Antisense Therapy in Neurology



1
Department of Medical Genetics, Faculty of Medicine and Dentistry, University of Alberta, 8812-112 St, Edmonton T6G 2H7, Canada
2
The Friends of Garrett Cumming Research and Muscular Dystrophy Canada HM Toupin Neurological Science Research Chair, 8812-112 St, Edmonton T6G 2H7, Canada
*
Author to whom correspondence should be addressed.
J. Pers. Med. 2013, 3(3), 144-176; https://doi.org/10.3390/jpm3030144
Submission received: 27 May 2013
/
Revised: 26 July 2013
/
Accepted: 29 July 2013
/
Published: 2 August 2013
Abstract
:Antisense therapy is an approach to fighting diseases using short DNA-like molecules called antisense oligonucleotides. Recently, antisense therapy has emerged as an exciting and promising strategy for the treatment of various neurodegenerative and neuromuscular disorders. Previous and ongoing pre-clinical and clinical trials have provided encouraging early results. Spinal muscular atrophy (SMA), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), Duchenne muscular dystrophy (DMD), Fukuyama congenital muscular dystrophy (FCMD), dysferlinopathy (including limb-girdle muscular dystrophy 2B; LGMD2B, Miyoshi myopathy; MM, and distal myopathy with anterior tibial onset; DMAT), and myotonic dystrophy (DM) are all reported to be promising targets for antisense therapy. This paper focuses on the current progress of antisense therapies in neurology.
Keywords:
Duchenne muscular dystrophy (DMD); Fukuyama congenital muscular dystrophy (FCMD); myotonic dystrophy (DM); spinal muscular atrophy (SMA); Huntington’s disease (HD); amyotrophic lateral sclerosis (ALS); limb-girdle muscular dystrophy 2B (LGMD2B); Miyoshi myopathy (MM); distal myopathy with anterior tibial onset (DMAT); antisense therapy1. Introduction
Antisense oligonucleotides (AOs) are short, synthetic nucleic acid sequences that selectively hybridize to target sequences in messenger RNA (mRNA). AOs can cause inhibition or redirection of splicing and inhibition of protein synthesis through various mechanisms, including disruption of the cell’s splicing machinery, interference with the ribosomal complex, and/or by activation of RNase H1-mediated degradation of the oligo-RNA heteroduplex [1]. Antisense therapy is an approach to fighting diseases using DNA-like molecules (AOs). After initially observing antisense-mediated RNA regulation in nature, investigations using model systems to test the feasibility of using synthetic AOs to reduce levels of specific mRNA transcripts quickly followed. Early experiments showed that AOs were effective in reducing target transcripts and protein synthesis [2]. However, despite promising early results, the use of AOs in disease therapy has been stymied by technical challenges and progress has been slow. Despite more than 20 years of research and clinical investigations, the United States Food and Drug Administration (FDA) has only ever approved two marketable AO drugs, Vitravene (Isis Pharmaceuticals, Carlsbad, CA, USA), for the treatment of cytomegalovirus retinitis in immunocompromized Acquired Immune Deficiency Syndrome (AIDS) patients with human immunodeficiency virus (HIV) infection [3], and, recently, Kynamro® (Isis Pharmaceuticals, Carlsbad, CA, USA) for the treatment of familial hypercholesterolemia. Although approved in 1998, Vitravene was removed from the market in 2004. Notwithstanding its slow progress, antisense remains a widely popular area of research in molecular biology, and with recent advancements in oligo chemistries and promising results from recent clinical trials it may well be that the day of AOs in the clinical arena in neurology is close at hand.
2. Challenges
Although promising, the headway of antisense therapy in the clinical realm has been quite slow. To better appreciate the current status of AO drug therapies, it is important to consider the hurdles that AOs have had to overcome. The first of these hurdles is drug delivery. First generation AOs do not easily cross the lipid bilayer of the cell, making intracellular potency via systemic delivery problematic since these AOs cannot readily penetrate to their intracellular targets at significant concentrations to be effective [4,5,6,7]. In the case of certain neurodegenerative diseases, such as Huntington’s disease and Alzheimer’s, the limited permeability of the blood-brain barrier further compounds the difficulty of effective drug administration to target cells of the central nervous system (CNS) [8]. Another problem associated with first generation AOs is off-target toxic effects [9]. DNA and RNA can be immunostimulatory, binding to and activating toll-like receptors or other receptors involved in innate immunity in a sequence- and chemistry-dependent manner [10]. Other biological barriers include uptake and sequestration of AOs in the reticuloendothelial system and intracellular sequestration in oligo-protein complexes and phagolysosomes [11]. Furthermore, to achieve biochemical efficacy, a large proportion of RNA targets must be hybridized and silenced—this number can vary widely, but can be as high as >90 percent [12]. To overcome these challenges, AOs have been designed such that the ribose backbone (normally present in RNA and DNA) is replaced with other chemistries. These constructs are so distinct from classical nucleic acid structures that they are not readily targeted by nucleases or DNA/RNA-binding proteins. These modifications result in increased stability and help prevent most off-target toxic effects. The various chemistries and modifications of AOs will be discussed in-depth in the next section. Regarding issues of delivery to CNS tissues, studies have shown the feasibility of AO-mediated RNA silencing in CNS tissues by AO drug administration into cerebrospinal fluid (CSF) via cerebral ventricles and intrathecal injection [13,14]. Drug administration into CSF via cerebral ventricles is a common medical practice in humans [15]. Studies involving administration of AOs into cerebral ventricles have shown significant oligonucleotide concentrations present not only in the brain and brainstem but also in many levels of the spinal cord after delivery in rats and nonhuman primates, providing evidence of delivery efficacy and sidestepping the hurdle of permeating the blood-brain barrier [16].
3. Comparative AO Chemistries
To avoid nuclease degradation, facilitate stronger base-pairing with target mRNA sequences, increase stability, and enable easier delivery into the cell, a variety of AO chemistries have been developed (Figure 1). One of the most widely used oligo chemistries is the 2'O-methylphosphorothioate- modified (2'OMePS) antisense oligo. These oligos contain a 2'-modification of the ribose ring as well as phosphorothioate linkages throughout their length (Figure 1C). The 2'OMePS AOs exhibit improved stability and increased cellular uptake via conventional delivery reagents. These AOs have also been shown to be very efficient in vivo [17,18]. The safety of this particular AO chemistry has been well characterized through a number of preclinical and clinical trials for several diseases [19,20,21]. Of note, the Prosensa/GlaxoSmithKline Duchenne muscular dystrophy (DMD) drug development program (Prosensa Therapeutics, Leiden, the Netherlands, and GlaxoSmithKline, London, UK), currently one of the leading bodies in antisense therapy research, employs this particular antisense chemistry [20,21].
Another oligo chemistry that is gaining in popularity is the phosphorodiamidate morpholino oligomer (PMO, morpholino). The PMO chemistry differs from traditional DNA/RNA chemistry in that the nucleic acid bases are bound to morpholine moieties as opposed to deoxyribose/ribose rings and the phosphodiester backbone is replaced by a phosphorodiamidate linkage [22] (Figure 1D). Like other oligos, the chemical modifications to PMOs render them sufficiently different from conventional nucleic acid chemistries so that they are not recognized by nucleases, making them very stable. Advantages of PMOs include increased binding efficiency to RNA targets and insusceptibility to metabolic degradation. Moreover, PMOs do not activate toll-like receptors, the nuclear factor (NF)-κB-mediated inflammatory response, or the interferon system [23]. Currently a phase 2 clinical trial involving PMOs for Duchenne muscular dystrophy is being conducted by Sarepta Therapeutics (Cambridge, MA, USA), and a significant improvement in 6-min walking distance (6-min walk test) has already been reported (MDA conference presentation, Washington, April 2013).
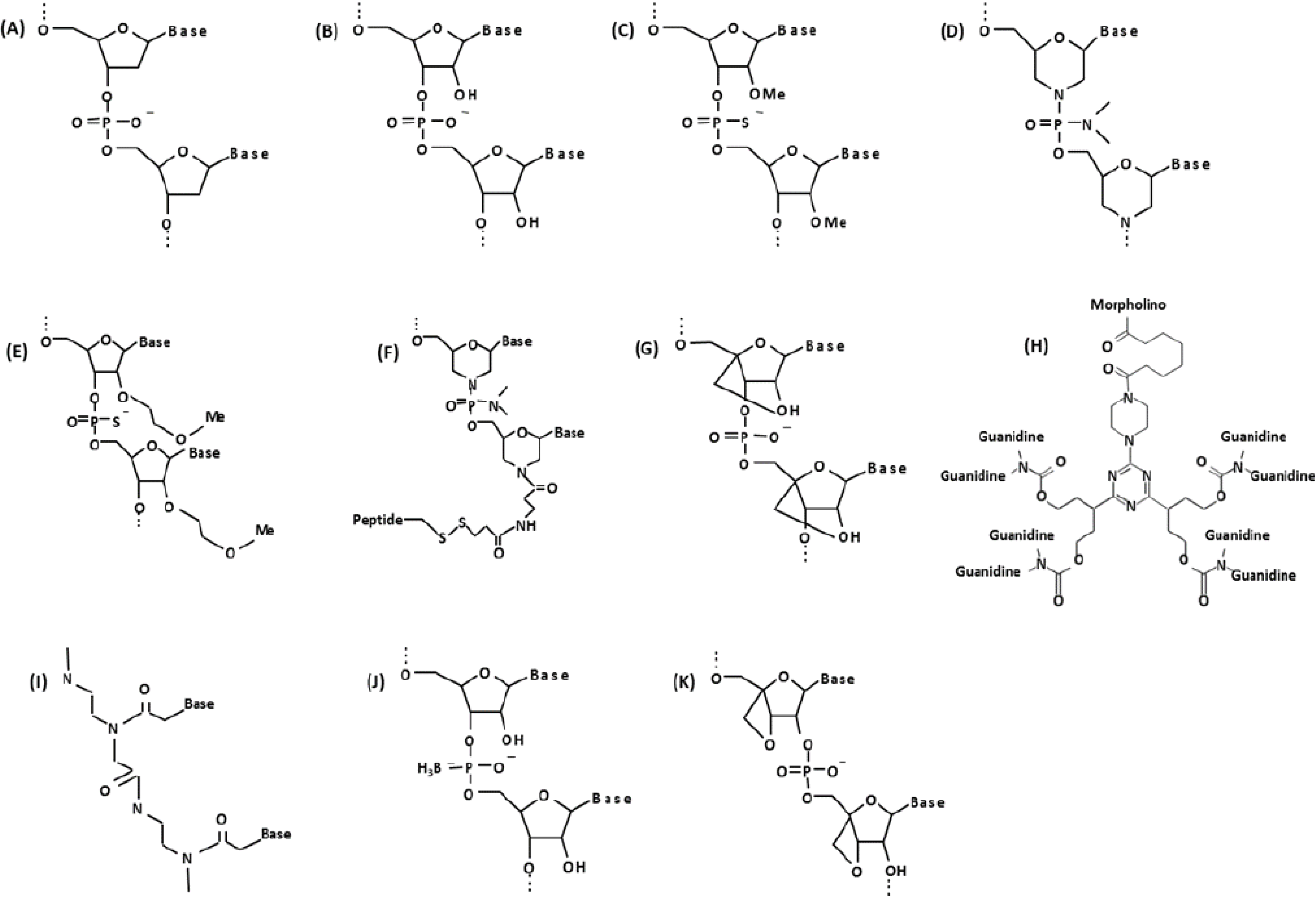
Figure 1.
Chemical structure of biological and synthetic oligonucleotides. (A) DNA; (B) RNA; (C) 2'O-methylphosphorothioate (2'O-MePS); (D) Morpholino (PMO); (E) 2'-methoxyethoxy (2'-MOE); (F) PMO with peptide conjugate (PPMO); (G) Locked nucleic acid (LNA); (H) Vivo-morpholino (vPMO); (I) Peptide nucleic acid (PNA); (J) Boranophosphate-oligodeoxy-nucleoside (BH3-ODN); (K) Oxetane-modified AO.
Figure 1.
Chemical structure of biological and synthetic oligonucleotides. (A) DNA; (B) RNA; (C) 2'O-methylphosphorothioate (2'O-MePS); (D) Morpholino (PMO); (E) 2'-methoxyethoxy (2'-MOE); (F) PMO with peptide conjugate (PPMO); (G) Locked nucleic acid (LNA); (H) Vivo-morpholino (vPMO); (I) Peptide nucleic acid (PNA); (J) Boranophosphate-oligodeoxy-nucleoside (BH3-ODN); (K) Oxetane-modified AO.

There are several groups of next generation antisense compounds that have shown very promising results in animal models. For example, 2'-methoxyethoxy (2'-MOE)-modified oligonucleotides containing lipophilic 2'-O-alkyl-substituted nucleobase modifications demonstrate high RNA binding affinity and metabolic stability, and can be used as gapmers to catalyze RNase H1-mediated degradation of target nucleic acids [24,25,26] (Figure 1E). 2'-MOE oligos have been used in vivo to target toxic mRNA triplet repeats in myotonic dystrophy [27]. Vivo-morpholinos (vPMOs) are octa guanidine (cell-penetrating moiety) conjugated PMOs (Figure 1H) and have shown very efficient splicing modulation in studies targeting the FCMD gene, DMD exons 6 and 8 multi skipping in dystrophic dogs, and exons 45–55 in mdx52 mice [28,29,30]. PMOs with peptide conjugates (PPMOs or PMOs with muscle targeting peptides; Figure 1F) act similarly to vPMOs and efficiently rescued cardiac muscle as well as skeletal muscles in mdx mice [31,32,33,34,35,36,37]. Peptide nucleic acids (PNAs) are another class of antisense oligo in which the phosphodiester-linked deoxyribose/ribose backbone is replaced by peptide-linked repeating N-(2-aminoethyl)-glycine units, to which the nucleobases are attached [38] (Figure 1I). PNAs exhibit greater binding strength than many other AOs and are extremely stable, though their solubility in water is much lower [39,40]. Locked nucleic acid (LNA) AOs contain a 2'-C, 4'-C-oxymethylene-linkage which “locks” the deoxyribo/ribo sugar structure in an N-type conformation [41] (Figure 1G). LNAs are stable against exonucleolytic degradation, exhibit high thermostability and hybridize strongly with target nucleic acids [42,43]. Several LNA analogs have been developed [42,44]. The characteristics of LNA constructs have made them the oligo of choice for several molecular applications, including microarrays [45], genotyping assays [46,47,48], and for the stabilization of DNA triplex formation in gene silencing [49]. In 1992, Sood et al. first reported an antisense oligo chemistry containing a boronated phosphate backbone (boranophosphate) [50]. Known as boranophosphate-oligodeoxy-nucleosides (BH3−-ODN), these AOs differ from classical DNA/RNA constructs in that they contain a borane group in place of a non-bridging oxygen species in the phosphodiester backbone (Figure 1J). Boranophosphates have been shown to activate RNase H1-mediated RNA cleavage [51]. Furthermore, experiments have demonstrated the highly lipophilic nature of boranophosphates [52], thus facilitating their transport across the bilipid membrane to target nucleic acids. This characteristic is likely due to the increased hydrophobicity of BH3 compared with oxygen. Boron-modified dNTPs have also been successfully employed in DNA sequencing assays—by taking advantage of the nuclease-resistant nature of boranophosphates [53,54], researchers are able to sequence resultant nucleic acid fragments following exonuclease digestion [55]. Oxetane-modified oligonucleotides (Figure 1K) are another form of AO which have proven their feasibly as antisense molecules by exhibiting resistance to nuclease digestion, the ability to activate RNase H1-mediated cleavage of the AO/RNA heteroduplex, tightly bind to their target nucleic acid sequences, and efficiently silence gene expression in vitro [56,57]. Development of more effective and less toxic AOs will be a key to the success of AO therapy.
4. Antisense Oligo Delivery
The method of delivery of antisense oligonucleotides in neurology is mainly predicated on the nature of the disease. There are two major targets of delivery: tissues of the central nervous system (CNS) and all other non-CNS tissues. In the case of neurodegenerative diseases such as Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and spinal muscular atrophy (SMA), direct targeting of CNS tissues is often desirable and can be accomplished via intrathecal injection, intracerebroventricular administration, and intraparenchymal delivery to the striatum [58,59,60,61]. This sidesteps the hurdle of the blood-brain barrier and increases the likelihood of oligo uptake to desired CNS tissues. A recently concluded phase I clinical trial involving Isis Pharmaceuticals’ antisense drug ISIS 333611 against SOD1 for the treatment of ALS reported no serious adverse effects following intrathecal injection [60].
For the antisense treatment of myopathic diseases, such as Duchenne muscular dystrophy (DMD), systemic administration via subcutaneous or intravenous injection, as well as direct intramuscular injection has been shown to facilitate widespread oligo distribution and effective intracellular uptake [62,63]. In the case of DMD, the preexisting pathology of the muscle tissues further enhances oligo uptake, as the plasma membranes of these muscle cells are unstable and contain small perforations, allowing AOs to more readily penetrate to their intracellular targets [64].
As previously mentioned, the intracellular delivery of AOs is further aided by chemical modifications which allow the oligos to more easily penetrate cell membranes. These modifications come in various forms, such as arginine-rich peptide conjugated morpholinos (PPMOs) or morpholinos linked to octa guanidine dendrimers (vPMOs), but each chemical adduct is designed to aid intracellular uptake.
In some instances, a dualistic targeting of both CNS and non CNS tissues is favorable, especially in cases involving multiorgan diseases. For example, Hua et al. demonstrated that liver plays an important role in SMA pathogenesis and were able to show a significant increase in survival in severely affected SMA mice following subcutaneous delivery of AOs. Increased survival following systemic AO administration was more pronounced than intracerebroventricular administration to CNS tissues alone and was further increased when both routes of oligo administration were coupled together [59].
5. Antisense Therapy in Neurology: Overview
In the second half of this article, the use of antisense oligos for Duchenne muscular dystrophy (DMD), Fukuyama congenital muscular dystrophy (FCMD), myotonic dystrophy (DM), spinal muscular atrophy (SMA), dysferlinopathy, Amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD) will be covered. Although they are all targeted by antisense therapy, therapeutic strategies for these disorders are quite different. For example, to target DMD, antisense-mediated exon skipping can remove nonsense mutations or frame-shifting mutations from mRNA [65,66,67]. To treat the mutation in the FCMD gene, a cocktail of vivo-morpholino AOs targeting splice enhancer sites and splice silencer sites led to correction of the aberrant splicing pattern in cell and mouse models [29]. RNase H1-mediated degradation of toxic RNA with 2'-MOE antisense for myotonic dystrophy type 1 showed very promising results in the mouse model [68]. A unique “knock up” approach (exon inclusion) targeting the SMN2 gene with 2'-MOE antisense or PMOs has been used to treat SMA cell and mouse models [69,70]. In the following sections, recent progress of antisense therapy in neurology and remaining challenges will be discussed.
6. Exon Skipping Therapy for DMD
DMD is an X-linked recessive form of muscular dystrophy, affecting around one in 3,500 boys worldwide, which leads to muscle degeneration and eventual death [71,72]. DMD is caused by mutations in the gene encoding dystrophin [73]. Recently, exon skipping has been heavily researched for the treatment of DMD [74,75]. Exon skipping employs antisense oligos as “DNA Band-Aids” to skip over the parts of the mutated gene that block the effective creation of proteins and restore the reading frame (Figure 2) [76]. In fact, such exon skipping of disease-causing mutations occurs spontaneously in DMD patients and animal models to some extent [77,78,79,80,81]. The efficacy of exon skipping was tested in several animal models including dystrophic mdx mice and dystrophic dogs as well as human DMD cells [30,35,82,83,84,85,86,87,88,89,90,91,92,93,94,95]. Systemic rescue of animal models with exon skipping has been demonstrated in dystrophic dogs (exons 6 and 8 multi-skipping), mdx mice (exon 23), and mdx52 mice (exon 51 and exons 45–55 multi-skipping) [17,28,82,96]. Currently, systemic clinical trials are being conducted targeting exon 51 in the DMD gene with PMOs and 2'OMePS antisense oligos, and very promising data have already been presented [20,97,98,99,100] (Table 1). Possibly, these antisense drugs will be approved by the Federal Drug Administration (FDA) in the near future. In addition, the first clinical trial of DMD targeting exon 53 skipping will start in Japan scheduled in 2013 (Nippon Shinyaku Co. Ltd. and National Center of Neurology and Psychiatry news release; UMIN-CTR Clinical Trial number UMIN000010964) (Table 1). Remaining challenges include: (1) limited efficacy of AOs, especially in the heart; (2) unknown long-term safety; (3) limited applicability (only approximately 10% of DMD patients can be treated with exon 51 and exon 53 skipping therapy, respectively).
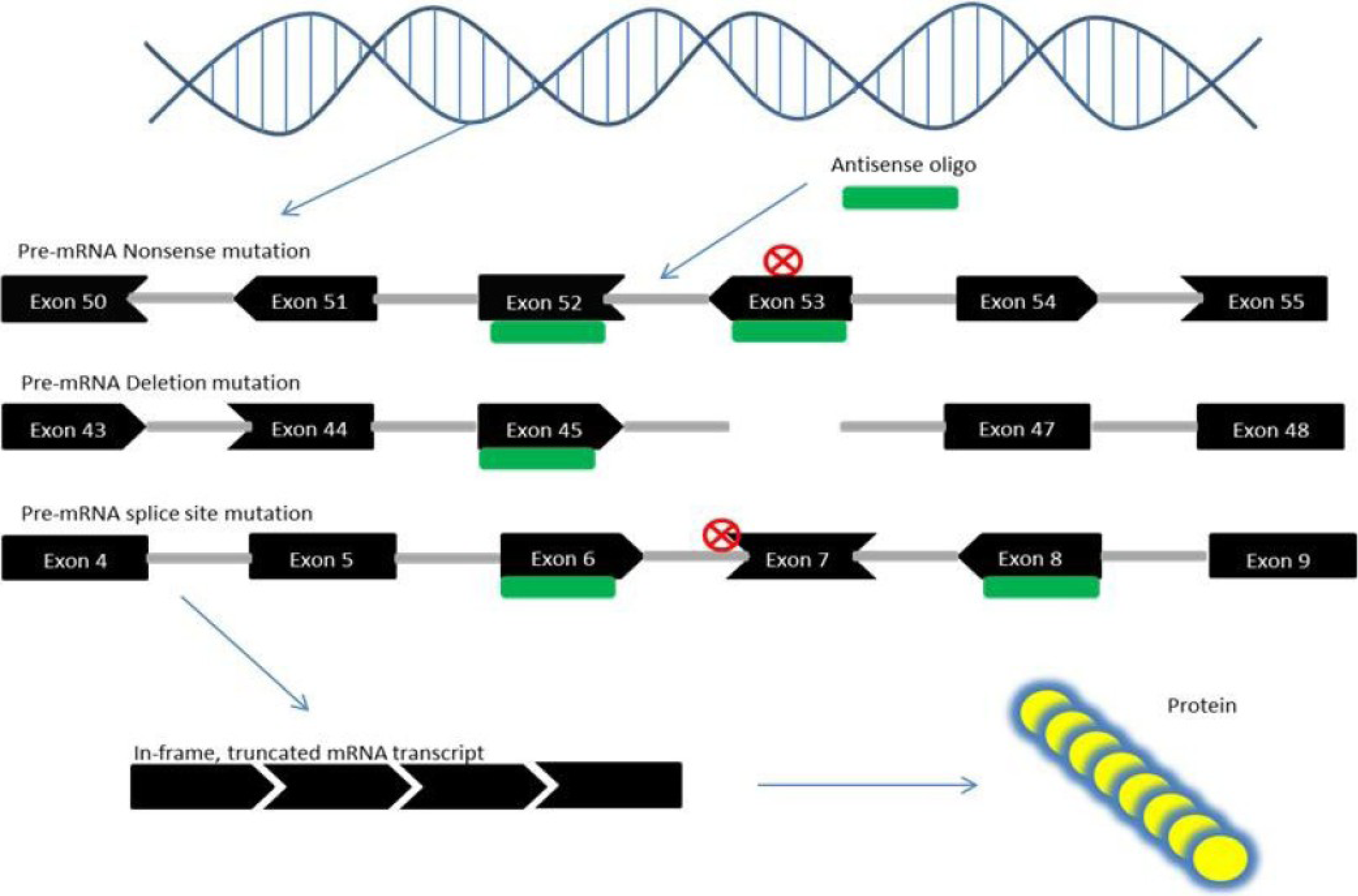
Figure 2.
Mechanism of exon skipping therapy for Duchenne muscular dystrophy (DMD). Nonsense mutations in the DMD gene can create a novel STOP codon which results in the loss of DMD protein. Exon skipping corrects this error when exons (black) that are bound to antisense oligos (green) are spliced out of the pre-mRNA, and the resulting exon sequences “fit together”, i.e., are in-frame (denoted by the shape of each exon—ends that fit together are in-frame). Out-of-frame mutations caused by the loss of exonic sequences, through deletion or splice site mutations, can also be corrected through exon skipping, which removes exons adjacent to the mutation site so that the remaining exons are in-frame. The result is a truncated yet partly functional protein, as in the case of Becker muscular dystrophy (BMD).
Figure 2.
Mechanism of exon skipping therapy for Duchenne muscular dystrophy (DMD). Nonsense mutations in the DMD gene can create a novel STOP codon which results in the loss of DMD protein. Exon skipping corrects this error when exons (black) that are bound to antisense oligos (green) are spliced out of the pre-mRNA, and the resulting exon sequences “fit together”, i.e., are in-frame (denoted by the shape of each exon—ends that fit together are in-frame). Out-of-frame mutations caused by the loss of exonic sequences, through deletion or splice site mutations, can also be corrected through exon skipping, which removes exons adjacent to the mutation site so that the remaining exons are in-frame. The result is a truncated yet partly functional protein, as in the case of Becker muscular dystrophy (BMD).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Current clinical trial status of antisense drugs for use in neurodegenerative and neuromuscular disorders.
| Disease | Drug | Chemistry | Mechanism of action | Target | Clinical Phase | Status | Sponsor | Clinicaltrials.gov ID |
|---|---|---|---|---|---|---|---|---|
| DMD | Eteplirsen (AVI-4658) | PMO | Exon Skipping | Exon 51 | Phase II | Completed | Sarepta Therapeutics | NCT01396239 |
| DMD | Drisapersen (PRO051/GSK2402968) | 2'OMePS | Exon Skipping | Exon 51 | Phase III | Recruiting | Prosensa Therapeutics/ GlaxoSmithKline | NCT01803412 |
| DMD | NS-065/NCNP-01 | PMO | Exon Skipping | Exon 53 | Phase I | Recruiting | Nippon Shinyaku Pharmaceuticals | NA |
| SMA | ISIS-SMNRx | 2'-MOE | Exon Inclusion | Exon 7 | Phase II | Recruiting | Isis Pharmaceuticals | NCT01839656 |
| ALS | ISIS-SOD1Rx/ISIS 333611 | 2'-MOE | Gapmer | Exon 1 | Phase I | Completed | Isis Pharmaceuticals | NCT01041222 |
| DM1 | PRO135 | NA | NA | CUG expansion | Preclinical | In progress | Prosensa Therapeutics | NA |
| HD | PRO289 | NA | NA | CAG expansion | Preclinical | In progress | Prosensa Therapeutics | NA |
7. Splicing Correction Therapy for FCMD
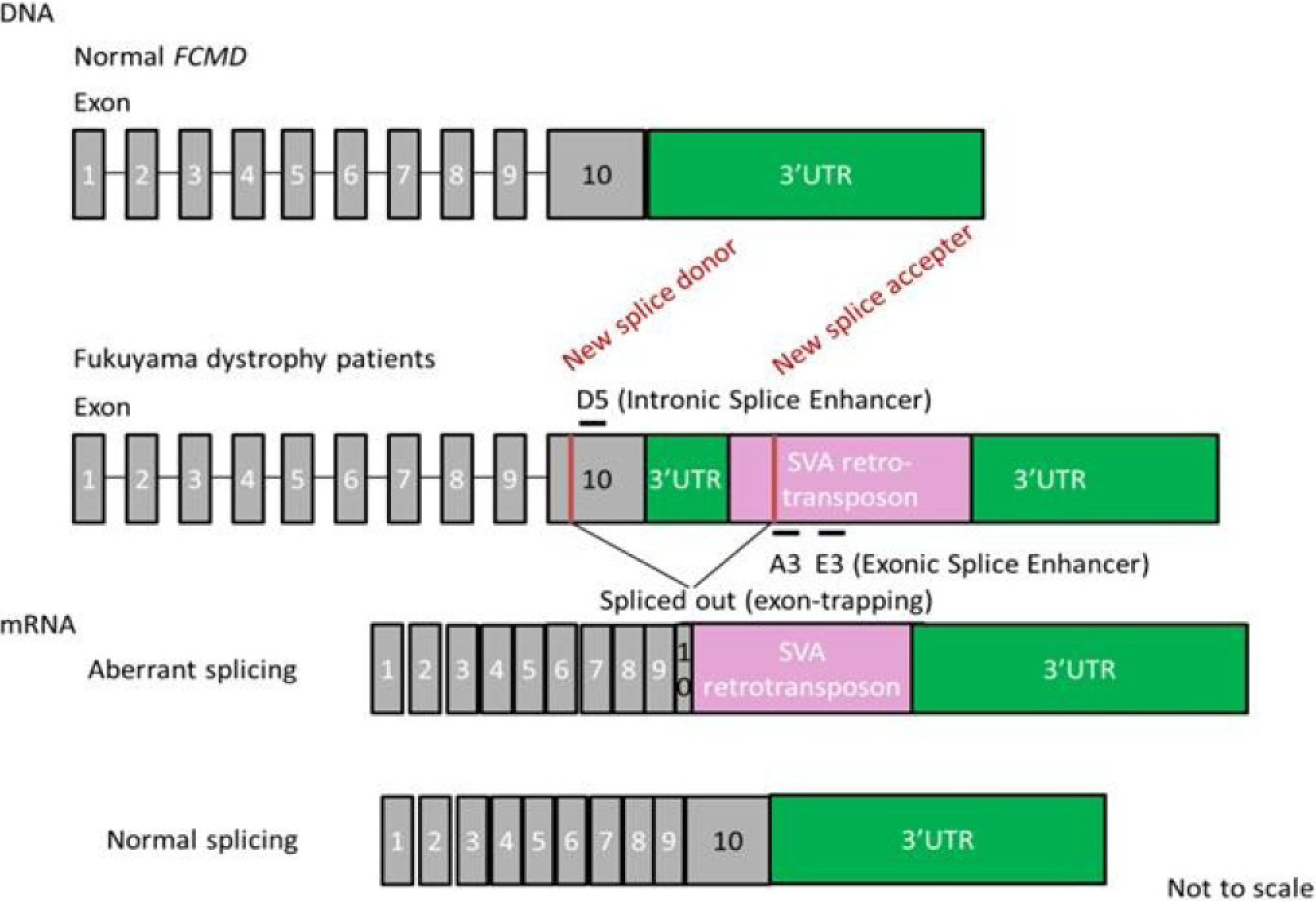
FCMD is an autosomal recessive form of muscular dystrophy mainly described in Japan [101]. The gene responsible for FCMD encodes a novel protein, fukutin [102]. Fukutin is believed to add chains of sugar molecules (glycosylation) to α-dystroglycan, a member of the dystrophin glycoprotein complex [103,104]. Interestingly, most patients (87%) with mutated FCMD gene bear chromosomes that have a 3-kb retrotransposon insertion into the 3'-untranslated region (UTR) of the gene derived from a single ancestral founder [105,106]. The aberrant mRNA splicing induced by the SINE-VNTR-Alu (SVA) retrotransposon exon-trapping is responsible for the pathogenesis of FCMD [29] (Figure 3). The insertion induces splicing errors and cryptic splice site activation with a new splice donor in exon 10 and a new splice accepter in the SVA insertion site. This results in aberrant splicing and truncation of exon 10. To rescue the mutated gene, a cocktail of at least three antisense oligos was required [91]. These oligos were targeted against intronic or exonic splicing enhancer sites (called ISE or ESE). These splicing enhancers are sites with consensus sequences that bind to splicing activator proteins [107,108]. They increase the probability that a nearby site will be used as a splice junction [109]. A cocktail of vPMOs led to normal fukutin mRNA expression and protein production in human patient cells as well as the mouse model in vivo [29].
Figure 3.
Strategy of antisense therapy for Fukuyama dystrophy. Retrotransposon insertion in the FCMD gene leads to aberrant splicing. An antisense vivo-morpholino cocktail (A3, E3 and D5) restores normal splicing.
Figure 3.
Strategy of antisense therapy for Fukuyama dystrophy. Retrotransposon insertion in the FCMD gene leads to aberrant splicing. An antisense vivo-morpholino cocktail (A3, E3 and D5) restores normal splicing.

8. Antisense Therapy for DM1
Myotonic dystrophy is the most common adult form of muscular dystrophy and is characterized by myotonia (slow relaxation of the muscles), progressive muscle weakness, and atrophy [110]. DM can also cause dysfunction of heart, eye, and brain tissues, as well as the gastrointestinal and endocrine systems [111,112]. Myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2) are multisystemic microsatellite expansion disorders caused by an expanded CTG tract in the 3' UTR of the dystrophia myotonica-protein kinase gene (DMPK) and an expanded CCTG tract in the first intron of the CCHC-type zinc finger, nucleic acid binding protein gene (CNBP, also known as ZNF9), respectively [113,114,115,116,117,118]. Disease phenotype (including age of onset and severity) is highly correlated with repeat number. In the case of DM1, unaffected individuals tend to have CTG repeats between 5 and 35 while DM1 patients often present with expansions between 50 and >2,000 [119,120]. DM follows an autosomal dominant pattern of inheritance and, although the precise molecular mechanisms are unknown, symptoms are thought to arise owing to the toxic gain-of-function of RNA transcripts containing expanded repeats, which causes the transcripts to be retained and accumulate in the nucleus [121]. Wang et al. have also provided evidence to suggest a possible dominant-negative effect of expansion-containing mutant RNA transcripts [122]. Protein-level gain-of-function is not likely, as the CTG expansion region lies outside of the DMPK coding region in the 3' UTR. Antisense-mediated suppression of DMPK RNA transcripts is, therefore, a promising therapeutic approach [123,124] (Figure 4). Importantly, there is considerable evidence implicating diminished DMPK transcripts in DM1 pathology, with a consensus among several studies that production and processing of DMPK mRNA is inhibited by expansion-containing mutant transcripts [125,126,127,128,129,130]. In their study utilizing homozygous DMPK-null mice, Reddy et al. showed that these mutants develop a progressive myopathy that is pathologically similar to DM, underscoring the importance of DMPK in maintaining proper skeletal muscle condition [131].
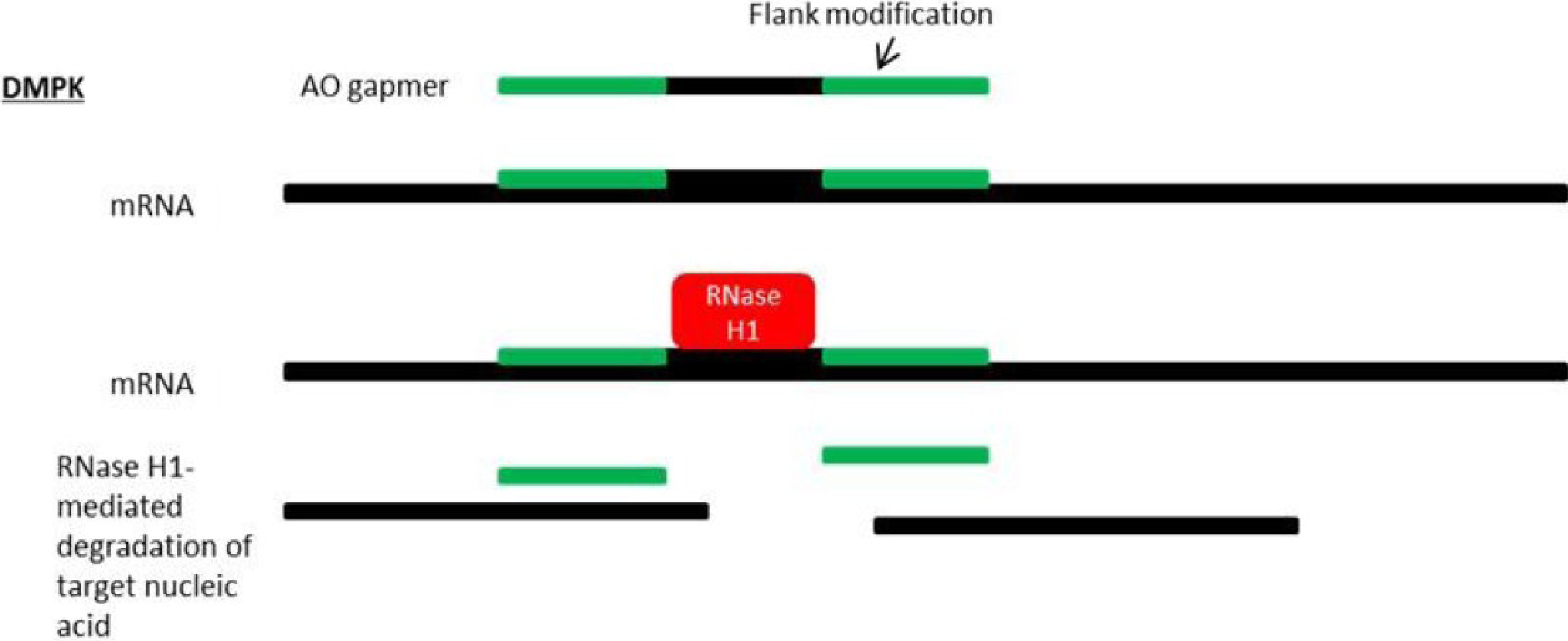
Figure 4.
Mechanism of antisense silencing via RNase H1 activity. Myotonic dystrophy (DM1) is caused by RNA gain-of-function due to an expanded CUG repeat in the dystrophia myotonica-protein kinase (DMPK) gene transcript.RNase H1-mediated degradation of target nucleic acids is facilitated by AO “gapmers”, composed of a central gap region which supports RNase H1 activity and flanking nucleotides at the 5' and 3'-ends which are resistant to RNase H1 degradation and display strong binding affinity for target RNA.
Figure 4.
Mechanism of antisense silencing via RNase H1 activity. Myotonic dystrophy (DM1) is caused by RNA gain-of-function due to an expanded CUG repeat in the dystrophia myotonica-protein kinase (DMPK) gene transcript.RNase H1-mediated degradation of target nucleic acids is facilitated by AO “gapmers”, composed of a central gap region which supports RNase H1 activity and flanking nucleotides at the 5' and 3'-ends which are resistant to RNase H1 degradation and display strong binding affinity for target RNA.

Recent in vitro studies have helped shed light on the therapeutic efficacy of several AO chemistries targeted against the microsatellite expansion of DM1 [132]. In vivo studies using 2'-MOE, LNA, and PPMO chemistries have provided evidence of efficient, long-lasting antisense-mediated knockdown of mutant RNA transcripts, as well as amelioration of physiological and transcriptomic abnormalities in DM1 mouse models [27,133,134]. Researchers from the University of Rochester and Isis Pharmaceuticals, Inc. have developed efficient methods to treat DM1 in a mouse model with systemically administered 2'-MOE modified antisense oligos [27]. They have successfully reversed symptoms of DM1 in these mice by eliminating toxic RNA in muscle fibers. Currently the group is working to improve their lead compound further, developing antisense oligos with stronger efficacy against the toxic RNA, but with minimal toxic effects.
Currently, no clinical trials are underway which involve AOs for the treatment of DM. Prosensa Therapeutics (Leiden, Netherlands), is currently in the pre-clinical stage of developing an antisense oligo, PRO135, which was shown to ameliorate toxic effects in vivo in DM1 preclinical models (Table 1).
9. Exon Inclusion Therapy for SMA
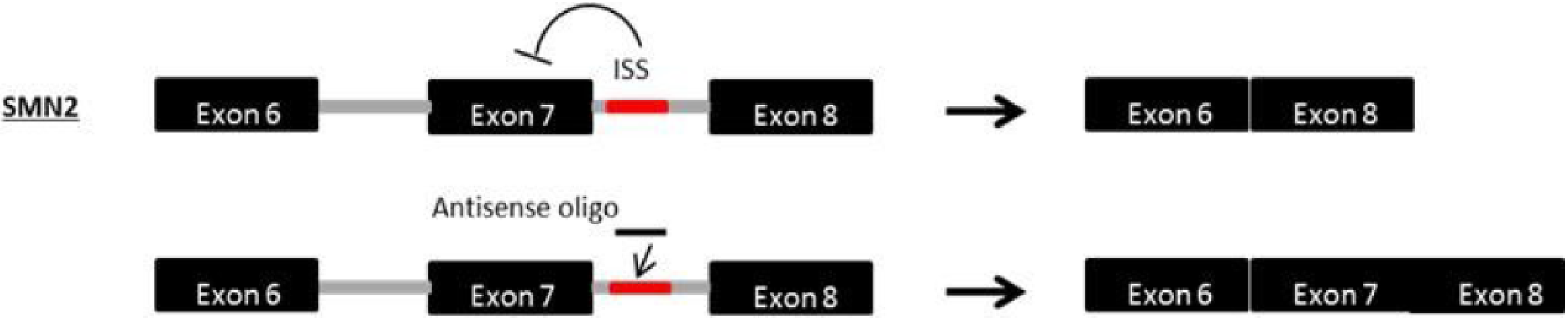
Spinal muscular atrophy (SMA) is a lethal autosomal recessive disease caused by a genetic defect in the SMN1 (survival motor neuron) gene [135,136]. SMA is characterized by the deterioration of spinal motor neurons, followed by weakness and wasting of the voluntary muscles in the arms and legs of infants and children, resulting in death during childhood [137]. Interestingly, SMA patients retain at least one copy of a highly homologous gene called SMN2 [138]. SMN2, an inverted duplicate copy nearly identical to SMN1, is unable to compensate for the loss of SMN1 due to a C-T transition in exon 7 which interferes with a splice modulator, causing exon 7 to be lost and rendering the resultant SMN protein nonfunctional; however, some full-length SMN transcripts (~10%) and functional SMN proteins are still produced. The SMN2 gene differs from SMN1 by only five base pair changes [139]. Consequently, upregulation of SMN by modification of SMN2 exon 7 splicing is a promising therapeutic approach (Figure 5), an approach that has already demonstrated favourable results in animal models [69,140,141,142,143]. Antisense PMOs targeting splice silencing motifs that promote exon 7 retention successfully rescued the phenotype in a severe mouse model of SMA after intracerebroventricular delivery [144]. In addition, the PMO injection led to longer survival after a single dosing by ICV injection.
Figure 5.
Mechanism of antisense exon 7 inclusion in SMN2. Spinal muscular atrophy (SMA) is caused by a loss-of-function mutation in the SMN1 gene. Within the SMN2 gene, a paralogue of SMN1, a single nucleotide substitution in exon 7 interferes with an exonic splicing enhancer, impairing production of normal SMN protein. AOs targeted to the intronic splice silencer site (ISS) in intron 7 of SMN2 facilitate the retention of exon 7 within the mature mRNA, increasing the production of functional SMN protein.
Figure 5.
Mechanism of antisense exon 7 inclusion in SMN2. Spinal muscular atrophy (SMA) is caused by a loss-of-function mutation in the SMN1 gene. Within the SMN2 gene, a paralogue of SMN1, a single nucleotide substitution in exon 7 interferes with an exonic splicing enhancer, impairing production of normal SMN protein. AOs targeted to the intronic splice silencer site (ISS) in intron 7 of SMN2 facilitate the retention of exon 7 within the mature mRNA, increasing the production of functional SMN protein.

Recently, Isis Pharmaceuticals, Inc. announced the commencement of an open-label, multiple-dose, dose-escalation Phase II clinical trial which utilizes their antisense oligo drug ISIS-SMNRx (Table 1). The study involves patients with infantile-onset SMA and is currently seeking to recruit eight participants between three weeks and seven months of age in the US and Canada. The aim of the study is to provide information regarding the safety and tolerability of ISIS-SMNRx. The results of this investigation will help lay the foundation for a future large-scale phase II/III clinical trial. The drug under investigation, ISIS-SMNRx, is a 2'-MOE modified AO designed to modulate SMN2 splicing, thereby increasing levels of SMN protein. A previously concluded Phase I trial evaluating ISIS-SMNRx (ClinicalTrials.gov identifier: NCT01494701) showed the drug to be well-tolerated across all doses and also reported a significant improvement in muscle function in several participants.
10. Exon Skipping Therapy for Dysferlinopathy
The dysferlinopathies are a category of muscular dystrophy arising due to mutations in the dysferlin (DYSF) gene [145,146]. Three clinically distinct autosomal recessive muscular dystrophies are attributed to DYSF mutations: limb-girdle muscular dystrophy type 2B (LGMD2B), Miyoshi myopathy (MM), and distal myopathy with anterior tibial onset (DMAT) [147,148,149,150,151,152]. Dysferlinopathy is characterized by progressive muscle weakness and atrophy with onset usually beginning in adulthood and commencing in either the proximal or distal muscles, defining the clinical phenotype. Although distinct initially, the clinical phenotypes of dysferlinopathy include a wide spectrum of pathology that becomes less divergent as the disease progresses, eventually including both proximal and distal muscle groups, becoming one indistinguishable disorder. The sarcolemmal protein dysferlin is a transmembrane protein that is ubiquitously expressed and is found abundantly in cardiac and skeletal muscle where it plays a pivotal role in plasma membrane re-sealing [147,153,154,155,156,157,158,159].
A promising therapeutic approach to treating dysferlinopathies is exon skipping, wherein AOs are used to selectively target exonic sequences and prevent their incorporation into the final mRNA transcript [65,160]. This process of splicing modulation restores the open reading frame and leads to the production of a truncated-yet functional protein and has already been demonstrated in vitro using dysferlinopathy patient-derived cells [161]. In addition, Sinnreich et al. reported a case wherein a mildly affected mother with two severely affected daughters, both having LGMD2B with homozygous DYSF null mutations, was found to carry a lariat branch point mutation that resulted in the in-frame exon skipping of exon 32. The action of the resulting dysferlin protein is thought to account for her mild phenotype [162]. Therefore, at least dysferlin exon 32 is thought to be a promising target of exon skipping therapy, although there are currently no ongoing or pending clinical trials involving AO-mediated therapy for dysferlinopathy.
11. Antisense Therapy for Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease affecting upper and lower motor neurons in the brain and spinal cord [163]. Though associated with some clinical heterogeneity, ALS typically manifests during adulthood and is characterized by progressive neuronal death, spasticity, muscle atrophy, paralysis, and death within ~5 years of diagnosis [164,165,166]. Most cases of ALS are sporadic; however, ~10% of cases are familial and follow an autosomal-dominant pattern of inheritance [167,168]—of these, 20% are caused by mutations in the Cu/Zn superoxide dismutase (SOD1) gene, resulting in a toxic gain-of-function via a currently unknown mechanism [169,170,171]. Although currently believed to be the result of a gain-of-function mechanism, initial investigations into the role of SOD1 in ALS supported a loss-of-function mechanism [172,173]. Belief in a loss-of-function model waned significantly following in vivo experiments involving transgenic mice expressing human SOD1 protein, which exhibited progressive neurodegeneration, mirroring human ALS clinical pathology [170,174,175]. Clinical observations which failed to support a connection between SOD1 activity and disease progression further eclipsed the idea of loss-of-function [176]. However, in their recent article, Saccon et al. compile previous and recent findings to provide a compelling argument for the existence of a possible modifying role of loss-of-function in ALS [177]. They note that SOD1 activity is significantly reduced in ALS patients and that SOD1-null mice exhibit neuropathology similar to human ALS. Although a loss of SOD1 activity does not appear directly responsible for ALS phenotype, these data support the idea of a possible synergistic relationship between gain-of-function and loss-of-function in ALS disease progression. The interplay between gain- and loss-of function has also been described in a host of other neurodegenerative disorders, including Huntington’s disease and Parkinson’s disease [178,179]. As such, the implications to antisense therapy in neurology, and especially the antisense-mediated reduction of SOD1, are profound. The long-term effects of downregulating SOD1, therefore, should be an important focus of future clinical trials.
Isis Pharmaceuticals recently concluded a Phase 1 placebo-controlled, double-blind, dose-escalation, safety and tolerability clinical trial for their antisense drug ISIS-SOD1Rx (Table 1). The oligo employed in this study, ISIS 333611, was a 2'-MOE modified antisense oligo targeted to the first exon (19th–38th bps) of SOD1 (regardless of mutation) and catalyzed RNase H1-mediated degradation [60,180]. The study involved patients from four US centers aged 18 years or older and carrying SOD1 mutations. Participants were given 12-h intrathecal infusions of ISIS 333611 at varying concentrations, or placebo. No clinically significant adverse effects associated with oligo administration were reported. Following administration, AO was detected in the CSF of all AO-treated participants and increased with dosage concentration. SOD1 concentrations in the CSF did not change significantly, though achieving SOD1 reduction was never an aim of the study.
In addition to the SOD1 gene, several other genes have also been implicated in ALS pathogenesis, including the TAR DNA binding protein (TARDBP), fused in sarcoma (FUS), angiogenin (ANG), ubiquilin 2 (UBQLN2), and valosin-containing protein (VCP) genes [181,182,183,184,185,186,187,188,189]. Most notably, it was recently discovered that a GGGGCC hexanucleotide repeat expansion in the first intron of the C9orf72 gene is the most common genetic cause of ALS pathogenesis, more common than all other known ALS gene mutations combined, accounting for between 37%–50% of familial ALS cases among studied cohorts [190,191,192,193,194,195,196,197]. Although both loss-of-function and gain-of-function mechanisms have been postulated, the underlying etiology by which these C9orf72 expanded repeats result in neurodegeneration is, as yet, unknown; however, evidence suggests a pathogenic threshold of hexanucleotide repeats may exist, though such a threshold has not yet been fully demarcated [191,192,195,198,199,200]. Because of the high prevalence of C9orf72 mutations in cases of ALS, and because mutations in C9orf72 have also been associated with other neurodegenerative disorders, such as Parkinson’s disease and frontotemporal dementia (FTD), C9orf72 is a promising candidate for targeted antisense therapy [191,195,201,202,203]. Research groups are currently working with ISIS Pharmaceuticals to develop an antisense strategy for C9orf72-based ALS, working under the hypothesis that reducing mutant C9orf72 transcripts using AOs will ameliorate toxic aggregations of expanded repeat mRNA, which present as nuclear foci in brain and spinal cord in affected patients [191,204]. Early investigations using AOs have yielded promising results, reducing the frequency of C9orf72 expanded repeat aggregates and stabilizing gene expression in vitro [204,205].
12. Antisense Therapy for Huntington’s Disease
Huntington’s disease (HD) is an adult-onset, lethal, progressive neurodegenerative disease that follows an autosomal dominant pattern of inheritance. Clinical manifestations of HD include cognitive decay, such as the diminished ability to perform executive functions, motor deficits, such as chorea (involuntary, spastic movements), the inability to manage prehensile controls, and psychiatric disturbances, such as dysphoria, anxiety, irritability, mania and psychosis [206,207,208,209,210,211]. Neuropathological features of HD include widespread neuronal atrophy and the formation of nuclear/intranuclear inclusions in neural tissues of the brain [208,212,213,214,215,216,217]. Although the precise etiology of HD is still unknown, the disease is caused by a trinucleotide CAG-expansion in the first exon of the Huntingtin (HTT) gene, which results in a toxic gain-of-function of the resultant mutant huntingtin protein (mHTT) [218,219]. The inclusion bodies are composed of aggregates of misfolded mHTT and their density is highly correlated with repeat length [220,221,222]. Wild-type huntingtin (HTT) is ubiquitously expressed and is found at high concentration in the brain [223,224,225]. HTT is vital to proper embryonic development and neurogenesis, and also plays a role in protecting CNS cells from apoptosis, vesicular trafficking, axonal transport, and synaptic transmission [224,226,227,228,229,230,231,232,233,234,235]. Because the loss of HTT is associated with several deleterious consequences, the allele-specific silencing of mHTT is a promising therapeutic approach to treating HD [58,61,179,236], although some studies have shown significant beneficial effects from the co-suppression of both mutant and wild-type alleles [237,238,239].
The two foremost therapeutic approaches to allele-specific silencing of mHTT are the targeting of single nucleotide polymorphisms (SNP) and direct targeting of the expanded CAG region [240,241,242,243,244,245]. In vivo studies have demonstrated successful selective reduction of mHTT and a concomitant amelioration of HD neuropathology and behavioral/motor dysfunctions in mouse models [246,247,248]. Since AOs were first used to downregulate the expression of HTT, much attention has been given to developing antisense strategies aimed at selectively reducing mHTT levels [249]. A variety of AO chemistries, including PNA, LNA, 2'-MOE, and morpholino chemistries have been used in vitro and in vivo to selectively reduce levels of mHTT [58,239,243,245,250,251,252]. Notably, similar to DM1, 2'-MOE modified antisense oligo infusion into the cerebrospinal fluid of HD mouse models successfully reversed the disease progression with RNase H1-mediated degradation of huntingtin mRNA [239]. No clinical trials involving antisense oligos for the therapeutic treatment HD are currently being pursued; however, Prosensa Therapeutics is currently conducting preclinical tests of their antisense drug PRO289, designed to reduce levels of mHTT by targeting the expanded CAG tract (Table 1). So far, PRO289 has been successful in reducing mutant transcripts in HD patient-derived fibroblasts.
13. Conclusions—What Does the Future Hold?
Lately, antisense therapies have moved one step closer to entrance into the clinical arena. The data from the Phase 2 DMD clinical trials are very promising. Isis Pharmaceuticals has recently started clinical trials of an antisense oligonucleotide therapy for SMA and ALS. Antisense drugs against FCMD, DM1, and Huntington’s disease are still in the preclinical stage of the development process but showed promising results in animal models. Some in vitro studies have demonstrated that dysferlinopathy is also a possible target for antisense therapy. Remaining challenges include limited delivery to the heart, potential off-target effects, lack of long term safety data, and limited applicability of each antisense oligo targeting each mutation (particularly in exon skipping therapy for DMD and dysferlinopathies). Unfortunately, the current regulatory process for drug development is not designed to handle these kinds of sequence-specific oligonucleotide therapies [253]. A re-evaluation of the current drug approval process, which takes into consideration the common characteristics of the same antisense chemistry and differences in the specific sequences, will help create a more efficient path for the development of antisense drugs and will benefit the progress of personalized medicine.
With the recent clinical success of several antisense-based therapies, and establishment of proof-of-concept efficacy in several disease models, antisense oligos have established themselves as a promising and rapidly-developing therapeutic strategy covering a wide range of genetic disorders. With such dramatic improvements in antisense technology in a relatively short time frame, and with the current frenzied pace of antisense research, new and enhanced AO designs will likely be forthcoming and will facilitate their widespread application in the clinical realm.
Acknowledgments
This work was supported by the University of Alberta Faculty of Medicine and Dentistry, Parent Project Muscular Dystrophy (USA), The Friends of Garrett Cumming Research Fund, HM Toupin Neurological Science Research Fund, Muscular Dystrophy Canada, Canada Foundation for Innovation, Alberta Advanced Education and Technology, and the Women’s and Children's Health Research Institute (WCHRI).
Conflict of Interest
The authors declare no conflict of interest.
References
- Kuzmiak, H.A.; Maquat, L.E. Applying nonsense-mediated mRNA decay research to the clinic: Progress and challenges. Trends Mol. Med. 2006, 12, 306–316. [Google Scholar] [CrossRef]
- Bennett, C.F.; Condon, T.P.; Grimm, S.; Chan, H.; Chiang, M.Y. Inhibition of endothelial cell adhesion molecule expression with antisense oligonucleotides. J. Immunol. 1994, 152, 3530–3540. [Google Scholar]
- Jiang, K. Biotech comes to its “antisenses” after hard-won drug approval. Nat. Med. 2013, 19. [Google Scholar] [CrossRef]
- Bendifallah, N.; Rasmussen, F.W.; Zachar, V.; Ebbesen, P.; Nielsen, P.E.; Koppelhus, U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjug. Chem. 2006, 17, 750–758. [Google Scholar] [CrossRef]
- Miller, P.S.; Braiterman, L.T.; Ts'o, P.O. Effects of a trinucleotide ethyl phosphotriester, Gmp(Et)Gmp(Et)U, on mammalian cells in culture. Biochemistry 1977, 16, 1988–1996. [Google Scholar] [CrossRef]
- Shiraishi, T.; Nielsen, P.E. Improved cellular uptake of antisense peptide nucleic acids by conjugation to a cell-penetrating peptide and a lipid domain. Methods Mol. Biol. 2011, 751, 209–221. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 2006, 8, 343–375. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef]
- Muntoni, F.; Wood, M.J. Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 2011, 10, 621–637. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Bronson, A.; Levin, A.A.; Takeda, S.; Yokota, T.; Baudy, A.R.; Connor, E.M. Restoring dystrophin expression in Duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathol. 2011, 179, 12–22. [Google Scholar] [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef]
- Broaddus, W.C.; Prabhu, S.S.; Gillies, G.T.; Neal, J.; Conrad, W.S.; Chen, Z.J.; Fillmore, H.; Young, H.F. Distribution and stability of antisense phosphorothioate oligonucleotides in rodent brain following direct intraparenchymal controlled-rate infusion. Neurosurg. Focus 1997, 3, e6. [Google Scholar]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Hokfelt, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug delivery to the brain. J. Cereb. Blood Flow Metab. 1997, 17, 713–731. [Google Scholar] [CrossRef]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006, 116, 2290–2296. [Google Scholar] [CrossRef]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef]
- Heemskerk, H.A.; de Winter, C.L.; de Kimpe, S.J.; van Kuik-Romeijn, P.; Heuvelmans, N.; Platenburg, G.J.; van Ommen, G.J.; van Deutekom, J.C.; Aartsma-Rus, A. In vivo comparison of 2'-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J. Gene Med. 2009, 11, 257–266. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef]
- van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef]
- Moulton, J.D.; Jiang, S. Gene knockdowns in adult animals: PPMOs and vivo-morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar] [CrossRef]
- Sazani, P.; Ness, K.P.; Weller, D.L.; Poage, D.W.; Palyada, K.; Shrewsbury, S.B. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int. J. Toxicol. 2011, 30, 313–321. [Google Scholar] [CrossRef]
- Altmann, K.H.; Fabbro, D.; Dean, N.M.; Geiger, T.; Monia, B.P.; Muller, M.; Nicklin, P. Second-generation antisense oligonucleotides: Structure-activity relationships and the design of improved signal-transduction inhibitors. Biochem. Soc. Trans. 1996, 24, 630–637. [Google Scholar]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2'-modified oligonucleotides containing 2'-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar]
- Prakash, T.P.; Bhat, B. 2'-Modified oligonucleotides for antisense therapeutics. Curr. Top. Med. Chem. 2007, 7, 641–649. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef]
- Taniguchi-Ikeda, M.; Kobayashi, K.; Kanagawa, M.; Yu, C.C.; Mori, K.; Oda, T.; Kuga, A.; Kurahashi, H.; Akman, H.O.; DiMauro, S.; et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature 2011, 478, 127–131. [Google Scholar] [CrossRef]
- Yokota, T.; Nakamura, A.; Nagata, T.; Saito, T.; Kobayashi, M.; Aoki, Y.; Echigoya, Y.; Partridge, T.; Hoffman, E.P.; Takeda, S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012, 22, 306–315. [Google Scholar]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.; Betts, C.; Wood, M. CPP-directed oligonucleotide exon skipping in animal models of Duchenne muscular dystrophy. Methods Mol. Biol. 2011, 683, 321–338. [Google Scholar] [CrossRef]
- Yin, H.; Lu, Q.; Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 2008, 16, 38–45. [Google Scholar] [CrossRef]
- Yin, H.; Betts, C.; Saleh, A.F.; Ivanova, G.D.; Lee, H.; Seow, Y.; Kim, D.; Gait, M.J.; Wood, M.J. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol. Ther. 2010, 18, 819–827. [Google Scholar] [CrossRef]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Karkare, S.; Bhatnagar, D. Promising nucleic acid analogs and mimics: Characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 2006, 71, 575–586. [Google Scholar] [CrossRef]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef]
- Pfundheller, H.M.; Lomholt, C. Locked nucleic acids: Synthesis and characterization of LNA-T diol. Curr. Protoc. Nucleic Acid Chem. 2002. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, R.; Wengel, J. Synthesis of Novel Bicyclo [2.2.1] Ribonucleosides: 2'-Amino- and 2'-Thio-LNA Monomeric Nucleosides. J. Org. Chem. 1998, 63, 6078–6079. [Google Scholar]
- McTigue, P.M.; Peterson, R.J.; Kahn, J.D. Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry 2004, 43, 5388–5405. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, S.K.; Koshkin, A.A.; Rajwanshi, V.K.; Meldgaard, M.; Wengel, J. The first analogues of LNA (locked nucleic acids): Phosphorothioate-LNA and 2'-thio-LNA. Bioorg. Med. Chem. Lett. 1998, 8, 2219–2222. [Google Scholar] [CrossRef]
- Tolstrup, N.; Nielsen, P.S.; Kolberg, J.G.; Frankel, A.M.; Vissing, H.; Kauppinen, S. Oligo Design: Optimal design of LNA (locked nucleic acid) oligonucleotide capture probes for gene expression profiling. Nucleic Acids Res. 2003, 31, 3758–3762. [Google Scholar] [CrossRef]
- Johnson, M.P.; Haupt, L.M.; Griffiths, L.R. Locked nucleic acid (LNA) single nucleotide polymorphism (SNP) genotype analysis and validation using real-time PCR. Nucleic Acids Res. 2004, 32, e55. [Google Scholar] [CrossRef]
- Latorra, D.; Campbell, K.; Wolter, A.; Hurley, J.M. Enhanced allele-specific PCR discrimination in SNP genotyping using 3' locked nucleic acid (LNA) primers. Hum. Mutat. 2003, 22, 79–85. [Google Scholar] [CrossRef]
- Simeonov, A.; Nikiforov, T.T. Single nucleotide polymorphism genotyping using short, fluorescently labeled locked nucleic acid (LNA) probes and fluorescence polarization detection. Nucleic Acids Res. 2002, 30, e91. [Google Scholar] [CrossRef]
- Petersen, M.; Wengel, J. LNA: A versatile tool for therapeutics and genomics. Trends Biotechnol. 2003, 21, 74–81. [Google Scholar]
- Sood, A.; Spielvogel, B.F.; Shaw, B.R.; Carlton, L.D.; Burnham, B.S.; Hall, E.S.; Hall, I.H. The synthesis and antineoplastic activity of 2'-deoxy-nucleoside-cyanoboranes in murine and human culture cells. Anticancer Res. 1992, 12, 335–343. [Google Scholar]
- Rait, V.K.; Shaw, B.R. Boranophosphates support the RNase H cleavage of polyribonucleotides. Antisense Nucleic Acid Drug Dev. 1999, 9, 53–60. [Google Scholar]
- Rait, V.; Sergueev, D.; Summers, J.; He, K.; Huang, F.; Krzyzanowska, B.; Shaw, B.R. Boranophosphate nucleic acids―A versatile DNA backbone. Nucleos. Nucleot. 1999, 18, 1379–1380. [Google Scholar]
- Li, P.; Shaw, B.R. Synthesis of prodrug candidates: Conjugates of amino acid with nucleoside boranophosphate. Org. Lett. 2002, 4, 2009–2012. [Google Scholar]
- Shaw, B.R.; Dobrikov, M.; Wang, X.; Wan, J.; He, K.; Lin, J.L.; Li, P.; Rait, V.; Sergueeva, Z.A.; Sergueev, D. Reading, writing, and modulating genetic information with boranophosphate mimics of nucleotides, DNA, and RNA. Ann. N. Y. Acad. Sci. 2003, 1002, 12–29. [Google Scholar] [CrossRef]
- Shaw, B.R.; Sergueev, D.; He, K.; Porter, K.; Summers, J.; Sergueeva, Z.; Rait, V. Boranophosphate backbone: A mimic of phosphodiesters, phosphorothioates, and methyl phosphonate. Meth. Enzymol. 2000, 313, 226–257. [Google Scholar]
- Opalinska, J.B.; Kalota, A.; Gifford, L.K.; Lu, P.; Jen, K.Y.; Pradeepkumar, P.I.; Barman, J.; Kim, T.K.; Swider, C.R.; Chattopadhyaya, J.; et al. Oxetane modified, conformationally constrained, antisense oligodeoxyribonucleotides function efficiently as gene silencing molecules. Nucleic Acids Res. 2004, 32, 5791–5799. [Google Scholar] [CrossRef]
- Opalinska, J.B.; Gewirtz, A.M. Rationally targeted, conformationally constrained, oxetane-modified oligonucleotides demonstrate efficient gene-silencing activity in a cellular syste. Ann. N. Y. Acad. Sci. 2005, 1058, 39–51. [Google Scholar] [CrossRef]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar]
- Hua, X.; Yu, L.; Huang, X.; Liao, Z.; Xian, Q. Expression and role of fibroblast activation protein-alpha in microinvasive breast carcinoma. Diagn. Pathol. 2011, 6, e111. [Google Scholar]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Sah, D.W.; Aronin, N. Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Invest. 2011, 121, 500–507. [Google Scholar]
- Heemskerk, H.; de Winter, C.; van Kuik, P.; Heuvelmans, N.; Sabatelli, P.; Rimessi, P.; Braghetta, P.; van Ommen, G.J.; de Kimpe, S.; Ferlini, A.; et al. Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 2010, 18, 1210–1217. [Google Scholar] [CrossRef]
- Kim, Y.; Tewari, M.; Pajeroski, D.J.; Sen, S.; Jason, W.; Sirsi, S.; Lutz, G.; Discher, D.E. Efficient nuclear delivery and nuclear body localization of antisense oligo-nucleotides using degradable polymersomes. In Proceedings of the Engineering in Medicine and Biology Society, 2006. EMBS'06. 28th Annual International Conference of the IEEE, New York, NY, USA, 30 August–3 September 2006; pp. 4350–4353.
- Hoffman, E.P. Skipping toward personalized molecular medicine. N. Engl. J. Med. 2007, 357, 2719–2722. [Google Scholar]
- Yokota, T.; Pistilli, E.; Duddy, W.; Nagaraju, K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2007, 7, 831–842. [Google Scholar]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: Too many mutations, too few patients? Expert Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef]
- Malerba, A.; Boldrin, L.; Dickson, G. Long-term systemic administration of unconjugated morpholino oligomers for therapeutic expression of dystrophin by exon skipping in skeletal muscle: Implications for cardiac muscle integrity. Nucleic Acid Ther. 2011, 21, 293–298. [Google Scholar]
- Wheeler, T.M. Myotonic dystrophy: Therapeutic strategies for the future. Neurotherapeutics 2008, 5, 592–600. [Google Scholar]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar]
- Duchenne, G.B. The pathology of paralysis with muscular degeneration (paralysie myosclerotique), or paralysis with apparent hypertrophy. Br. Med. J. 1867, 2, 541–542. [Google Scholar]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar]
- Lu, Q.L.; Yokota, T.; Takeda, S.; Garcia, L.; Muntoni, F.; Partridge, T. The status of exon skipping as a therapeutic approach to Duchenne muscular dystrophy. Mol. Ther. 2011, 19, 9–15. [Google Scholar]
- Aartsma-Rus, A. Antisense-mediated modulation of splicing: Therapeutic implications for Duchenne muscular dystrophy. RNA Biol. 2010, 7, 453–461. [Google Scholar]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar]
- Yokota, T.; Lu, Q.L.; Morgan, J.E.; Davies, K.E.; Fisher, R.; Takeda, S.; Partridge, T.A. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. J. Cell Sci. 2006, 119, 2679–2687. [Google Scholar]
- Hoffman, E.P.; Morgan, J.E.; Watkins, S.C.; Partridge, T.A. Somatic reversion/suppression of the mouse mdx phenotype in vivo. J. Neurol. Sci. 1990, 99, 9–25. [Google Scholar] [CrossRef]
- Klein, C.J.; Coovert, D.D.; Bulman, D.E.; Ray, P.N.; Mendell, J.R.; Burghes, A.H. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): Evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am. J. Hum. Genet. 1992, 50, 950–959. [Google Scholar]
- Lu, Q.L.; Morris, G.E.; Wilton, S.D.; Ly, T.; Artem'yeva, O.V.; Strong, P.; Partridge, T.A. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 2000, 148, 985–996. [Google Scholar]
- Echigoya, Y.; Lee, J.; Rodrigues, M.; Nagata, T.; Tanihata, J.; Nozohourmehrabad, A.; Panesar, D.; Miskew, B.; Aoki, Y.; Yokota, T. Mutation types and aging differently affect revertant fiber expansion in dystrophic Mdx and Mdx52 mice. PLoS One 2013, in press. [Google Scholar]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar]
- Saito, T.; Nakamura, A.; Aoki, Y.; Yokota, T.; Okada, T.; Osawa, M.; Takeda, S. Antisense PMO found in dystrophic dog model was effective in cells from exon 7-deleted DMD patient. PLoS One 2010, 5, e12239. [Google Scholar]
- Aartsma-Rus, A.; de Winter, C.L.; Janson, A.A.; Kaman, W.E.; van Ommen, G.J.; den Dunnen, J.T.; van Deutekom, J.C. Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: Indication for steric hindrance of SR protein binding sites. Oligonucleotides 2005, 15, 284–297. [Google Scholar]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar]
- Bertoni, C.; Lau, C.; Rando, T.A. Restoration of dystrophin expression in mdx muscle cells by chimeraplast-mediated exon skipping. Hum. Mol. Genet. 2003, 12, 1087–1099. [Google Scholar]
- Bremmer-Bout, M.; Aartsma-Rus, A.; de Meijer, E.J.; Kaman, W.E.; Janson, A.A.; Vossen, R.H.; van Ommen, G.J.; den Dunnen, J.T.; van Deutekom, J.C. Targeted exon skipping in transgenic hDMD mice: A model for direct preclinical screening of human-specific antisense oligonucleotides. Mol. Ther. 2004, 10, 232–240. [Google Scholar]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Steinhaus, J.P.; Moulton, H.M.; Iversen, P.L.; Wilton, S.D. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 2007, 15, 1587–1592. [Google Scholar]
- Mann, C.J.; Honeyman, K.; Cheng, A.J.; Ly, T.; Lloyd, F.; Fletcher, S.; Morgan, J.E.; Partridge, T.A.; Wilton, S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 42–47. [Google Scholar]
- McClorey, G.; Fall, A.M.; Moulton, H.M.; Iversen, P.L.; Rasko, J.E.; Ryan, M.; Fletcher, S.; Wilton, S.D. Induced dystrophin exon skipping in human muscle explants. Neuromuscul. Disord. 2006, 16, 583–590. [Google Scholar]
- McClorey, G.; Moulton, H.M.; Iversen, P.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef]
- van Deutekom, J.C.; Bremmer-Bout, M.; Janson, A.A.; Ginjaar, I.B.; Baas, F.; den Dunnen, J.T.; van Ommen, G.J. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 2001, 10, 1547–1554. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Takeshima, Y.; Yagi, M.; Wada, H.; Matsuo, M. Intraperitoneal administration of phosphorothioate antisense oligodeoxynucleotide against splicing enhancer sequence induced exon skipping in dystrophin mRNA expressed in mdx skeletal muscle. Brain Dev. 2005, 27, 488–493. [Google Scholar] [CrossRef]
- Yokota, T.; Hoffman, E.; Takeda, S. Antisense oligo-mediated multiple exon skipping in a dog model of Duchenne muscular dystrophy. Methods Mol. Biol. 2011, 709, 299–312. [Google Scholar] [CrossRef]
- Cirak, S.; Feng, L.; Anthony, K.; Arechavala-Gomeza, V.; Torelli, S.; Sewry, C.; Morgan, J.E.; Muntoni, F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 462–467. [Google Scholar] [CrossRef]
- Muntoni, F.; Bushby, K.; van Ommen, G. 128th ENMC international workshop on “preclinical optimization and phase I/II clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy” 22–24 October 2004, Naarden, The Netherlands. Neuromuscul. Disord. 2005, 15, 450–457. [Google Scholar] [CrossRef]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Kamoshita, S.; Konishi, Y.; Segawa, M.; Fukuyama, Y. Congenital muscular dystrophy as a disease of the central nervous system. Arch. Neurol. 1976, 33, 513–516. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef]
- Michele, D.E.; Barresi, R.; Kanagawa, M.; Saito, F.; Cohn, R.D.; Satz, J.S.; Dollar, J.; Nishino, I.; Kelley, R.I.; Somer, H.; et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002, 418, 417–422. [Google Scholar] [CrossRef]
- Hayashi, Y.K.; Ogawa, M.; Tagawa, K.; Noguchi, S.; Ishihara, T.; Nonaka, I.; Arahata, K. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001, 57, 115–121. [Google Scholar] [CrossRef]
- Colombo, R.; Bignamini, A.A.; Carobene, A.; Sasaki, J.; Tachikawa, M.; Kobayashi, K.; Toda, T. Age and origin of the FCMD 3'-untranslated-region retrotransposal insertion mutation causing Fukuyama-type congenital muscular dystrophy in the Japanese population. Hum. genet. 2000, 107, 559–567. [Google Scholar] [CrossRef]
- Kobayashi, K.; Sasaki, J.; Kondo-Iida, E.; Fukuda, Y.; Kinoshita, M.; Sunada, Y.; Nakamura, Y.; Toda, T. Structural organization, complete genomic sequences and mutational analyses of the Fukuyama-type congenital muscular dystrophy gene, fukutin. FEBS Lett. 2001, 489, 192–196. [Google Scholar] [CrossRef]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeo, G.W.; Yeh, R.; Goldstein, P.; Mawson, M.; Sharp, P.A.; Burge, C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004, 32, W187–W190. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef]
- Bhagavati, S.; Leung, B.; Shafiq, S.A.; Ghatpande, A. Myotonic dystrophy: Decreased levels of myotonin protein kinase (Mt-PK) leads to apoptosis in muscle cells. Exp. Neurol. 1997, 146, 277–281. [Google Scholar] [CrossRef]
- Meola, G. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle Nerve 2000, 23, 1789–1799. [Google Scholar] [CrossRef]
- Meola, G. Myotonic dystrophies. Curr. Opin. Neurol. 2000, 13, 519–525. [Google Scholar] [CrossRef]
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genomics 2010, 11, 77–90. [Google Scholar] [CrossRef]
- Cho, D.H.; Tapscott, S.J. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim. Biophys. Acta 2007, 1772, 195–204. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G., Jr.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O'Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3' untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar]
- Hamshere, M.G.; Harley, H.; Harper, P.; Brook, J.D.; Brookfield, J.F. Myotonic dystrophy: The correlation of (CTG) repeat length in leucocytes with age at onset is significant only for patients with small expansions. J. Med. Genet. 1999, 36, 59–61. [Google Scholar]
- Harper, P. Myotonic Dystrophy, 3rd ed.; W B Saunders: London, UK, 2001. [Google Scholar]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef]
- Wang, J.; Pegoraro, E.; Menegazzo, E.; Gennarelli, M.; Hoop, R.C.; Angelini, C.; Hoffman, E.P. Myotonic dystrophy: Evidence for a possible dominant-negative RNA mutation. Hum. Mol. Genet. 1995, 4, 599–606. [Google Scholar] [CrossRef]
- Magana, J.J.; Cisneros, B. Perspectives on gene therapy in myotonic dystrophy type 1. J. Neurosci. Res. 2011, 89, 275–285. [Google Scholar] [CrossRef]
- Foff, E.P.; Mahadevan, M.S. Therapeutics development in myotonic dystrophy type 1. Muscle Nerve 2011, 44, 160–169. [Google Scholar] [CrossRef]
- Carango, P.; Noble, J.E.; Marks, H.G.; Funanage, V.L. Absence of myotonic dystrophy protein kinase (DMPK) mRNA as a result of a triplet repeat expansion in myotonic dystrophy. Genomics 1993, 18, 340–348. [Google Scholar] [CrossRef]
- Hofmann-Radvanyi, H.; Lavedan, C.; Rabes, J.P.; Savoy, D.; Duros, C.; Johnson, K.; Junien, C. Myotonic dystrophy: Absence of CTG enlarged transcript in congenital forms, and low expression of the normal allele. Hum. Mol. Genet. 1993, 2, 1263–1266. [Google Scholar] [CrossRef]
- Koga, R.; Nakao, Y.; Kurano, Y.; Tsukahara, T.; Nakamura, A.; Ishiura, S.; Nonaka, I.; Arahata, K. Decreased myotonin-protein kinase in the skeletal and cardiac muscles in myotonic dystrophy. Biochem. Biophys. Res. Commun. 1994, 202, 577–585. [Google Scholar] [CrossRef]
- Krahe, R.; Ashizawa, T.; Abbruzzese, C.; Roeder, E.; Carango, P.; Giacanelli, M.; Funanage, V.L.; Siciliano, M.J. Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics 1995, 28, 1–14. [Google Scholar]
- Maeda, M.; Taft, C.S.; Bush, E.W.; Holder, E.; Bailey, W.M.; Neville, H.; Perryman, M.B.; Bies, R.D. Identification, tissue-specific expression, and subcellular localization of the 80- and 71-kDa forms of myotonic dystrophy kinase protein. J. Biol. Chem. 1995, 270, 20246–20249. [Google Scholar]
- Novelli, G.; Gennarelli, M.; Zelano, G.; Pizzuti, A.; Fattorini, C.; Caskey, C.T.; Dallapiccola, B. Failure in detecting mRNA transcripts from the mutated allele in myotonic dystrophy muscle. Biochem. Mol. Biol. Int. 1993, 29, 291–297. [Google Scholar]
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef]
- Gonzalez-Barriga, A.; Mulders, S.A.; van de Giessen, J.; Hooijer, J.D.; Bijl, S.; van Kessel, I.D.; van Beers, J.; van Deutekom, J.C.; Fransen, J.A.; Wieringa, B.; et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol. Ther. Nucleic Acids 2013, 2, e81. [Google Scholar] [CrossRef]
- Lee, J.E.; Bennett, C.F.; Cooper, T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2012, 109, 4221–4226. [Google Scholar]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Rudnik-Schoneborn, S.; Wienker, T.; Zerres, K. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef]
- Zellweger, H. The genetic heterogeneity of spinal muscular atrophy (SMA). Birth Defects Orig. Artic. Ser. 1971, 7, 82–89. [Google Scholar]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Khoo, B.; Krainer, A.R. Splicing therapeutics in SMN2 and APOB. Curr. Opin. Mol. Ther. 2009, 11, 108–115. [Google Scholar]
- Markowitz, J.A.; Singh, P.; Darras, B.T. Spinal muscular atrophy: A clinical and research update. Pediatr. Neurol. 2012, 46, 1–12. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef]
- Lorson, C.L.; Rindt, H.; Shababi, M. Spinal muscular atrophy: Mechanisms and therapeutic strategies. Hum. Mol. Genet. 2010, 19, R111–R118. [Google Scholar] [CrossRef]
- van Meerbeke, J.P.; Sumner, C.J. Progress and promise: The current status of spinal muscular atrophy therapeutics. Discov. Med. 2011, 12, 291–305. [Google Scholar]
- Mitrpant, C.; Porensky, P.; Zhou, H.; Price, L.; Muntoni, F.; Fletcher, S.; Wilton, S.D.; Burghes, A.H. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: Towards a treatment for spinal muscular atrophy. PLoS One 2013, 8, e62114. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Aoki, M.; Liu, J.; Richard, I.; Bashir, R.; Britton, S.; Keers, S.M.; Oeltjen, J.; Brown, H.E.; Marchand, S.; Bourg, N.; et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology 2001, 57, 271–278. [Google Scholar]
- Anderson, L.V.; Davison, K.; Moss, J.A.; Young, C.; Cullen, M.J.; Walsh, J.; Johnson, M.A.; Bashir, R.; Britton, S.; Keers, S.; et al. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum. Mol. Genet. 1999, 8, 855–861. [Google Scholar] [CrossRef]
- Argov, Z.; Sadeh, M.; Mazor, K.; Soffer, D.; Kahana, E.; Eisenberg, I.; Mitrani-Rosenbaum, S.; Richard, I.; Beckmann, J.; Keers, S.; et al. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain 2000, 123, 1229–1237. [Google Scholar] [CrossRef]
- Foxton, R.M.; Laval, S.H.; Bushby, K.M. Characterisation of the dysferlin skeletal muscle promoter. Eur. J. Hum. Genet. 2004, 12, 127–131. [Google Scholar] [CrossRef]
- Guglieri, M.; Bushby, K. How to go about diagnosing and managing the limb-girdle muscular dystrophies. Neurol. India 2008, 56, 271–280. [Google Scholar] [CrossRef]
- Guglieri, M.; Straub, V.; Bushby, K.; Lochmuller, H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008, 21, 576–584. [Google Scholar] [CrossRef]
- Illa, I.; Serrano-Munuera, C.; Gallardo, E.; Lasa, A.; Rojas-Garcia, R.; Palmer, J.; Gallano, P.; Baiget, M.; Matsuda, C.; Brown, R.H. Distal anterior compartment myopathy: A dysferlin mutation causing a new muscular dystrophy phenotype. Ann. Neurol. 2001, 49, 130–134. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar]
- Han, R.; Bansal, D.; Miyake, K.; Muniz, V.P.; Weiss, R.M.; McNeil, P.L.; Campbell, K.P. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 2007, 117, 1805–1813. [Google Scholar] [CrossRef]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar]
- Matsuda, C.; Aoki, M.; Hayashi, Y.K.; Ho, M.F.; Arahata, K.; Brown, R.H., Jr. Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology 1999, 53, 1119–1122. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Singh, K.H.; Fokkema, I.F.; Ginjaar, I.B.; van Ommen, G.J.; den Dunnen, J.T.; van der Maarel, S.M. Therapeutic exon skipping for dysferlinopathies? Eur. J. Hum. Genet. 2010, 18, 889–894. [Google Scholar] [CrossRef]
- Wein, N.; Avril, A.; Bartoli, M.; Beley, C.; Chaouch, S.; Laforet, P.; Behin, A.; Butler-Browne, G.; Mouly, V.; Krahn, M.; et al. Efficient bypass of mutations in dysferlin deficient patient cells by antisense-induced exon skipping. Hum. Mutat. 2010, 31, 136–142. [Google Scholar] [CrossRef]
- Sinnreich, M.; Therrien, C.; Karpati, G. Lariat branch point mutation in the dysferlin gene with mild limb-girdle muscular dystrophy. Neurology 2006, 66, 1114–1116. [Google Scholar] [CrossRef]
- Rothstein, J.D. ALS―Motor neuron disease: Mechanism and development of new therapies. J. Vis. Exp. 2007, e245. [Google Scholar]
- Turner, M.; Al-Chalabi, A. Early symptom progression rate is related to ALS outcome: A prospective population-based study. Neurology 2002, 59, 2012–2013. [Google Scholar] [CrossRef]
- Cleveland, D.W. From Charcot to SOD1: Mechanisms of selective motor neuron death in ALS. Neuron 1999, 24, 515–520. [Google Scholar] [CrossRef]
- Cheah, B.C.; Vucic, S.; Krishnan, A.V.; Boland, R.A.; Kiernan, M.C. Neurophysiological index as a biomarker for ALS progression: Validity of mixed effects models. Amyotroph. Lateral Scler. 2011, 12, 33–38. [Google Scholar] [CrossRef]
- Morariu, M.A. A new classification of amyotrophic lateral sclerosis (ALS) and familial amyotrophic lateral sclerosis (FALS). Dis. Nerv. Syst. 1977, 38, 468–469. [Google Scholar]
- Armani, M.; Pierobon-Bormioli, S.; Mostacciuolo, M.L.; Cacciavillani, M.; Cassol, M.A.; Candeago, R.M.; Angelini, C. Familial ALS: Clinical, genetic and morphological features. Adv. Exp. Med. Biol. 1987, 209, 109–110. [Google Scholar]
- Penco, S.; Schenone, A.; Bordo, D.; Bolognesi, M.; Abbruzzese, M.; Bugiani, O.; Ajmar, F.; Garre, C. A SOD1 gene mutation in a patient with slowly progressing familial ALS. Neurology 1999, 53, 404–406. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar]
- Ratovitski, T.; Corson, L.B.; Strain, J.; Wong, P.; Cleveland, D.W.; Culotta, V.C.; Borchelt, D.R. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 1999, 8, 1451–1460. [Google Scholar] [CrossRef]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Devos, S.L.; Miller, T.M. Antisense oligonucleotides: Treating neurodegeneration at the level of RNA. Neurotherapeutics 2013, 10, 486–497. [Google Scholar] [CrossRef]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Koppers, M.; van Blitterswijk, M.M.; Vlam, L.; Rowicka, P.A.; van Vught, P.W.; Groen, E.J.; Spliet, W.G.; Engelen-Lee, J.; Schelhaas, H.J.; de Visser, M.; et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 837.e7–837.e13. [Google Scholar]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Mosca, L.; Lunetta, C.; Tarlarini, C.; Avemaria, F.; Maestri, E.; Melazzini, M.; Corbo, M.; Penco, S. Wide phenotypic spectrum of the TARDBP gene: Homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson’s disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol. Aging 2012, 33, 1846.e1–1846.e4. [Google Scholar]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- van Es, M.A.; Diekstra, F.P.; Veldink, J.H.; Baas, F.; Bourque, P.R.; Schelhaas, H.J.; Strengman, E.; Hennekam, E.A.; Lindhout, D.; Ophoff, R.A.; et al. A case of ALS-FTD in a large FALS pedigree with a K17I ANG mutation. Neurology 2009, 72, 287–288. [Google Scholar] [CrossRef] [Green Version]
- van Langenhove, T.; van der Zee, J.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Gijselinck, I.; van den Broeck, M.; Mattheijssens, M.; Peeters, K.; ve Deyn, P.P.; et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 2010, 74, 366–371. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; de Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Byrne, S.; Elamin, M.; Bede, P.; Shatunov, A.; Walsh, C.; Corr, B.; Heverin, M.; Jordan, N.; Kenna, K.; Lynch, C.; et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: A population-based cohort study. Lancet Neurol. 2012, 11, 232–240. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Harms, M.B.; Cady, J.; Zaidman, C.; Cooper, P.; Bali, T.; Allred, P.; Cruchaga, C.; Baughn, M.; Libby, R.T.; Pestronk, A.; et al. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 2234.e13–2234.e19. [Google Scholar]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Mok, K.Y.; Koutsis, G.; Schottlaender, L.V.; Polke, J.; Panas, M.; Houlden, H. High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol. Aging 2012, 33, 1851.e1–1851.e5. [Google Scholar]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013, 21, 102–108. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar] [CrossRef]
- Garcia-Redondo, A.; Dols-Icardo, O.; Rojas-Garcia, R.; Esteban-Perez, J.; Cordero-Vazquez, P.; Munoz-Blanco, J.L.; Catalina, I.; Gonzalez-Munoz, M.; Varona, L.; Sarasola, E.; et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum. Mutat. 2013, 34, 79–82. [Google Scholar] [CrossRef]
- Rutherford, N.J.; DeJesus-Hernandez, M.; Baker, M.C.; Kryston, T.B.; Brown, P.E.; Lomen-Hoerth, C.; Boylan, K.; Wszolek, Z.K.; Rademakers, R. C9ORF72 hexanucleotide repeat expansions in patients with ALS from the Coriell Cell Repository. Neurology 2012, 79, 482–483. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Heckman, M.G.; Dejesus-Hernandez, M.; Baker, M.C.; Soto-Ortolaza, A.I.; Rayaprolu, S.; Stewart, H.; Finger, E.; Volkening, K.; Seeley, W.W.; et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol. Aging 2012, 33, 2950.e5–2950.e7. [Google Scholar]
- Cruts, M.; Gijselinck, I.; van Langenhove, T.; van der Zee, J.; van Broeckhoven, C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013, in press. [Google Scholar]
- Nuytemans, K.; Bademci, G.; Kohli, M.M.; Beecham, G.W.; Wang, L.; Young, J.I.; Nahab, F.; Martin, E.R.; Gilbert, J.R.; Benatar, M.; et al. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Rademakers, R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012, 11, 297–298. [Google Scholar] [CrossRef]
- Dance, A. Alzheimer Research Forum. In Proceedings of 23rd Annual International Symposium on ALS/MND, Chicago, IL, USA, 5–7 December 2012.
- Donnelly, C.J.; Ostrow, L.W.; Zhang, P.; Vidensky, S.; Hoover, B.N.; Balasubramanian, U.; Li, Y.; Maragakis, N.J.; Tienari, P.; Traynor, B.J.; et al. Development of a C9ORF72 ALS Antisense Therapy and a Therapeutic Biomarker. In Presented at the 2012 Neuroscience Meeting Planner, New Orleans, LA, USA, 17 October 2012.
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Frank, S.; Ondo, W.; Fahn, S.; Hunter, C.; Oakes, D.; Plumb, S.; Marshall, F.; Shoulson, I.; Eberly, S.; Walker, F.; et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clin. Neuropharmacol. 2008, 31, 127–133. [Google Scholar] [CrossRef]
- Arnulf, I.; Nielsen, J.; Lohmann, E.; Schiefer, J.; Wild, E.; Jennum, P.; Konofal, E.; Walker, M.; Oudiette, D.; Tabrizi, S.; et al. Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol. 2008, 65, 482–488. [Google Scholar] [CrossRef]
- Carlock, L.; Walker, P.D.; Shan, Y.; Gutridge, K. Transcription of the Huntington disease gene during the quinolinic acid excitotoxic cascade. Neuroreport 1995, 6, 1121–1124. [Google Scholar] [CrossRef]
- Burns, A.; Folstein, S.; Brandt, J.; Folstein, M. Clinical assessment of irritability, aggression, and apathy in Huntington and Alzheimer disease. J. Nerv. Ment. Dis. 1990, 178, 20–26. [Google Scholar] [CrossRef]
- Marder, K.; Zhao, H.; Myers, R.H.; Cudkowicz, M.; Kayson, E.; Kieburtz, K.; Orme, C.; Paulsen, J.; Penney, J.B., Jr.; Siemers, E.; et al. Rate of functional decline in Huntington’s disease. Neurology 2000, 54, 452–458. [Google Scholar] [CrossRef]
- Reiner, A.; Albin, R.L.; Anderson, K.D.; D'Amato, C.J.; Penney, J.B.; Young, A.B. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA 1988, 85, 5733–5737. [Google Scholar]
- Rosas, H.D.; Hevelone, N.D.; Zaleta, A.K.; Greve, D.N.; Salat, D.H.; Fischl, B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 2005, 65, 745–747. [Google Scholar] [CrossRef]
- Cha, J.H.; Kosinski, C.M.; Kerner, J.A.; Alsdorf, S.A.; Mangiarini, L.; Davies, S.W.; Penney, J.B.; Bates, G.P.; Young, A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6480–6485. [Google Scholar] [CrossRef]
- Ross, C.A.; Shoulson, I. Huntington disease: Pathogenesis, biomarkers, and approaches to experimental therapeutics. Parkinsonism Relat. Disord. 2009, 15, S135–S138. [Google Scholar] [CrossRef]
- Liu, J.P.; Zeitlin, S.O. The long and the short of aberrant ciliogenesis in Huntington disease. J. Clin. Invest. 2011, 121, 4237–4241. [Google Scholar] [CrossRef]
- Urbaniak Hunter, K.; Yarbrough, C.; Ciacci, J. Gene- and cell-based approaches for neurodegenerative disease. Adv. Exp. Med. Biol. 2010, 671, 117–130. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Barton, D.E.; Davison, B.C.; Ferguson-Smith, M.A. Analysis of the huntingtin gene reveals a trinucleotide-length polymorphism in the region of the gene that contains two CCG-rich stretches and a correlation between decreased age of onset of Huntington’s disease and CAG repeat number. Hum. Mol. Genet. 1993, 2, 1713–1715. [Google Scholar] [CrossRef]
- Aronin, N.; Chase, K.; Young, C.; Sapp, E.; Schwarz, C.; Matta, N.; Kornreich, R.; Landwehrmeyer, B.; Bird, E.; Beal, M.F.; et al. CAG expansion affects the expression of mutant Huntingtin in the Huntington’s disease brain. Neuron 1995, 15, 1193–1201. [Google Scholar] [CrossRef]
- Becher, M.W.; Kotzuk, J.A.; Sharp, A.H.; Davies, S.W.; Bates, G.P.; Price, D.L.; Ross, C.A. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar]
- Myers, R.H.; Vonsattel, J.P.; Stevens, T.J.; Cupples, L.A.; Richardson, E.P.; Martin, J.B.; Bird, E.D. Clinical and neuropathologic assessment of severity in Huntington’s disease. Neurology 1988, 38, 341–347. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Gutekunst, C.A.; Persichetti, F.; McNeil, S.M.; Kowall, N.W.; Gusella, J.F.; MacDonald, M.E.; Beal, M.F.; Hersch, S.M. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J. Neurosci. 1997, 17, 3052–3063. [Google Scholar]
- Hoogeveen, A.T.; Willemsen, R.; Meyer, N.; de Rooij, K.E.; Roos, R.A.; van Ommen, G.J.; Galjaard, H. Characterization and localization of the Huntington disease gene product. Hum. Mol. Genet. 1993, 2, 2069–2073. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O'Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- White, J.K.; Auerbach, W.; Duyao, M.P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; MacDonald, M.E. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 1997, 17, 404–410. [Google Scholar] [CrossRef]
- Rigamonti, D.; Bauer, J.H.; De-Fraja, C.; Conti, L.; Sipione, S.; Sciorati, C.; Clementi, E.; Hackam, A.; Hayden, M.R.; Li, Y.; et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 2000, 20, 3705–3713. [Google Scholar]
- Zhang, Y.; Leavitt, B.R.; van Raamsdonk, J.M.; Dragatsis, I.; Goldowitz, D.; MacDonald, M.E.; Hayden, M.R.; Friedlander, R.M. Huntingtin inhibits caspase-3 activation. EMBO J. 2006, 25, 5896–5906. [Google Scholar] [CrossRef]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; de Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef]
- Velier, J.; Kim, M.; Schwarz, C.; Kim, T.W.; Sapp, E.; Chase, K.; Aronin, N.; DiFiglia, M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef]
- Smith, R.; Brundin, P.; Li, J.Y. Synaptic dysfunction in Huntington’s disease: A new perspective. Cell. Mol. Life Sci. 2005, 62, 1901–1912. [Google Scholar] [CrossRef]
- Parker, J.A.; Metzler, M.; Georgiou, J.; Mage, M.; Roder, J.C.; Rose, A.M.; Hayden, M.R.; Neri, C. Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity. J. Neurosci. 2007, 27, 11056–11064. [Google Scholar] [CrossRef]
- Dragatsis, I.; Levine, M.S.; Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000, 26, 300–306. [Google Scholar] [CrossRef]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef]
- Lombardi, M.S.; Jaspers, L.; Spronkmans, C.; Gellera, C.; Taroni, F.; di Maria, E.; Donato, S.D.; Kaemmerer, W.F. A majority of Huntington’s disease patients may be treatable by individualized allele-specific RNA interference. Exp. Neurol. 2009, 217, 312–319. [Google Scholar] [CrossRef]
- Pfister, E.L.; Kennington, L.; Straubhaar, J.; Wagh, S.; Liu, W.; DiFiglia, M.; Landwehrmeyer, B.; Vonsattel, J.P.; Zamore, P.D.; Aronin, N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 2009, 19, 774–778. [Google Scholar] [CrossRef]
- van Bilsen, P.H.; Jaspers, L.; Lombardi, M.S.; Odekerken, J.C.; Burright, E.N.; Kaemmerer, W.F. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum. Gene Ther. 2008, 19, 710–719. [Google Scholar] [CrossRef]
- Hu, J.; Matsui, M.; Corey, D.R. Allele-selective inhibition of mutant huntingtin by peptide nucleic acid-peptide conjugates, locked nucleic acid, and small interfering RNA. Ann. N. Y. Acad. Sci. 2009, 1175, 24–31. [Google Scholar] [CrossRef]
- Hu, J.; Matsui, M.; Gagnon, K.T.; Schwartz, J.C.; Gabillet, S.; Arar, K.; Wu, J.; Bezprozvanny, I.; Corey, D.R. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol. 2009, 27, 478–484. [Google Scholar] [CrossRef]
- Gagnon, K.T.; Pendergraff, H.M.; Deleavey, G.F.; Swayze, E.E.; Potier, P.; Randolph, J.; Roesch, E.B.; Chattopadhyaya, J.; Damha, M.J.; Bennett, C.F.; et al. Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry 2010, 49, 10166–10178. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef]
- Yamamoto, A.; Lucas, J.J.; Hen, R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [Google Scholar] [CrossRef]
- Boado, R.J.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Pardridge, W.M. Antisense-mediated down-regulation of the human huntingtin gene. J. Pharmacol. Exp. Ther. 2000, 295, 239–243. [Google Scholar]
- Hu, J.; Dodd, D.W.; Hudson, R.H.; Corey, D.R. Cellular localization and allele-selective inhibition of mutant huntingtin protein by peptide nucleic acid oligomers containing the fluorescent nucleobase [bis-o-(aminoethoxy)phenyl]pyrrolocytosine. Bioorg. Med. Chem. Lett. 2009, 19, 6181–6184. [Google Scholar] [CrossRef]
- Nellemann, C.; Abell, K.; Norremolle, A.; Lokkegaard, T.; Naver, B.; Ropke, C.; Rygaard, J.; Sorensen, S.A.; Hasholt, L. Inhibition of Huntington synthesis by antisense oligodeoxynucleotides. Mol. Cell. Neurosci. 2000, 16, 313–323. [Google Scholar] [CrossRef]
- Fiszer, A.; Olejniczak, M.; Switonski, P.M.; Wroblewska, J.P.; Wisniewska-Kruk, J.; Mykowska, A.; Krzyzosiak, W.J. An evaluation of oligonucleotide-based therapeutic strategies for polyQ diseases. BMC Mol. Biol. 2012, 13, e6. [Google Scholar] [CrossRef]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Lee, J.J.A.; Yokota, T. Antisense Therapy in Neurology. J. Pers. Med. 2013, 3, 144-176. https://doi.org/10.3390/jpm3030144
AMA Style
Lee JJA, Yokota T. Antisense Therapy in Neurology. Journal of Personalized Medicine. 2013; 3(3):144-176. https://doi.org/10.3390/jpm3030144
Chicago/Turabian StyleLee, Joshua J. A., and Toshifumi Yokota. 2013. "Antisense Therapy in Neurology" Journal of Personalized Medicine 3, no. 3: 144-176. https://doi.org/10.3390/jpm3030144