Preparation of Mullite-Silica Composites Using Silica-Rich Monophasic Precursor Obtained as a Byproduct of Mineral Carbonation of Blast-Furnace Slag

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Materials
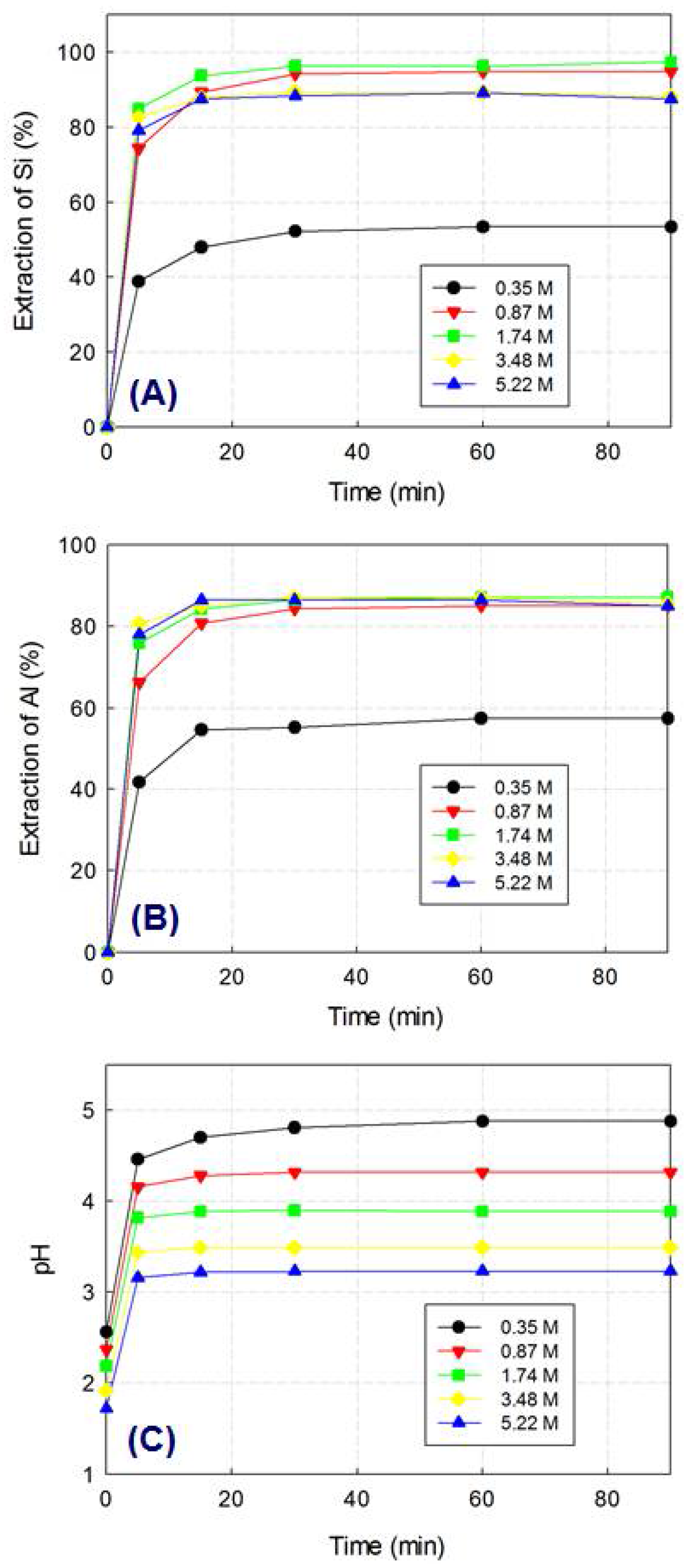
2.2. Optimization of Leaching Parameters
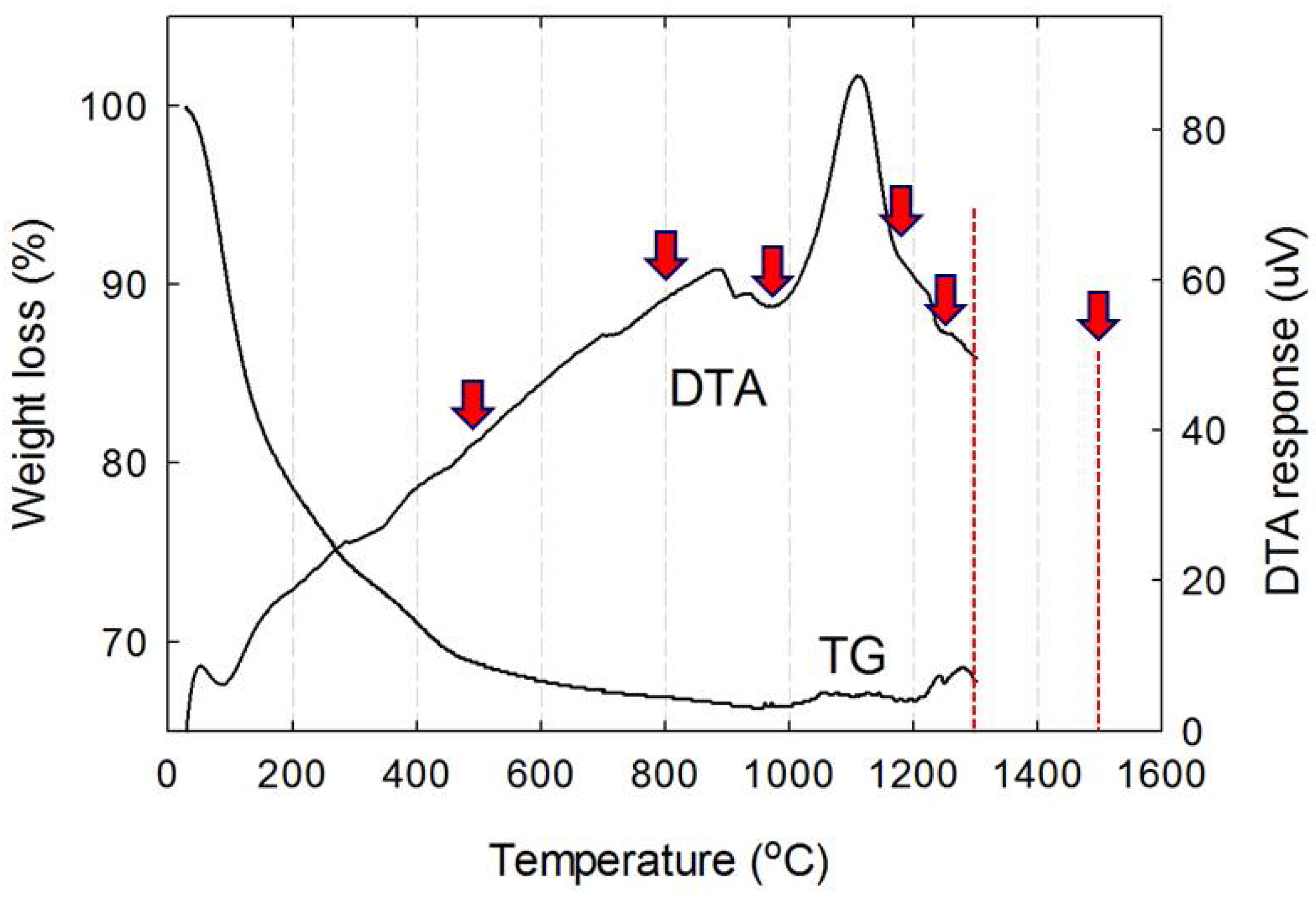
2.3. Synthesis and Calcination of the Precursor
2.4. Characterization
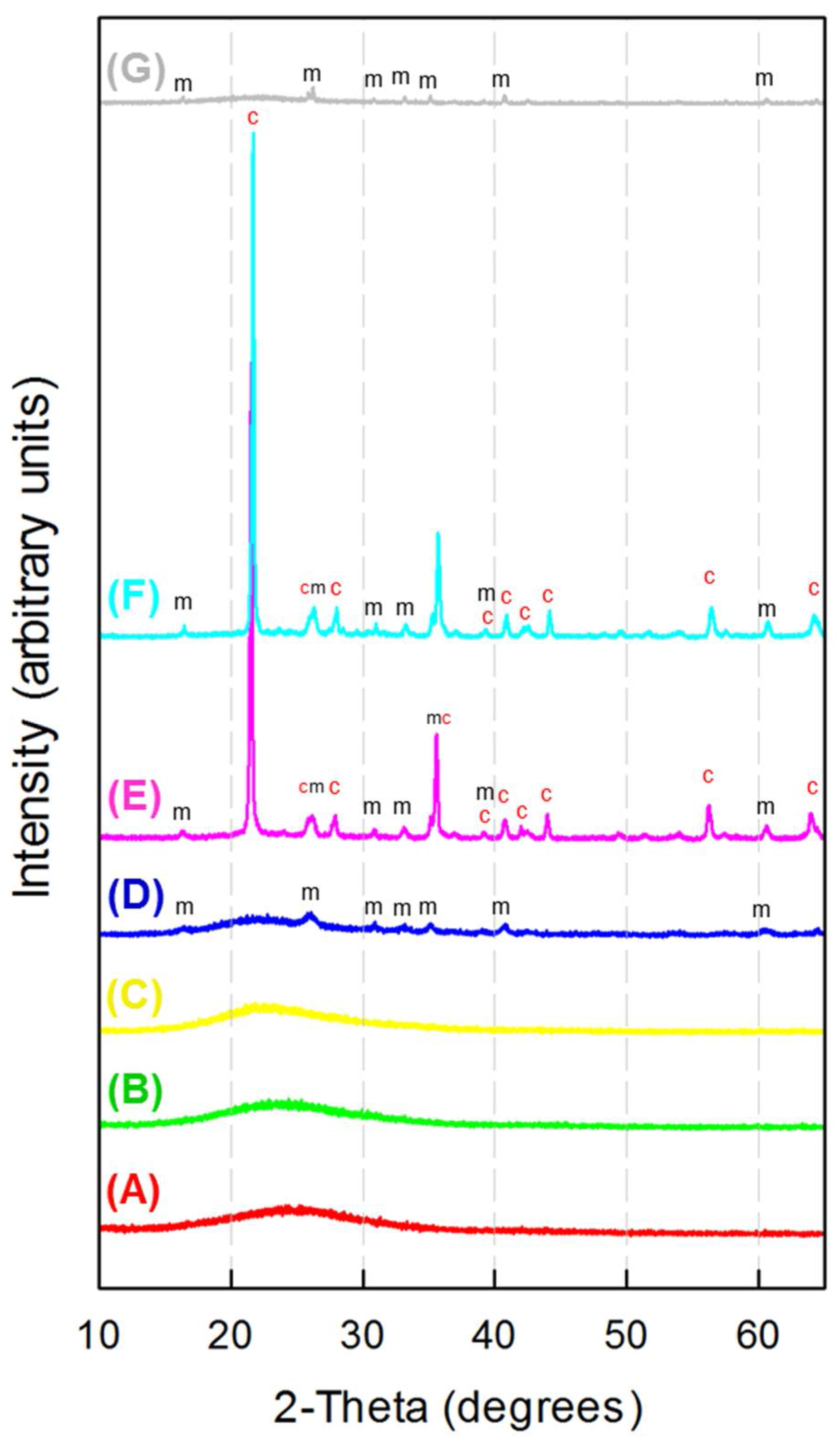
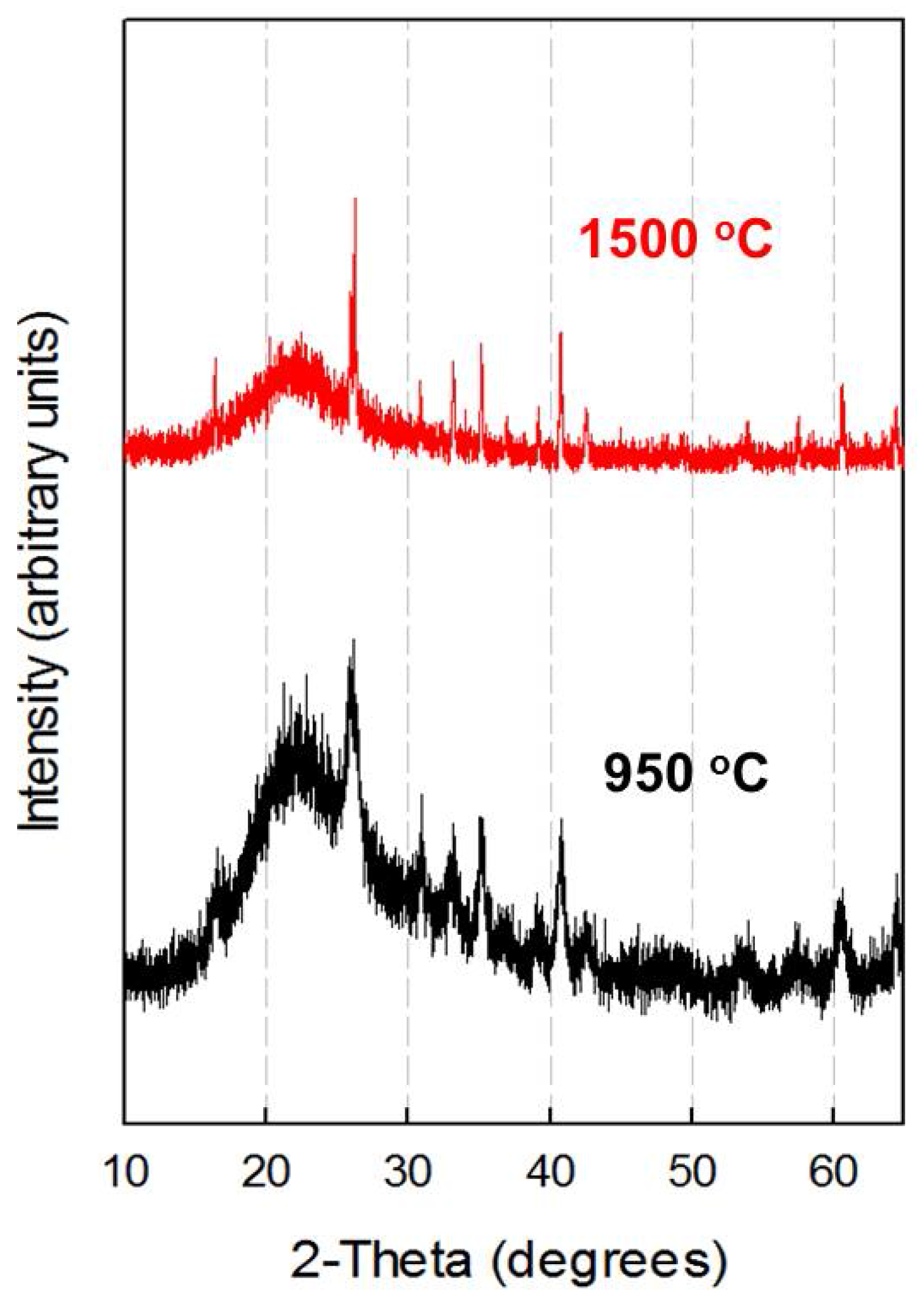
3. Results and Discussion
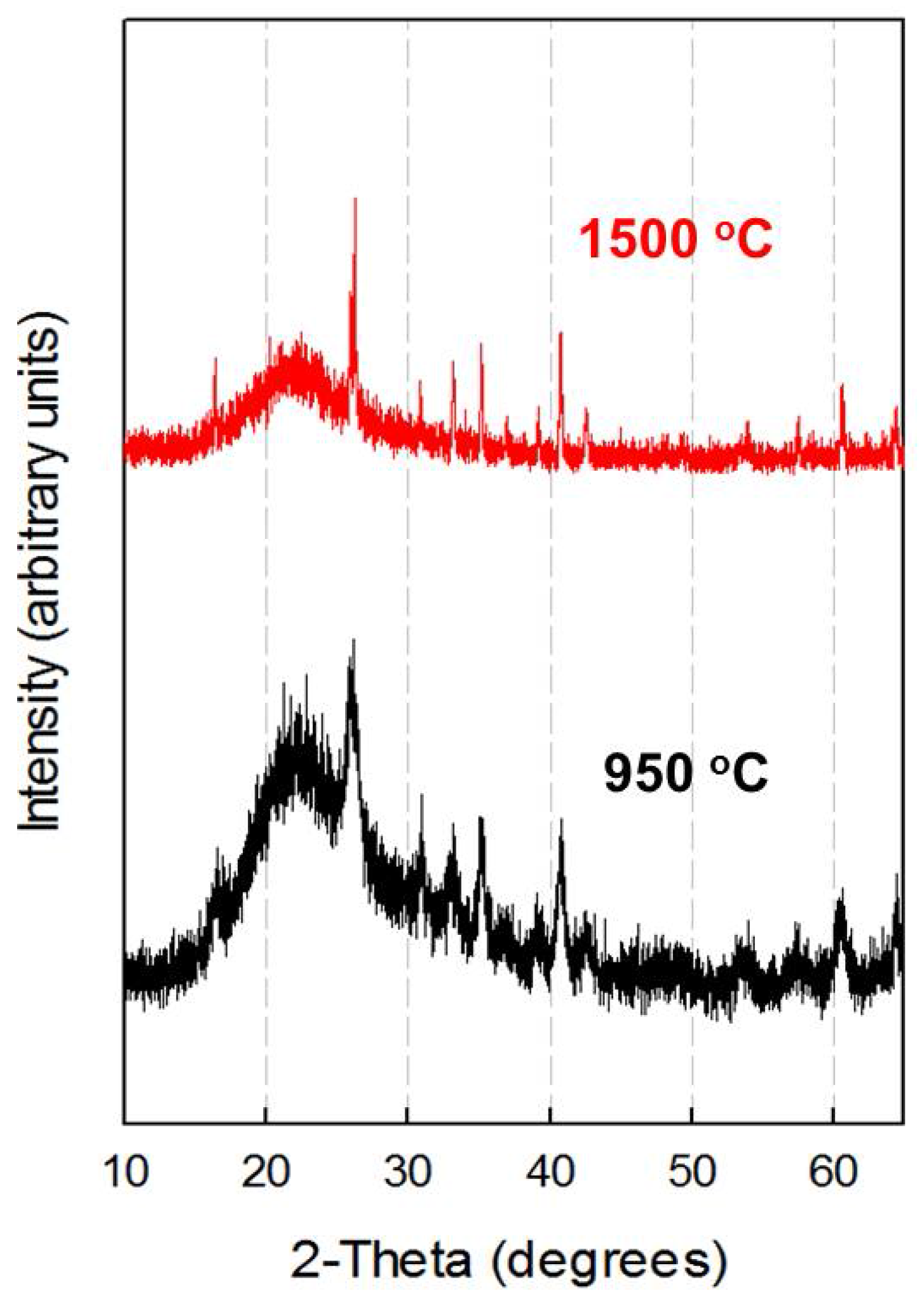
3.1. Preparation of the Monophasic Precursor from BFS
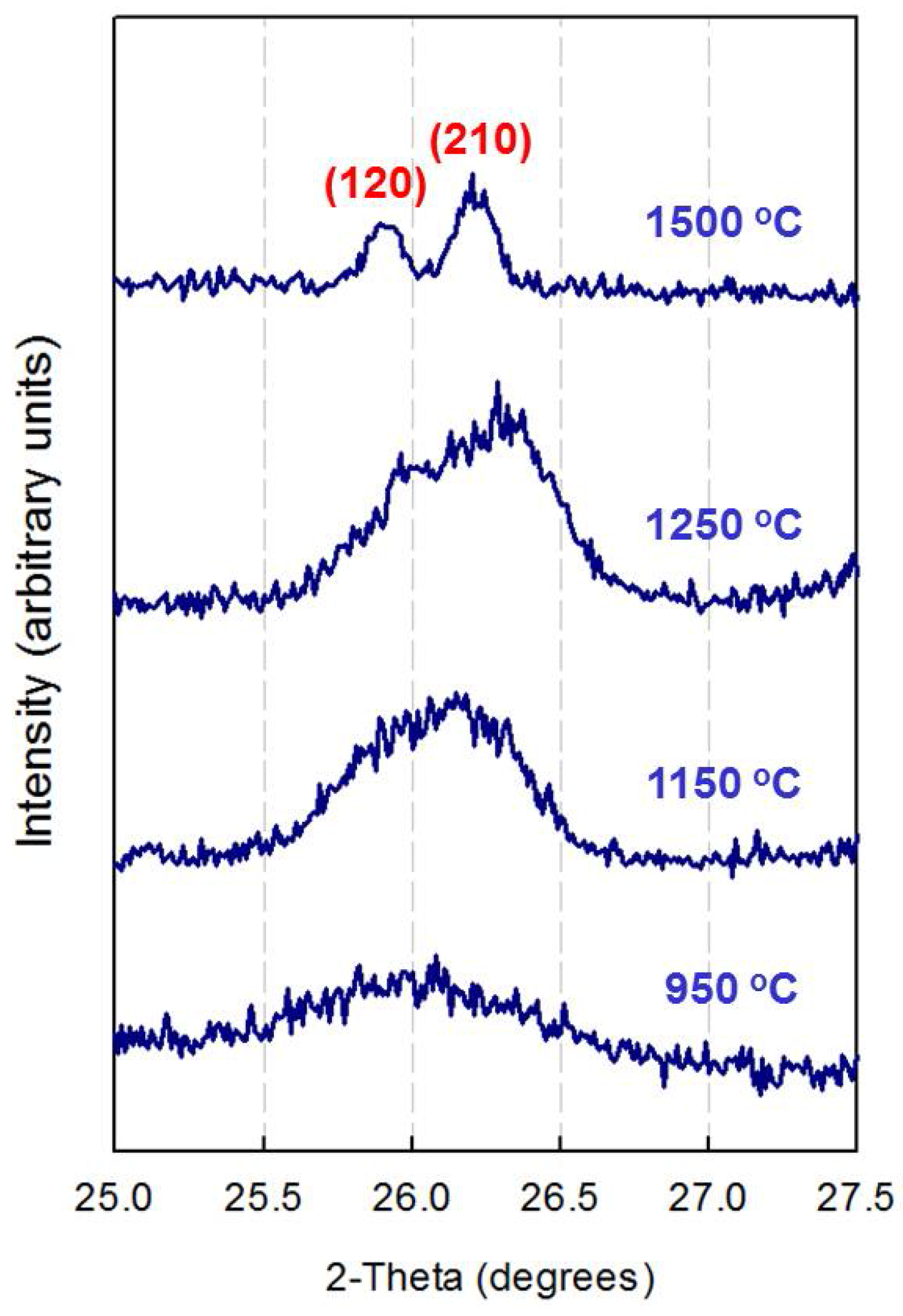
3.2. X-ray Diffraction Analysis
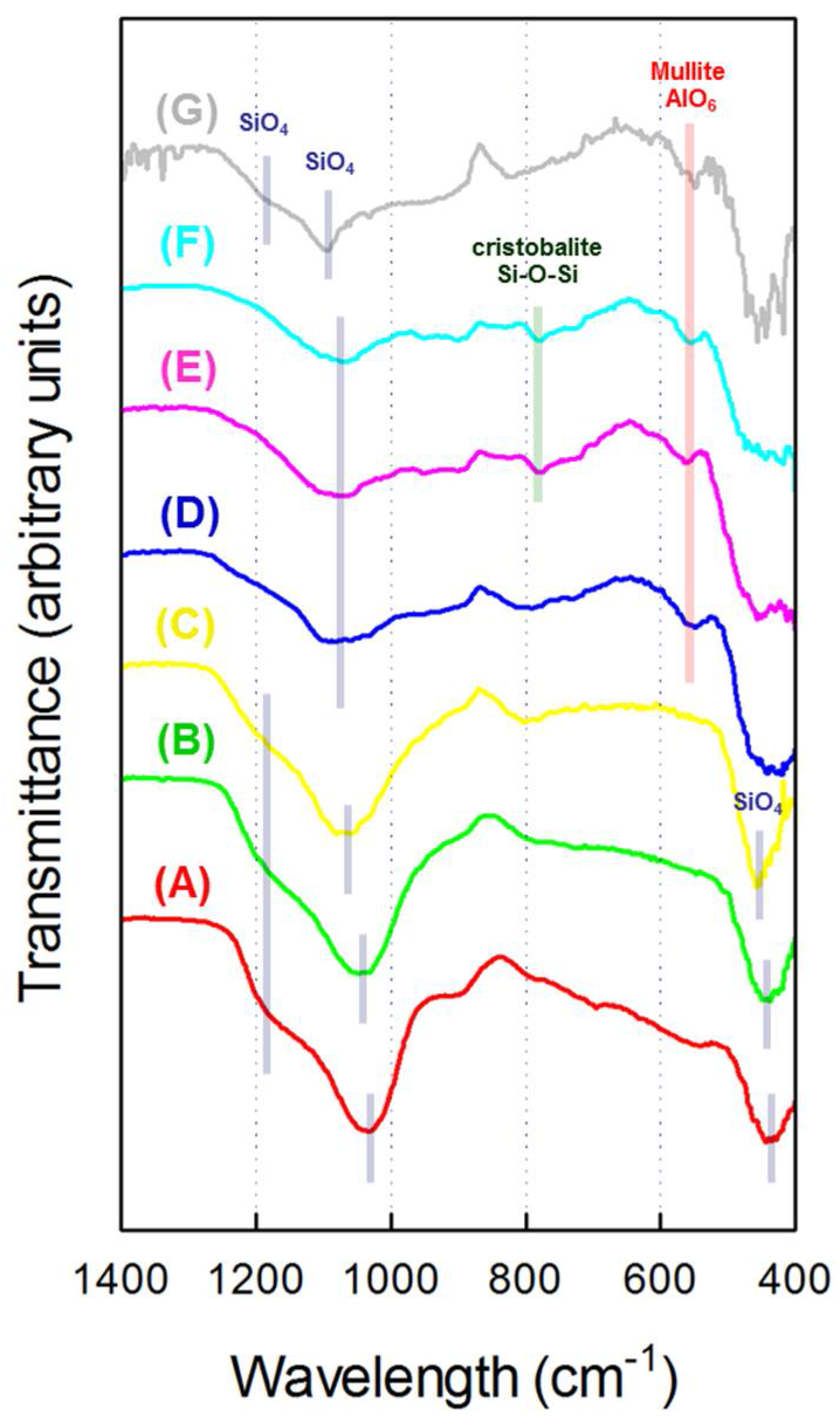
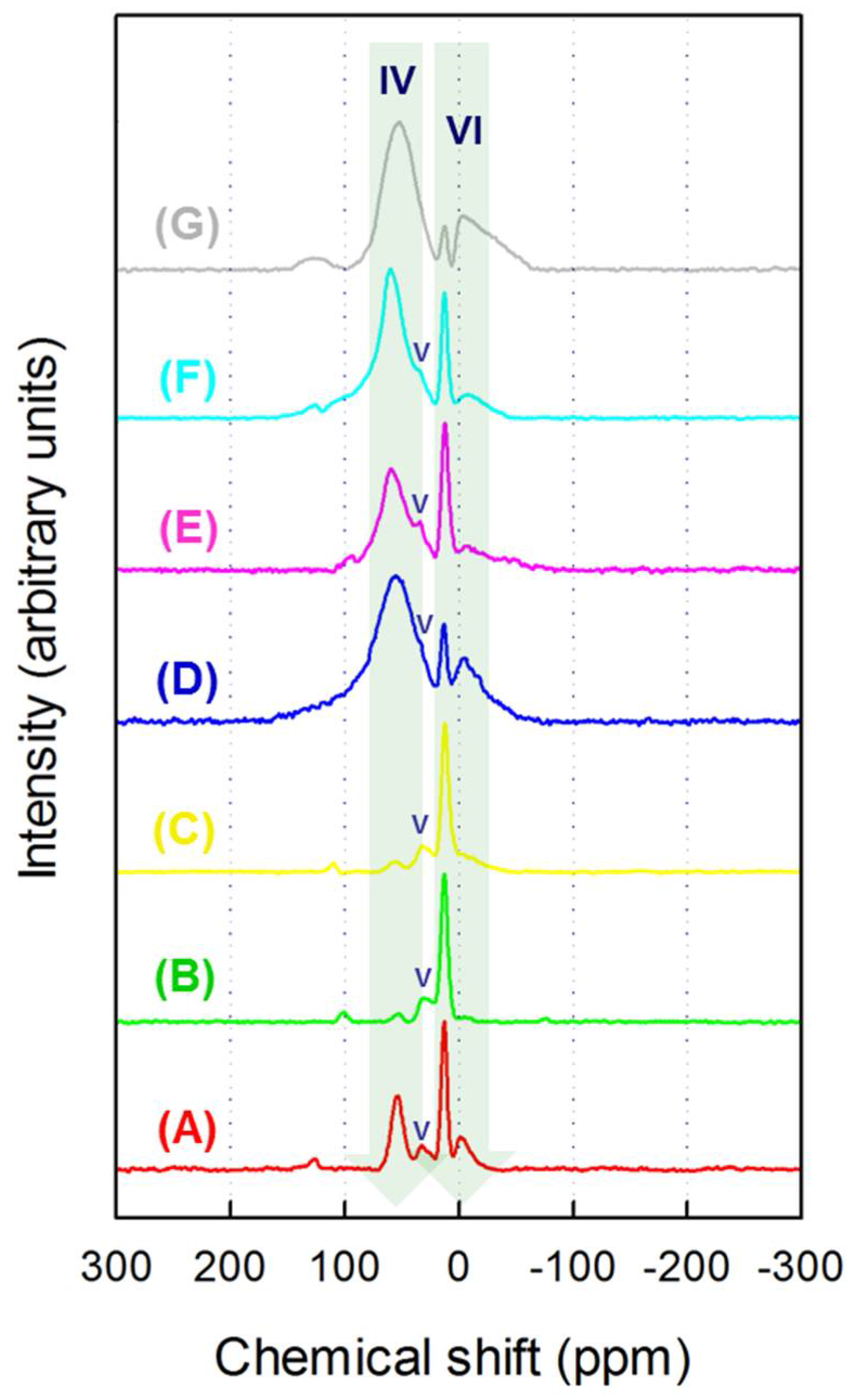
3.3. FT-IR and 27Al MAS NMR Study
3.4. Microstructural Development of Mullite-Silica Composites
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Das, B.; Prakash, S.; Reddy, P.S.R.; Misra, V.N. An overview of utilization of slag and sludge from steel industries. Resour. Conserv. Recycl. 2007, 50, 40–57. [Google Scholar] [CrossRef]
- Seifritz, W. CO2 disposal by means of silicates. Nature 1990, 345, 486. [Google Scholar] [CrossRef]
- Huijgen, W.J.J.; Comans, R.N.J. Mineral CO2 sequestration by steel slag carbonation. Environ. Sci. Technol. 2005, 39, 9676–9682. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Li, H.; Zhang, Y. Selective leaching of steelmaking slag for indirect CO2 mineral sequestration. Ind. Eng. Chem. Res. 2010, 49, 2055–2063. [Google Scholar] [CrossRef]
- De Crom, K.; Chiang, Y.W.; Van Gerven, T.; Santos, R.M. Purification of slag-derived leachate and selective carbonation for high-quality precipitated calcium carbonate synthesis. Chem. Eng. Res. Des. 2015, 104, 180–190. [Google Scholar] [CrossRef]
- Teir, S.; Eloneva, S.; Fogelholm, C.-J.; Zevenhoven, R. Dissolution of steelmaking slags in acetic acid for precipitated calcium carbonate production. Energy 2007, 32, 528–539. [Google Scholar] [CrossRef]
- Chiang, Y.W.; Santos, R.M.; Elsen, J.; Meesschaert, B.; Martens, J.A.; Van Gerven, T. Towards zero-waste mineral carbon sequestration via two-way valorization of ironmaking slag. Chem. Eng. J. 2014, 249, 260–269. [Google Scholar] [CrossRef]
- Sadik, C.; El Amrani, I.-E.; Albizane, A. Recent advances in silica-alumina refractory: A review. J. Asian Ceram. Soc. 2014, 2, 83–96. [Google Scholar] [CrossRef]
- Aksay, İ.A.; Pask, J.A. The silica-alumina system: Stable and metastable equilibria at 1.0 atmosphere. Science 1974, 183, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Sainz, M.A.; Serrano, F.J.; Amigo, J.M.; Bastida, J.; Caballero, A. XRD microstructural analysis of mullites obtained from kaolinite-alumina mixtures. J. Eur. Ceram. Soc. 2000, 20, 403–412. [Google Scholar] [CrossRef]
- Tezuka, N.; Low, I.-M.; Davies, I.J.; Prior, M.; Studer, A. In situ neutron diffraction investigation on the phase transformation sequence of kaolinite and halloysite to mullite. Phys. B 2006, 385–386, 555–557. [Google Scholar] [CrossRef]
- Dong, Y.; Feng, X.; Feng, X.; Ding, Y.; Liu, X.; Meng, G. Preparation of low-cost mullite ceramics from natural bauxite and industrial waste fly ash. J. Alloys Compd. 2008, 460, 599–606. [Google Scholar] [CrossRef]
- Cividanes, L.S.; Campos, T.M.B.; Rodrigues, L.A.; Brunelli, D.D.; Thim, G.P. Review of mullite synthesis routes by sol-gel method. J. Sol-Gel Sci. Technol. 2010, 55, 111–125. [Google Scholar] [CrossRef]
- Song, K.; Park, S.; Kim, W.; Jeon, C.; Ahn, J.-W. Effects of experimental parameters on the extraction of silica and carbonation of blast furnace slag at atmospheric pressure in low-concentration acetic acid. Metals 2017, 7, 199. [Google Scholar] [CrossRef]
- Song, K.; Kim, W.; Park, S.; Bang, J.-H.; Kim, J.; Ahn, J.-W. Preparation of silica-alumina nanoparticles via blast-furnace slag dissolution in low-concentration acetic acid for carbonation. Minerals 2017, 7, 206. [Google Scholar] [CrossRef]
- Kanzaki, S.; Kurihara, T.; Iwai, S.-I.; Ohashi, M.; Tabata, H. Sintering of mullite-silica ceramics and some properties for insulating substrate material. J. Ceram. Assoc. Jpn. 1987, 95, 1213–1218. [Google Scholar] [CrossRef]
- Donkai, N.; Miyamoto, T.; Kokubo, T.; Tanei, H. Preparation of transparent mullite-silica film by heat-treatment of imogolite. J. Mater. Sci. 1992, 27, 6193–6196. [Google Scholar] [CrossRef]
- Terry, B. The acid decomposition of silicate minerals part II. Hydrometallurgical applications. Hydrometallurgy 1983, 10, 151–171. [Google Scholar] [CrossRef]
- Park, H.K.; Bae, M.W.; Nam, I.H.; Kim, S.-G. Acid leaching of CaO-SiO2 resources. J. Ind. Eng. Chem. 2013, 19, 633–639. [Google Scholar] [CrossRef]
- Macdowell, J.F.; Beall, G.H. Immiscibility and crystallization in A12O3-SiO2 glasses. J. Am. Ceram. Soc. 1969, 52, 17–25. [Google Scholar] [CrossRef]
- Anilkumar, G.M.; Mukundan, P.; Warrier, K.G.K. Low-temperature mullitization in boehmite-tetraethoxysilane gel precursor containing γ-alumina and mullite nucleating seeds. Chem. Mater. 1998, 10, 2217–2220. [Google Scholar] [CrossRef]
- Li, D.X.; Thomson, W.J. Mullite formation from nonstoichiometric diphasic precursors. J. Am. Ceram. Soc. 1991, 74, 2382–2387. [Google Scholar] [CrossRef]
- Hou, P.; Basu, S.N.; Sarin, V.K. Structure and high-temperature stability of compositionally graded CDV mullite coatings. Int. J. Refract. Met. Hard Mater 2001, 19, 467–477. [Google Scholar] [CrossRef]
- Jiao, D.; Zheng, S.; Wang, Y.; Guan, R.; Cao, B. The tribology properties of alumina/silica composite nanoparticles as lubricant additives. Appl. Surf. Sci. 2011, 257, 5720–5725. [Google Scholar] [CrossRef]
- Shoval, S.; Boudeulle, M.; Yariv, S.; Lapides, I.; Panczer, G. Micro-Raman and FT-IR spectroscopy study of the thermal transformations of St. Claire dickite. Opt. Mater. 2001, 16, 319–327. [Google Scholar] [CrossRef]
- Beran, A.; Voll, D.; Schneider, H. Dehydration and structural development of mullite precursors: An FTIR spectroscopic study. J. Eur. Ceram. Soc. 2001, 21, 2479–2485. [Google Scholar] [CrossRef]
- Padmaja, P.; Anilkumar, G.M.; Mukundan, P.; Aruldhas, G.; Warrier, K.G.K. Characterisation of stoichiometric sol-gel mullite by fourier transform infrared spectroscopy. Int. J. Inorg. Mater. 2001, 3, 693–698. [Google Scholar] [CrossRef]
- Tang, C.; Zhu, J.; Li, Z.; Zhu, R.; Zhou, Q.; Wei, J.; He, H.; Tao, Q. Surface chemistry and reactivity of SiO2 polymorphs: A comparative study on α-quartz and α-cristobalite. Appl. Surf. Sci. 2015, 355, 1161–1167. [Google Scholar] [CrossRef]
- Schmücker, M.; MacKenzie, K.J.D.; Schneider, H.; Meinhold, R. NMR studies on rapidly solidified SiO2-Al2O3 and SiO2-Al2O3-Na2O-glasses. J. Non-Cryst. Solids 1997, 217, 99–105. [Google Scholar] [CrossRef]
- Omegna, A.; Prins, R.; van Bokhoven, J.A. Effect of temperature on aluminum coordination in zeolites H−Y and H−USY and amorphous silica-alumina: An in situ Al K-edge XANES Study. J. Phys. Chem. B 2005, 109, 9280–9283. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, K.; Kim, W.; Suh, C.-Y.; Bang, J.-H.; Ahn, J.-W. Preparation of Mullite-Silica Composites Using Silica-Rich Monophasic Precursor Obtained as a Byproduct of Mineral Carbonation of Blast-Furnace Slag. Minerals 2018, 8, 219. https://doi.org/10.3390/min8050219
Song K, Kim W, Suh C-Y, Bang J-H, Ahn J-W. Preparation of Mullite-Silica Composites Using Silica-Rich Monophasic Precursor Obtained as a Byproduct of Mineral Carbonation of Blast-Furnace Slag. Minerals. 2018; 8(5):219. https://doi.org/10.3390/min8050219
Chicago/Turabian StyleSong, Kyungsun, Wonbaek Kim, Chang-Yul Suh, Jun-Hwan Bang, and Ji-Whan Ahn. 2018. "Preparation of Mullite-Silica Composites Using Silica-Rich Monophasic Precursor Obtained as a Byproduct of Mineral Carbonation of Blast-Furnace Slag" Minerals 8, no. 5: 219. https://doi.org/10.3390/min8050219