A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Genotyping and Data Quality Control Analysis
2.3. Principal Component Analysis
2.4. Heritability Analysis
2.5. Imputation
2.6. GWAS Association Analysis and Post GWAS Quality Control
2.7. Pathway Analysis
3. Results
3.1. Heritability Analysis
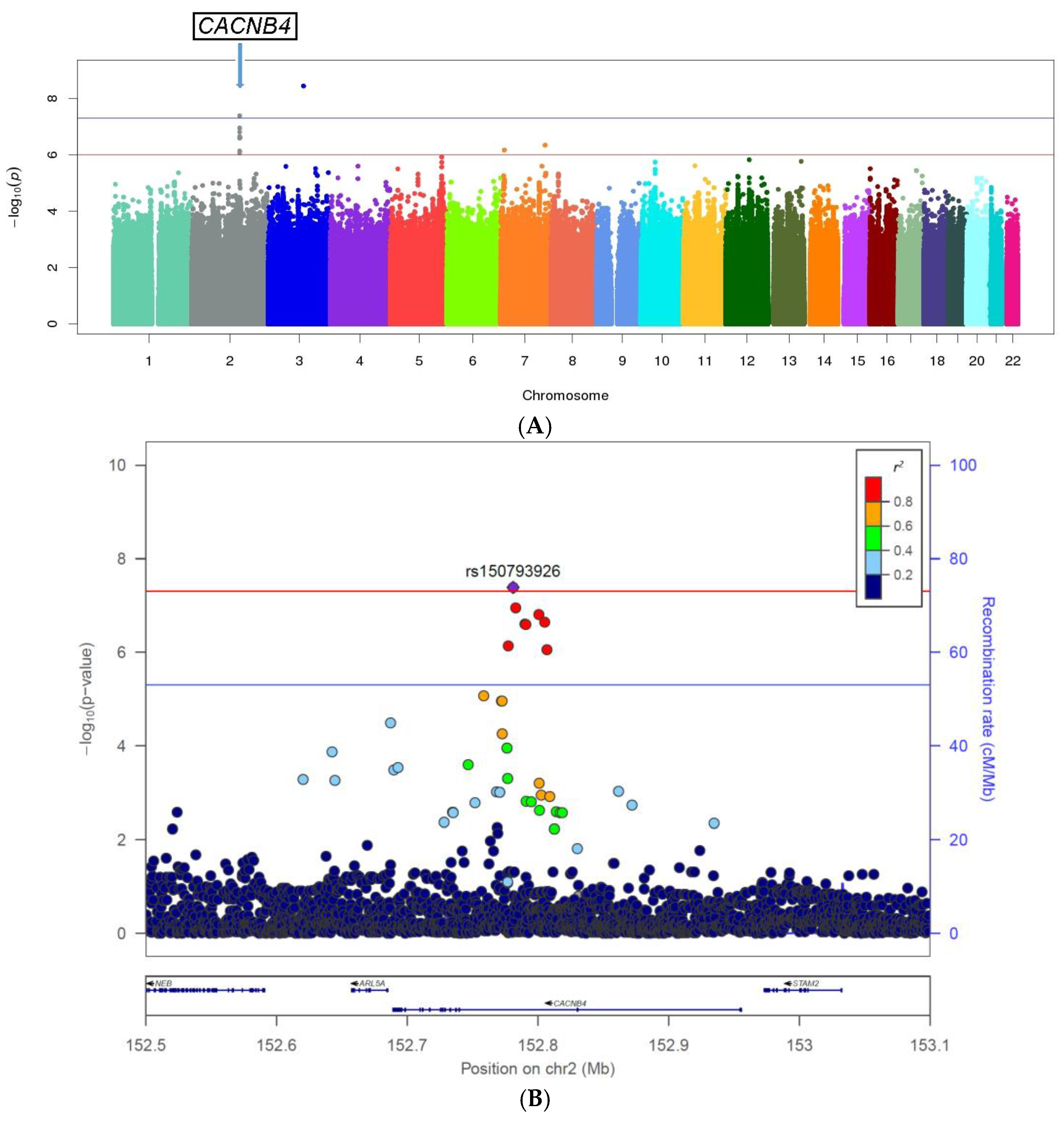
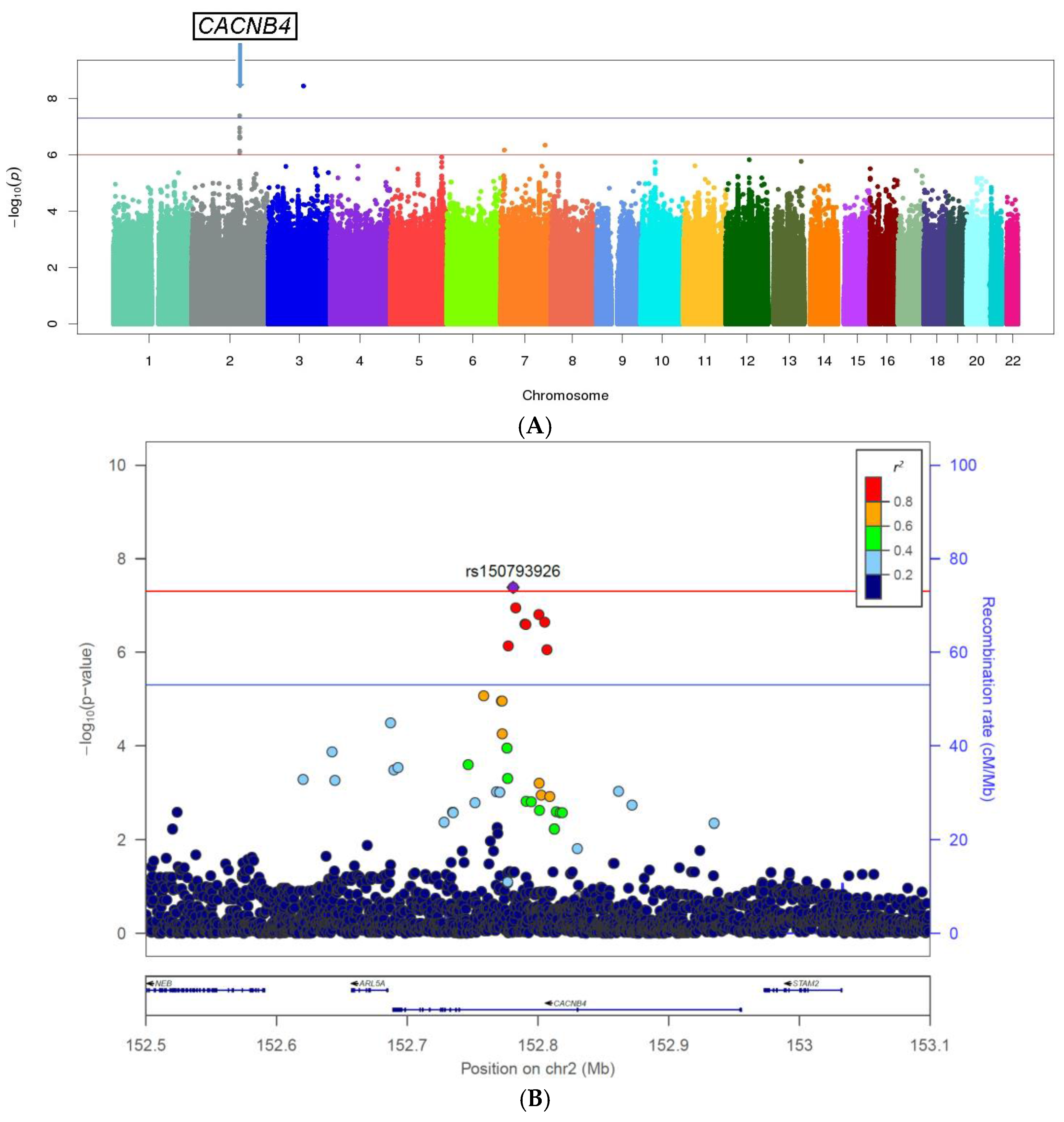
3.2. Genome-Wide Single Variant Association Analysis
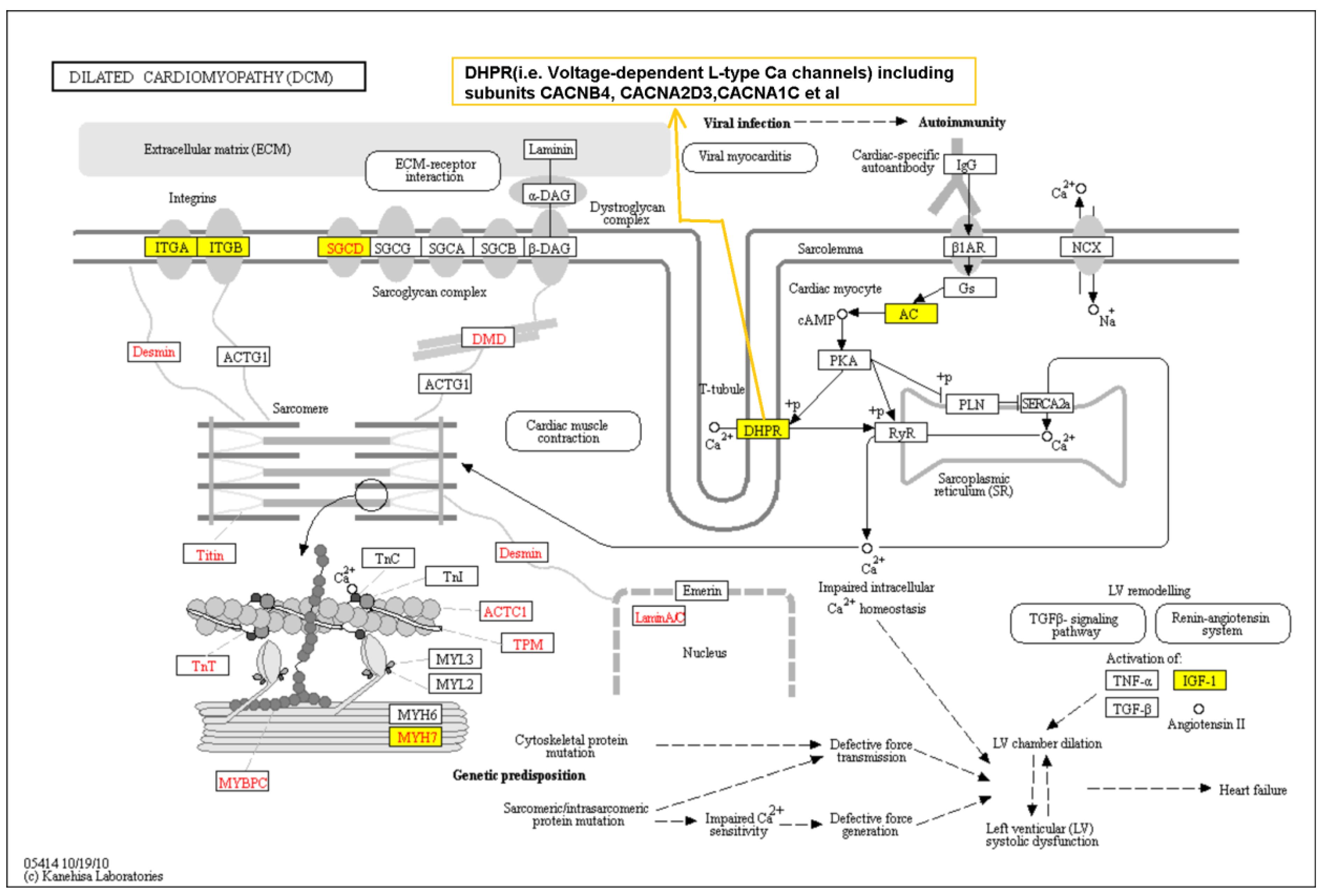
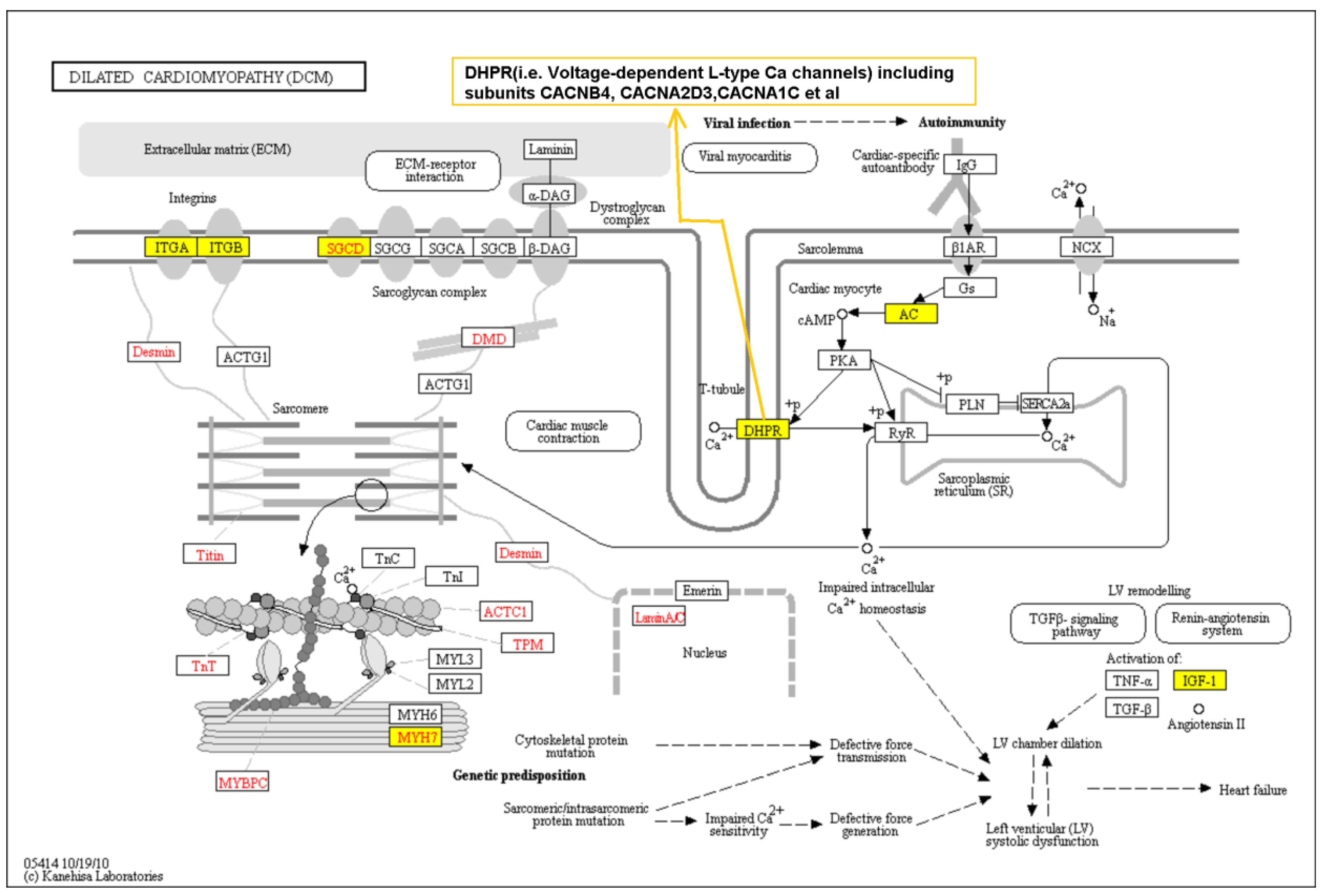
3.3. Pathway Analysis Based on Genome-Wide Single Variant Association Analysis
3.4. Gene-Based Association Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Writing Group Members; Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; et al. Executive summary: Heart disease and stroke statistics—2016 update: A report from the american heart association. Circulation 2016, 133, 447–454. [Google Scholar]
- Adams, K.F., Jr.; Fonarow, G.C.; Emerman, C.L.; LeJemtel, T.H.; Costanzo, M.R.; Abraham, W.T.; Berkowitz, R.L.; Galvao, M.; Horton, D.P.; Committee, A.S.A.; et al. Characteristics and outcomes of patients hospitalized for heart failure in the united states: Rationale, design, and preliminary observations from the first 100,000 cases in the acute decompensated heart failure national registry (adhere). Am. Heart J. 2005, 149, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Wexler, R.K.; Elton, T.; Pleister, A.; Feldman, D. Cardiomyopathy: An overview. Am. Fam. Phys. 2009, 79, 778–784. [Google Scholar]
- Petretta, M.; Pirozzi, F.; Sasso, L.; Paglia, A.; Bonaduce, D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am. J. Cardiol. 2011, 108, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Seidelmann, S.B.; Laur, O.; Hwa, J.; Depasquale, E.; Bellumkonda, L.; Sugeng, L.; Pomianowski, P.; Testani, J.; Chen, M.; McKenna, W.; et al. Familial dilated cardiomyopathy diagnosis is commonly overlooked at the time of transplant listing. J. Heart Lung Transplant. 2016, 35, 474–480. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Barefield, D.Y.; Puckelwartz, M.J. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015, 21, 174–182. [Google Scholar] [PubMed]
- Sisakian, H. Cardiomyopathies: Evolution of pathogenesis concepts and potential for new therapies. World J. Cardiol. 2014, 6, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.S.; Labenberg, J.R.; Tefft, M.C. Black-white differences in idiopathic dilated cardiomyopathy: The Washington DC dilated cardiomyopathy study. Epidemiology 1993, 4, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Kinnamon, D.D.; Morales, A.; Bowen, D.J.; Burke, W.; Hershberger, R.E.; DCM Consortium. Toward genetics-driven early intervention in dilated cardiomyopathy: Design and implementation of the DCM precision medicine study. Circ. Cardiovasc. Genet. 2017, 10, pii: e001826. [Google Scholar]
- Carnethon, M.R.; Pu, J.; Howard, G.; Albert, M.A.; Anderson, C.A.M.; Bertoni, A.G.; Mujahid, M.S.; Palaniappan, L.; Taylor, H.A., Jr.; Willis, M.; et al. Cardiovascular health in african americans: A scientific statement from the american heart association. Circulation 2017, 136, e393–e423. [Google Scholar] [CrossRef] [PubMed]
- Franciosa, J.A.; Ferdinand, K.C.; Yancy, C.W.; Consensus Statement on Heart Failure in African Americans Writing Group. Treatment of heart failure in African Americans: A consensus statement. Congest. Heart Fail. 2010, 16, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Colvin-Adams, M.; Yancy, C.W. Heart failure in African Americans: Disparities can be overcome. Cleve Clin. J. Med. 2014, 81, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Basavarajaiah, S.; Boraita, A.; Whyte, G.; Wilson, M.; Carby, L.; Shah, A.; Sharma, S. Ethnic differences in left ventricular remodeling in highly-trained athletes relevance to differentiating physiologic left ventricular hypertrophy from hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2008, 51, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Peprah, E.; Xu, H.; Tekola-Ayele, F.; Royal, C.D. Genome-wide association studies in Africans and African Americans: Expanding the framework of the genomics of human traits and disease. Public Health Genom. 2015, 18, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.D.; Fornage, M.; McArdle, P.F.; Cheng, Y.C.; Pulit, S.L.; Wong, Q.; Dave, T.; Williams, S.R.; Corriveau, R.; Gwinn, K.; et al. Using previously genotyped controls in genome-wide association studies (GWAS): Application to the stroke genetics network (SiGN). Front. Genet. 2014, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.A.; Lange, E.M. Using public control genotype data to increase power and decrease cost of case-control genetic association studies. Hum. Genet. 2010, 128, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Shete, S.; Hosking, F.J.; Robertson, L.B.; Dobbins, S.E.; Sanson, M.; Malmer, B.; Simon, M.; Marie, Y.; Boisselier, B.; Delattre, J.Y.; et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009, 41, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bernardo, M.C.; Crowther-Swanepoel, D.; Broderick, P.; Webb, E.; Sellick, G.; Wild, R.; Sullivan, K.; Vijayakrishnan, J.; Wang, Y.; Pittman, A.M.; et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 2008, 40, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Network, N.S.G.; International Stroke Genetics, C. Loci associated with ischaemic stroke and its subtypes (SiGN): A genome-wide association study. Lancet Neurol. 2016, 15, 174–184. [Google Scholar]
- Cheng, Y.C.; O'Connell, J.R.; Cole, J.W.; Stine, O.C.; Dueker, N.; McArdle, P.F.; Sparks, M.J.; Shen, J.; Laurie, C.C.; Nelson, S.; et al. Genome-wide association analysis of ischemic stroke in young adults. G3 2011, 1, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. Genome-wide complex trait analysis (GCTA): Methods, data analyses, and interpretations. Methods Mol. Biol. 2013, 1019, 215–236. [Google Scholar] [PubMed]
- Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011, 88, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [Green Version]
- Delaneau, O.; Zagury, J.F.; Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 2013, 10, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Howie, B.; Marchini, J.; Stephens, M. Genotype imputation with thousands of genomes. G3 2011, 1, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Marchini, J.; Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010, 11, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Ruhle, F.; Weis, T.; Homuth, G.; Keller, A.; Franke, J.; Peil, B.; Lorenzo Bermejo, J.; Frese, K.; Huge, A.; et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur. Heart J. 2014, 35, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Esslinger, U.B.; Reinhard, W.; Petrov, G.; Winkler, T.; Komajda, M.; Isnard, R.; Charron, P.; Villard, E.; Cambien, F.; et al. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genet. 2010, 6, e1001167. [Google Scholar] [CrossRef] [PubMed]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–462. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Zhu, Z.; Vinkhuyzen, A.A.; Hill, W.D.; McRae, A.F.; Visscher, P.M.; Yang, J. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci. Rep. 2016, 6, 32894. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Consortium, G.T.; Nicolae, D.L.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Horstick, E.J.; Hirata, H.; Kuwada, J.Y. Identification and expression of voltage-gated calcium channel β subunits in Zebrafish. Dev. Dyn. 2008, 237, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Haase, H. Ahnak, a new player in β-adrenergic regulation of the cardiac L-type Ca2+ channel. Cardiovasc. Res. 2007, 73, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Foell, J.D.; Balijepalli, R.C.; Delisle, B.P.; Yunker, A.M.; Robia, S.L.; Walker, J.W.; McEnery, M.W.; January, C.T.; Kamp, T.J. Molecular heterogeneity of calcium channel β-subunits in canine and human heart: Evidence for differential subcellular localization. Physiol. Genom. 2004, 17, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Buraei, Z.; Yang, J. The β subunit of voltage-gated Ca2+ channels. Physiol. Rev. 2010, 90, 1461–1506. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, G.; Feng, J.; Zhang, L.; Li, J. Identifying ion channel genes related to cardiomyopathy using a novel decision forest strategy. Mol. Biosyst. 2014, 10, 2407–2414. [Google Scholar] [CrossRef] [PubMed]
- Fretheim, A.; Odgaard-Jensen, J.; Brors, O.; Madsen, S.; Njolstad, I.; Norheim, O.F.; Svilaas, A.; Kristiansen, I.S.; Thurmer, H.; Flottorp, S. Comparative effectiveness of antihypertensive medication for primary prevention of cardiovascular disease: Systematic review and multiple treatments meta-analysis. BMC Med. 2012, 10, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Yang, M.; Guo, J.; Zhu, C.; Yang, J.; Wang, Y.; Yang, X.; He, L. Calcium channel blockers versus other classes of drugs for hypertension. Cochrane Database Syst. Rev. 2010, CD003654. [Google Scholar] [CrossRef] [PubMed]
- Materson, B.J.; Reda, D.J.; Cushman, W.C.; Massie, B.M.; Freis, E.D.; Kochar, M.S.; Hamburger, R.J.; Fye, C.; Lakshman, R.; Gottdiener, J.; et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The department of veterans affairs cooperative study group on antihypertensive agents. N. Engl. J. Med. 1993, 328, 914–921. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Weber, M.; Baker, M.B.; Patel, R.S.; Quyyumi, A.A.; Bao, G.; Searles, C.D. MicroRNA expression profile in CAD patients and the impact of ACEI/ARB. Cardiol. Res. Pract. 2011, 2011, 532915. [Google Scholar] [CrossRef] [PubMed]
- Mopidevi, B.; Ponnala, M.; Kumar, A. Human angiotensinogen +11525 C/A polymorphism modulates its gene expression through microrna binding. Physiol. Genomics 2013, 45, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Fernando Garcia, G.W.; Young, N.; Foster, B.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movsesian, M.A.; Bristow, M.R. Alterations in cAMP-mediated signaling and their role in the pathophysiology of dilated cardiomyopathy. Curr. Top. Dev. Biol. 2005, 68, 25–48. [Google Scholar] [PubMed]
- Li, J. Alterations in cell adhesion proteins and cardiomyopathy. World J. Cardiol. 2014, 6, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Concato, J.; Brophy, M.; Fiore, L.; Pyarajan, S.; Breeling, J.; Whitbourne, S.; Deen, J.; Shannon, C.; Humphries, D.; et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 2016, 70, 214–223. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Genes | #SNPs | CHR | SNP ID | Position | Minor Allele | Reference Allele | MAF Cases | MAF Controls | OR | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|
| (intronic) CACNB4 | rs150793926 | 152781063 | GTA | G | 0.04 | 0.07 | 0.49 | 4.10 × 10−8 | ||
| 8 | 2 | rs113760736 | 152800750 | C | T | 0.03 | 0.07 | 0.50 | 1.50 × 10−7 | |
| rs12623883 | 152790117 | A | G | 0.03 | 0.06 | 0.50 | 2.45 × 10−7 | |||
| (intronic) MGAM | 1 | 7 | rs4341082 | 141759846 | T | C | 0.38 | 0.47 | 0.70 | 4.50 × 10−7 |
| (intergenic) THSD7A, THEM106B | 1 | 7 | rs74676849 | 11889492 | G | A | 0.1 | 0.05 | 2.03 | 6.80 × 10−7 |
| Classification | Canonical Pathways $ | Rank | p-Value | Corrected p-Value * | Genes in the Pathway That Are Associated with IDC |
|---|---|---|---|---|---|
| Cardiovascular system | Role of NFAT in cardiac hypertrophy | 1 | 4.3 × 10−5 | 0.01 | GNAI1, PLCB1, CAMK2B, SRC, CALM1, PIK3R4, MRAS, CAMK1D, PPP3CC, IGF1, PLCG2, IGF1R, CAMK2G, ADCY2, CAMK1G, PLCL1, ADCY8, MEF2A, ADCY9 |
| Cellular effects of sildenafil (Viagra) | 6 | 3.4 × 10−4 | 0.02 | PLCB1, MYH7, CALM1, PRKG2, PRKG1, PLCG2, CACNA1C, ADCY2, PDE3A, ACTA1, KCNQ3, PLCL1, ADCY8, ADCY9 | |
| Cardiac hypertrophy signaling | 17 | 4.4 × 10−3 | 0.10 | GNAI1, PLCB1, CALM1, PIK3R4, MRAS, ADRA1D, PPP3CC, IGF1, PLCG2, IGF1R, CACNA1C, ADCY2, ADRA2A, PLCL1, ADCY8, MEF2A, ADCY9 | |
| Cellular signals | cAMP-mediated signaling | 2 | 7.8 × 10−5 | 0.01 | GNAI1, CAMK2B, SRC, CALM1, CHRM3, CAMK1D, PPP3CC, PDE8A, CAMK2G, GRM7, ADCY2, PDE3A, CAMK1G, ADRA2A, OPRM1, ADCY8, AKAP6, ADCY9, AKAP1, HTR4, XCR1 |
| Chemokine signaling | 5 | 3.0 × 10−4 | 0.02 | GNAI1, PLCB1, CAMK2B, SRC, CALM1, MRAS, CAMK2G, CAMK1D, CAMK1G, PLCG2 | |
| Gap junction signaling | 8 | 7.2 × 10−4 | 0.03 | GNAI1, PLCB1, SRC, PIK3R4, MRAS, PRKG2, PRKG1, PPP3CC, PLCG2, TUBB2B, ADCY2, ACTA1, PLCL1, ADCY8, ADCY9 | |
| RhoA signaling | 11 | 2.0 × 10−3 | 0.07 | IGF1R, PLXNA1, DLC1, SEPT9, ARHGAP1, ANLN, CDC42EP3, ACTA1, ARHGAP12, RHPN2, IGF1, RAPGEF2 | |
| RhoGDI signaling | 12 | 2.2 × 10−3 | 0.07 | GNAI1, CDH13, CDH2, ARHGEF3, SRC, CDH12, MRAS, CDH4, ARHGAP1, WASL, DLC1, CDH10, ACTA1, CDH8, ARHGAP12 | |
| Calcium signaling | 13 | 2.9 × 10−3 | 0.07 | CAMK2B, TP63, MYH7, CALM1, CAMK1D, PPP3CC, NFATC3, GRIK1, CAMK2G, CAMK1G, CAMKK2, ACTA1, GRIN3A, MEF2A, CHRNA9 | |
| Protein kinase A signaling | 14 | 2.9 × 10−3 | 0.07 | CAMK2B, GNAI1, CALM1, PPP3CC, DCC, PDE3A, ADCY2, ADCY8, PLCL1, AKAP6, PTPRS, AKAP1, PLCB1, PTPRM, CDC25A, PTPRT, DUSP22, PLCG2, NFATC3, PDE8A, PTPN12, CAMK2G, PTPRE, CDC23, PTPN7, ADCY9 | |
| Dopamine-DARPP32 feedback in cAMP signaling | 15 | 3.0 × 10−3 | 0.07 | GNAI1, PLCB1, CALM1, PRKG2, PRKG1, PPP3CC, PLCG2, CACNA1C, ADCY2, CAMKK2, GRIN3A, ADCY8, PLCL1, ADCY9 | |
| G-protein coupled receptor signaling | 16 | 3.6 × 10−3 | 0.09 | GNAI1, PLCB1, CAMK2B, SRC, PIK3R4, MRAS, CHRM3, ADRA1D, PDE8A, CAMK2G, GRM7, ADCY2, PDE3A, ADRA2A, OPRM1, ADCY8, ADCY9, HTR4, XCR1 | |
| Neuronal/neuromuscular system | Neuropathic pain signaling in dorsal horn neurons | 3 | 8.9 × 10−5 | 0.01 | PLCB1, CAMK2B, SRC, TACR1, PIK3R4, CAMK1D, PLCG2, CAMK2G, GRM7, CAMK1G, GRIN3A, KCNQ3, PLCL1 |
| Glioma signaling | 4 | 2.2 × 10−4 | 0.02 | IGF1R, CAMK2B, CALM1, MRAS, PIK3R4, CAMK2G, CAMK1D, CAMK1G, E2F3, IGF1, IGF2R, PLCG2 | |
| Netrin signaling | 7 | 5.4 × 10−4 | 0.03 | NFATC3, UNC5C, ABLIM3, NCK2, PRKG1, DCC, PPP3CC | |
| Synaptic long-term potentiation | 9 | 1.7 × 10−3 | 0.07 | PLCB1, CAMK2B, CALM1, MRAS, CAMK2G, GRM7, CACNA1C, PPP3CC, PLCL1, ADCY8, GRIN3A, PLCG2 | |
| CREB signaling in neurons | 10 | 1.9 × 10−3 | 0.07 | GNAI1,PLCB1,CAMK2B,CALM1,PIK3R4,MRAS, GRIK2,PLCG2,GRIK1,CAMK2G,GRM7, ADCY2,PLCL1,ADCY8,ADCY9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Dorn II, G.W.; Shetty, A.; Parihar, A.; Dave, T.; Robinson, S.W.; Gottlieb, S.S.; Donahue, M.P.; Tomaselli, G.F.; Kraus, W.E.; et al. A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans. J. Pers. Med. 2018, 8, 11. https://doi.org/10.3390/jpm8010011
Xu H, Dorn II GW, Shetty A, Parihar A, Dave T, Robinson SW, Gottlieb SS, Donahue MP, Tomaselli GF, Kraus WE, et al. A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans. Journal of Personalized Medicine. 2018; 8(1):11. https://doi.org/10.3390/jpm8010011
Chicago/Turabian StyleXu, Huichun, Gerald W. Dorn II, Amol Shetty, Ankita Parihar, Tushar Dave, Shawn W. Robinson, Stephen S. Gottlieb, Mark P. Donahue, Gordon F. Tomaselli, William E. Kraus, and et al. 2018. "A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans" Journal of Personalized Medicine 8, no. 1: 11. https://doi.org/10.3390/jpm8010011