The Epigenetic Landscape of Pancreatic Cancer Stem Cells



1
Department of Surgery and Cancer, Division of Cancer, Imperial College London, Imperial Centre for Translational and Experimental Medicine (ICTEM), London W12 0NN, UK
2
Department of Biochemistry, Cancer Stem Cell and Tumor Microenvironment Group, Universidad Autónoma de Madrid (UAM), 28029 Madrid, Spain
3
Biomarkers and Therapeutic Targets Group, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Red de Investigación Renal (RedinRen) Instituto de Salud Carlos III (ISCIII), 28034 Madrid, Spain
4
Department of Cancer Biology, Instituto de Investigaciones Biomédicas “Alberto Sols” (IIBM), 28029 Madrid, Spain
5
Chronic Diseases and Cancer Area, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), 28034 Madrid, Spain
*
Author to whom correspondence should be addressed.
Epigenomes 2018, 2(2), 10; https://doi.org/10.3390/epigenomes2020010
Submission received: 31 May 2018
/
Revised: 8 June 2018
/
Accepted: 11 June 2018
/
Published: 14 June 2018
(This article belongs to the Special Issue Epigenetics of Pancreatic Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Data now indicates that in addition to genetic alterations/mutations, human cancer cells exhibit important changes in their epigenome. In the context of this review, we define the epigenome as the chemical compounds and/or proteins that can interact with nuclear DNA to direct the specific and localized activation or silencing of genes to control the production of cellular proteins (directly or indirectly) in a given cell. Our ever-growing knowledge of how the epigenome can affect cellular processes has largely changed our view of cancer being a solely genetic disease. Nowadays, cancer is largely defined and characterized by the dynamic changes in both the genome and epigenome, which function together and contribute concomitantly to cancer initiation and progression. Since epigenetic modifications are crucial processes involved in controlling cellular identity and lineage fate, perturbations in this layer of gene regulation can contribute to the acquisition of new cellular characteristics different than those that were “initially” intended. For example, aberrant epigenetic alterations may transform normal non-cancer cells into cancer stem cells (CSCs), endowing them with the loss of differentiation and the acquisition of stem-like characteristics. In this review, we will focus our discussion on CSCs in the context of pancreatic ductal adenocarcinoma (PDAC). We will discuss how different epigenetic modifications create a landscape that can impact CSC identity and the way this small sub-population of cells contributes to tumor initiation, progression, and resistance to therapy. Moreover, we will highlight the latest discoveries in epigenetic-based therapies as a means of targeting CSCs.
1. Introduction
The general model of cancer progression has been viewed as a multistep process of transformation of normal cells into malignant cells driven by genetic alterations [1]. Nowadays, however, we know that cancer is not solely a genetic disease [2,3], and over the past decade, a vast amount of research data has unequivocally shown that solid tumors are also not simply a ‘mass’ of homogeneous tumor cells, but rather a very complex ecosystem consisting of tumor cells and other cell types, such as endothelial, hematopoietic, stromal and immune cells, all of which can influence the biology and overall status of the tumor as a whole. Importantly, there exists both heterogeneity across tumor cells and a defined hierarchical structure within the tumor, with a small sub-population of cells with stem-like properties, known as cancer stem cells (CSCs), residing at the apex of this hierarchy. Just as with normal stem cells, CSCs possess the ability to self-renew and divide both symmetrically and asymmetrically. Moreover, CSCs can differentiate giving rise to more differentiated progenies, including progenitor cells, transient amplifying cells and more differentiated cells, all of which form part of the tumor bulk. Lastly, and more importantly, these cells possess high tumor-initiating capacity and can form tumors that recapitulate the heterogeneity of the original tumor from which they were derived when injected in vivo in mouse models. Hence, by definition, self-renewal and differentiation of CSCs leads to the production of all cell types present within the tumor bulk, thereby driving both tumor hierarchy and heterogeneity [4]. It is important to note that the idea that CSCs are the source of tumor heterogeneity represents the main principle of the CSC model. This contrasts with the stochastic model (or clonal evolution model), which argues against particular cell populations driving tumor heterogeneity and claims that all tumors are biologically homogeneous. Thus, the stochastic model would argue that functional heterogeneity in tumor cells would be due to random or stochastic influences that alter the behavior of individual cells in the tumor [5]. Despite the underlying differences between both models, research over the past two decades has shown that these two models are not mutually exclusive and can be used in combination to explain tumor heterogeneity [6,7,8]. For example, Navin et al. provide an elegant explanation for tumor heterogeneity by suggesting that the tumor mass is continuously evolving, with some CSC clones becoming more dominant than others in terms of proliferation, adaptation, metastatic potential or chemoresistance at given times during the evolution of the tumor [9]. Thus, the representation of CSC clones at a particular time is just a snapshot of the state of tumor at a specific time during its evolution.
Studies in hematopoietic malignancies have provided the foundation for our understanding of CSCs. As early as 1935, Furth and Kahn [10] would be the first to suggest that CSC existed by showing that single leukemic cells could confer systemic disease when transplanted into recipient mice. This study, together with later ones, pointed towards the idea that functional heterogeneity exists within tumors. That is, not every tumor cell is able to proliferate to form a colony in vitro or to give rise to a tumor when transplanted in vivo. These concepts would inevitably lead to the birth of the CSC concept. In the early 1990s, Dick and colleagues, using limiting dilution in vivo transplantation assays together with fluorescence-associated cell sorting, showed that only a small fraction of tumor cells isolated from acute myeloid leukemia (AML) patients, with a characteristic cell-surface marker signature, were able to establish leukemia in recipient immunocompromised mice [11]. Following their identification in hematopoietic cancers, CSCs would soon be discovered in solid tumors, the first being breast cancer. In 2003, Al-Hajj et al., using the mammary stem cell markers CD44 and CD24, would isolate for the first time breast CSCs [12]. Soon after, CSCs would be identified and isolated from several solid tumors including brain, liver, ovary, prostate, lung, melanoma, colon and pancreatic ductal adenocarcinoma (PDAC), by using different cell-surface markers or via side population (SP) functional analysis [12,13,14,15]. For PDAC, Hermann et al., demonstrated that CD133-positive cells could form more tumors than their CD133-negative counterparts [16]. While large numbers of CD133-negative cells could not induce tumor formation, small numbers of CD133-positive cells were found to be very tumorigenic. In liver cancer, such as hepatocellular carcinoma (HCC), several cell surface markers such as EpCAM, CD133, cytokeratin 7 and 19 have been used as specific markers for liver CSCs. Even though cell surface markers are widely used for CSC identification in different tumors, there exist inherent limitations linked to the use of markers for the identification and isolation of CSCs. For example, the expression of CSC-associated cell surface markers is not exclusive to CSCs, they can change after chemotherapy or their levels can be modulated by the microenvironment. Thus, additional experiments, such as assessing in vivo tumor-initiating capacity or in vitro self-renewal ability, should always be conducted to support the stemness features of putative “CSC” populations.
2. Epigenetic Landscape of CSCs
Several studies have attempted to decipher the genetic and epigenetic mechanisms behind the establishment and maintenance of CSCs. Understanding that epigenetic mechanisms regulate key transcriptional programs in adult stem cells, such as those involved in controlling self-renewal and differentiation, several studies have suggested that similar mechanisms likely also govern CSC-genesis and the maintenance of key CSC features [17,18,19]. The observation that the CSC population is rare across many different cancers implies that epigenetic rather than genetic differences are the underlying drivers for why CSCs are functionally different from their non-CSC counterparts. Moreover, while it was generally believed that epigenetic differences would have to be largely irreversible to prevent non-CSCs from efficiently reverting to a CSC state, this concept has been recently challenged. Using mouse models of colorectal cancer (CRC), two groups recently showed that non-CSCs can convert into CSCs when the CSC pool is eliminated [20,21]. Thus, the regulation of epigenetic marks and the epigenome of CSCs and non-CSCs may be more complex and plastic than previously believed.
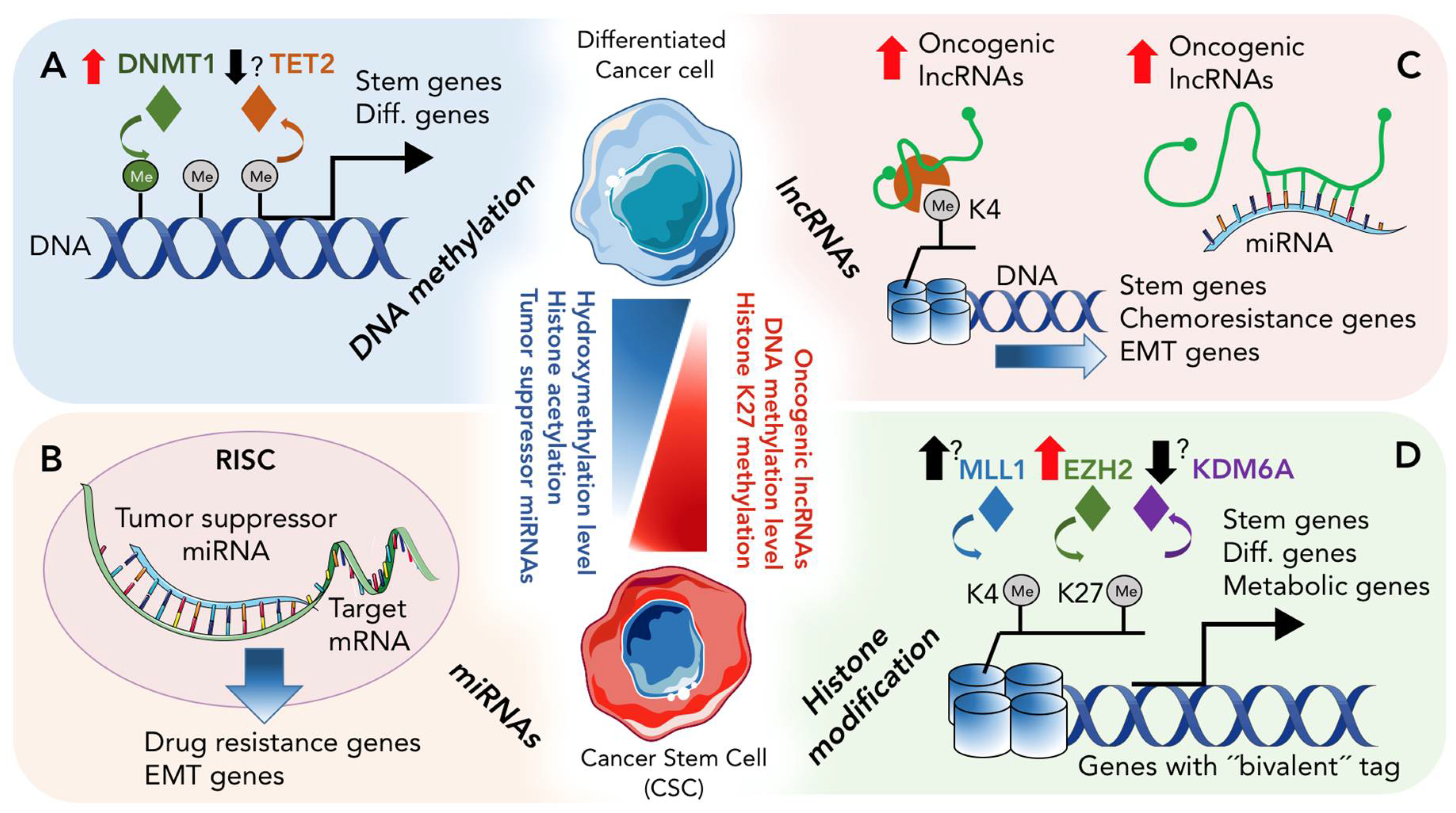
The epigenetic landscape of any cell represents the combination of 4 different mechanisms of non-genetic gene regulation. These mechanisms include (1) DNA methylation and/or de-methylation, which represent a covalent modification of DNA; (2) Histone modification, representing covalent modification of histone tails; (3) non-coding RNA molecules and (4) Nucleosome remodeling. Below and in Figure 1, we discuss how the first three processes participate in CSC biology, specifically in pancreatic CSCs.
2.1. DNA Methylation and De-Methylation in CSCs
Of the epigenetic modifications known, DNA methylation is the most well-studied. In general, DNA methylation, together with histone modifications and chromatin-associated proteins, mediates stable gene silencing, regulates gene expression and functions in chromatin architecture [22]. While non-CpG methylation has been observed in induced pluripotent stem cells (iPSCs) [23], DNA methylation occurs predominantly at CpG dinucleotides in mammals [24]. CpG dinucleotides are DNA regions where a cytosine is followed by a guanine. They are not evenly distributed across the genome but rather are concentrated in CpG-rich stretches of DNA, known as CpG islands, typically stretches of 500–1500 base pairs of DNA with CG:GC ratios of greater than 0.6 [25].
The class of enzymes known as DNA methyltransferases (DNMTs) mediate the methylation reaction of cytosine by catalyzing the transfer of the methyl group from S-adenosyl-methionine onto cytosine. This family of enzymes consists of 5 members: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L [26]. Of these 5 members, DNMT2 functions as an RNA methyltransferase rather than a methyltransferase for DNA [27], and DNMT3L does not contain a methyltransferase catalytic domain. Thus, only DNMT1, DNMT3A and DNMT3B are considered true functional DNA methyltransferases [28]. After DNA replication, the maintenance of cytosine methylation at CpG dinucleotides is carried our primarily by DNMT1 [29]. DNMT1 copies the methylation patterns from the parental strand of DNA to the newly synthesized DNA strand [30]. While DNMT3A and DNMT3B are also capable of methylating hemimethylated DNA, these two methyltransferases function more during embryogenesis as de-novo methyltransferases [31].
DNA methylation can contribute to gene silencing in various ways, such as creating a “physical barrier” whereby transcription factors cannot access their target-binding sites. Such “blocking” has been observed for the genes MYC and MLTF. Alternatively, DNA methylation can facilitate the binding of methyl-binding domain proteins to DNA, mediating gene repression via interactions with histone deacetylases (HDACs) [32,33]. Thus, DNA methylation can silence genes and even non-coding genomic regions via a variety of different mechanisms.
The literature has demonstrated that methylation changes follow cancer development and progression [34]. Likewise, studies in embryonic stem cells (ESCs) show that during differentiation and cell fate decision, a major switch in the methylation landscape occurs [35], reinforcing the idea that methylation is an important epigenetic process linked to cellular differentiation. Adult stem cells also exhibit similar methylation changes in specific tissues under both normal and pathological conditions, including cancer [36]. Thus, methylation likely plays a very important role in cancer development and/or progression. Indeed, many studies have already shown that in different tumor cells populations, differential DNA methylation could account for the expression of key genes, such as stem cell marker genes [37]. In breast cancer, for example, it was shown that the CSC genes CD44, CD133 and Musashi-1 (MSI1) are regulated by the methylation of CpG regions in their promoters, and hypomethylation can activate these CSC genes in clinically more aggressive subtypes (triple- negative breast cancer) [38,39,40]. Moreover, a study by Sun et al. comparing methylation profiles in invasive and non-invasive pancreatic cancer cells revealed a significant correlation between the methylation profile and the expression of key pathways, such as NF-κB signaling, in the two cell populations studied. For example, genes such as BIMP, TNFR and CD49 were demethylated in the invasive population but methylated in the non-invasive fraction [41]. In addition, the authors also found BMP4, GATA6 and SOX9 to be differentially methylated among invasive and non-invasive cells. This is of particular interest as these genes have also been shown to play important roles in multiple cancer types.
Proteins involved in the establishment and maintenance of DNA methylation have also been identified as drivers of CSC formation. Trowbridge et al. found that the development of leukemia was blocked by abrogation of DNMT1; haploinsufficiency of DNMT1 induced re-expression of tumor suppressor genes, impairing CSC self-renewal and attenuating leukemia progression [42]. In breast cancer, mammary stem and progenitor cell populations were significantly reduced in Dnmt1-knockout mice, suggesting a critical role for DNMT1 in the expansion and maintenance of mammary stem cells [43]. Also, in lung cancer, up regulation of DNMT1 by Interleukin (IL)-6 has been shown to be associated with an increase in the CSC population, which the authors show to be due to methylation of TP53 and p21 (WAF1/CIP1) [44]. Similar to breast cancer and leukemia, lack of DNMT1 also impaired the stemness of lung CSCs. In PDAC, our recent publication [45] indicates that pancreatic CSCs are dependent on DNMT1 for their in vivo and in vitro stemness properties. Specifically, pancreatic CSCs show high expression of DNMT1, and abrogation of DNMT1 impaired their self-renewal and promoted their differentiation toward non-stem populations. Thus, our and other studies published to date indicate that inhibition of DNMT1 may prove to be a therapeutic option to modulate and subsequently eliminate CSCs in various tumor types.
In general DNA methylation was considered to be a very stable chromatin modification process; however, studies examining global DNA methylation throughout embryonic development have shown, in early zygotes, that global loss of DNA methylation occurs, especially in the male pronucleus [46]. High-resolution genome-wide mapping of methylation has also confirmed that DNA methylation is not stable and can also be modified in iPSCs and differentiated cells [47]. Together, these studies, indicate an active enzymatic process within cells that can erase or alter DNA methylation. The ten-eleven translocation (TET) family proteins, which include TET1, TET2 and TET3, actively participate in DNA demethylation, via iron and α-ketoglutarate dependent 5-methylcytosine dioxygenase activity, converting 5-methylcytosine (5mC) bases to 5-hydroxymethylcytosine (5-hmC) bases [48,49]. Based on their distinct expression patterns, it is believed that the three TET proteins have non-overlapping functions. For example, TET1 and TET2 are highly expressed in ESCs, TET2 is also abundantly expressed in hematopoietic cells, and TET3 expression has been detected in oocytes [50]. In addition, emerging evidence suggests that apart from their well-studied DNA modification roles in ESC and neuronal systems, TET-mediated DNA modifications may also be important mediators of tumorigeneses, as detailed below.
Somatic mutations in TET proteins have been detected in CRC, and mutations and/or deletions of TET2 have been described in clear-cell renal cell carcinoma [51] and metastatic castration-resistant prostate cancer [52]. The observation that the decrease in 5-hmC levels and/or TET expression correlates with aggressive tumor growth, shown in several studies, supports the idea that TET proteins might play important tumor suppressor roles in certain types of solid tumors [53,54]. A 2013 study using the HpaII tiny fragment Enrichment by Ligation-mediated PCR beta-glucosyl transferase (β-GT) assay, known as HELP-GT, found redistribution of 5-hmC sites in PDAC cells [55]. Interestingly, the authors observed that the enrichment was more variable in promoters, CpG islands and shores, and redistribution of many 5-hmC sites occurred in promoters known to play a role in PDAC tumorigenesis and metastasis, such as GATA6, whose expression has been directly linked to EMT [56], a CSC-associated phenotype. These results suggest that redistribution of 5-hmC peaks in PDAC correlates with sites of active transcription allowing activation of cancer-promoting oncogenes that are linked to these differently hemi-methylated sites. Interestingly, we showed that at the DNA level, pancreatic CSCs display a hypermethylated phenotype, which is most probably due to the higher expression levels of methyltransferase proteins such as DNMT1 [45]. These cells may very well display lower levels of hydroxymethyl marks and/or their distribution could vary compared to their non-CSC counterparts. Unfortunately, very few reports exist regarding 5-hmC and TET proteins in pancreatic cancer and pancreatic CSCs, thus future studies are needed to shed more light on if and how these processes are involved in pancreatic CSC biology.
2.2. Histone Modification in PDAC and CSCs
Post-translational modification of histone tails is an evolutionary conserved mechanism that plays a critical role in regulating the chromatin state and gene activation [57,58]. These modifications can structurally change the chromatin to facilitate or exclude protein complexes from interacting with specific DNA regions. Thus, histone modifications can influence gene transcription, by altering their activity, which could potentially contribute to oncogenic transformation and cancer progression.
Five types of histones are known and include histone 2A (H2A), H2B, H3, and H4, and one linker histone, H1 (or H5), that is not present in the nucleosome bead, but helps to secure the DNA that is wound around each histone octamer. Approximately 146 base pairs of DNA are wrapped, in 1.67 left-handed super helical turns, around each nucleosome bead, consisting of two copies of each of the core histones H2A, H2B, H3 and H4 [57,58,59].
The amino acid residues located on the N- and C-terminal tails of histones can be post-translationally modified via process such as methylation, acetylation, phosphorylation, ubiquitination and SUMOylation. All these modifications are generally referred to as histone marks and they form a special kind of language that nowadays we call “histone code”. Similar to genetic code, epigenetic “tagging” represents a fundamental regulatory mechanism that has an impact on most, if not all, chromatin processes [22]. The dynamic addition or removal of post-translational modifications from histone tails is mediated by several different histone-modifying enzymes. The “writers” and “erasers” of histone marks include many enzymes, such as histone acetyltransferases, HDACs, histone methyltransferases, histone demethylases, histone ubiquitinating enzymes, and histone deubiquitinating enzymes, all of which vary in their ability to recognize and alter the amino acid residues of histone tails [60,61,62].
Of all modifications, one of the most widely studied are alterations of lysine modifications on the histone H3 N-terminal tail, including lysine methylation, acetylation, and phosphorylation. Lysine methylation marks on chromatin associated with promoter regions have been shown to be correlated with transcriptional activation (e.g., histone 3, lysine 4 trimethylation) or silencing (e.g., H3K9me3 or H3K27me3), usually through recruitment of co-effectors or polycomb group proteins [63]. Furthermore, the histone code can be very complex as a specific regulatory region may be associated with not only one type of mark but with multiple marks. For example, genes whose promoters are marked with H3K4me3 and H3K27me3 are called bivalent genes. These genes are expressed at low levels and are said to be “poised” to be activated upon a stimulating signal [64]. Bivalent genes are a frequent feature in stem cells and pluripotent cells, where these genes can be activated by loss of H3K27me3 (silencing histone mark) or repressed by loss of H3K4me3 (activating histone mark), upon signals for differentiation or development [65]. For instance, in AML CSCs, genes involved in stem cell identity, proliferation and metabolic reprogramming were shown to be bivalently marked with H3K4me3/H3K27me3, while in non-CSCs, H3K4me3 marks are lost to repress stem cell identity genes [66]. Similarly, in glioblastoma, CSCs possess more open chromatin conformation due to reduction of the silencing histone mark, H3K27me3, compared to non-CSCs, leading to a de-repression of genes involved in tumor initiation [67].
In the case of PDAC, few studies have looked at the differences in the chromatin landscape between CSCs and non-CSCs. Instead, the focus has more frequently been on deciphering the role of particular chromatin writers, readers, or erasers in the pathogenesis of this disease. For example, EZH2, a component of the Polycomb Repressive Complex 2 (PRC2), is a well-studied trimethylation writer of H3K27 whose increased expression in pancreatic cancer patients has been associated with not only a greater incidence of positive nodes but also significantly larger tumors [68]. Chen et al. [69], for example, used RNA interference to knockdown EZH2 and showed that EZH2 silencing decreased tumor growth and the incidence of liver metastasis in a PDAC model. In terms of CSCs, van Vlerken et al. isolated EpCAM+/CD44+/CD24+CSCs from the pancreatic cancer cell lines HPAC and Panc-1 and found elevated levels of H3K27me3 compared with the non-CSC control population [70]. Similarly, EZH2 levels were also elevated in the CSC population. Interestingly, when they downregulated EZH2, a decrease in CSC frequency followed by a significant gain in the non-CSC population was observed, which the authors attributed to EZH2 loss driving the CSCs into a more differentiated state. It is possible that apart from bearing more H3K27me3 histone marks, due to an increase in EZH2 protein levels, the distribution of histone methylation in PDAC CSCs may differ from non-CSCs, which would result in differential regulation of specific genes. That being said, it would be of interest to investigate which are the most preferred regions occupied by H3K27me3 histone mark and if they show the characteristic bivalent trait. In the aforementioned study, the authors also hypothesized that EZH2 expression could serve as a reporter assay to detect CSC activity. In line with this hypothesis, they observed an increase or decrease in the percentage of EZH2high cells upon treatment with Gemcitabine or Salinomycin, respectively, which mirrored changes in the percentage of EpCAM+CD44+CD24+ cells; however, more comprehensive experiments including transplantation assays in vivo would be necessary to confirm whether EZH2 expression could be exploited as a marker to detect pancreatic and other types of CSCs [70].
Apart from EZH2, which as previously mentioned is often overexpressed in cancer, another histone methylation regulatory gene has been found to be frequently mutated in PDAC [71,72]. KDM6A (also known as UTX) encodes for a histone H3K27me3 demethylase [73,74,75]. KDM6A is an integral component of the complex of proteins associated with Set1 (COMPASS)-like complex, which apart from the core proteins WDR5, RBBP5, DPY30, and ASH2L, also contains the methyltransferases KMT2C or KMT2D, which mono-methylate H3K4 [76,77,78,79]. Data from The Cancer Cell Line Encyclopedia [79] interrogating the copy-number and gene-expression changes in over 1000 human cancer cell lines revealed selective copy-number losses and downregulation of KDM6A in PDAC. Interestingly, loss of KDM6A expression or mutations were found more frequently in the more aggressive squamous subtype of pancreatic cancer [72]. While ablation of KDM6A expression in the pancreas of Pdx1Cre; Kdm6anull and Ptf1αCre; Kdm6anull mice did not change the global level of H3K27me3, H3K27ac, H3K4me1 or EZH2, and these mice exhibited normal pancreatic histology, when KDM6A loss was combined with mutant KrasG12D expression, mice developed aggressive, metastatic squamous-like pancreatic tumors. Furthermore, KDM6A loss resulted in gene expression changes independent of H3K27me3 that promoted squamous and quasi-mesenchymal differentiation in female mice. Mechanistically, loss of KDM6A deregulated the COMPASS-like complex, disrupting the super enhancer landscape to promote aberrant activation of oncogenes including TP63, MYC, and RUNX3 [80]. Interestingly, demethylase-independent roles for KDM6A have been described as important for stem cell homeostasis and developmental processes [81,82]. While this would indicate an important role for KDM6A in regulating normal cell identity, in cancer this aspect has not been extensively explored. In CSCs, a study performed by Taube et al., reported that fast-cycling, differentiated subpopulations from multiple mammary cell lines showed significantly higher KDM6A staining compared to their slow-cycling, stem cell-enriched counterparts [83]. Unfortunately, similar studies in pancreatic CSCs are still lacking. Thus, considering the role of KDM6A in PDAC progression, investigating the potential role of KDM6A in PDAC CSCs is warranted.
A crucial role for histone modifications in the progression of PDAC have been highlighted by two recent studies [84,85]. While investigating the source of genetic heterogeneity across metastatic sub-clones, Makohon-Moore et al., found that the genetic landscape of metastatic PDAC tumors largely reflected that of the primary tumor [84]. In a follow-up study, Mc Donald, Li, and colleagues interrogated the same group of samples focusing their attention on histone marks [85]. Surprisingly, they discovered major differences in the epigenetic landscape between primary tumor sub-clones that seed regional sites (peritoneal) and those that seed distant sites (liver and lung). More specifically, in peritoneal metastasis, histone 3 lysine 9 dimethylation (H3K9me2) was strongly enriched across large block-like domains of heterochromatin (LOCKs), whereas the same regions displayed global reduction in H3K9me2 in the distant metastasis and primary tumor precursors. Furthermore, the authors observed differential gene expression and reduction in DNA methylation in the LOCKs from distant metastases versus peritoneal metastases. They also looked at gene-rich euchromatin domains (ECDs), defined by enrichment for acetylated H3K27 (H3K27ac) and H3K36 trimethylation (H3K36me3), and found that distant metastases and primary tumor sub-clones displayed local reprograming of H3K27Ac and H3K36me3 specifically over differently expressed genes within ECDs. Thus, this study discovered that substantial epigenetic reprograming occurs at sites of heterochromatin and euchromatin and that this differs between regional and distant metastasis. Later, in the same year, Roe and colleagues also studied chromatin regulation in PDAC metastasis, but with a focus on enhancer regions [86]. In paired PDAC organoids derived from primary KPC mouse (KRas+/LSLG12D; TP53R172H/+; Pdx1-Cre) tumors and metastases, genome-wide profiling revealed regions with increased levels of the active enhancer mark H3K27ac (GAIN regions) and regions with decreased H3K27ac (LOSS regions), although global H3K27ac levels were similar across the samples. The large majority of GAIN and LOSS regions occurred in enhancers, and GAIN enhancers exhibited enrichment of H3K4me1 in metastatic organoids without changes in chromatin accessibility. Furthermore, the FOXA1 pioneer factor was enriched in GAIN regions and cooperated with GATA5 to remodel enhancer histone marks to promote PDAC progression and metastasis in vivo.
Taken together, both studies have contributed substantially to furthering our understanding of the molecular and epigenetic mechanisms that drive PDAC metastasis, but both studies lacked any interrogation of the role of CSCs in this process. Multiple studies have shown that circulating tumors cells (CTCs) can intravasate into the bloodstream promoting the generation of micro-metastatic reservoirs, some of which can progress to macro-metastatic disease [87,88]. In theory, a small fraction of these CTCs should have CSC activity and/or properties. While additional studies would still need to be performed to better understand the relationship between CSCs and CTC populations in PDAC, the aforementioned studies may offer an attractive idea for looking at chromatin patterns in different cell populations from primary and metastatic tumor sites to reveal if epigenetic reprograming is occurring ubiquitously or if it is a unique feature of a particular population or pool of cells.
3. Role of Non-Coding RNAs in CSCs
Data from the Human Genome Project indicate that only around 20,000 genes of the human genome encode for proteins [89]. Thus, the vast majority of transcribed RNAs are not translated into proteins and are known as non-coding RNAs (ncRNAs) [90,91,92]. ncRNAs used to be considered “junk RNAs”; however, an increasing body of evidence now suggests that ncRNAs are critical for epigenetic, transcriptional, posttranscriptional, and translational regulation of gene expression in both physiological and pathological conditions [93,94,95]. Similarly, it has been shown that ncRNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), can regulate cancer cell stemness and are necessary for the maintenance of the CSC pool. Therefore, a more comprehensive understanding of the role of ncRNAs in CSC regulation could reveal novel strategies to eradicate this population of cells, responsible for tumor relapse and metastasis.
Based on their size, ncRNAs can be divided in two major groups: small ncRNAs refer to ncRNAs shorter than 200 nucleotides, while long ncRNAs (lncRNAs) refer to ncRNAs composed of 200 or more nucleotides. Small ncRNAs can be further categorized into subcategories based on their length, function, and subcellular localization such as microRNAs (miRNAs), short interfering RNAs (siRNAs), piwi-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), short hairpin RNAs (shRNA), and other short RNAs [96]. These small ncRNAs have been implicated in various aspects of CSCs biology. Of the small ncRNAs, in this review we will primarily discuss the role of miRNAs in CSCs biology.
3.1. Role of miRNAs in CSCs
miRNAs are small (20–25 nucleotides) ncRNAs and key players in the post-transcriptional regulation of genes. More than 80% of mammalian genes are under their direct or indirect control, highlighting that almost every cellular function is tightly regulated by miRNAs. Indeed, miRNAs show very specific patterns of expression across different tissues and cell types, and approximately 2000 miRNAs have already been identified in the human genome. Their mechanism of action is based on the recognition of small (6–8 nt) sequences present in their target mRNAs [97]. Due to the small size of these recognition sites, one miRNA can potentially recognize and regulate several mRNA targets, and a single mRNA can be regulated by more than one miRNA [98]. This dynamic regulation has revealed miRNAs as critical regulators of a multitude of cellular processes, and thus, their deregulation can have significant pathological consequences, contributing to disease onset and development.
Mature miRNAs are loaded into the interference multiprotein complex (RISC), where translational repression takes place. Inside RISC, miRNAs recognize their target mRNAs by base-pair complementarity of the miRNA seed sequence with the 3′ untranslated region (UTR) of the mRNA. When complementarity between the seed sequence and mRNA target sequence mRNA is perfect, cleaving inside the RISC complex occurs. When complementarity is only partial, mRNA translational repression or degradation occurs, involving the recruitment of deadenylase complexes tasked with the removal or shortening of the mRNA poly-A tail [99]. miRNA binding sites have also been identified in the 5′ UTR and open reading frame regions.
It has been widely demonstrated that miRNAs can also be secreted to the extracellular environment/milieu, where they can travel to distant sites and exert their regulatory effects on new target cells. miRNAs have been detected in a wide range of cell-free body fluids such as urine, serum, or saliva. Their secretion is believed to not only be a highly regulated process, but the selection of which miRNAs will be secreted is also believed to be tightly controlled and not a random process [100]. Thus, secreted miRNAs may be as functionally relevant as their non-secreted counterparts. As stated above, the deregulation of miRNAs (over-expression or silencing) has been associated with several different pathologies, thus physiological or pathological regulation of intracellular miRNAs may give rise to different profiles of secreted miRNAs. Indeed, changes in miRNA serum profiles have been used as biomarkers for a wide-range of diseases including cancer [101], cardiovascular diseases [102] or neurodegeneration [103], among others.
In the last 10 years, miRNAs have also provided new insights into gene regulation implicated in tumorigenesis and CSC biology [104]. Several miRNAs involved in CSC biology in different solid tumors have been identified: (i) miR-5703, miR-630, miR-1246, miR-424-5p and miR-320b were all deregulated in ovarian CSCs [105]; (ii) miR-34a, miR-200b/c, miR-203 and miR-137 appear to be critical in CRC CSCs [106]; (iii) the miR-200 family and miR-let7 family are crucial for CSC dynamics in breast cancer [107]; (iv) miR181-5p, miR153 and the miR17-92 cluster are relevant for the pancreatic CSC sub-population [45,108,109]. Remarkably, secreted exosomes from CSCs also orchestrate autocrine and paracrine functions, which alter the tumor microenvironment, as well as the growth and progression of the tumor. CSC-derived exosomes contain stemness-specific proteins, miRNAs capable of promoting self-renewal, and survival factors, all of which can contribute to tumor heterogeneity and tumor progression [110].
As previously mentioned, pancreatic CSCs undergo EMT, contributing to relapse and chemoresistance, and miRNAs are one of the major epigenetic mechanisms involved in the EMT process. The first association between a miRNA and EMT was described for the miR-200 family, which consists of miR-200a/b/c, miR-141 and miR-429. In 2008, Gregory et al. showed that decreased expression of the miR-200 family led to enhanced ZEB1 and ZEB2 expression [111]. Via a reciprocal feedback loop, ZEB1 and miR-200 family members repress the expression of each other. Later, Brabletz et al. would show that miR-200 members also target Jagged1 and the mastermind-like coactivators MAML2 and MAML3, important components of the Notch pathway. Specifically, the authors showed that in PDAC, ZEB1-mediated reduced miR-200 expression promoted Notch activation and indirectly induced stemness maintenance [112]. Additional miRNAs regulating EMT transcription factors have also been described and include miR-29b, miR-30a and miR-205 [113,114,115]. Moreover, relevant EMT molecules are directly regulated by miRNAs, including: E-cadherin (miR-9), N-cadherin (miR-194), Nestin and Star1 (miR-661), pulmonary adenoma resistance 3 protein (miR-491-5p), which is engaged in tight junction alteration and p120 (catenin δ1) (miR-197) [116,117,118,119,120]. Thus, miRNAs are necessary for cells to enter EMT, and consequently they may also act as regulators of de novo CSC-genesis as it has been shown that EMT activation can give rise to CSCs [121].
Several miRNAs have been directly linked to pancreatic CSC biology. For example, we reported that the miR17-92 cluster plays an anti-CSC role in PDAC, thus pancreatic CSCs downregulate the expression of this cluster, via DNMT1-mediated methylation, to maintain their stem state by preventing degradation of key CSC mRNAs [109]. Nalls et al., demonstrated that the miR-34a regulates CSC characteristics and therefore pancreas cancer progression [122]. miR-34 regulates Notch pathway proteins and BCL-2, and it can counteract the tumor suppressing function of p53 in p53-deficient human PDAC tumors [123]. miRNAs associated to RAS proteins, altered in the majority of PDAC tumors, have also been identified. For example, miR-217 specifically targets the KRAS oncogene [124]. miR-96 is also a potent negative regulator of KRAS signaling, directly targeting KRAS and having anti-proliferative, pro-apoptotic and anti-metastatic effects [125]. Downregulation of miR-126 and let-7d contributes to PDAC transformation by post-transcriptional upregulation of KRAS [126]. miR-21 has also recently been linked to RAS, through activation of AP-1 in response to RAS [127], and activated/mutated KRAS (G12D) stimulates the promoter of miR-21 in human PDAC cells.
While the cell of origin in PDAC is unknown, it is believed that PDAC arises from a stepwise process from low-grade to high-grade pancreatic intraepithelial neoplasias (PanINs) with accumulation of specific genetic mutational events (e.g., mutations in KRAS, p16, p53, etc.). Initial PDAC lesions exhibit different miRNA expression profiles depending on severity. For example, miR-155 has been shown to be significantly overexpressed in PanIN-2/PanIN-3 lesions, and the levels of miR-155 increased from PanIN-2 to PanIN-3 lesions, strongly suggesting that miR-155 activation could be an initial event in tumor progression, perhaps at the level of the cell of origin [128]. Moreover, Yu et al., identified several miRNAs including miR-378, miR-130b, miR-133a, miR-151-5p, miR-148a/b, miR-185, miR-331-3p/5p, miR-200c, miR-330-3p, miR-34c-5p, miR-129-3p, and miR-423-5p to be overexpressed in low-grade PanINs (PanIN-1 or PanIN-2), while others have shown that different miRNAs, such as miR-196b, are exclusively expressed in advanced PanIN-3 lesions and undetectable in low-grade PanIN lesions [129]. Thus, it is interesting to hypothesize that miRNA expression in PDAC precursors could cooperatively function with genetic mutations to give rise to the CSC population.
Apart from their tumorigenic and self-renewal capacity, CSCs also significantly contribute to the radio and chemoresistance inherent of PDAC. Gemcitabine is the standard of care for PDAC, and several miRNAs have been described to be related to Gemcitabine response. miR-145, acting as sponge for the long intergenic ncRNAs (lincRNAs) linc-DYNC2H1-4, regulates EMT and CSC properties, impacting resistance to Gemcitabine in PDAC cells [130]. miR-205, a miRNA associated to CSC phenotypes in PDAC, is highly downregulated in Gemcitabine-resistant cells, and a reduction in CSCs, EMT and chemoresistance markers is observed when miR-205 is overexpressed, strongly suggesting that miR-205 re-sensitizes Gemcitabine-resistant PDAC cells to Gemcitabine [131]. miR-200c confers therapy resistance in several solid tumors, including PDAC. Chemoresistance, targeted therapy resistance and radiotherapy resistance is observed when miR-200c is deregulated as it impacts EMT processes, affecting important signaling cascades such as TGF-β, PI3K/Akt, NOTCH, VEGF, and NF-κB signaling [132]. The miR-17-92 cluster, mentioned above, is also downregulated in chemoresistant CSCs. In fact, overexpression of miR-17-92 reduced chemoresistance to Gemcitabine, CSC self-renewal and tumorigenicity in animal models, through inhibition of NODAL/ACTIVIN/TGF-β1 signaling and targeting of p21, p57 and TBX3 [109]. miR-1246 is also related with tumor-initiation and drug resistance induction targeting CCNG2 expression. Indeed, higher levels of miR-1246 have been correlated with a worse prognosis in Gemcitabine-treated patients, correlating with lower CCNG2 expression in primary PDAC tumors [133]. Therefore, modulation of miRNAs in PDAC and in PDAC CSCs has emerged as an attractive approach for restoring chemosensitivity to Gemcitabine and potentially other chemotherapies [134].
As previously mentioned, miRNAs can be secreted to the extracellular space and body fluids, including blood; therefore, the easy detection of miRNAs in serum or plasma represents a very useful non-invasive methods for early diagnosis and monitoring of treatment response. Until now, the carbohydrate antigen 19-9 is the only US Food and Drug Administration (FDA) approved marker for the diagnosis of PDAC and evaluation of treatment response; however, it is widely known that its utility as a biomarker is limited due to its restricted sensitivity and specificity. Therefore, there is an unmet and urgent clinical need for novel and more precise biomarkers in PDAC. miRNA levels in the blood could provide very valuable information in the early diagnosis of PDAC, stratification and treatment response, as observed in other solid tumors, allowing for more efficient management of PDAC patients in the context of personalized medicine.
3.2. Role of lncRNA in CSCs
lncRNAs are a class of ncRNAs that are longer than 200 nucleotides, do not encode for proteins and are found to have limited expression across different tissues. As reported in the literature, dysregulation of lncRNAs has been found in various types of cancers, such as leukemia, breast cancer, gastric cancer, CRC, HCC, and lung cancer [135,136,137,138,139]. Most lncRNAs are transcribed by RNA polymerase II, and around ~13,000 lncRNAs have been confirmed to exist and more than half of them have been found between genes, known as lincRNAs [140,141]. Others lncRNAs include overlapping, antisense, and intronic lncRNAs [89].
LncRNAs can regulate the expression of genes via four mechanisms of action [142,143]. They can function as (1) Signals: functioning as a molecular signal or indicator of transcriptional activity, thus reflecting the biological outcome of gene expression; (2) Decoys: lncRNA can bind and titrate away proteins or RNA targets; (3) Guides: lncRNA can bind RNA-binding proteins and guide them to either a near or distant target gene locus and/or (4) Scaffolds: helping to assemble different proteins to form RNA:protein complexes important for the initiation of specific biological functions.
Multiple studies have revealed that lncRNAs regulate pluripotency via several mechanisms including histone modifications, working as scaffolds or by forming competing endogenous RNAs (ce-RNA) for miRNAs that repress expression of pluripotency-associated gene. Moreover, lncRNAs can influence CSC-genesis and plasticity by altering signaling pathways related to self-renewal or pluripotency or by modulating chemoresistance and EMT processes [144,145].
In the case of PDAC, the field has recently started to explore how lncRNA molecules could contribute to the development and progression of this cancer, and more specifically, how lncRNAs can influence pancreatic CSC biology. Among the first lncRNAs explored in PDAC CSCs was metastasis-associated lung adenocarcinoma transcript 1 (MALAT1). Feng et al., found higher expression of MALAT1 in CD133-positive PDAC CSCs when compared to their CD133-negative counterparts [146]. Moreover, when they knocked-down MALAT1 in PDAC cells, these cells showed a reduction in both in vitro and in vivo stemness properties. Specifically, the authors observed a decrease in the expression of pluripotency genes SOX2, BMI1 and NANOG, less ability to form spheres and colonies, and MALAT1 knock-down cells showed decreased in vivo xenograft growth capacity. This study, however, did not deeply explore the exact mechanism(s) by which MALAT1 regulated pancreatic CSCs. Since then, several studies have investigated the molecular role of MALAT1 in other tumor entities and in CSCs. As with most factors, MALAT1 expression varies across different tumor types. For example, its over-expression was observed in liver, cervical, colon and gallbladder cancer, and its expression has been shown to be a predictive marker in stage I non-small cell lung cancer patients and in stage II/III CRC patients [147,148,149,150,151]. In contrast, MALAT1 levels have been shown to be low in glioma tissue. At the molecular levels, Han et al. showed that low levels of MALAT1 promoted cancer cell proliferation and metastasis by activating the ERK/MAPK signaling pathway in the glioma stem cell line SHG139S [152]. In liver cancer stem cells, Wu et al. showed that MALAT1 overexpression resulted in more RNA polII, P300, CREPT loading onto the promoter region of telomere repeat-binding factor 2 (TRF2), enhancing TRF2 expression at the level of transcription and its phosphorylation and SUMOylation. TRF2 is involved in telomere maintenance and protection, thus in this study MALAT1 promoted liver CSCs through telomere regulation [150]. Whether MALAT1 regulates ERK/MAPK signaling or telomere stability in pancreatic CSCs is still unknown.
HOTTIP (HOXA transcript at the distal tip) is another lncRNA, shown to regulate stemness and tumorigenicity in pancreatic cancer. It is a lncRNA located near chromosome (chr) 7p15.2 and transcribed from the 5′ tip of the HOXA locus. Alteration of HOTTIP in PDAC CSCs affected sphere formation, expression of the pluripotency genes NANOG, OCT4 and SOX2, expression of known CSC markers ALDH1, CD44 and CD133 and tumor growth. In their study, Fu et al. proposed a very interesting mechanism for how HOTTIP could exert its role on CSCs, which included its direct binding to the adapter WD Repeat Domain 5 (WDR5) protein [153]. This protein can contribute to histone modification, as part of the mixed-lineage leukemia 1 (MLL1)/MLL complex that is involved in methylation and dimethylation at Lys-4 of histone H3, a specific tag for epigenetic transcriptional activation. HOTTIP could target WDR5/MLL complexes across the HOXA9 locus, driving histone H3 lysine 4 trimethylation and HOXA9 gene transcription. Moreover, HOXA9 regulation by HOTTIP could further mediate the activation of the Wnt pathway in pancreatic CSCs by promoting the expression of Wnt genes. Thus, the HOTTIP/HOXA9 axis may regulate PDAC CSCs by modulating the Wnt/β-catenin signaling pathway.
An increasing number of lncRNAs have also been implicated in the regulation of EMT acting as either promoters (pro-EMT) or antagonizers (anti-EMT), often by functioning as competing endogenous RNAs (ceRNAs) for miRNAs involved in EMT regulation or by mediating epigenetic silencing via the recruitment of PRC2. The lncRNA-activated by TGF-β (lncRNA-ATB) is an excellent example of how a lncRNA can modulate different CSC pathways to trigger an EMT program to enhance cell migration and invasiveness. In the study by Yuan and collaborators, the authors found the expression of lncRNA-ATB to be induced following TGF-β treatment in HCC cells [154]. Overexpression of wild-type lncRNA-ATB was able to induced EMT and tumor cell invasion and facilitated disseminated tumor cell colonization, thus mimicking the pro-metastatic role of TGF-β. Mechanistically, this lncRNA-ATB was shown to sequester miR-200, a known repressor of EMT and tumor invasion, which can target the 3′UTRs of ZEB1 and ZEB2 [111,155]. In this context, lncRNA-ATB functions as a ceRNA, freeing ZEB1 and ZEB2 mRNAs from miR-200 post-translational inhibition. The resulting increase in ZEB1 and ZEB2 levels trigger the EMT program, ultimately leading to an enhanced invasive potential of HCC cells in vitro and in vivo. The role of lncRNA-ATB in cancer metastasis; however, goes beyond its role in tumor invasion because the same study showed it to be an equally important player in the colonization of the metastatic site. This effect was not dependent on sponging miR-200, but rather it involved additional pathways crucial for the maintenance of the CSC state, including JAK/STAT signaling. By physically interacting with the IL-11 mRNA, lncRNA-ATB could increase its stability, translation, and secretion, all of which activated STAT3 signaling in an autocrine manner. The activation of this autocrine loop is required for enhancing the effect of lncRNA-ATB on HCC cell colonization. Interestingly, in a study by Shibin et al., the authors found lncRNA-ATB expression levels to be significantly downregulated in PDAC tissues versus paired adjacent normal tissues [156]. Additionally, the decrease in expression levels positively correlated with lymphatic metastasis and clinical stage, suggesting that in PDAC, this lncRNA can function in a manner opposite of that described for HCC, suppressing invasion and metastasis. Further studies, however, are still needed to explore the exact mechanism by which lncRNA-ATB functions in PDAC.
An example of lncRNAs found to be both overexpressed in PDAC and involved in EMT are lncRNA plasmacytoma variant translocation 1 gene (lncRNA-PVT1) and lncRNA- taurine up-regulated 1 (lncRNA-TUG1) [157,158]. Both lncRNAs play important roles in regulating PDAC cell proliferation and migration. lncRNA-PVT1 works partially by regulating p21 expression, and in the case of lncRNA-TUG1, this lncRNA is similar to lncRNA-ATB, in that it acts as a ceRNA, sponging miR-382. When TUG1 was overexpressed in PDAC cells, miR-382 was significantly downregulated; however, TUG1 knockdown significantly increased the level of miR-382, impairing PDAC cell migration and the expression of EMT markers. This sponging effect allowed expression of miR-382 target genes, including EZH2, which can promote EMT, invasiveness and metastasis via silencing of the E-cadherin promoter by H3K27 trimethylation [159,160]. As mentioned above, EZH2 is highly expressed in PDAC CSCs and its levels could serve as a readout of CSC activity, thus it would be interesting to see if lncRNA-TUG1 could also serve as a surrogate marker for PDAC CSC activity.
Finally, Arnes et al. [161], recently generated a catalogue of PDAC-associated lncRNAs and showed that lncRNAs can be used to associate patients into two predominant mutant KRAS allele clusters. Interestingly, these clusters correlated with the two prominent subtypes categorized to date for PDAC tumors, the epithelial (Cluster 1) and squamous (Cluster 2) subtypes. In addition, a clear reduction in disease-free survival was observed for those patients from Cluster 2 relative to the other clusters. Of the lncRNAs identified, the authors functionally evaluated FAM83H-AS1 and LINC0067, as both were found in recurrent amplified genomic regions. Interestingly, LINC00673 was shown to be a major regulator of the epithelial state of PDAC cells, as its loss promoted acquisition of mesenchymal markers such as vimentin, loss of epithelial markers and enhanced tumor cell migration both in vitro and in vivo, demonstrating that modulation alone of LINC00673 can affect PDAC cell plasticity, which is a key feature of CSCs. Interestingly, it was recently shown by Zheng et al., that a G>A change in exon 4 of LINC00673 (at rs1111655237) in a percentage of PDAC patients creates a new miR-1231 binding site [162]. This results in LINC00673 sequestration, preventing it from promoting PTPN11/SHP2 phosphatase degradation via ubiquitination. The subsequent stabilization and increase in SHP2 levels is believed to confer susceptibility to tumorigenesis. Indeed, SHP2 is overexpressed in PDAC, and Algül and collaborators recently showed that mutant KRAS is dependent on SHP2 during carcinogenesis, and this interaction can be abrogated with dual SHP2/MEK inhibition [163]. Together, these data show that lncRNAs in PDAC are biologically relevant, their interacting partners can play important roles in tumorigenesis, and targeting lncRNAs or their protein partners can be therapeutically beneficial and could potentially affect PDAC cell plasticity.
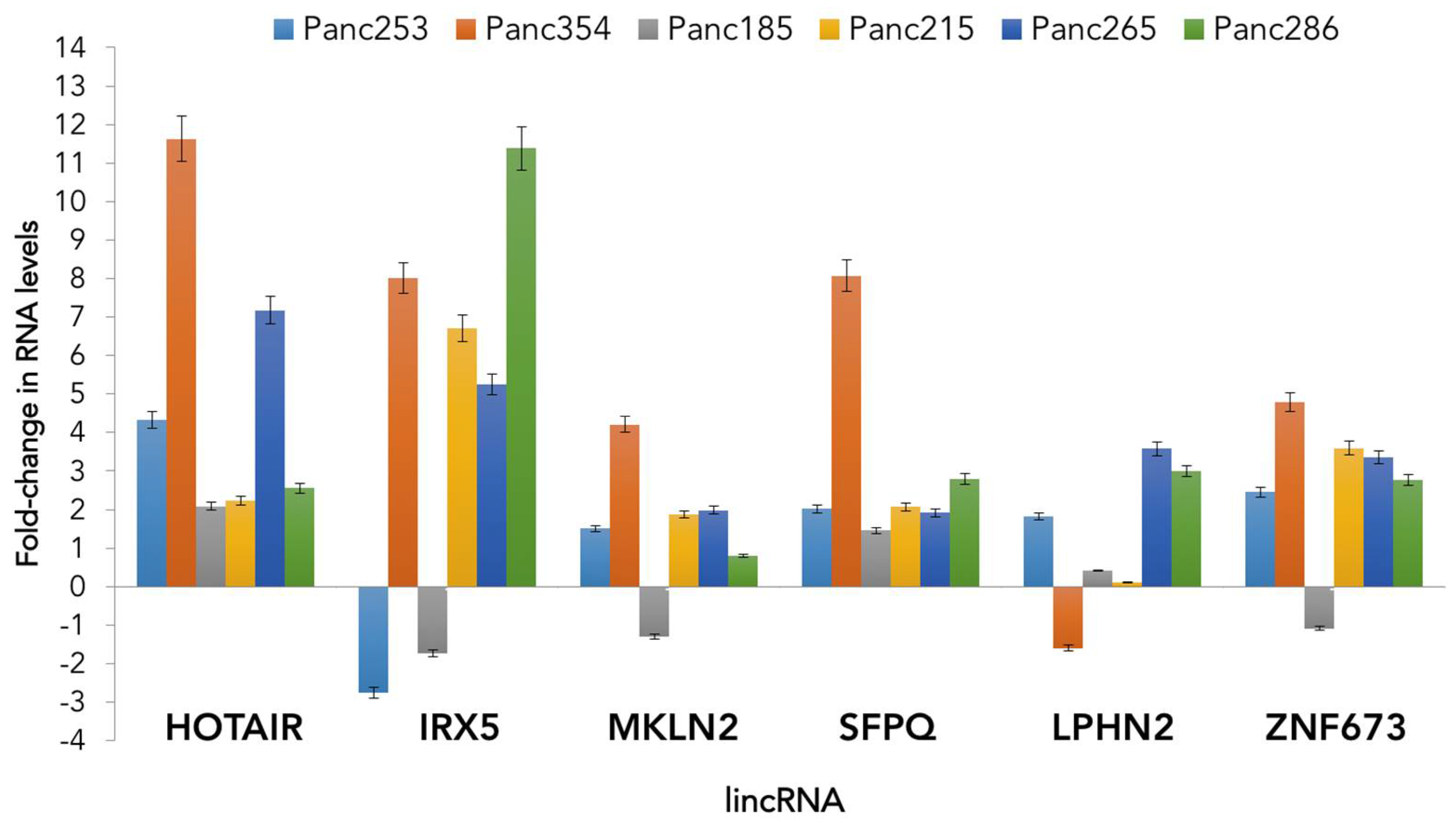
While more research is required to fully understand the role of lncRNAs in tumor biology, CSC biology and in PDAC, we performed an analysis of the expression of several published lincRNAs in CSCs versus non-CSCs derived from several low-passage PDAC patient-derived xenografts. Our initial analyses of the expression of HOTAIR, IRX5, MKLN2, SFPQ, LPHN2 and ZNF673 demonstrate that CSCs generally over-express these lncRNAs compared to their non-CSCs counterparts, although differences were observed across PDAC tumors, indicating patient-to-patient variability and perhaps sub-type-specific differences (Figure 2). While preliminary, these unpublished results demonstrate that lincRNAs are differentially expressed in pancreatic CSCs and their role in CSC stemness may be more important than previously recognized.
4. Epigenetic Therapy in Cancer
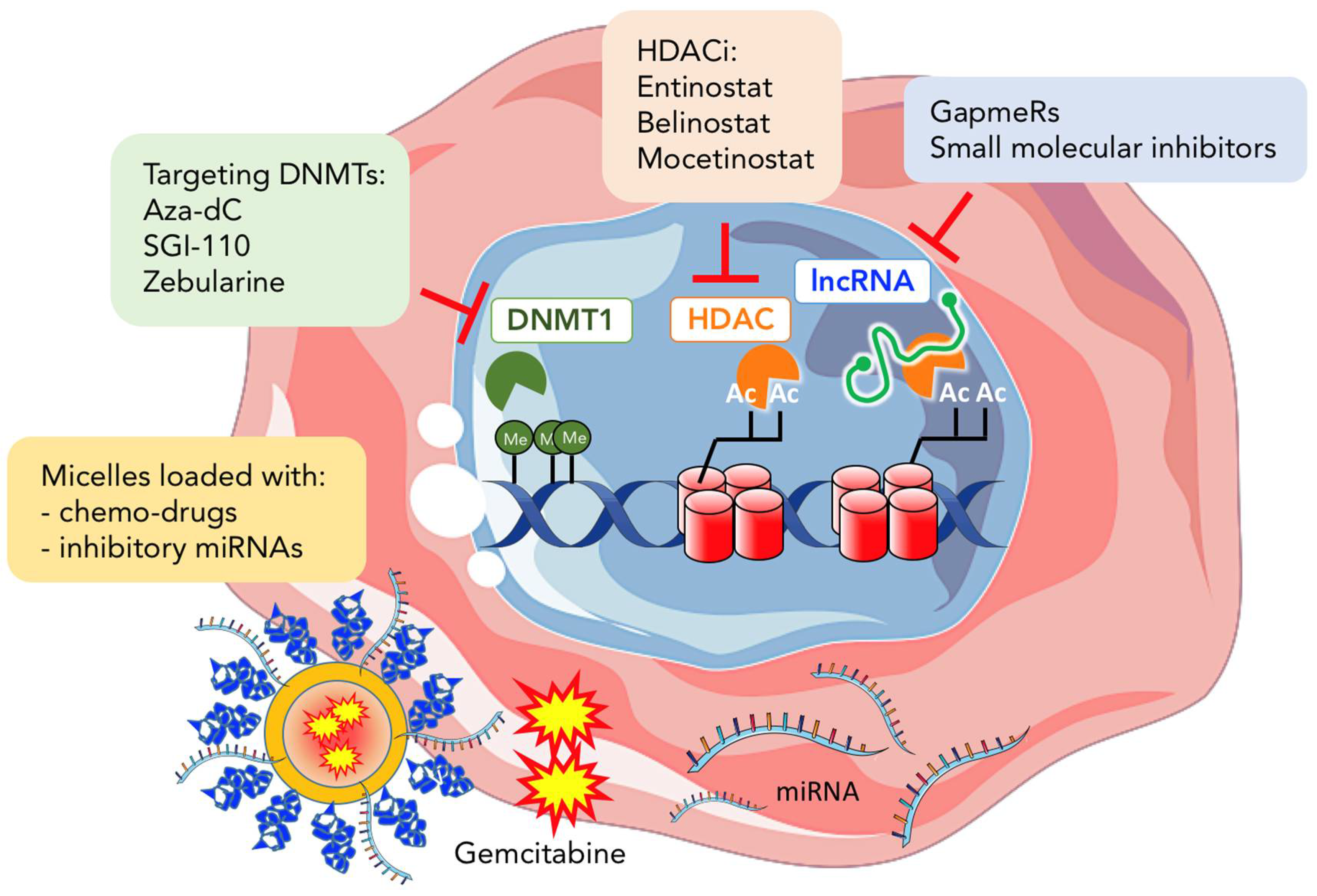
Unlike genetic mutations, which are considered irreversible, epigenetic changes are reversible. Thus, drugs that alter epigenetic modifications or marks could, in theory, restore the epigenetic balance in cancer (Figure 3). Epigenetic therapy therefore aims to reverse the genetic modifications/alterations present in cancer to restore the “normal epigenome landscape”. Among the first epigenetic drugs tested to treat cancer were DNA methylation inhibitors [22]. Azacitidine (5-azacitidine) and Decitabine (5-aza-2′-deoxycytidine), are the two most widely studied DNMT inhibitors that act as analogues of cytosine. During DNA replication, both inhibitors are incorporated into the DNA forming covalent bonds with the DNA methyltransferase DNMT1, preventing its methyltransferase capacity [164,165] and leading to its subsequent degradation [166]. Initially these drugs showed promising results in hematological malignancies, leading to their approval by the US FDA for the treatment of myelodysplastic syndromes; however, their use in solid tumors remains limited due to their high cytotoxicity [167,168,169]. Interestingly, recent studies in leukemia and epithelial tumor cells have demonstrated that low doses of DNMT inhibitors are more effective in maintaining decreased DNA methylation and associated re-expression of silenced genes [170]. Moreover, low doses of DNMT inhibitors also reduced overall tumorigenicity and the percentage of CSCs within the tumor. Nalls et al. showed that pancreatic CSCs are sensitive to Decitabine. Treatment with Decitabine strongly inhibited cell proliferation, self-renewal, invasion, and induced apoptosis of CSCs through re-expression of the tumor suppressor miRNA-34 [122]. In addition to Decitabine-mediated induced apoptosis, Decitabine can also induce differentiation of PDAC progenitor cell lines, including MIA PaCa-2 [171] and Panc-1 [172]. Indeed, drug-triggered differentiation is an attractive approach in epigenetic therapy and has long been considered a strategy for targeting CSCs. For example, in prostate cancer, CSCs treated with the Decitabine showed decreased expression of the pluripotency-associated genes OCT4 and NANOG and increased expression of the differentiation markers CK5, CK8, Nkx3.1, and PSA/PSP94. Re-activation of androgen receptor, found to be methylated in prostate CSCs, was determined to be the mechanism behind differentiation induction mediated by Decitabine. Decitabine-induced differentiation led to a significant decrease in the self-renewal and tumorigenic capacity of the treated prostate CSCs. In addition, low doses of a newer DNMT1 inhibitor, SGI-110, was recently shown to reprogram ovarian CSCs to a more differentiated non-CSC state [173]. Lastly, we have also shown that the less toxic DNMT1 inhibitor Zebularine can potently abrogate the in vivo and in vitro pluripotency and tumorigenicity of PDAC CSCs by promoting differentiation of CSCs toward less tumorigenic non-CSCs [45]. Since current available chemotherapeutics can effectively target fast proliferating bulk tumor cells, leaving slow-cycling CSCs unaffected, therapies based on DNMT inhibitors that push CSCs to a more differentiated state could offer promising alternatives to force CSCs into a state where they are more permissive and sensitive to standard therapies.
HDAC inhibitors (HDACIs) are another group of epigenetic drugs that can act on both CSCs and bulk non-CSCs. HDACIs can induce CSC differentiation, inhibit their self-renewal capacity, reverse their chemo/radiotherapy resistance, or promote their death. For example, it was recently shown in triple-negative breast cancer that the selective inhibitor of class I HDACs, Entinostat, can decrease the CSC population and abrogate their EMT phenotype [174]. In pancreatic cancer, Trichostatin A (TSA) can strongly inhibit the growth of pancreatic cancer cell lines via cell-cycle arrest at the G2 phase and induced-apoptotic cell death [175]. Chien et al. showed that Belinostat, another HDACI, can suppress human PDAC cells via multiple pathways [176]. Specifically, Belinostat treatment decreased PDAC cell growth and increased apoptosis, which was associated with blocking the AKT/mTOR pathway. Interestingly, Belinostat also blocked hypoxic growth-related signals. Alone or in combination with Gemcitabine, a significant decrease in the size of human pancreatic tumor growth was observed in immunodeficient mice. Furthermore, treatment of the pancreatic cancer cell line Panc-1 with Mocetinostat reduced ZEB1 expression, causing up regulation of the stemness inhibiting miRNA-203 molecule, resulting in reduced expression of CSC-associated markers, such as CD133, as well as the sphere-forming capacity and sensitization of undifferentiated ZEB1-expressing cells to Gemcitabine [177]. Thus, all these studies together suggest that targeting histone deacetylases could be a very promising strategy for treating PDAC, and while HDACIs clearly affect PDAC cells in preclinical settings, HDACIs have yet to show relevant antitumor activity in clinical studies in PDAC patients. This certainly reflects the very poor state of PDAC patients at the time of diagnosis, when tumors in most patients have already locally advanced and spread to distant organs, making treatment with HDACIs relatively ineffective. It remains to be determined if HDACIs would be more effective if initiated at early times. In addition, the complexity of the actions of HDACs in PDAC and in PDAC CSCs may be particularly unique, limiting even further the efficacy of HDACIs. Interestingly, a 2012 study by Woodward WA and colleagues showed that the HDACIs Suberoylanilide Hydroxamic Acid (SAHA) or Valproic acid, could induce the dedifferentiation of ALDH1-negative non-CSCs resulting in the de novo production of quiescent ALDH1-positive CSCs [178]. While potentially alarming, the same group would later show that dedifferentiation of cancer cells to CSCs was concomitant with metabolic reprograming, such as upregulation of G6PD, a rate-limiting enzyme in the pentose phosphate pathway. The authors exploited this metabolic reprogramming induced by HDACIs by using two G6PD inhibitors, 6-aminonicotinamide and dehydroepiandrosterone, to successfully target Valproic acid-induced ALDH-positive cells, decreasing sphere formation efficiency and ALDH activity [179]. Thus, these studies highlight that HDACIs could have cell population specific effects and could potentially reprogram non-CSCs into CSCs. Therefore, more comprehensive analyses of the effect(s) of HDACIs on CSCs and on different sub-populations of CSCs is necessary before HDACIs are considered as possible treatment options for cancer patients, including pancreatic cancer patients. A better understanding of the exact mechanism(s) of their action and their related effects on non-CSCs will help predict treatment outcome and potential harmful side effects.
As a class of molecules, miRNAs hold particular therapeutic potential because they can regulate various gene targets belonging to a specific pathway, or they can modulate several target genes across several independent pathways [180,181]. Over the past years, the delivery method for miRNAs has been substantially improved allowing for the efficient introduction of miRNAs into pancreatic cancer cells and subsequent suppression of these cells in animal models. For example, liposomal nanoparticles carrying miR-34a were able to significantly inhibit the growth of orthotopic xenografts models where reduction of CSC markers was also observed [182]. miR-145-loaded magnetic nanoparticle formulations (miR-145-MNPF) have also been successfully delivered to PDAC cells, demonstrating a functional inhibitory effect on growth, invasion and motility via inhibition of MUC13-associated oncogenic protein, HER2, pAKTSer473, and restoring p53 levels [183]. Furthermore, co-administration of antisense oligonucleotides (ASOs) of miR-21 and miR-221 reduced primary tumor growth and metastasis of pancreatic cancer [184]. Other studies have revealed that co-administration of miRNA therapeutics and anti-cancer drugs can improve response and/or overcome chemoresistance. The co-delivery of ASO-miR-21 and Gemcitabine induced more cell apoptosis, and the administration of miR-205 and Gemcitabine enhanced chemosensitivity of Gemcitabine-resistant pancreatic cell lines [185,186]. In addition, micelles encapsulating the Hedgehog inhibitor GDC-044 and complexed with let-7b effectively inhibited pancreatic tumor growth in vivo, by decreasing tumor cell proliferation and promoting apoptosis [187].
Similar to miRNAs, lncRNAs are another attractive therapeutic target considering their tightly controlled transcriptional regulation, tissue-specific expression and frequent dysregulation in disease. One of the first evidences that lncRNAs could have therapeutic value came from studies performed in bladder cancer. Intratumoral injection of plasmids encoding for the A subunit of diphtheria toxin, under the control of lncRNA H19 regulatory sequences, led to tumor reduction in bladder cancer xenograft mouse models [188]. Following this work, several phase I/II clinical trials were initiated in different cancer types, including one in PDAC, showing that local administration of BC-819, in combination with systemic chemotherapy, may prove therapeutically beneficial for the treatment of this disease [189]. Although lncRNAs represent appealing pharmacological targets, their inhibition in vivo remains a challenge. Many limitations still need to be overcome, such as their extensive secondary structure hindering the design of effective small molecule inhibitors, in vivo toxicity and efficient intratumor delivery. Nonetheless, one amenable approach could be the use of small molecules that disrupt the interactions between lncRNAs and proteins to alter their steady state levels in target cells. It is well accepted that lncRNAs can bind EZH2 [190] or β-catenin [191], and small molecules could be developed to block these interactions and prevent lncRNA/protein binding. Similar approaches have been used to disrupt the miRNA processing machinery [192,193]. Taken together, these delivery efforts, along with further elucidation of lncRNA regulatory mechanisms, will ultimately lead to the development of effective therapeutic strategies that target lncRNAs in vivo.
5. Concluding Remarks
In the past, it has been common to define CSCs as cells with the “fixed” ability to constantly “maintain” their so-called “CSC-phenotype”; however, several recent studies have challenged this view by demonstrating a very dynamic conversion between cancer cell populations (non-CSCs) through trans-differentiation and reprograming events. These trans-conversion events likely occur when CSCs are directly targeted and the tumor (including the tumor microenvironment) senses the loss of the CSC pool. For example, in glioblastoma it has been shown that therapeutic doses of Temozolomide (TMZ) can increase the glioblastoma stem cell (GSC) pool, and the increase was not due to an enrichment in GSCs due to the elimination of non-GSCs, but rather due to the capacity of non-GSC cells to “phenotypically shift” to a GSC-like state, resulting in de novo GSC-like cells that expressed stem and pluripotency markers, including CD133, NESTIN, SOX2 and OCT4 [194]. Moreover, these new GSCs served as a reservoir for initiating tumor relapse. A similar effect was observed in breast cancer cells following treatment with the HDACIs SAHA and Valproic acid, as mentioned above. Moreover, two recent studies targeting LGR5+ colon cancer cells in vivo have shown that non-CSCs can be a pulled out of the tumor bulk population to replenish the CSC compartment when CSCs are eliminated [20,21]. Both studies highlight that tumors can be maintained by proliferative non-CSCs that respond to the loss of the CSC pool, leading to rapid tumor re-growth when treatments targeting the CSCs are stopped. Thus, stemness is likely not a hardwired or fixed trait as previously believed but rather a dynamic property fueled by the heterogeneity of the tumor and the inherent and perhaps unappreciated cellular plasticity of non-CSCs. From a therapeutic perspective, this implies that merely targeting the CSC will not be curative as non-CSCs can quickly replenish the CSC pool. The dynamic nature of stemness implies that this process must be fast enough to allow for rapid acquisition of specific CSC traits, such as aggressiveness, ability to metastasize, drug resistance, etc.—all of which are important for tumor growth and relapse. Many stem cell-related genes are found in a bivalent state, with activating and repressing histone marks which can allow for rapid activation or inhibition. Consequently, it is very probable that epigenetic changes and modifications are the determining factor in this fast and rapid plastic process. Thus, in the new age of the CSC concept and with our ever-growing understanding of cellular plasticity, it is even more important that we dissect the mechanism by which epigenetic modifications play a role in trans-conversion/plasticity to make future anti-CSC strategies more effective. We envision the future of cancer therapies consisting of inhibitors that target highly proliferating cells, active CSCs, quiescent CSC and the epigenetic mechanisms regulating cancer cell plasticity.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Pogribny, I.P. Epigenetic events in tumorigenesis: Putting the pieces together. Exp. Oncol. 2010, 32, 132–136. [Google Scholar] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Looking ahead in cancer stem cell research. Nat. Biotechnol. 2009, 27, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Khiabanian, H.; Rossi, D.; Fabbri, G.; Gattei, V.; Forconi, F.; Laurenti, L.; Marasca, R.; Del Poeta, G.; Foa, R.; et al. Tumor evolutionary directed graphs and the history of chronic lymphocytic leukemia. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michor, F.; Polyak, K. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. 2010, 3, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.C.; Hollingsworth, R.E.; Hurt, E.M. Cancer stem cell plasticity and tumor hierarchy. World J. Stem. Cells 2015, 7, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furth, J. Transmission of myeloid leukemia of mice: Its relation to myeloma. J. Exp. Med. 1935, 61, 423–446. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Hadnagy, A.; Gaboury, L.; Beaulieu, R.; Balicki, D. Sp analysis may be used to identify cancer stem cell populations. Exp. Cell Res. 2006, 312, 3701–3710. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, H.; Ouzounova, M.; Le Calvez-Kelm, F.; Lambert, M.P.; McKay-Chopin, S.; Tavtigian, S.V.; Puisieux, A.; Matar, C.; Herceg, Z. Methylome analysis reveals jak-stat pathway deregulation in putative breast cancer stem cells. Epigenetics 2011, 6, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Mani, S.; Cros, M.P.; Scoazec, J.Y.; Chemin, I.; Hainaut, P.; Herceg, Z. Epigenetic silencing of SFRP1 activates the canonical WNT pathway and contributes to increased cell growth and proliferation in hepatocellular carcinoma. Tumour Biol. 2012, 33, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Schwarzenbacher, D.; Stanzer, S.; Heitzer, E.; Auer, M.; Geigl, J.B.; Cote, R.J.; Datar, R.H.; Dandachi, N. Genetic and epigenetic analysis of putative breast cancer stem cell models. BMC Cancer 2013, 13, 358. [Google Scholar] [CrossRef] [PubMed]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for LGR5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of cpg and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNA(AsP) by the DNA methyltransferase homolog DNMT2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Kawasaki, K.; Scott, H.S.; Ollila, J.; Vihinen, M.; Heino, M.; Shintani, A.; Kawasaki, K.; Minoshima, S.; Krohn, K.; et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000, 65, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Gopisetty, G.; Xu, J.; Sampath, D.; Colman, H.; Puduvalli, V.K. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by SP1/MYC and promoter methylation. Oncogene 2013, 32, 3119–3129. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kagara, N.; Huynh, K.T.; Kuo, C.; Okano, H.; Sim, M.S.; Elashoff, D.; Chong, K.; Giuliano, A.E.; Hoon, D.S. Epigenetic regulation of cancer stem cell genes in triple-negative breast cancer. Am. J. Pathol. 2012, 181, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Tsai, H.C.; Glockner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the nf-kappab signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Sinha, A.U.; Zhu, N.; Li, M.; Armstrong, S.A.; Orkin, S.H. Haploinsufficiency of DNMT1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012, 26, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 6910. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lin, J.H.; Hsu, T.W.; Su, K.; Li, A.F.; Hsu, H.S.; Hung, S.C. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int. J. Cancer 2015, 136, 547–559. [Google Scholar] [PubMed]
- Zagorac, S.; Alcala, S.; Fernandez Bayon, G.; Bou Kheir, T.; Schoenhals, M.; Gonzalez-Neira, A.; Fernandez Fraga, M.; Aicher, A.; Heeschen, C.; Sainz, B., Jr. Dnmt1 inhibition reprograms pancreatic cancer stem cells via upregulation of the miR-17-92 cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of tet proteins in 5MC to 5HMC conversion, es-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of TET3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Im, K.M.; Misner, K.J.; Tan, W.; Lou, H.; Gold, B.; Wells, D.W.; Bravo, H.C.; Fredrikson, K.M.; Harkins, T.T.; et al. Somatic alterations contributing to metastasis of a castration-resistant prostate cancer. Hum. Mutat. 2013, 34, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, L.; Mao, S.Q.; Li, Z.; Chen, J.; Zhang, R.R.; Wu, H.P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and tdg mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Yu, Y.; Suzuki, M.; Campbell, N.; Mazdo, J.; Vasanthakumar, A.; Bhagat, T.D.; Nischal, S.; Christopeit, M.; Parekh, S.; et al. Genome-wide hydroxymethylation tested using the help-gt assay shows redistribution in cancer. Nucl. Acids Res. 2013, 41, e157. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B., Jr.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. Gata6 regulates emt and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2016, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; McHale, C.M.; Skibola, C.F.; Smith, A.H.; Smith, M.T.; Zhang, L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect. 2011, 119, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Snapshot: Histone-modifying enzymes. Cell 2007, 128, 802. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell. Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Olguin, P.; Recillas-Targa, F. Chromatin structure of pluripotent stem cells and induced pluripotent stem cells. Brief. Funct. Genom. 2011, 10, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, J.; Estecio, M.R.; Lu, Y.; Long, H.; Malouf, G.G.; Graber, D.; Huo, Y.; Ramagli, L.; Liang, S.; Kornblau, S.M.; et al. The epigenome of aml stem and progenitor cells. Epigenetics 2013, 8, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Lee, H.; Yoon, J.G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toll, A.D.; Dasgupta, A.; Potoczek, M.; Yeo, C.J.; Kleer, C.G.; Brody, J.R.; Witkiewicz, A.K. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum. Pathol. 2010, 41, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, D.; Yin Li, W.; Man Cheung, C.; Yao, H.; Chan, C.Y.; Chan, C.Y.; Xu, F.P.; Liu, Y.H.; Sung, J.J.; et al. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett. 2010, 297, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. Ezh2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. Utx and JMJD3 are histone H3K27 demethylases involved in hox gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Bayliss, P.E.; Rinn, J.L.; Whetstine, J.R.; Wang, J.K.; Chen, S.; Iwase, S.; Alpatov, R.; Issaeva, I.; Canaani, E.; et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007, 449, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Welstead, G.G.; Creyghton, M.P.; Bilodeau, S.; Cheng, A.W.; Markoulaki, S.; Young, R.A.; Jaenisch, R. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc. Natl. Acad. Sci. USA 2012, 109, 13004–13009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. Ptip associates with mll3- and mll4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 branches of the compass family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by polycomb and compass families. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. Utx and uty demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Starmer, J.; Yee, D.; Pohlers, M.; Magnuson, T. Kdm6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014, 10, e1004507. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Sphyris, N.; Johnson, K.S.; Reisenauer, K.N.; Nesbit, T.A.; Joseph, R.; Vijay, G.V.; Sarkar, T.R.; Bhangre, N.A.; Song, J.J.; et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget 2017, 8, 65548–65565. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Rack, B.; Schindlbeck, C.; Juckstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. Emt and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. Gencode: The reference human genome annotation for the encode project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Wilk, K.; Vargas, D.; Shirato, Y.; Bilinski, S.; Etkin, L.D. Potential structural role of non-coding and coding RNAs in the organization of the cytoskeleton at the vegetal cortex of xenopus oocytes. Development 2005, 132, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Deniz, E.; Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 2017, 17, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, K. Long noncoding RNAs: Pivotal regulators in acute myeloid leukemia. Exp. Hematol. Oncol. 2015, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med. 2012, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying miRNAs, targets and functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Odenthal, M.; Fries, J.W. Exosomes as miRNA carriers: Formation-function-future. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Dittmar, R.L.; Sen, S. Secretory microRNAs as biomarkers of cancer. Semin. Cell Dev. Biol. 2018, 78, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Nouraee, N.; Mowla, S.J. MiRNA therapeutics in cardiovascular diseases: Promises and problems. Front. Genet. 2015, 6, 232. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Zhao, B.; Zhao, J.; Li, S. Potential roles of exosomal microRNAs as diagnostic biomarkers and therapeutic application in alzheimer’s disease. Neural Plast. 2017, 2017, 7027380. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.Y.; Choi, Y.H.; Hwang, S.; Jeong, J.Y.; An, H.J. Clinical impact of microRNAs associated with cancer stem cells as a prognostic factor in ovarian carcinoma. J. Cancer 2017, 8, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Mukohyama, J.; Shimono, Y.; Minami, H.; Kakeji, Y.; Suzuki, A. Roles of microRNAs and RNA-binding proteins in the regulation of colorectal cancer stem cells. Cancers 2017, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Salvador, M.A.; Birnbaum, D.; Charafe-Jauffret, E.; Ginestier, C. Breast cancer stem cells programs: Enter the (non)-code. Brief. Funct. Genom. 2016, 15, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Moriya, C.; Igarashi, H.; Saitoh, A.; Yamamoto, H.; Adachi, Y.; Imai, K. Cancer stem cells in human gastrointestinal cancer. Cancer Sci. 2016, 107, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y.; Miranda-Lorenzo, I.; Lonardo, E.; Dorado, J.; Reis Vieira, C.; Ramirez, J.C.; Hidalgo, M.; Aicher, A.; et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut 2015, 64, 1936–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A. Role of stem cell derived exosomes in tumor biology. Int. J. Cancer 2018, 142, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Ru, P.; Steele, R.; Newhall, P.; Phillips, N.J.; Toth, K.; Ray, R.B. MiRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol. Cancer Ther. 2012, 11, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Liu, J.; Tu, X.; Zang, Y.; Zhu, J.; Chen, J.; Dong, L.; Zhang, J. miR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting snail1. Biochem. Biophys. Res. Commun. 2012, 417, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Giannoni, E.; Casamichele, A.; Taddei, M.L.; Callari, M.; Piovan, C.; Valdagni, R.; Pierotti, M.A.; Zaffaroni, N.; Chiarugi, P. miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antioxid. Redox Signal. 2014, 20, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Fu, X.; Chen, X.; Zeng, S.; Tian, Y.; Jove, R.; Xu, R.; Huang, W. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology 2010, 52, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, G.; Saumet, A.; Moes, M.; Vallar, L.; Le Bechec, A.; Laurini, C.; Sabbah, M.; Arar, K.; Theillet, C.; Lecellier, C.H.; et al. miR-661 expression in SNAI1-induced epithelial to mesenchymal transition contributes to breast cancer cell invasion by targeting Nectin-1 and StarD10 messengers. Oncogene 2010, 29, 4436–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Fan, J.; Ding, X.; Peng, W.; Yu, X.; Chen, Y.; Nie, J. TGF-{β}-induced miR-491-5p expression promotes par-3 degradation in rat proximal tubular epithelial cells. J. Biol. Chem. 2010, 285, 40019–40027. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Satoh, K.; Miura, S.; Hirota, M.; Kanno, A.; Masamune, A.; Kikuta, K.; Kume, K.; Unno, J.; Egawa, S.; et al. miR-197 induces epithelial-mesenchymal transition in pancreatic cancer cells by targeting p120 catenin. J. Cell Physiol. 2013, 228, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34A for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE 2009, 4, e6816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.G.; Yu, S.N.; Lu, Z.H.; Ma, Y.H.; Gu, Y.M.; Chen, J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting kras. Carcinogenesis 2010, 31, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lu, Z.; Liu, C.; Meng, Y.; Ma, Y.; Zhao, W.; Liu, J.; Yu, J.; Chen, J. miRNA-96 suppresses kras and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010, 70, 6015–6025. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.R.; Frampton, A.E.; Jacob, J.; Pellegrino, L.; Krell, J.; Giamas, G.; Tsim, N.; Vlavianos, P.; Cohen, P.; Ahmad, R.; et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS ONE 2012, 7, e32068. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Cimmino, A.; Matarazzo, M.R.; Casalino, L.; De Vita, G.; D’Esposito, M.; Di Lauro, R.; Verde, P. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in ras transformation. Oncogene 2009, 28, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Hong, S.M.; Karikari, C.A.; Hruban, R.H.; Goggins, M.G.; Maitra, A. Aberrant microRNA-155 expression is an early event in the multistep progression of pancreatic adenocarcinoma. Pancreatology 2010, 10, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, A.; Hong, S.M.; Hruban, R.H.; Goggins, M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin. Cancer Res. 2012, 18, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Z.; Li, K.; Gong, L.; Yang, Q.; Huang, X.; Hong, C.; Ding, M.; Yang, H. Linc-DYNC2H1-4 promotes emt and csc phenotypes by acting as a sponge of miR-145 in pancreatic cancer cells. Cell Death Dis. 2017, 8, e2924. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.K.; Mondal, G.; Kumar, V.; Kattel, K.; Mahato, R.I. Chemosensitization and inhibition of pancreatic cancer stem cell proliferation by overexpression of microRNA-205. Cancer Lett. 2017, 402, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.; Raza, U.; Saatci, O.; Eyupoglu, E.; Yurdusev, E.; Sahin, O. miR-200C: A versatile watchdog in cancer progression, emt, and drug resistance. J. Mol. Med. 2016, 94, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Eguchi, H.; Nagano, H.; Konno, M.; Tomimaru, Y.; Wada, H.; Hama, N.; Kawamoto, K.; Kobayashi, S.; Nishida, N.; et al. MicroRNA-1246 expression associated with CCNG2-mediated chemoresistance and stemness in pancreatic cancer. Br. J. Cancer 2014, 111, 1572–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Chitkara, D.; Kumar, V.; Behrman, S.W.; Mahato, R.I. MiRNA profiling in pancreatic cancer and restoration of chemosensitivity. Cancer Lett. 2013, 334, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, L.; Yu, B.; Zhuang, Q.Y.; Wang, Y.P. Expression signatures of long noncoding RNAs in adolescent idiopathic scoliosis. Biomed. Res. Int. 2015, 2015, 276049. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.D.; Chen, W.M.; Qi, F.Z.; Xia, R.; Sun, M.; Xu, T.P.; Yin, L.; Zhang, E.B.; De, W.; Shu, Y.Q. Long non-coding RNA anril is upregulated in hepatocellular carcinoma and regulates cell proliferation by epigenetic silencing of KLF2. J Hematol. Oncol. 2015, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Qu, X.; Li, W.; Zhong, X.; Li, P.; Yang, S.; Chen, X.; Shao, M.; Zhang, L. The long non-coding RNA, GAS5, enhances gefitinib-induced cell death in innate egfr tyrosine kinase inhibitor-resistant lung adenocarcinoma cells with wide-type egfr via downregulation of the IGF-1r expression. J. Hematol. Oncol. 2015, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.P.; Huang, M.D.; Xia, R.; Liu, X.X.; Sun, M.; Yin, L.; Chen, W.M.; Han, L.; Zhang, E.B.; Kong, R.; et al. Decreased expression of the long non-coding RNA fendrr is associated with poor prognosis in gastric cancer and fendrr regulates gastric cancer cell metastasis by affecting FIBRONECTIN1 expression. J. Hematol. Oncol. 2014, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Tian, J.; Tang, X.; Ma, J.; Wang, S. Long non-coding RNAs in the regulation of myeloid cells. J. Hematol. Oncol. 2016, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Peng, G. Non-coding RNAs: An emerging player in DNA damage response. Mutat. Res. Rev. Mutat. Res. 2015, 763, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Liao, Y.W.; Chen, P.Y.; Hsieh, P.L.; Fang, C.Y.; Wu, C.Y.; Yen, M.L.; Peng, B.Y.; Wang, D.P.; Cheng, H.C.; et al. Targeting lncRNA hotair suppresses cancer stemness and metastasis in oral carcinomas stem cells through modulation of emt. Oncotarget 2017, 8, 98542–98552. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lv, Y.; Jin, F.; Liu, Y.; Ma, Y.; Xiong, Y.; Liu, L.; Zhang, S.; Sun, Y.; Tipoe, G.L.; et al. LncRNA hanr promotes tumorigenesis and increase of chemoresistance in hepatocellular carcinoma. Cell Physiol. Biochem. 2017, 43, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Hu, H.; Han, T.; Yuan, C.; Wang, L.; Jin, Z.; Guo, Z.; Wang, L. Long noncoding RNA MALAT-1 enhances stem cell-like phenotypes in pancreatic cancer cells. Int. J. Mol. Sci. 2015, 16, 6677–6693. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Maeda, S.; Liu, C.; Karin, M.; Edgington, T.S. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 2007, 26, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, M.; Tian, J.; Wang, X.; Li, Z. Malat-1: A long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011, 39, 169–175. [Google Scholar] [PubMed]
- Wu, M.; Lin, Z.; Li, X.; Xin, X.; An, J.; Zheng, Q.; Yang, Y.; Lu, D. Hulc cooperates with MALAT1 to aggravate liver cancer stem cells growth through telomere repeat-binding factor 2. Sci. Rep. 2016, 6, 36045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.T.; Shi, D.B.; Wang, Y.W.; Li, X.X.; Xu, Y.; Tripathi, P.; Gu, W.L.; Cai, G.X.; Cai, S.J. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3174–3181. [Google Scholar] [PubMed]
- Han, Y.; Zhou, L.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Zhou, Y.; Du, Z. Downregulation of lncRNA-MALAT1 affects proliferation and the expression of stemness markers in glioma stem cell line SHG139s. Cell Mol. Neurobiol. 2016, 36, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. lncRNA hottip modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by tgf-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Yang, X.; Song, W.; Sun, W.; Li, X.; Wang, J.; Zhong, Y.; Shang, R.; Ruan, B.; Zhang, Z.; et al. Downregulation of lncRNA-ATB correlates with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2016, 37, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.Q.; Jiang, Y.; Zhu, F.; Sun, D.L.; He, X.Z. Long noncoding RNA PVT1 promotes emt and cell proliferation and migration through downregulating p21 in pancreatic cancer cells. Technol. Cancer Res. Treat. 2017, 16, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, H.; Kong, H.; Chen, Z.; Chen, B.; Zhou, M. The lncRNA-TUG1/EZH2 axis promotes pancreatic cancer cell proliferation, migration and emt phenotype formation through sponging miR-382. Cell Physiol. Biochem. 2017, 42, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Zhao, J.; Vieth, E.; Nephew, K.P.; Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 2016, 7, 84453–84467. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of e-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Arnes, L.; Liu, Z.; Wang, J.; Carlo Maurer, H.; Sagalovskiy, I.; Sanchez-Martin, M.; Bommakanti, N.; Garofalo, D.C.; Balderes, D.A.; Sussel, L.; et al. Comprehensive characterisation of compartment-specific long non-coding RNAs associated with pancreatic ductal adenocarcinoma. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Huang, X.; Tan, W.; Yu, D.; Du, Z.; Chang, J.; Wei, L.; Han, Y.; Wang, C.; Che, X.; et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016, 48, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-AZA-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-AZA-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the ken box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Abele, R.; Clavel, M.; Dodion, P.; Bruntsch, U.; Gundersen, S.; Smyth, J.; Renard, J.; van Glabbeke, M.; Pinedo, H.M. The eortc early clinical trials cooperative group experience with 5-AZA-2′-deoxycytidine (NSC 127716) in patients with colo-rectal, head and neck, renal carcinomas and malignant melanomas. Eur. J. Cancer Clin. Oncol. 1987, 23, 1921–1924. [Google Scholar] [CrossRef]
- Clavel, M.; Monfardini, S.; Fossa, S.; Smyth, J.; Renard, J.; Kaye, S.B. 5-AZA-2′-deoxycytidine (NSC 127716) in non-seminomatous testicular cancer. Phase ii from the eortc early clinical trials cooperative group and genito-urinary group. Ann. Oncol. 1992, 3, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A phase ii study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer 2006, 5, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ohwada, S.; Saitoh, F.; Adachi, M.; Morishita, Y.; Hozumi, M. Induction of ley antigen by 5-AZA-2′-deoxycytidine in association with differentiation and apoptosis in human pancreatic cancer cells. Anticancer Res. 1996, 16, 735–740. [Google Scholar] [PubMed]
- Lefebvre, B.; Belaich, S.; Longue, J.; Vandewalle, B.; Oberholzer, J.; Gmyr, V.; Pattou, F.; Kerr-Conte, J. 5′-AZA induces NGN3 expression and endocrine differentiation in the PANC-1 human ductal cell line. Biochem. Biophys. Res. Commun. 2010, 391, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef] [PubMed]
- Schech, A.; Kazi, A.; Yu, S.; Shah, P.; Sabnis, G. Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol. Cancer Ther. 2015, 14, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Morales, P.; Gomez-Martinez, A.; Carrato, A.; Martinez-Lacaci, I.; Barbera, V.M.; Soto, J.L.; Carrasco-Garcia, E.; Menendez-Gutierrez, M.P.; Castro-Galache, M.D.; Ferragut, J.A.; et al. Histone deacetylase inhibitors induced caspase-independent apoptosis in human pancreatic adenocarcinoma cell lines. Mol. Cancer Ther. 2005, 4, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, W.; Lee, D.H.; Zheng, Y.; Wuensche, P.; Alvarez, R.; Wen, D.L.; Aribi, A.M.; Thean, S.M.; Doan, N.B.; Said, J.W.; et al. Growth inhibition of pancreatic cancer cells by histone deacetylase inhibitor belinostat through suppression of multiple pathways including HIF, NFkB, and mTOR signaling in vitro and in vivo. Mol. Carcinog. 2014, 53, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through wnt/beta-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Debeb, B.G.; Lacerda, L.; Larson, R.; Wolfe, A.R.; Krishnamurthy, S.; Reuben, J.M.; Ueno, N.T.; Gilcrease, M.; Woodward, W.A. Histone deacetylase inhibitor-induced cancer stem cells exhibit high pentose phosphate pathway metabolism. Oncotarget 2016, 7, 28329–28339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs-microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R776. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Setua, S.; Khan, S.; Yallapu, M.M.; Behrman, S.W.; Sikander, M.; Khan, S.S.; Jaggi, M.; Chauhan, S.C. Restitution of tumor suppressor microRNA-145 using magnetic nanoformulation for pancreatic cancer therapy. J. Gastrointest. Surg. 2017, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, L.; Ischenko, I.; Bao, Q.; Schwarz, B.; Niess, H.; Wang, Y.; Renner, A.; Mysliwietz, J.; Jauch, K.W.; et al. Antisense inhibition of microRNA-21 and microRNA-221 in tumor-initiating stem-like cells modulates tumorigenesis, metastasis, and chemotherapy resistance in pancreatic cancer. Target. Oncol. 2015, 10, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Chitkara, D.; Behrman, S.W.; Mahato, R.I. Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials 2014, 35, 7077–7087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Li, J.; Zhang, Z.; Huang, C.; Lian, G.; Yang, K.; Chen, S.; Lin, Y.; Wang, L.; et al. Co-delivery of microRNA-21 antisense oligonucleotides and gemcitabine using nanomedicine for pancreatic cancer therapy. Cancer Sci. 2017, 108, 1493–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Mondal, G.; Slavik, P.; Rachagani, S.; Batra, S.K.; Mahato, R.I. Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol. Pharm. 2015, 12, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, M.C.; Davies, B.J. BC-819, a plasmid comprising the H19 gene regulatory sequences and diphtheria toxin a, for the potential targeted therapy of cancers. Curr. Opin. Mol. Ther. 2010, 12, 607–616. [Google Scholar] [PubMed]
- Hanna, N.; Ohana, P.; Konikoff, F.M.; Leichtmann, G.; Hubert, A.; Appelbaum, L.; Kopelman, Y.; Czerniak, A.; Hochberg, A. Phase 1/2A, dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene. Ther. 2012, 19, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Bonasio, R.; Saldana-Meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 2014, 53, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, N.; Ohira, T.; Kataoka, M.; Inaoka, D.; Tanabe, H.; Nakayama, Y.; Oshimura, M.; Kugoh, H. Regulation of functional KCNQ1OT1 lncRNA by β-catenin. Sci. Rep. 2016, 6, 20690. [Google Scholar] [CrossRef] [PubMed]
- Paris, O.; Ferraro, L.; Grober, O.M.; Ravo, M.; De Filippo, M.R.; Giurato, G.; Nassa, G.; Tarallo, R.; Cantarella, C.; Rizzo, F.; et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor β in hormone-responsive breast cancer. Oncogene 2012, 31, 4196–4206. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, Y. Small molecule targeting miR-34A for cancer therapy. Mol. Cell Oncol. 2015, 2, e977160. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Epigenetic regulation of Cancer Stem Cells. Graphical summary of the epigenetic mechanisms regulating CSCs. DNA methylation, histone modifications and non-coding RNA molecules (lncRNAs and miRNAs) work together to modify CSC biology and plasticity. (A) The DNMT1 methyltransferase methylates CpG sites contributing to differential methylation of genes important for stemness and differentiation (Diff.). It is likely that in parallel, TET proteins generate 5hmC conferring active or passive demethylation. While several studies have shown that DNMT1 is over expressed in CSCs, the level of TET proteins in CSCs is yet to be determined. Non-coding RNA molecules, specifically miRNA and lncRNA molecules add an additional layer of complexity and regulation; (B) miRNAs can target several mRNAs that play important roles as pro- or anti-CSC regulators. For example, the miR17-92 cluster targets several genes important for pancreatic CSC-ness and chemoresistance, and as such the expression of this cluster is downregulated in CSCs. On the other hand, miRNAs that promote EMT features, such as the loss of cell adhesion, are upregulated in CSCs. Examples include miR-9 (target: E-cadherin), miR-194 (target N-cadherin), miR-661 (target: Nestin and Star1). (C) lncRNAs can represent a bridge between the many layers of epigenetic gene regulation, interacting with, for example, histone modifiers or serving as ceRNAs for miRNAs; (D) Genes important for stemness, differentiation (Diff.) and metabolic reprogramming are found with active and inhibiting histone tags, and histone-modifying enzymes are expressed at different levels between CSCs and their non-CSC counterparts, such as MLL1, EZH2 and KDM6A; (Center) The plasticity of differentiated cancer cells and CSCs and their ability to quickly transdifferentiate could be a due to a balance between, high and/or low levels of DNA methylation, hydroxymethylation, histone acetylation or methylation, together with tumor-suppressive/oncogenic miRNAs and lncRNAs.
Figure 1.
Epigenetic regulation of Cancer Stem Cells. Graphical summary of the epigenetic mechanisms regulating CSCs. DNA methylation, histone modifications and non-coding RNA molecules (lncRNAs and miRNAs) work together to modify CSC biology and plasticity. (A) The DNMT1 methyltransferase methylates CpG sites contributing to differential methylation of genes important for stemness and differentiation (Diff.). It is likely that in parallel, TET proteins generate 5hmC conferring active or passive demethylation. While several studies have shown that DNMT1 is over expressed in CSCs, the level of TET proteins in CSCs is yet to be determined. Non-coding RNA molecules, specifically miRNA and lncRNA molecules add an additional layer of complexity and regulation; (B) miRNAs can target several mRNAs that play important roles as pro- or anti-CSC regulators. For example, the miR17-92 cluster targets several genes important for pancreatic CSC-ness and chemoresistance, and as such the expression of this cluster is downregulated in CSCs. On the other hand, miRNAs that promote EMT features, such as the loss of cell adhesion, are upregulated in CSCs. Examples include miR-9 (target: E-cadherin), miR-194 (target N-cadherin), miR-661 (target: Nestin and Star1). (C) lncRNAs can represent a bridge between the many layers of epigenetic gene regulation, interacting with, for example, histone modifiers or serving as ceRNAs for miRNAs; (D) Genes important for stemness, differentiation (Diff.) and metabolic reprogramming are found with active and inhibiting histone tags, and histone-modifying enzymes are expressed at different levels between CSCs and their non-CSC counterparts, such as MLL1, EZH2 and KDM6A; (Center) The plasticity of differentiated cancer cells and CSCs and their ability to quickly transdifferentiate could be a due to a balance between, high and/or low levels of DNA methylation, hydroxymethylation, histone acetylation or methylation, together with tumor-suppressive/oncogenic miRNAs and lncRNAs.

Figure 2.
LncRNAs are differentially expressed in pancreatic CSCs. RTqPCR analysis of the RNA expression levels of the lincRNAs HOTAIR, IRX5, MKLN2, SFPQ, LPHN2 and ZNF673 in sphere-derived cultures (CSCs-enriched) versus adherent non-CSC cultures established from a panel of 6 patient-derived xenografts Panc253, 354, 185, 215, 265 and 286). RNA expression levels for each target gene are normalized to β-actin levels, and data are represented as fold change compared to adherent control cultures (set at 1.0). n = 4 samples per group.
Figure 2.
LncRNAs are differentially expressed in pancreatic CSCs. RTqPCR analysis of the RNA expression levels of the lincRNAs HOTAIR, IRX5, MKLN2, SFPQ, LPHN2 and ZNF673 in sphere-derived cultures (CSCs-enriched) versus adherent non-CSC cultures established from a panel of 6 patient-derived xenografts Panc253, 354, 185, 215, 265 and 286). RNA expression levels for each target gene are normalized to β-actin levels, and data are represented as fold change compared to adherent control cultures (set at 1.0). n = 4 samples per group.

Figure 3.
Targeting the epigenome of Cancer Stem Cells. The variability of the epigenetic mechanisms within cells offers a wide array of targeting strategies. Differentiation therapy is one attractive approach that, in combination with chemotherapeutics, can potentially eliminate both fast proliferating cancer cells and CSCs, once pushed into a more differentiated state. DNMTs can be targeted with several drugs, such as Aza-dC (Decitabine), SGI-110 or Zebularine, decreasing DNMT1 protein levels, decreasing global methylation and impairing CSCs biology. HDAC inhibitors (HDACI), such as Entinostat, Belinostat and Mocetinostat, have also shown promising results but these drugs could have population specific side-effects. Both miRNAs and lncRNAs also represent interesting target molecules. miRNAs can be complexed with drug-loaded micelles and delivered successfully to cancer cells. lncRNAs can be targeted with GapmeRs or small molecule inhibitors that disrupt lncRNA-protein interactions. Importantly the field is progressing to improve delivery methods and in vivo applicability for these molecules.
Figure 3.
Targeting the epigenome of Cancer Stem Cells. The variability of the epigenetic mechanisms within cells offers a wide array of targeting strategies. Differentiation therapy is one attractive approach that, in combination with chemotherapeutics, can potentially eliminate both fast proliferating cancer cells and CSCs, once pushed into a more differentiated state. DNMTs can be targeted with several drugs, such as Aza-dC (Decitabine), SGI-110 or Zebularine, decreasing DNMT1 protein levels, decreasing global methylation and impairing CSCs biology. HDAC inhibitors (HDACI), such as Entinostat, Belinostat and Mocetinostat, have also shown promising results but these drugs could have population specific side-effects. Both miRNAs and lncRNAs also represent interesting target molecules. miRNAs can be complexed with drug-loaded micelles and delivered successfully to cancer cells. lncRNAs can be targeted with GapmeRs or small molecule inhibitors that disrupt lncRNA-protein interactions. Importantly the field is progressing to improve delivery methods and in vivo applicability for these molecules.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zagorac, S.; Garcia-Bermejo, L.; Sainz, B., Jr. The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes 2018, 2, 10. https://doi.org/10.3390/epigenomes2020010
AMA Style
Zagorac S, Garcia-Bermejo L, Sainz B Jr. The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes. 2018; 2(2):10. https://doi.org/10.3390/epigenomes2020010
Chicago/Turabian StyleZagorac, Sladjana, Laura Garcia-Bermejo, and Bruno Sainz, Jr. 2018. "The Epigenetic Landscape of Pancreatic Cancer Stem Cells" Epigenomes 2, no. 2: 10. https://doi.org/10.3390/epigenomes2020010