bulk | Si | 0.57 | 1.07 | 1.64 | +2.36 |
| O | 1.83 | 5.35 | 7.18 | −1.18 | |
| Si termination | Si1 | 1.66 | 1.14 | 2.80 | +1.20 |
| O1 | 1.84 | 5.33 | 7.17 | −1.17 | |
| O2 | 1.84 | 5.33 | 7.17 | −1.17 | |
| Si2 | 0.57 | 1.09 | 1.66 | +2.34 | |
| O3 | 1.83 | 5.36 | 7.19 | −1.19 | |
| O-middle termination | O1 | 1.94 | 5.10 | 7.04 | −1.04 |
| Si1 | 0.59 | 1.21 | 1.80 | +2.20 | |
| O2 | 1.85 | 5.34 | 7.19 | −1.18 | |
| O3 | 1.83 | 5.34 | 7.17 | −1.18 | |
| Si2 | 0.57 | 1.06 | 1.63 | +2.37 | |
| O-rich termination | O1 | 1.95 | 4.67 | 6.62 | −0.63 |
| O2 | 1.95 | 4.69 | 6.64 | −0.64 | |
| Si1 | 0.54 | 1.00 | 1.54 | +2.46 | |
| O3 | 1.83 | 5.36 | 7.19 | −1.19 | |
| O4 | 1.83 | 5.37 | 7.20 | −1.20 | |
| Termination | Bond | Surface Population (Bulk) | Bond Length (Bulk)/Å |
|---|---|---|---|
| Si | Si1–O1 | 0.27 (0.52) | 1.67 (1.624) |
| Si1–O2 | 0.52 (0.53) | 1.61 (1.632) | |
| O1–Si2 | 0.4 (0.51) | 1.61 (1.625) | |
| O2–Si2 | 0.5 (0.63) | 1.62 (1.622) | |
| O3–Si3 | 0.5 (0.52) | 1.62 (1.617) | |
| O-middle | O1–Si1 | 0.9 (0.52) | 1.51 (1.620) |
| Si1–O2 | 0.4 (0.53) | 1.60 (1.618) | |
| Si1–O3 | 0.4 (0.53) | 1.62 (1.628) | |
| O2–Si2 | 0.5 (0.53) | 1.61 (1.631) | |
| O3–Si2 | 0.5 (0.52) | 1.62 (1.624) | |
| O-rich | O1–Si1 | 0.3 (0.53) | 1.62 (1.624) |
| O2–Si1 | 0.3 (0.52) | 1.62 (1.626) | |
| Si1–O3 | 0.4 (0.53) | 1.60 (1.616) | |
| Si1–O4 | 0.5 (0.53) | 1.61 (1.625) | |
| O3–Si2 | 0.5 (0.51) | 1.61 (1.625) |
| Atom | Adsorption | s | p | Total | Charge/e |
|---|---|---|---|---|---|
| H1 | Before | 0.48 | 0 | 0.48 | 0.52 |
| After | 0.46 | 0 | 0.46 | 0.54 | |
| H2 | Before | 0.48 | 0 | 0.48 | 0.52 |
| After | 0.50 | 0 | 0.50 | 0.50 | |
| Ow | Before | 1.89 | 5.15 | 7.05 | −1.05 |
| After | 1.85 | 5.24 | 7.09 | −1.09 | |
| O1 | Before | 1.94 | 5.10 | 7.04 | −1.04 |
| After | 1.86 | 5.26 | 7.12 | −1.12 | |
| O2 | Before | 1.85 | 5.34 | 7.19 | −1.18 |
| After | 1.83 | 5.36 | 7.19 | −1.19 | |
| O3 | Before | 1.83 | 5.34 | 7.17 | −1.18 |
| After | 1.84 | 5.36 | 7.20 | −1.20 | |
| O4 | Before | 1.94 | 5.10 | 7.04 | −1.04 |
| After | 1.91 | 5.14 | 7.06 | −1.06 | |
| Si1 | Before | 0.59 | 1.21 | 1.80 | 2.20 |
| After | 0.58 | 1.05 | 1.63 | 2.37 |
| Bond | Population | Bond Length/Å | ||
|---|---|---|---|---|
| Before | After | Before | After | |
| Si1–O1 | 0.96 | 0.51 | 1.513 | 1.637 |
| Si1–O2 | 0.42 | 0.45 | 1.600 | 1.630 |
| Si1–O3 | 0.43 | 0.45 | 1.625 | 1.645 |
| Ow–H2 | 0.51 | 0.53 | 0.978 | 1.000 |
| Si1–Ow | - | 0.59 | - | 1.594 |
| O1–H1 | - | 0.54 | - | 0.972 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Q.; Li, X.; Ye, J.; Li, L. Structural and Electronic Properties of Different Terminations for Quartz (001) Surfaces as Well as Water Molecule Adsorption on It: A First-Principles Study. Minerals 2018, 8, 58. https://doi.org/10.3390/min8020058
Wang X, Zhang Q, Li X, Ye J, Li L. Structural and Electronic Properties of Different Terminations for Quartz (001) Surfaces as Well as Water Molecule Adsorption on It: A First-Principles Study. Minerals. 2018; 8(2):58. https://doi.org/10.3390/min8020058
Chicago/Turabian StyleWang, Xianchen, Qin Zhang, Xianbo Li, Junjian Ye, and Longjiang Li. 2018. "Structural and Electronic Properties of Different Terminations for Quartz (001) Surfaces as Well as Water Molecule Adsorption on It: A First-Principles Study" Minerals 8, no. 2: 58. https://doi.org/10.3390/min8020058