
Driver or Passenger: Epigenomes in Alzheimer’s Disease

Max Planck Institute of Psychiatry, Translational Psychiatry, Munich 80805, Germany
*
Author to whom correspondence should be addressed.
Epigenomes 2017, 1(1), 5; https://doi.org/10.3390/epigenomes1010005
Submission received: 17 March 2017
/
Revised: 26 April 2017
/
Accepted: 26 April 2017
/
Published: 30 April 2017
(This article belongs to the Special Issue Epigenetics of the Nervous System)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Alzheimer’s disease (AD) is a fatal neurodegenerative disease which is on the rise worldwide. Despite a wealth of information, genetic factors contributing to the emergence of AD still remain incompletely understood. Sporadic AD is polygenetic in nature and is associated with various environmental risks. Epigenetic mechanisms are well-recognized in the mediation of gene environment interactions, and analysis of epigenetic changes at the genome scale can offer new insights into the relationship between brain epigenomes and AD. In fact, recent epigenome-wide association studies (EWAS) indicate that changes in DNA methylation are an early event preceding clinical manifestation and are tightly associated with AD neuropathology. Further, candidate genes from EWAS interact with those from genome-wide association studies (GWAS) that can undergo epigenetic changes in their upstream gene regulatory elements. Functionally, AD-associated DNA methylation changes partially influence transcription of candidate genes involved in pathways relevant to AD. The timing of epigenomic changes in AD together with the genes affected indicate a critical role, however, further mechanistic insight is required to corroborate this hypothesis. In this respect, recent advances in neuronal reprogramming of patient-derived cells combined with new genome-editing techniques offer unprecedented opportunities to dissect the functional and mechanistic role of epigenomic changes in AD.
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease leading to fatal dementia and death [1]. Currently, there is no established treatment that efficiently delays the onset or progression of AD [2]. The prevalence of dementia in subjects 60 years and older is approximately 5–8% in the general population, with Alzheimer’s disease (AD) accounting for two-thirds of cases [3]. Other common causes of dementia include cerebrovascular disease, Lewy body formation, and frontotemporal dementia. Worldwide, 47.5 million people suffer from dementia, with 7.7 million new cases diagnosed every year, and the total number of people with dementia is projected to be 75.6 million in 2030 and almost triple the current number by 2050 at 135.5 million [3]. This development largely reflects an increase in life expectancy and a decline in fertility over the past century; a demographic trend that will continue throughout the 21st century with similar trends anticipated for developing countries in the latter half of this century.
AD manifests three cardinal symptoms. Firstly, there is cognitive dysfunction characterized by memory loss, language difficulties, executive dysfunction (i.e., decline of intellectual coordination skills, problem-solving abilities, and higher level planning), and disorientation. Memory loss is common, and in most cases the first symptoms, due to their insidious but relentless onset, are often attributed to normal ageing and only recognized once they interfere significantly with social and work activities [4]. Population studies of ageing and cognition indicate that impairment in multiple cognitive domains is detectable several years before clinical diagnosis of AD and is not qualitatively different from that seen in normal ageing, indicating a continuous shift from normal ageing to preclinical dementia [5,6]. Secondly, AD patients show psychiatric and behavioral disturbances encompassing mood swings, anxiety (17–27%), depression (24–33%), loss of motivation and apathy (up to 41%), and productive symptoms (23% of the cases showing hallucinations, delusions, and agitation) [4]. Thirdly, performance of activities of daily living progressively deteriorates. These activities include complex activities such as driving and shopping and basic activities such as managing self-care (e.g., dressing and eating unaided) [4].
The neuropathological hallmarks of AD were firstly described in 1906 by the psychiatrist Alois Alzheimer and at about the same time by Oskar Fischer [7]. They comprise a gross atrophy of the brain, with key features of amyloid plagues and neurofibrillary tangles, and widespread cell loss [8]. Pathological processes contributing to AD have been extensively studied over the last decades and involve above all amyloid-β and tau proteins, [9,10,11,12], neurovascular and inflammatory pathways [13,14], lipid, insulin, calcium, and G protein-coupled signaling pathways [15,16,17,18] as well as neural circuit and network abnormalities [19] among others [8].
Despite this wealth of information, the primary cause(s) of AD is still incompletely understood. Mendelian forms of AD are rare and heritability estimates about 60% for most cases of sporadic AD, suggesting an important role of environmental risk factors [20]. Epigenetic mechanisms (see Section 4) are well-known for their role in the mediation of gene environment interaction [21,22]; specifically, they are thought to cause long-lasting regulatory changes in genes involved in cognition, memory, and mood [23,24] and could contribute to AD [25]. With respect to this hypothesis, we will first reconsider the genetic architecture of AD (Section 2), concepts of epigenomics, and current guidelines for the design and analysis of epigenome-wide association studies (EWAS) (Section 3). Following this, new insights on different forms of DNA methylation and dynamic transitions during critical developmental windows will be presented (Section 4). Then, we will discuss recent findings on the role of genome-wide epigenetic changes in AD and their potential contribution to disease onset and progression (Section 5). Interestingly, these epigenomic changes seem to intersect with risk genes identified from genome-wide association studies (GWAS), suggesting a potential role in the modulation of genetic risk in AD.
2. The Genetic Architecture of AD
AD can be subdivided into two major categories; Mendelian AD and sporadic AD, each exhibiting a characteristic clinical course. Mendelian forms of AD typically show an early onset with heritability estimates of 92–100% and account for less than 1% of the disease [26]. Causal, rare autosomal dominant mutations in the amyloid beta precursor protein (APP), and presilin 1 (PSEN1), and 2 (PSEN2) lead to early-onset familial Alzheimer’s disease [27], in support of the amyloid cascade hypothesis [12]. Since these genes show large effect sizes, they can be detected by conventional candidate gene association and genome-wide linkage studies.
In contrast, the more common late-onset form of AD (termed LOAD) develops in individuals over 65 years and is genetically complex, with 56–79% heritability as inferred from a large twin study that also indicates a crucial role of familial factors [25]. Different approaches ranging from genetic epidemiology and segregation analysis to candidate gene associations, genome-wide linkage and GWAS have been applied to elucidate the genetics of LOAD. Overall, these studies support a role of both rare and common risk variants in AD, which show different prevalence and confer varying degrees of risks for disease development. Since the early 1990s, candidate association studies have pointed to a role of apolipoprotein E (APOE) as a rare susceptibility gene for LOAD with an unusually large effect size (p ≤ 1 × 10−295) [28,29]. The APOE locus comprises three allelic variants (ApoE ε2, ApoE ε3, and ApoE ε4) and heterozygosity or homozygosity of ApoE ε4 are associated with a 3- and 8-fold increased risk, respectively, for the development of AD.
The understanding of common genetic variations in AD has greatly progressed with the advent of high-throughput genotyping technologies. The most frequent forms of genetic variation are single nucleotide polymorphisms (SNPs) that make up 95% of all known sequence variants. More than 85 million SNPs have been detected in the human population [30], whereby GWAS arrays typically incorporate only tag SNPs that represent all SNPs in the same linkage disequilibrium (LD) block. Risk-associated SNPs are statistically overrepresented in disease populations and multiple associations are thought to all tag a single causal variant. In order to deduce potential risk genes, physical boundaries are statistically inferred for the found SNP associations.
Since 2009, five well-powered GWAS have detected 10 risk loci in AD that account for ≈20% of the total variation in risk or ≈33% of the risk attributable to genetic effect, with a major causative role for APOE [28,31,32,33,34]. With the merger of these consortia in the International Genomics of Alzheimer’s Project (IGAP), a large meta-analysis (17,008 AD cases versus 37,154 controls and a replication sample of 8572 AD cases versus 11,312 controls) identified a total of 19 risk loci that consistently fulfilled the accepted threshold value for genome-wide significance (p < 5 × 10−8) [35]. Eleven new susceptibility loci were highlighted in addition to the already known ABCA7, APOE, BIN1, CLU, CR1, CD2AP, EPHA1, MS4A6A-MS4A4E, and PICALM candidate loci and strengthened the relevance of some previously suggested pathways such as APP (SORL1, INPP5D, and CASS4) and tau (CASS4 and FERMT2) in AD neuropathology. Specifically, SORL1 encodes a lipid transporter and regulator of endocytosis, INPP5D encodes an inositol polyphosphate-5-phosphatase known to interact with the previously identified candidate CD2AP (a critical scaffolding protein that facilitates intracellular junctions and modulates APP metabolism), CASS4 encodes a protein with a scaffolding function possibly relevant to actin dynamics, and FERMT2 encodes a fermitin family member with a likely role in matrix adhesion structures and integrin activation [35]. Other loci contain candidate genes acting in pathways well-known to be enriched in AD, such as immune response and inflammation (HLA-DRB5-DRB1, encoding major histocompatibility complex class II, DRβ5 and DRβ1, respectively), neuronal signaling (PTK2B, encoding protein tyrosine kinase 2β increasing calcium influx and long-term potentiation in the hippocampal CA1 region), and synapse formation (MEF2C, encoding myocyte enhancer factors 2 that adjusts excessive synapse formation during activity-dependent refinement of synaptic activity).
Conspicuously though, a large fraction of the common genetic risk variants minor allele frequency ≥ 0.05%) in AD remains still unexplained in present GWAS. Although many of them are plausible candidates for pathology, it is important to remember that GWAS are not able to identify per se the causal variant underlying a disease. In fact, tag SNPs capture all the other SNPs mapping to the risk-associated haplotype block and are on their own unlikely to be the causal genetic variant that bring about the association.
At the same time, the advent of massively parallel sequencing-based technologies has laid the groundwork for genome-wide maps of functional elements and has taken the study of epigenetic mechanisms (see Section 4) from the local to the genome scale [36] and thus leveraged epigenomics as a possible inroad to missing heritability in AD.
3. Epigenomics—Beyond Sequence Variation
Epigenomics refers to the study of epigenetic mechanisms [37] involving DNA methylation (see Section 4), posttranslational modifications of core histones, nucleosome positioning, and non-coding RNA (ncRNA) at the genome scale [38]. All of these mechanisms operate in “the structural adaptation of chromosomes so as to register, signal, or perpetuate altered activity states” (see p. 398 in [39]).
Epigenomics can provide important insights into the functional relationships of genes in health and disease by means of investigating complex phenotypes that result from genetically induced epigenetic alterations and/or environmentally induced epigenetic alterations, which in turn can be modulated by genetic effects. The epigenome of a cell is highly dynamic due to the complex interplay between genetic and environmental factors mediated by epigenetic mechanisms [40]. Consequently, for any human complex disease the extent of interindividual phenotypic variation is unknown and can be only determined by large-scale, systematic epigenome-wide associations studies (EWAS). Through this approach, EWAS represents the equivalent to GWAS.
EWAS are thought to identify new regulatory mechanisms at the population scale that are sensitive to environmental and lifestyle factors underlying susceptibility to disease. The full range of epigenetic marks is still uncharted but certainly vast considering that the human epigenome comprises >108 cytosine nucleotide bases of which >107 are CpGs (see Section 4) and >108 histone tails that carry multifaceted posttranslational modifications. The best studied epigenetic mark is DNA methylation (see Section 4 and Section 5), whereby variation at a single CpG site corresponds to an epigenetic equivalent of a SNP [41]. This view needs to be cautioned, however, in light of the dynamic nature of epigenetic signatures that are modifiable throughout life course, even in postmitotic neuronal cells (see Section 4), and make the design and interpretation of EWAS conceptually different from GWAS.
At the single molecule level, DNA methylation is a binary mark, whereas thousands of DNA copies are averaged in measurement at the tissue level and are reported as the percentage of cells that are methylated at any given CpG site. Accordingly, DNA methylation levels are continuous when measured across a large number of cells as opposed to the categorical nature of genotype information and differences in mean methylation rate between cases and controls provide a common summary of effect size. Genetic variants that affect the variability of DNA methylation, rather than the mean DNA methylation, cannot be assessed by bulk tissue preparations [42].
4. A Multifaceted Brain Methylome
4.1. Canonical DNA Methylation
Methylation of cytosines has been commonly analyzed in the context of cytosine-guanine dinucleotides (CG) and ≈80% of all CGs in the human brain are methylated similarly to other tissues [43]. In the mammalian brain, mCG exists in neural and glial cells from prenatal life to old age [44] (Figure 1), is broadly distributed across various brain regions and the genome [44,45,46,47], and guides cell differentiation and identity [21]. De novo DNA methyltransferases (DNMTs) comprise DNMT3A and DNMT3B which catalyze mCG and once established, this mark is maintained by DNMT1 following genome replication. Interestingly, both DNMT1 and DNMT3a are required for synaptic plasticity, learning, and memory through their overlapping roles in maintaining DNA methylation and modulating neuronal gene expression in adult hippocampal neurons [48].
CGs are usually depleted in the genome and cluster in regions called CG islands (CGIs), of which less than half are located at human promoters and stay methylation-free [49,50]. Inactive promoter CGIs frequently recruit Polycomb complexes that impose repressive chromatin marks. Only few promoter CGIs undergo DNA methylation, which is associated with a closed, transcriptionally inactive, chromatin state [51,52]. Instead, predominantly promoters devoid of CGIs are regulated by DNA methylation [53]. In this regard, the conventional view that CpG methylation confers an all-purpose repressive mark needs to be re-considered in light of recent reports that gene body methylation frequently occurs at ubiquitously expressed genes where it enhances prolongation efficiency, inhibits spurious transcription initiation, and positively correlates with gene expression [49]. Additionally, the effects of DNA methylation depend on genomic location, pre-existing transcriptional activity, and a primary sequence that jointly regulate gene repression and activation.
The advent of high-throughput sequencing of sodium bisulfite-converted DNA has paved the way to map CG methylation at the genome-scale and to identify the exact sites, sequence context, and levels of DNA methylation with unprecedented accuracy. At the same time, this progress has also evidenced the unique properties of the human brain methylome when compared to peripheral tissues. First, non-canonical DNA methylation in the CH context (mCH, where H = A, C, or T) has been detected in both neural and glial cells [44,46,47,54] as well as in embryonic stem cells but is much less present in peripheral differentiated tissues [51,55]. Second, brain cells contain a sizeable amount of 5-hydroxymethlatin (5hmC) [51,56].
4.2. Non-CG Methylation
mCH is largely absent in most tissue of the human body and has so far been only detected in human pluripotent stem cells and adult brain tissue with high abundance [51,57]. In the fetal and early-infant brain methylome mCH is also absent, but rapidly increases postnatally and over the first 2 years of life (Figure 1). This period is characterized by extensive synaptogenesis and synaptic pruning that shape neural network formation and remodeling. Part of these processes may be regulated by mCG that gradually accumulates to the level of canonical DNA methylation by adulthood and accounts then for more than half of all neuronal methylcytosines.
Knockdown of DNMT3A and DNMT3B in human embryonic stem cells (ESCs) results in a global decline of mCG [57]. Consistent with this result, conditional neuronal knockout of all three Dnmts or of Dnmt3a alone reduces mCH [45,46]. Collectively, these findings support that canonical and non-canonical DNA methylation share the same catalytic mechanism for de novo DNA methylation though, puzzlingly, a mechanism(s) for faithful duplication of mCH patterns during cell division, similar to Dnmt1-catalyzed CG methylation, remains still to be identified.
Both mCG and mCH strongly anti-correlate with gene expression, pointing to an inhibitory role. Glial genomes contain only ≈10–20% of neuronal mCG, whereby mCH hypermethylation within gene bodies associates with repressed genes that are specifically expressed in neuronal cells. In turn, the same genes show mCH hypomethylation in a neuronal context corroborating a cell type-specific role for mCH in gene repression.
In neuronal and glial cells, mCH prevails at CAC positions as opposed to the preferred CAG context in various pluripotent cell types [57,58]. A key question arising from these findings is whether mCH is differentially abundant across brain cell types and represents a substrate for functional epigenomic changes in mental and neurodegenerative diseases.
4.3. Hydroxymethylcytosine
A second unique feature of the brain methylome is the high enrichment of 5-hydroxymethylcytosine (5hmC) that particularly prevails in cerebellar Pukinje cells, where it reaches ≈40% of mCG levels [56,59]. Similarly to mCH, 5hmC gradually accumulates in various brain regions during development [60]. More recently, the discovery of a specific pathway for active demethylation via iterative oxidation has raised new interest in this type of cytosine modification. Specifically, the family of ten-eleven translocation enzymes has been shown to catalyze the conversion of mCG to 5hmC and further oxidation coupled with mechanisms such as DNA glycosylase-initiated base-excision can restore the final product into unmodified cytosine [61]. Functionally, recent findings suggest for DNA methylation a more dynamic role than originally thought and indicate that in mature neurons 5hmC could be a transient, intermediate state that is eliminated in a temporally and spatially highly constrained manner in small regions of the genome harboring regulatory function [62,63,64,65]. Independent from demethylation, neuronal 5hmC could also represent a stable mark associated with the loss of repressive chromatin structure characteristic of Polycomb-mediated repression [66].
Concluding, it is important to note that dynamic changes in DNA methylation in postmitotic neural cells can offer a mechanistic basis for the interaction of environmental and societal risk factors with the genetic blueprint and the subsequent development of disease. Defined exposures can trigger neuronal activation that directly couples to the epigenetic machinery or, more commonly, to transcriptional regulators that subsequently recruit the epigenetic machinery. Such neuronal activity-dependent factors can impose local and genome-wide long-lasting marks that persist beyond the initial stimulus and lead to changes in gene expression patterns pertinent to higher (cognition, emotion, and mood) and basic (neuronal survival, synapse formation, and neural connectivity) brain functions [67,68,69].
5. Epigenomics Landscapes in Alzheimer’s Disease
Two recent large-scale EWAS [70,71] provide new insight into epigenomic changes in AD and their relation with GWAS risk genes using Infinium® HumanMethylation450 BeadChip arrays (Illumina, Inc., San Diego, USA; hereafter referred to as 450K beadchip array).
De Jager et al. investigated CG methylation in the gray matter from dorsolateral prefrontal cortices (PFC) dissected from 708 subjects at the time of death [70]. Donors originated from two prospective cohort studies of aging representing a random selection of the older population. While non-impaired at study entry, some subjects declined cognitively over time and showed several signs of amyloid pathology at the time of death, with ≈61% fulfilling the diagnostic criteria for AD.
Adopting a three-stage strategy, De Jager et al. first correlated methylome analysis with AD pathology. Second, significant CG methylation changes were assessed in an independent replication study. Third, the role of replicated CG methylation changes for gene expression was assessed by RNA measurements in an independent case-control design (Figure 2).
In the discovery study, methylation levels of 137 autosomal CpGs significantly correlated (p < 1.20 × 10−7) with quantitative measurements of neuritic amyloid plaques (NP), a hallmark of AD neuropathology [70]. Following secondary analysis 71 CpGs remained significant, which mapped to 60 distinct differentially methylated regions (DMRs) throughout the genome (Figure 2). Importantly, each of these 71 CpGs accounted for 5% (range = 3.7–9.7%) of the variance in NP burden and exceeded the proportion of variance accounted for by present GWAS risk variants in AD exempt APOE. Taken together, all known AD risk variants including APOE explained 13.9% of the variance in NP burden as opposed to 28.7% explained by the aggregate effect of all 71 CpGs. This result suggests that CpG methylation levels at defined genomic regions correlate with each other and are a key feature of AD neuropathology [70].
Interestingly, two of the AD-associated CpGs mapped to known GWAS risk loci (Figure 2), namely BIN1 and ABCA7 (see Section 2). At BIN1, the CpG is located 5 kb from the 5’ end and 92 kb from the tag SNP, of which neither influenced CpG methylation nor the effect size of the NP association. In contrast, the tag SNP of ABCA7 associated with NP burden but not with the extent of CpG methylation. Together, these findings suggest that AD risk may be influenced by genetic and epigenetic variation contributing independently of each other to disease [70].
De Jager et al. further assessed the relevance of NP-associated CpGs for a postmortem neuropathological diagnosis of AD. Among 71 NP-associated CpGs, 22 also associated with a diagnosis of AD at a genome-wide level of significance (Figure 2) and 18 showed higher levels of DNA methylation in AD patients, suggesting a pathogenic role of AD-associated CpGs.
The 71 significantly associated CpGs were further tested in an independent replication cohort of 117 subjects diagnosed AD by postmortem Braak staging (Figure 2). This quantitative measure of AD pathology reflects the presence of neurofibrillary tangles (NF) across distinct brain areas comprising the transentorhinal and limbic regions, the hippocampus, and the neocortex. Methylation profiling of PFC samples with the 450K beadchip array revealed that 12 CpGs were significantly associated with AD among which two were detected in the same DMR near RHBDF2 (see also below) as in the discovery sample. Moreover, when evaluating the entire set of 71 CpGs, many showed trends towards significance (including ABCA7 and BIN1), with similar effect sizes across the two studies, suggesting that with more samples they would also be validated. Collectively, these results support a robust association of selected CpGs with AD irrespectively of the neuropathological criteria applied in postmortem AD diagnosis [70].
To discern between a potential role of epigenomic changes as cause or effect in AD, De Jager et al. limited NP analysis to those individuals free of cognitive impairment at the time of death. Interestingly, altered CpG methylation appeared to be an early sign of AD pathology that precedes overt manifestation and does not reflect secondary events along later disease stages. If this is the case, this result resembles findings from neuropathological and imaging studies showing that a large proportion of non-impaired, older individuals can accumulate asymptomatically amyloid pathology as an early sign of AD pathology [1].
Lastly, to gain insight into potential transcriptional effects, AD-associated CpGs were traced by genome-wide mapping of histone modifications in two postmortem PFC samples collected from cognitively unimpaired individuals. The 71 CpGs were enriched in predicted weak enhancer regions or weakly transcribed regions concomitant to a depletion at strong promoter regions. Since the latter regions mostly drive fundamental cellular processes in neuronal and glial cells, this result suggests that AD-associated CpGs preferentially reside in weakly transcribed or even inactive regions in healthy aged brains.
Going beyond histones, the researchers next focused on the 12 CpGs validated in the replication study and measured levels of nearby gene expression in a 50-kb window on either side in an independent cohort of 202 AD and 197 control samples. Here, 7 of the 21 neighboring genes showed changes in messenger RNA (mRNA) expression in the PFC that significantly associated with AD (Figure 2). These genes comprised ankyrin1 (ANK1, involved in compartmentalization of the neuronal plasma membrane [72]), cadherin 23 (CDK23, an extracellular adhesion protein [73]), disco-interacting protein 2 (DIP2A, a potential cell surface protein [74]), inactive rhomboid protein 2 (RHBDF2, a pseudoprotease regulating receptor-kinase signaling [75]), ribosomal protein L13 (RPL13, playing a role in ribosome assembly), and serine peptidase inhibitors 1 and 2 (SERPINF1 and SERPINF2, with a possible role in synaptic activity and behavior [76]). Finally, protein–protein interaction network analysis showed that these seven differentially methylated genes connected to an AD susceptibility network derived from GWAS risk variants and Mendelian genes in AD. Taken together, these findings indicate that epigenetic and genetic changes may intersect in AD pathology.
In the second large-scale EWAS study carried out at the same time, Lunnon et al. [71] conducted a cross-tissue analysis of methylomic variation in postmortem AD brains from four independent cohorts using a sequential replication design.
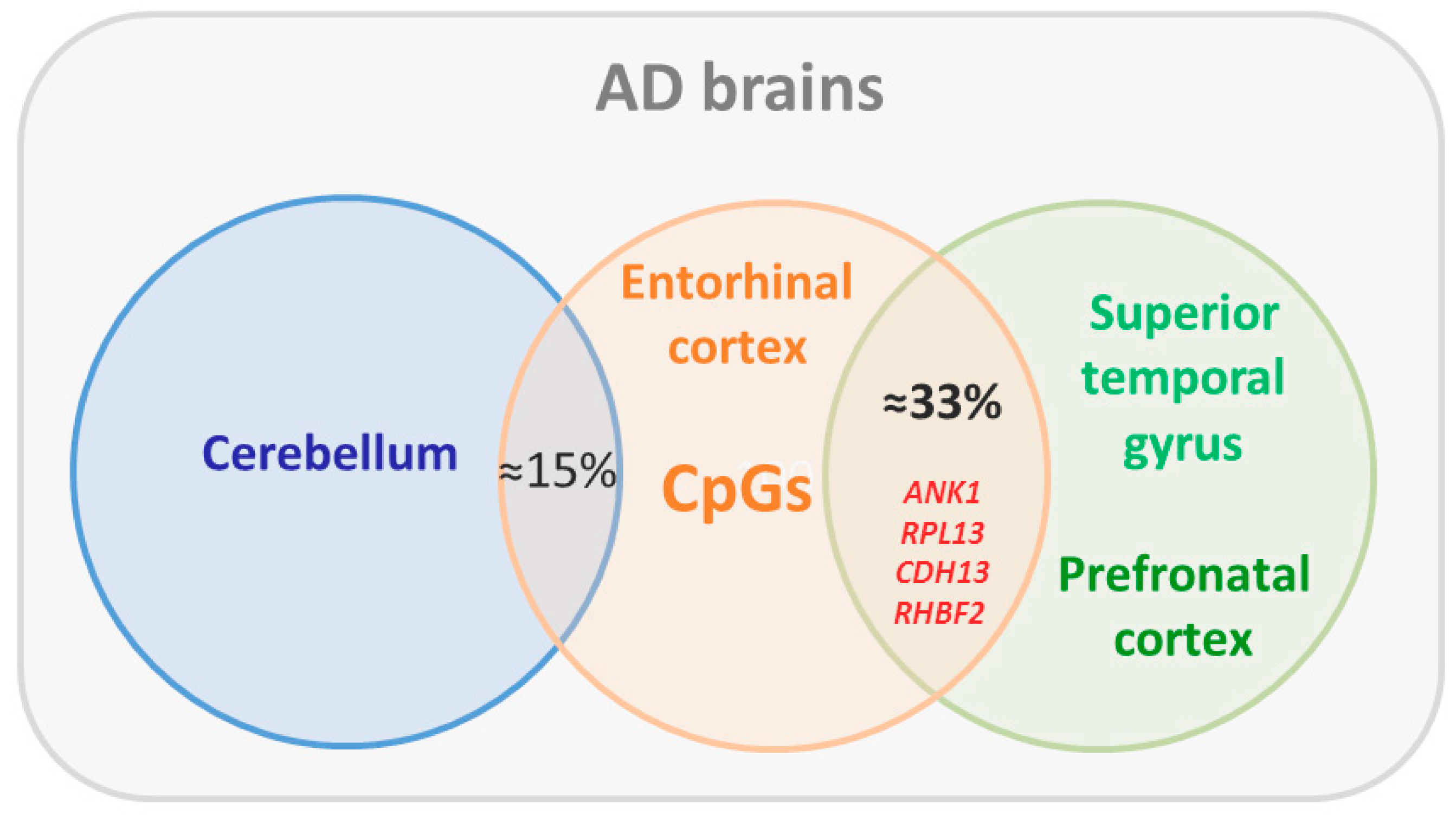
In the discovery study, genomic DNA (London brain bank, n = 122) isolated from four brain regions comprising the entorhinal cortex (a primary site in AD), the superior temporal gyrus, the prefrontal cortex, and the cerebellum (a poorly affected site in AD) was assessed for CpG methylation using 450K beadchip arrays (Figure 3). Among the differentially methylated positions associated with NF burden (Braak staging) in the entorhinal cortex, two of the top-ranked and the four-ranked CpGs were located 91 bp away from each other at ANK1. The association between Braak staging and increased methylation at both top-ranked CpGs remained significant following correction for cellular heterogeneity and extended also to the superior temporal gyrus and the prefrontal cortex but not to the cerebellum, indicating that ANK1 hypermethylation associates with neuropathology. Importantly, about one-third of the 100 top-ranked differentially methylated CpGs in the entorhinal cortex were methylated in the same direction in the superior temporal gyrus and the prefrontal cortex, including those close to RHBFDF2, RPL13-C10orf54, and CDH23 that had been previously identified by De Jager et al. (Figure 3). Among these, ANK1, RPL3, and RHBFDF2 are biologically linked to PTK2B, a known AD-associated risk gene. In contrast, top-ranked CpGs in the cerebellum were not differentially methylated in cortical regions, whereas ≈15% of the top-ranked cortical CpGs were also differentially methylated in the cerebellum despite little AD neuropathology [71].
Notably, in the replication study (Mount Sinai brain bank, n = 144) Braak-associated DNA methylation scores for the 100 top-ranked cross-cortex differentially methylated CpGs from the discovery study were also strongly correlated with neuropathology-associated differences at the same probes, including the two top-ranked CpG sites at ANK1, and thus validated AD-associated CpGs from the discovery sample. To strengthen a role in AD, Lunnon et al. further assessed differential DNA methylation at eight CpGs straddling ANK1 by bisulfite-pyrosequencing in an independent cohort of matched cortical samples (Thomas Willis brain bank, n = 62). This experiment showed that average DNA methylation across this region was significantly elevated in cortical regions from diseased subjects in support of previous findings from De Jager et al. Prompted by this result, Lunnon et al. analyzed the role of AD-associated DNA methylation for isoform-specific ANK1 expression in the entorhinal cortex (n = 36) [71]. This experiment suggested a significant association between the abundance of ANK1 isoforms 5-7-10, but not 1-2-3-4-9, and AD-associated neuropathology.
Overall, Lunnon et al. showed robust cross-tissue differences in differentially methylated NF-associated CpGs in AD whereby methylomic changes faithfully mirrored known patterns of neuropathology. Well-fitting methylomic changes were most prevalent in the entorhinal cortex and may translate in altered transcription of weakly expressed genes in aged healthy brains. In support of this view, AD-associated cortex-specific hypermethylation at ANK1 led to altered gene expression patterns.
These findings have been recently strengthened by Watson et al. [77], who analyzed superior temporal cortices samples from 34 AD patients and 34 controls by 450K beadchip arrays. Here, cell proportions were estimated based on a reference subset of neuronal and glial cells obtained from 59 adult individuals. Multiple statistically significant CpGs were clustered into DMRs using a 1-kb sliding window and exhibited concordant disease-associated changes in methylation. Of the top 100 CpGs reported by Lunnon et al., 22 were found to be differentially methylated with the same directional change in AD patients [71]. This extent of replication matches well the one initially reported by Lunnon et al. To increase power in this small-sized EWAS, concordant statistical signals from neighboring CpGs were leveraged generating 479 autosomal DMRs. The majority of these showed hypermethylation in AD cases compared to controls, whereby disease-associated average methylation differences were relatively modest in agreement with previous results [70,71]. At the genome level, DMRs preferentially localized within genes driving brain-relevant transcription factors. Specifically, hypermethylated DMRs were enriched in poised promoters characterized by the simultaneous presence of H3K27me3 (histone 3 trimethylation of lysine 27, a repressive chromatin mark) and H3K4me3 (histone 3 trimethylation of lysine 4, an active chromatin mark). Such bivalent chromatin structures occur at regulatory regions driving expression of developmental genes with key roles in cellular differentiation and pluripotency [53,78].
Furthermore, poised promoters also preferentially contained CpGs that undergo hypermethylation with age [79,80]. Interestingly, Watson et al. detected a strong bias for CpG hypermethylation among DMRs in AD and enrichment in age-associated methylation changes in control samples when compared to CpGs within hypomethylated DMRs [77]. Collectively, these findings highlight a potential interaction between disease-associated epigenetic alterations and aging in AD compatible with a continuous shift from normal ageing to dementia.
Lastly, a recent study has specifically explored a potential interaction between epigenomic changes and GWAS risk genes [81]. CpG methylation levels from the discovery sample by De Jager et al. were analyzed at 11 GWAS susceptibility gene regions contained on the 450K beadchip array and assessed for their association with NP formation. Interestingly, in the ABCA7, CD2AP, CLU, and MS4A6A risk loci, greater methylation of certain CpGs related to AD pathology appeared to reflect a single event, and developed prior to cognitive impairments and independent from genetic variation. Similarly, at the BIN1 locus, the CpG site with the strongest association was only partly influenced by the tag SNP. These results suggest that in some AD vulnerability loci factors acting through CpG methylation and SNPs operate independently of each other in the mediation of disease risk [81]. Moreover, the association of multiple, independent CpGs with NP in loci such as BIN1, CLU, and MS4A6A indicates that epigenomic changes in AD are not driven by a unifying mechanism such as a simple increase in NP burden but rather by different risk factors whose effects on pathology converge on different genomic sites or by different molecular processes mediating pathophysiology.
Overall, this study collaborates that epigenetic variation can apply to a substantial fraction of genetic risk variants in AD and mediate pathology independent from the underlying haplotype. If this is the case, different sources of variation—genetic and epigenomic—can converge in the brain of aging individuals and contribute to the accumulation of AD pathology.
6. Conclusions
Current evidence indicates that early epigenomic changes associated with neuropathological hallmarks of AD may precede clinical symptoms and influence the onset and development of AD. Following disease manifestation, a sizeable fraction of epigenomic changes is shared across brain tissues and targets genes that appear unlikely to be actively transcribed in healthy aged brains. Additionally, AD-associated epigenomic changes can extend to candidate genes that functionally intersect with potential risk genes from GWAS with the latter undergoing as well epigenetic changes that are largely independent of the respective tag SNP association. Collectively, epigenomic and genetic risk factors seem to converge independently of each other on neuropathology in AD.
While the impact of epigenomics on mental health and disease is increasingly recognized, this field still faces a number of challenges due to postmortem tissue heterogeneity, temporal and spatial effects of epigenomic changes, and eventually functional causality. Crucially, differences in DNA methylation in case-control studies can reflect changes in the prevalence of different cell types following from immune activation during the disease process rather than the impact of the disease on cells of primary interest. As a corrective, statistical deconvolution techniques can improve DNA methylation analysis from mixed cell samples [82,83].
Pertinent to an improved understanding of AD neuropathology and eventual therapy, the role of epigenomic changes for molecular and cellular brain functions needs to be further analyzed. AD-associated epigenomic changes are modest and effects on gene transcription have been either dismissed or studied for few candidate genes so far. In this respect the current use of homogenized bulk tissues is a confounding factor due to the generation of averages across large numbers of cells that are likely to dilute cell-type-specific signals from complex brain tissues and biologically relevant variability at the level of single cells. This concern relates to changes in either DNA methylation or mRNA levels. Implementation of innovative single-cell assays covering genome, epigenome, and transcriptional analysis [84,85] may refine detection of cell type-specific changes. Of further note, subtle changes in AD-associated CpGs could also influence cell type-specific alternative RNA splicing [86] that is thought to represent a primary link between genetic variation and disease [87]. Finally, even if a DMR does not impact transcription and gene expression, it may have some other yet unknown effect or serve as a marker of the exposures (for example maintaining a memory of exposure over lifetime) or outcome (for examples, biomarker of disease) of interest.
Epigenomic changes in AD are enriched in regulatory noncoding domains at which canonical and non-canonical CpG methylation could coexist, with each having distinct functions. Such domains are underrepresented by present array systems, which are gene biased and do not account for non-canonical CpG methylation. The latter modification could couple as well to environmental exposures and life experiences that act as risk factors in AD. For example, the neuronal activity-dependent transcriptional regulator methyl-CpG-binding protein (MeCP2) [88,89] has been recently shown to recognize mCH and to confer transcriptional repression [90]. Intriguingly, MeCP2’s role may extend well beyond early development [91] and mediate between different environmental exposures and the epigenome [92].
In turn, dynamic changes in gene expression and DNA methylation are thought to decode neuronal activity [22] and thus contribute to the regulation of protein synthesis and synaptic connectivity with a role in learning-induced neuronal activity [93]. AD-associated changes in DNA methylation could weaken the ability of neurons to respond to later stimuli and compromise neuronal plasticity. Moreover, sustained changes in DNA methylation without changes in gene expression could preset gene expression potential following further neuronal activation [94]. This kind of latent responsivity may offer a plausible explanation for the low correlation between AD-associated methylation changes and resting gene expression levels in current EWAS.
Conceptually, EWAS map differences in genome-wide DNA methylation levels between control and diseased individuals but do not per se identify causal variants; a limitation shared with classical GWAS. Any changes associated with a concrete disease phenotype can be either through direct cause or, in the case of epigenetic changes, a direct consequence. In this regard, retrospective studies cannot determine whether the identified epigenetic variants are due to disease-associated genetic differences, post-disease processes or disease-related drug intervention. Likewise, temporality can shed light on, but does not resolve the puzzle of “driver or passenger” for AD-associated epigenomic changes. Indicative of a potential role in the onset of AD, CpG associations to pathology colocalized with genetic susceptibility and occurred prior to disease manifestation in cognitively non-impaired subjects. Still, this does not answer whether the observed changes are causal to or simply a consequence of AD.
Establishing function and causation in EWAS will require human neuronal models in which site-specific introduction of epigenetic marks at AD-associated DMRs confer (de-) silencing of candidate genes in combination with a functional disease-relevant readout. Induced pluripotent stem cells (iPSC) [95] from AD patients and controls provide an opportunity to identify and explore epigenomic changes driven by different genetic make-ups. iPSCs can be differentiated into various cell types [96] and types of organoids (i.e., 3D cultures) resembling early developmental brain structures [97,98]. It is important to note that gene expression profiles from differentiating iPSCs closely match embryonic human tissue, especially from the first human trimester, and present a potential barrier to the study of late onset diseases [99] although some aspects of AD and Parkinson disease have been successfully modeled in these systems [100]. Moreover, data obtained from 2D versus 3D organoid culture systems show that mimicking normal tissue architecture does not critically advance differentiation [101,102]. These limitations have motivated the development of strategies for directing cellular age in vitro. Some studies rely on the induction of age-like features in iPSCs using various stress paradigms such as exposure to toxins, including compounds that trigger mitochondrial stress or reactive oxygen production [103,104,105,106,107,108]. These approaches raise the question whether the applied treatments faithfully mimic age-related disease susceptibility. Alternatively, expression of progerin, a truncated form of lamin A associated with premature aging, has been reported to induce multiple aging-related markers and characteristics in iPSCs, including dopamine-specific phenotypes such as neuromelanin accumulation [109].
As a promising alternative to iPSCs, directly converted induced neurons (iNs) have been recently shown to retain donor age-dependent transcriptome signatures [110,111]. In this approach, donor fibroblasts (or other somatic cells) are directly converted into neurons by the expression of reprogramming factors without undergoing any cell division that might dilute or induce repair of macromolecular damage. Thus, aging phenotypes present in cultured primary fibroblasts are translated to a neuronal context that can consist of different neuronal subtypes comprising glutamatergic, Gamma-aminobutyric acid-positive neurons [111] or striatal medium spiny neurons [110]. Importantly, Mertens et al. found that less than 4% of the iN aging genes are shared with corresponding fibroblast aging genes, whereas 7-fold more are shared with postmortem human cortex, suggesting that iNs can recapitulate aging transcriptome signatures pertinent to the aging brain. This enrichment included genes contributing to learning and memory, AD, intracellular transport, and other categories. Similar findings were made by Huh et al., who showed that genes differentially expressed with age in reprogrammed neurons were enriched in age-related biological processes including vesicle-mediated transport, NF-kappaB transcription, nervous system development, apoptosis, and inflammation [110]. Further, biological age can be accurately scored by the analysis of age-linked DNA-methylation markers known as the epigenetic clock, that faithfully estimates cellular age across human organs, tissues, and cell types [112]. Interestingly, the epigenetic clock was unperturbed in iNs when compared to the corresponding aged fibroblasts or age-matched postmortem cortices, thus strengthening the hypothesis that age is maintained during direct neuronal conversion [110]. This finding is in sharp contrast to iPSCs, where the epigenetic clock is reset to an embryonic state with a DNA methylation age that is negative or close to zero [112].
Taking all these approaches into account, recent improvements open up the perspective that iPSC- and iN-derived models of cellular aging can substantially advance insight into the impact and interplay of genetic risk factors and age-associated epigenomic changes in AD.
Until lately, investigation of the functional significance of AD-associated DMRs in iPSC- or iN-derived cellular models has been a challenge due to the lack of appropriate molecular tools that enable efficient editing of DNA methylation in a targeted manner. In this respect, hybridization of the key enzymes in the DNA methylation pathway with reprogrammable sequence-specific DNA-targeting molecular machinery has been recently shown to provide efficient editing of DNA methylation in a site-specific manner [113,114,115,116]. Specifically, fusion of Tet1 or Dnmt3a with a catalytically inactive Cas9 (CRISPR-associated) allowed to establish in vitro and in vivo a causal relationship between (de-) methylation and (de-) silencing of a target gene [114].
Moving beyond mechanistic and functional insight into AD-associated epigenomic changes in induced aged iPSCs/iNs, future strategies may aim for systematic genome-wide editing of DNA methylation to trigger iPSC/iN aging smoothly and/or disease specifically. In proof of concept, genome-wide gene editing in the turquoise killfish has been recently suggested as comprehensive genotype-phenotype platform for the rapid exploration of ageing and disease in this short-lived vertebrate species [117]. Likewise, targeted manipulation of epigenetic marks on a genome-wide scale recapitulating AD-associated epigenomic changes may be an important tool to establish age-related cellular phenotypes in different genetic backgrounds. If this is the case, the role of epigenomic changes as a driver or passenger in AD neuropathology is anticipated to be revisited soon.
Acknowledgments
We thank our groups for thoughtful discussions. Michael Ziller is supported by the BMBF grant 01ZX1504.
Author Contributions
All authors jointly contributed to this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ames, D.; O’Brien, J.; Burns, A. Dementia, 5th ed.; CRC Pr I Llc: Boca Raton, FL, USA, 2016. [Google Scholar]
- Selkoe, D.J. The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann. Neurol. 2013, 74, 328–336. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia Fact Sheet N°362; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Brayne, C. The elephant in the room—Healthy brains in later life, epidemiology and public health. Nat. Rev. Neurosci. 2007, 8, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.; Mormino, E.; Johnson, K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 2014, 84, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Oskar Fischer and the study of dementia. Brain 2009, 132, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and “wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Thathiah, A.; De Strooper, B. The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl.), 245–254. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Sweatt, J.D. Epigenetic mechanisms in cognition. Neuron 2011, 70, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. The lasting legacy of social stress on the epigenome of the hypothalamic-pituitary-adrenal axis. Epigenomics 2012, 4, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 2012, 69, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L. Epigenetics; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Beck, S.; Olek, A.; Walter, J. From genomics to epigenomics: A loftier view of life. Nat. Biotechnol. 1999, 17, 1144. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Hildmann, T.; Novik, K.L.; Lewin, J.; Tost, J.; Cox, A.V.; Andrews, T.D.; Howe, K.L.; Otto, T.; Olek, A.; et al. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the human epigenome project. PLoS Biol. 2004, 2, e405. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, E.; Feinberg, A.P. Regulated noise in the epigenetic landscape of development and disease. Cell 2012, 148, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science (New York, N.Y.) 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.R.; Urich, M.A.; Chen, H.; et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Müller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.-X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Shao, N.; Szulwach, K.E.; Vialou, V.; Huynh, J.; Zhong, C.; Le, T.; Ferguson, D.; Cahill, M.E.; Li, Y.; et al. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat. Neurosci. 2015, 18, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.-L.; King, J.R.; Song, H.; Sweatt, J.D. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, W.; Zhao, Q.-Y.; Widagdo, J.; Baker-Andresen, D.; Flavell, C.R.; D’Alessio, A.; Zhang, Y.; Bredy, T.W. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 7120–7125. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Su, Y.; Shin, J.; Zhong, C.; Guo, J.U.; Weng, Y.-L.; Gao, F.; Geschwind, D.H.; Coppola, G.; Ming, G.; Song, H. Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat. Neurosci. 2015, 18, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.-G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. DNA memories of early social life. Neuroscience 2014, 264, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Labonté, B.; Suderman, M.; Maussion, G.; Lopez, J.P.; Navarro-Sánchez, L.; Yerko, V.; Mechawar, N.; Szyf, M.; Meaney, M.J.; Turecki, G. Genome-wide methylation changes in the brains of suicide completers. Am. J. Psychiatry 2013, 170, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Wu, Y.; Bockmühl, Y.; Spengler, D. Genes learn from stress: How infantile trauma programs us for depression. Epigenetics 2010, 5, 194–199. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Boiko, T.; Vakulenko, M.; Ewers, H.; Yap, C.C.; Norden, C.; Winckler, B. Ankyrin-dependent and -independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J. Neurosci. 2007, 27, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-B.; Gillespie, P.G. Unraveling cadherin 23’s role in development and mechanotransduction. Proc. Natl. Acad. Sci. USA 2009, 106, 4959–4960. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Murakami, K.; Ozaki, S.; Imura, Y.; Tong, X.-P.; Watanabe, T.; Sawaki, T.; Kawanami, T.; Kawabata, D.; Fujii, T.; et al. DIP2 disco-interacting protein 2 homolog A (Drosophila) is a candidate receptor for follistatin-related protein/follistatin-like 1—Analysis of their binding with TGF-β superfamily proteins. FEBS J. 2010, 277, 4278–4289. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Grieve, A.; Xu, H.; Bambrough, P.; Christova, Y.; Freeman, M. Genetic interaction implicates iRhom2 in the regulation of EGF receptor signalling in mice. Biol. Open 2014, 3, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Almonte, A.G.; Sweatt, J.D. Serine proteases, serine protease inhibitors, and protease-activated receptors: roles in synaptic function and behavior. Brain Res. 2011, 1407, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Disanto, G.; Sandve, G.K.; Breden, F.; Giovannoni, G.; Ramagopalan, S.V. Age-associated hyper-methylated regions in the human brain overlap with bivalent chromatin domains. PLoS ONE 2012, 7, e43840. [Google Scholar] [CrossRef] [PubMed]
- Chibnik, L.B.; Yu, L.; Eaton, M.L.; Srivastava, G.; Schneider, J.A.; Kellis, M.; Bennett, D.A.; De Jager, P.L. Alzheimer’s loci: Epigenetic associations and interaction with genetic factors. Ann. Clin. Transl. Neurol. 2015, 2, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Guintivano, J.; Aryee, M.J.; Kaminsky, Z.A. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 2013, 8, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Farlik, M.; Sheffield, N.C.; Nuzzo, A.; Datlinger, P.; Schönegger, A.; Klughammer, J.; Bock, C. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015, 10, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.I.; van de Geijn, B.; Raj, A.; Knowles, D.A.; Petti, A.A.; Golan, D.; Gilad, Y.; Pritchard, J.K. RNA splicing is a primary link between genetic variation and disease. Science 2016, 352, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Bellini, E.; Pavesi, G.; Barbiero, I.; Bergo, A.; Chandola, C.; Nawaz, M.S.; Rusconi, L.; Stefanelli, G.; Strollo, M.; Valente, M.M.; et al. MeCP2 post-translational modifications: A mechanism to control its involvement in synaptic plasticity and homeostasis? Front. Cell. Neurosci. 2014, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Q. Regulation and function of stimulus-induced phosphorylation of MeCP. Front. Biol. 2014, 9, 367–375. [Google Scholar]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A.; Shaw, C.A.; Li, W.; Zoghbi, H.Y. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.A.; Hoffmann, A.; Raabe, F.; Spengler, D. Role of MECP2 in experience-dependent epigenetic programming. Genes (Basel) 2015, 6, 60–86. [Google Scholar] [CrossRef] [PubMed]
- Zovkic, I.B.; Guzman-Karlsson, M.C.; Sweatt, J.D. Epigenetic regulation of memory formation and maintenance. Learn. Mem. 2013, 20, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Baker-Andresen, D.; Ratnu, V.S.; Bredy, T.W. Dynamic DNA methylation: A prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 2013, 36, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, N.; Studer, L. Pluripotent stem cell-based disease modeling: Current hurdles and future promise. Curr. Opin. Cell Biol. 2015, 37, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2015, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.-D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.-P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, P.; Young-Pearse, T.L. Stem cells on the brain: Modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J. Neurogenet. 2014, 28, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2016, 5, e18648. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Paquola, A.C. M.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.-L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Harel, I.; Benayoun, B.A.; Machado, B.; Singh, P.P.; Hu, C.-K.; Pech, M.F.; Valenzano, D.R.; Zhang, E.; Sharp, S.C.; Artandi, S.E.; et al. A platform for rapid exploration of aging and diseases in a naturally short-lived vertebrate. Cell 2015, 160, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
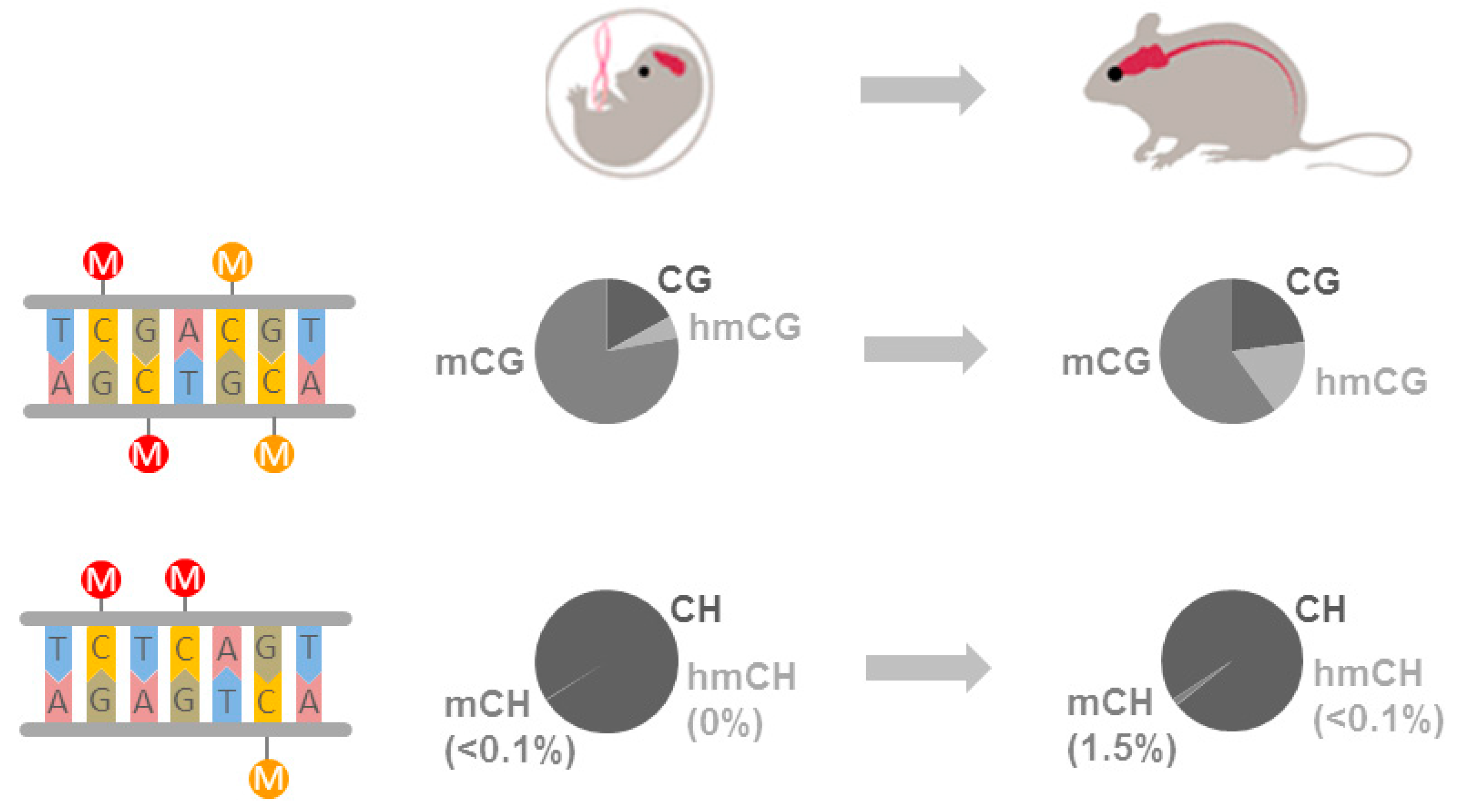
Figure 1.
The cortical methylome in fetal and adult mice. Canonical CpG methylation and non-CpG methylation are schematically depicted with methylcytosine or hydroxymethylcytosine symbolized by red and orange lollipops, respectively (left). The diagrams show the proportion of unmethylated (CG or CH), methylated (mCG or mCH), and hydroxymethylated (hmCG or hmCH) cytosines of fetal and adult frontal cortex. The proportion of hydroxymethylcytosine increases with age at the cost of methylcytosine compatible with a Ten-eleven translocation (Tet)-enzyme catalyzed conversion of methylcytosine into hydroxymethylcytosine. mCH is barely detectable at fetal stages but strongly increases during development.
Figure 1.
The cortical methylome in fetal and adult mice. Canonical CpG methylation and non-CpG methylation are schematically depicted with methylcytosine or hydroxymethylcytosine symbolized by red and orange lollipops, respectively (left). The diagrams show the proportion of unmethylated (CG or CH), methylated (mCG or mCH), and hydroxymethylated (hmCG or hmCH) cytosines of fetal and adult frontal cortex. The proportion of hydroxymethylcytosine increases with age at the cost of methylcytosine compatible with a Ten-eleven translocation (Tet)-enzyme catalyzed conversion of methylcytosine into hydroxymethylcytosine. mCH is barely detectable at fetal stages but strongly increases during development.
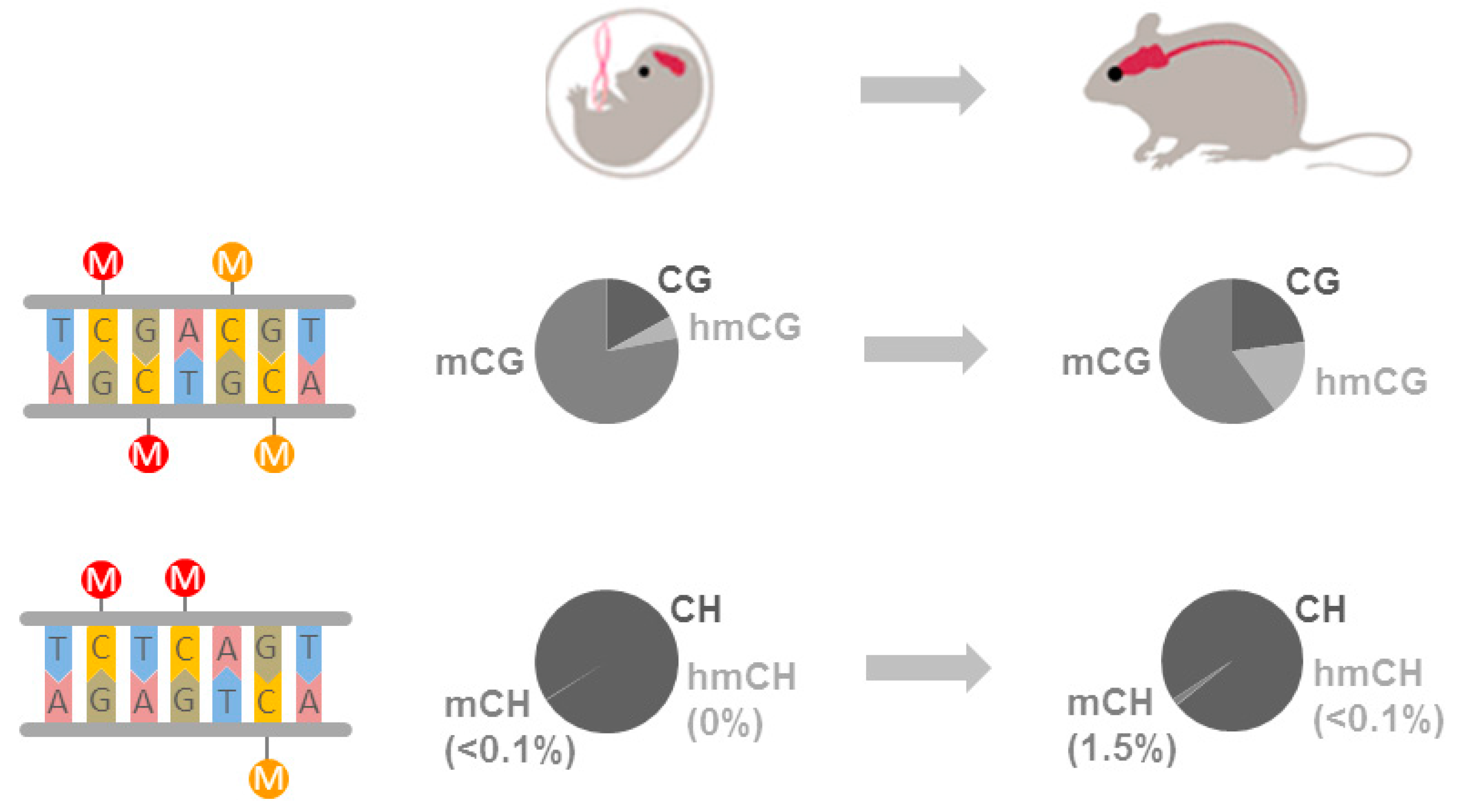
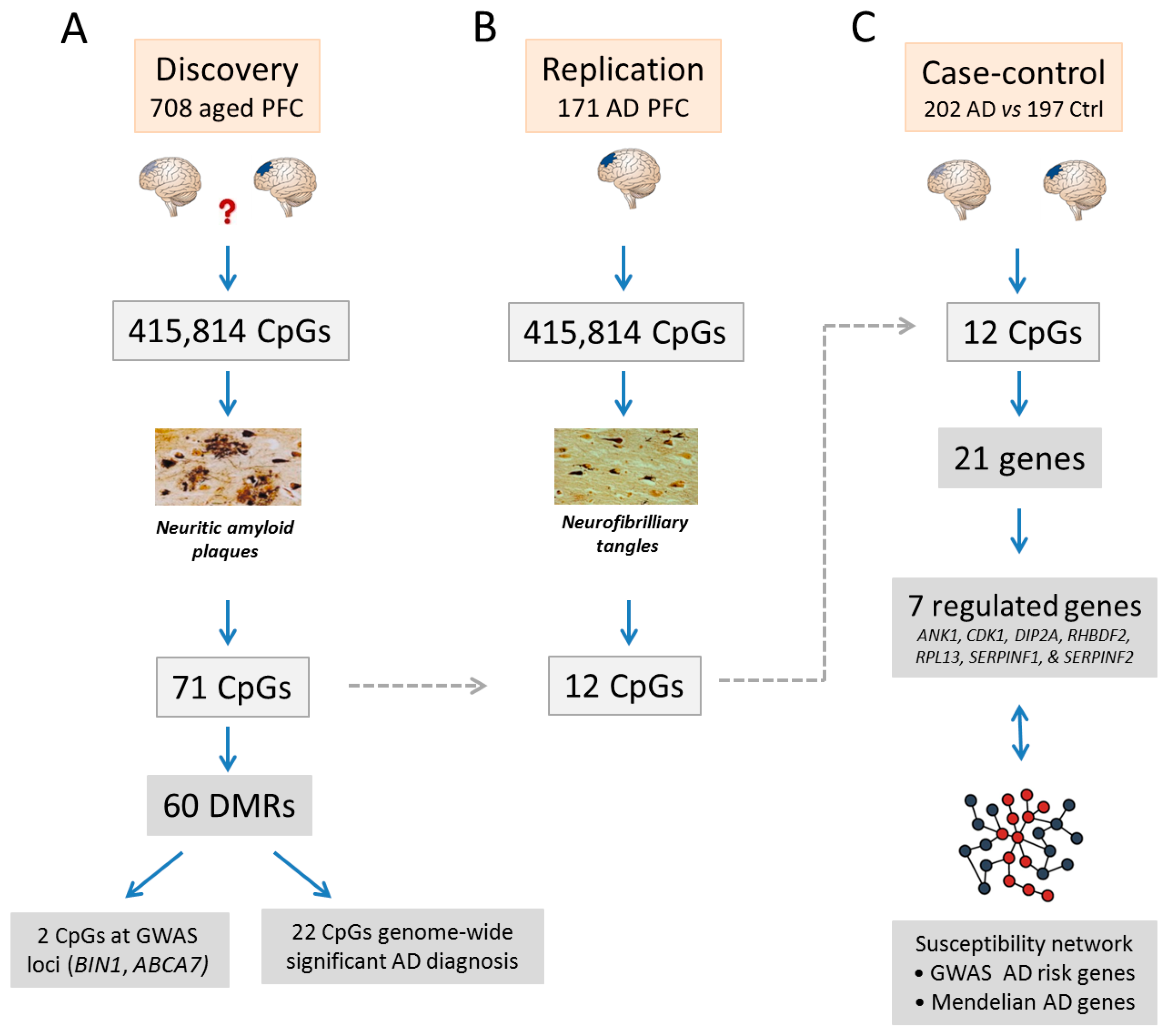
Figure 2.
Findings on epigenomic changes in Alzheimer’s disease (AD). The discovery study (A) represents a random selection of the older population unselected for. De Jager et al. [70] analyzed dorsolateral prefrontal cortices (PFC) from normal aged (light blue PFC) and AD (dark blue PFC) individuals. A total of 71 CpGs associated with neuritic amyloid plaque burden (NP). These AD-associated CpGs map to 60 differentially methylated regions (DMRs). Among the 71 NP-associated CpGs, two mapped to genome-wide association studies (GWAS) risk genes in AD and twenty-two were associated with a diagnosis of AD at the genome-wide level. In the replication study (B) comprising 171 AD PFC, 12 CpGs from the 71 NP-associated CpGs associated with neurofibrilliary tangles (NF), another primary pathology in AD. In a case-control study (C) comprising 202 AD versus 197 control PFCs, the role of these 12 CpGs was assessed for the expression of 23 neighboring genes. The 7 genes that were regulated by NP-associated CpGs additionally intersect with a susceptibility network consisting of GWAS risk genes and Mendelian genes in AD.
Figure 2.
Findings on epigenomic changes in Alzheimer’s disease (AD). The discovery study (A) represents a random selection of the older population unselected for. De Jager et al. [70] analyzed dorsolateral prefrontal cortices (PFC) from normal aged (light blue PFC) and AD (dark blue PFC) individuals. A total of 71 CpGs associated with neuritic amyloid plaque burden (NP). These AD-associated CpGs map to 60 differentially methylated regions (DMRs). Among the 71 NP-associated CpGs, two mapped to genome-wide association studies (GWAS) risk genes in AD and twenty-two were associated with a diagnosis of AD at the genome-wide level. In the replication study (B) comprising 171 AD PFC, 12 CpGs from the 71 NP-associated CpGs associated with neurofibrilliary tangles (NF), another primary pathology in AD. In a case-control study (C) comprising 202 AD versus 197 control PFCs, the role of these 12 CpGs was assessed for the expression of 23 neighboring genes. The 7 genes that were regulated by NP-associated CpGs additionally intersect with a susceptibility network consisting of GWAS risk genes and Mendelian genes in AD.

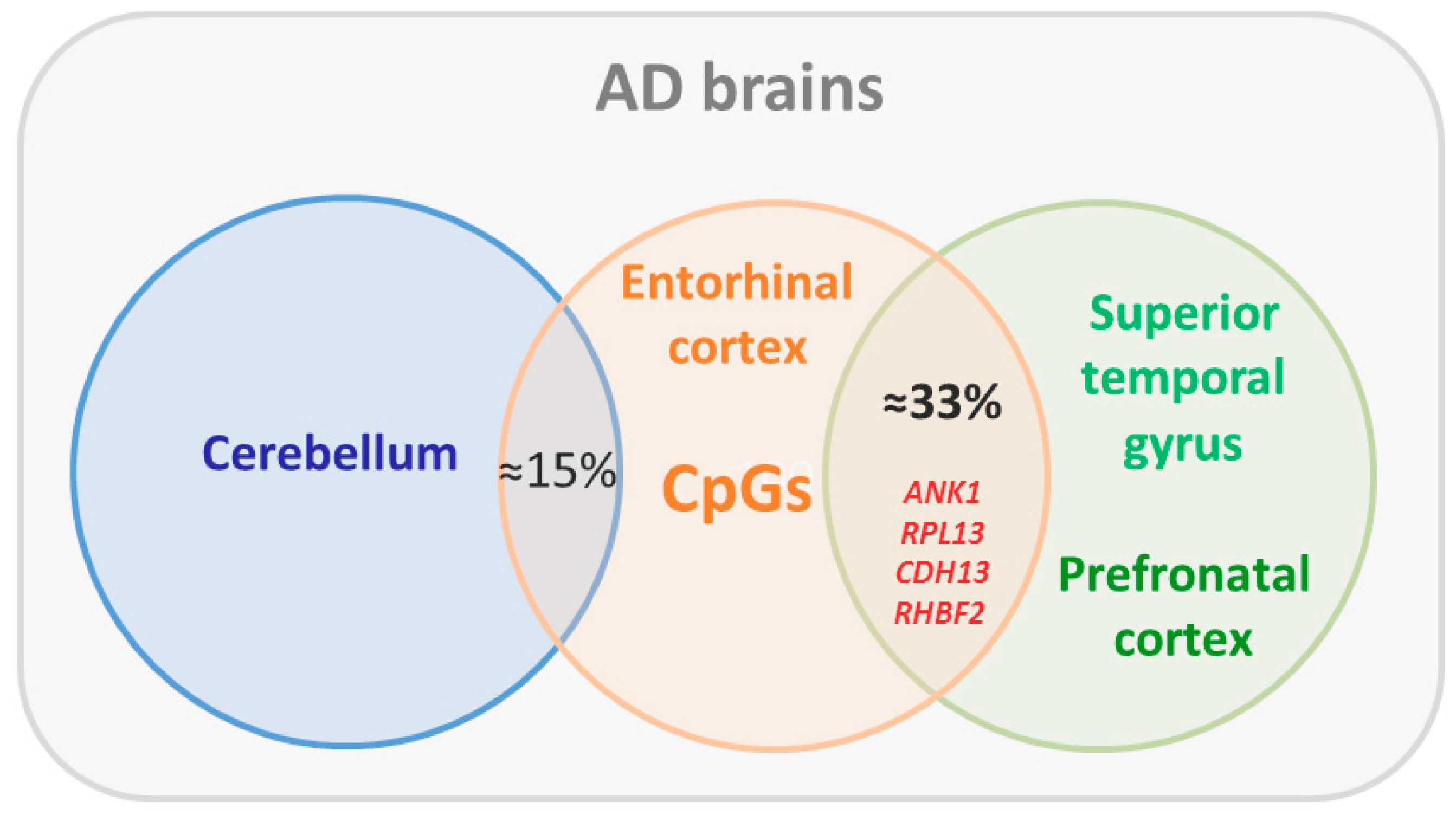
Figure 3.
Findings on cross-tissue epigenomic changes in Alzheimer’s disease (AD). Differentially methylated CpGs associated with neurofibrilliary tangle burden in the entorhinal cortex were shared by one-third with the superior temporal gyrus and prefrontal cortex, but only by ≈15% with the cerebellum, that is poorly affected in AD. Cross-tissue conserved CpGs comprised those mapping to ANK1, RPL1, CDH13, and RHBF2 previously identified by De Jager et al. [70].
Figure 3.
Findings on cross-tissue epigenomic changes in Alzheimer’s disease (AD). Differentially methylated CpGs associated with neurofibrilliary tangle burden in the entorhinal cortex were shared by one-third with the superior temporal gyrus and prefrontal cortex, but only by ≈15% with the cerebellum, that is poorly affected in AD. Cross-tissue conserved CpGs comprised those mapping to ANK1, RPL1, CDH13, and RHBF2 previously identified by De Jager et al. [70].
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Driver or Passenger: Epigenomes in Alzheimer’s Disease. Epigenomes 2017, 1, 5. https://doi.org/10.3390/epigenomes1010005
AMA Style
Hoffmann A, Sportelli V, Ziller M, Spengler D. Driver or Passenger: Epigenomes in Alzheimer’s Disease. Epigenomes. 2017; 1(1):5. https://doi.org/10.3390/epigenomes1010005
Chicago/Turabian StyleHoffmann, Anke, Vincenza Sportelli, Michael Ziller, and Dietmar Spengler. 2017. "Driver or Passenger: Epigenomes in Alzheimer’s Disease" Epigenomes 1, no. 1: 5. https://doi.org/10.3390/epigenomes1010005