Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures

Department of Pharmacology, Institute of Biomedical Sciences, University of São Paulo, São Paulo 05508-900, Brazil
Epigenomes 2018, 2(3), 13; https://doi.org/10.3390/epigenomes2030013
Submission received: 21 June 2018
/
Revised: 16 July 2018
/
Accepted: 16 July 2018
/
Published: 20 July 2018
{kind=link}
{kind=link}
Abstract
:Exposure to pathogen infection, and occupational and environmental agents, contributes to induction of most types of cancer through different mechanisms. Cancer is defined and characterized by accumulation of mutations and epimutations that lead to changes in the cellular genome and epigenome. According to a recent Bad Luck Hypothesis, random error mutations during DNA replication in a small population of stem cells may be implicated in two-thirds of variation of cancer risk in 25 organs and tissues. What determines stem cell vulnerability and risk of malignancy across the spectrum of organs, such as the brain, bone marrow, skeletal muscles, skin, and liver? Have stem cells pooled in particular tissues or organs evolved some critical ability to deal with DNA damage in the presence of extrinsic environmental factors? This paper describes how the complex replication and repair DNA systems control mutational events. In addition, recent advances on cancer epigenomic signatures and epigenetic mechanisms are discussed, which will guide future investigation of the origin of cancer initiating cells in tissue and organs in a clinical setting.
1. Introduction
The genetic principle and pathways for generation of a cancer cell are still widely discussed [1,2]. There is no longer doubt that mutations in the DNA molecule lead to mitotic catastrophic events and disordered proliferation observed in malignant cells. Anatomic sites of tissue and organs in the human body, and life exposure to intrinsic and extrinsic (environmental) factors, may explain the vulnerability of skin epidermis, epithelia of internal organs, and blood-circulating lymphoid cells to certain types of cancer [3]. Long time exposure to UV light leads to most common cancers, such as keratinocyte carcinomas, squamous cell carcinoma, and melanomas. However, this does not explain cases among smokers, although infrequent, who remain free of lung cancer despite undergoing daily bombardments with thousands of potential carcinogens. Would this be due to a matter of personal luck or another factor not yet known? Tomasetti and Vogelstein showed that cancers are most often caused neither by intrinsic genetic defects nor by extrinsic environmental factors, but by random bad luck mutations during cell divisions of a small population of cells named stem cells [4]. The analysis of the proliferative behavior of this type of cell within their anatomical sites may account for most variation of cancer risk through life. The large majority of cancers may be initiated by not yet fully-understood chemical processes denominated carcinogenesis [3]. Previous epidemiological studies have not found any relationship between cancer incidence, body size, and lifespan across species [5,6]. Over 15 types of key chemical and metabolic events have been associated with human carcinogens [3]. A single base mutation rate of 17.0 (0.2) × 10−9, on a per-cell division basis, has been estimated in human cells [7]. Genotoxic agents that induce DNA damage, mutation, or both, can alter DNA repair, which, in turn, causes instability of the genome and disrupts chromatin organization, thereby enabling development of hallmarks of cancer [8]. The intimate relationships between DNA repair, stem cells, aging, and cancer, have been interrogated for many years [9,10,11]. Stem cell populations within tissues and organs have the ability of self-renewal and differentiate into different functional cell types. To persist for long life-spans, they have developed highly efficient DNA repair processes which allow their natural co-evolution in the presence of high exposure to chemical carcinogens and cytotoxic drugs [1,9]. There are over 220 different types of tumors, in which genetically (or epigenetically) distinct sub-clones arising through successive intercellular variations (e.g., chromosome copy number, somatic point mutations, or epigenetic modifications) result in phenotypic diversity through a genetic recombination not yet fully understood [11,12,13]. The current models of tumor progression cannot encompass all the known complexity of cancer to make predictions about cancer epidemiology [2]. Mammalian cancer stem cells, as modeled, appear to be reliable predictors of hematopoietic tumors, but not of solid tumors that are organized in niches, or of clonal evolution that depends on interaction with variable microenvironments [2,14,15]. We now know that epigenetic changes are commonly found in cancer cells, as initially revealed by methylation of the tumor suppressor gene promoters [16,17]. Epigenetic imprinting, rather than catastrophic mutational processes, may be an important factor in dictating the evolutionary trajectory of single cancer cells to multiple cancer clones [18]. Therefore, the order of events that leads to carcinogenicity is not so obvious.
2. Tomasetti and Vogelstein Hypothesis
In their first study, Tomasetti and Vogelstein estimated the number of divisions of stem cells in the tissues that give rise to the most common types of cancer, such as lung, intestine and breast [4]. They found highly positive correlations between the lifetime number of stem cell divisions occurring in 31 tissues and the risk of cancer incidence in those tissues. The equation for this calculation is simple: They took into account the number (N) of stem cells in the tissue where this cancer originated, and the rate (b) at which these stem cells divide (i.e., to produce progenitor cells and to differentiate into normal or malignant daughter cells). They then calculated the incidence rate (D) for each cancer type, taking into account the risk of incidence (R) for the development of a life-long cancer and the total number of divisions of stem cells. To reach this number, they used the equation D = NbT, in which T is equal to the estimated life (in years) for a given population. The authors found a strong linear correlation (r = 0.81) between log R and log D, and concluded that 65% of variation in cancer risk between different tissues can be explained by the lifetime number of stem cell divisions when random mutations are acquired due to intrinsic factors [4]. Cells of the large intestine divide frequently, and 5% of people develop colorectal cancer in that tissue. Cells of the small intestine divide rarely, and only 0.2% of people develop small intestine adenocarcinoma. For pancreatic cancer, it was found that 77% of the mutations were due to random copy errors (DNA replication), 18% due to environmental factors and 5% due to heredity, whereas for cancers such as those of the prostate, bone and brain, more than 95% of the mutations were due to random copy errors. For lung adenocarcinoma, the researchers reported that 65% of the mutations were due to environmental factors (most likely smoking), and 35% due to random copy errors [4].
Tomasetti and Vogelstein determined absolute and relative risk and concluded that “bad luck” could explain cancer variation within cancer types as calculated by different mathematical models [19]. They took into account recent next-generation DNA sequencing data that revealed gene signatures (cancer fingerprints) of distinct types of neoplasms and patient epidemiological data to estimate the fraction of mutations caused by endogenous factors, and by environmental (external) and hereditary factors. Their interpretations took into account a number of events and factors. These included: (i) The number of hits (two or more) of driver mutated target genes (oncogenes or tumor suppressors); (ii) the number of additional mutations required for cancer progression; (iii) different rates of cell division and apoptosis; (iv) immunological surveillance; and, (v) exposure to environmental agents. Thus, they concluded that different factors can interfere with the risk of incidence (R), but result in the same incidence rate (D), and only some of those factors are “extrinsic” (i.e., caused by the environment or a hereditary factor (e.g., women carrying the mutation in the BRCA gene)). In a second study, the results were confirmed by analyzing oncogenomic and epidemiological databases from 32 cancer types of patients treated in 69 different countries [20]. Based on the overall data, they estimated that 66% of the mutations found among all cancer types were due to random error mutation, 29% to environmental factors, and 5% to heredity. The proportions varied with cancer types. In prostate, brain, and bone cancers, for example, 95% of the mutations were attributed to random error mutation [20]. Mutation rate per cell division is generally much greater in somatic cells, which commonly accumulate mutations 4 to 25 times more rapidly than germline cells [10]. The stem cell properties that could explain their potential role in carcinogenesis are discussed next.
3. Stem Cells and Cancer
The ability to both self-renew and differentiate into all specialized cell types is the definition of stem cells [9,21]. At different moments in prenatal and postnatal life, stem cells within their special niches divide asymmetrically to increase the pool of progenitor stem cells that give rise to differentiated cells that make up the tissues and organs. Stem cells lose their ability to self-renew due to replicative exhaustion, and then enter into senescence and die. In somatic cells, the cell-cycle limit (i.e., the Hayflick limit) and activity of telomerase (TERT), a reverse transcriptase that elongates telomeric repeats, determine the cell death ending cycle. However, the level of telomerase activity is low or absent in the majority of stem cells, even within tissues with a low cell turnover, such as those of the brain and bone [22]. Nonetheless, TERT DNA promoter methylation results in TERT expression [23]. Thus, telomerase activity and telomere maintenance can lead to the immortalization of germline cells, embryonic stem cells, and cancer initiating cells [24]. On the other hand, the long-term accumulation of senescent stem cells within their microenvironment niches can promote tissue damage and active innate immunity, and this increases their susceptibility to transformation [19,22].
Over the past few years, a number of studies have indicated that both the clonal evolution models (monoclonal, polyclonal, and self-seeding) and cancer stem cell models may contribute to clonal evolution of cancer [2]. In 1935, Furth was the first to introduce the concept of cancer stem cells (CSC) by showing that single leukemic cells could induce systemic myeloid leukemia in mice [25]. There are convincing data supporting cancer stem cell origin in many types of cancer [26], but the underlying mechanisms remain unclear [1]. A carcinogenic process may start early in life or later in adulthood. Many genomic and biochemical methods have been used to define and classify a carcinogenic process [27]. Pre-malignant, self-renewing neoplastic stem cells are slowly cycling or dormant at early stages and require additional drivers of somatic lesions to convert into an aggressive malignancy [27]. Cancer stem cell clones may provide genetic diversity that determines the cancer evolution [9,28,29]. Normal and malignant non-stem cells, due to their intrinsic plasticity, acquire stem cell features and continue to proliferate to form new subpopulations [30]. Thus, it is important to determine the underlying differences in cancer cells from both models, and their efficiency in fixing random errors during DNA replication that could explain their transformation.
4. DNA Repair and Cancer Stem Cells
Oxidative damage is considered a major factor in the generation of mutations in DNA, and over 100 different types of oxidative DNA damage have been identified [3,10,11]. There are various cellular repair systems that can remove DNA adducts that disturb normal DNA structure and functionality [3,10,11]. These systems include: direct repair of DNA bases by alkyltransferases (AGT); the excision of DNA damage by base and nucleotide excision repair (BER and NER, respectively); mismatch repair (MMR); and, double-strand break repair (DSBR) [11,31,32]. Two pathways exist for DSBRs: Homologous recombination (HR) and non-homologous end-joining (NHEJ). HR occurs during DNA replication in the S and G2 phases, whereas NHEJ occurs during G0 and G1 phases; how molecular mediators switch between NHEJ and HR is not known. If repair enzymes are overwhelmed by DNA damage, or for other reasons cannot function efficiently, DNA adducts may persist and increase the probability of developing somatic mutations. However, this depends on inter-individual variations in DNA repair capacity. Inherited polymorphic variants in some DNA repair enzymes are, in fact, closely associated with decreased DNA repair activity and a potentially higher probability of developing cancer. Many proteins involved in the DNA damage response are mutated in cancer [31,32,33]. The absence or lack of fidelity in NHEJ-driven repair increases the diversification of mutational lesions.
DNA repair enzymes play a major role in inducing point mutations [11,31,32,33]. The production of a new cell depends on the duplication of the DNA that is promoted by the DNA polymerases with different efficiency and fidelity. During this process, several types of mutations occur (e.g., exchange of bases, insertions, and deletions) that are corrected precisely by DNA repair enzymes [11,31,32,33]. Random mistakes are fixed during cell divisions and any failure in this repair can eliminate or disable genes, which leads to gain-of-function or inactivation of their normal function. If, for example, the mutated gene is involved in a cell cycle check point program (e.g., the p53 gene), anomalous division may occur during mitosis and leading to the formation of defective daughter cells or clones. These mutants (i.e., tumor-initiating cells with stem cell properties), rarely divide (by asymmetric division), and only a few clones will survive in a process of natural selection (i.e., Darwin’s theory of evolution). Survivors then lose their original characteristics (i.e., recognition antigens) and thus are no longer monitored by immune defense cells (i.e., immune surveillance). TP53 gene mutations do not occur at random along the coding sequence. They are typically clustered at mutation “hot spots”, which are within the DNA binding domain of the TP53 protein and span codons 120 to 300 of the gene. Random (accidental) mutations are technically difficult to predict or prevent, and so the hypothesis that cancer is caused by the accumulation of these types of errors (or lesions) in the DNA is called bad luck tumorigenesis [4,34].
DNA crosslinks and oxidative DNA damage are repaired by NER, BER, HR, or NHEJ systems [11,30,31,33]. Human ES cells display very low levels of mitochondrial mass and oxidative phosphorylation [30,31,33]. They obtain energy preferentially through non-oxidative glycolysis. Moreover, ES cells have a higher level of proteins involved in MMR (MLH-1, MSH-2, MSH-6), HR (MRE11, NBS1, and RAD52), and NHEJ (XRCC4 and ligase IV), when compared to differentiated cells [31,32]. The expression of MMR genes allows them to resist apoptosis and proliferate after injury, while high expression of NHEJ genes may cause higher levels of genomic alterations. NER is crucial in epidermal stem cells of the skin [31,32,33]. Patients with XPC (xeroderma pigmentosum group C-complementing protein) and XPA (xeroderma pigmentosum group A-complementing protein) deletions, key enzymes for NER, are highly susceptible to squamous cell carcinoma [33]. Mutations in the BRCA1 gene, thereby leading to impairment of HR, increases risk of luminal progenitor cells in breast cancer tumors. The most common mutational signatures observed in many types of cancer are associated with MMR and HR deficiencies or upregulation of cytosine deaminase enzymes known as APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family enzymes (APOBEC1, APOBEC2, and APOBEC3A-H), which display various physiological functions. APOBECs cause hypermutation, termed “kataegis”, which usually occurs exclusively in regions with dC>dT transitions at TpC dinucleotides [35]. The HR-deficiency signature is commonly observed in breast, ovarian and pancreatic tumors. Likewise, tumors caused by lifestyle factors such as diet, alcohol consumption, cigarette smoking, limited exercise, and UV light, have generated specific mutational signatures [36]. The tobacco smoke signature is characterized by the presence of C>A conversion, whereas the UV light signature is characterized by C>T conversion, which differs from the DNA repair deficiency observed in other types of extrinsic factors [31,36]. The presence of these mutational signatures has been detected at late stages and may not discriminate early molecular events in tumourigenesis. Thus, one extrinsic factor (benzene, for example) and one intrinsic factor (DNA repair machinery, for example), could act as the initiator agents in one stem cell target. Thus, both could contribute to somatic mutation rates in derived tumor clones. Future investigations are required to precisely define different subtypes of mutational signatures, as well as their amalgamations in the heterogeneous cancer cell populations.
5. Cancer Stem Cell and Epigenomic Reprogramming
DNA methylation is a primary mechanism of specific gene silencing and of inactivation of one copy of the X chromosome that determines allelic imprinting of mother and father genes, and during tissue differentiation and aging [37]. The epigenetic inheritance, as with classical genetic inheritance (i.e., DNA sequence), can fail, and mutations or epigenetic mutations (epialleles) can be transmitted to offspring [38]. DNA methylation, histone modifications and microRNA changes without real changes in the genomic DNA sequence are non-genomic mechanisms that lead to dynamic reprogramming of DNA methylation of germ cell lines following fertilization [37]. The Polycomb repression complexes (PRC1 and PRC2) are involved in many epigenetic pathways [39]. They play a key role in epigenetic silencing and thereby in the expression of the Hox genes involved in development, as well as inactivation, of the X chromosome [37,38,39]. The PRC1/2 complex has histone methyltransferase activity, primarily of trimethylated histone H3 on lysine 27 (i.e., H3K27me3). This reaction is required for transcriptional inactivation or silencing of the chromatin [39]. Environmentally induced epigenome changes during early life may be an important regulator of many phenotypes at adulthood [37]. The methylation of cytosine residues in CpG-rich regions within promoters occurs at carbon 5 of the pyrimidine ring (5mC). This chemical reaction is catalyzed by the DNA methyltransferases DNMT1, DNMT3A, and DNMT3B [37,40]. De-methylation is a process that erases methylation, which occurs passively through DNA replication, or actively through oxidization of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) or 5-carboxylcytosine (5caC). These reactions are catalyzed by DNMT1, DNMT3A, and DNMT3B, and by the Ten-eleven translocation methylcytosine dioxygenases TET1, TET2 and TET3 [40]. Abnormal epimodifications are most commonly observed during in vitro reprogramming of fibroblasts in pluripotent stem cells (iPSs). This somatic cell-induced reprograming is driven by the transcriptions factors: Oct4 (octamer-binding transcription factor 4); Sox2 (sex determining region Y-box 2); and, Klf4 (Kruppel-like factor 4) in combination with c-Myc [40,41]. One hallmark of cancer cells is the presence of focal aberrant hypermethylation in CpG islands (stretches of 500–1500 base pairs of DNA) and widespread DNA hypomethylation in other genomic regions [42]. Most aberrant hypermethylation occurs at promoter regions of tumor suppressor genes [42].
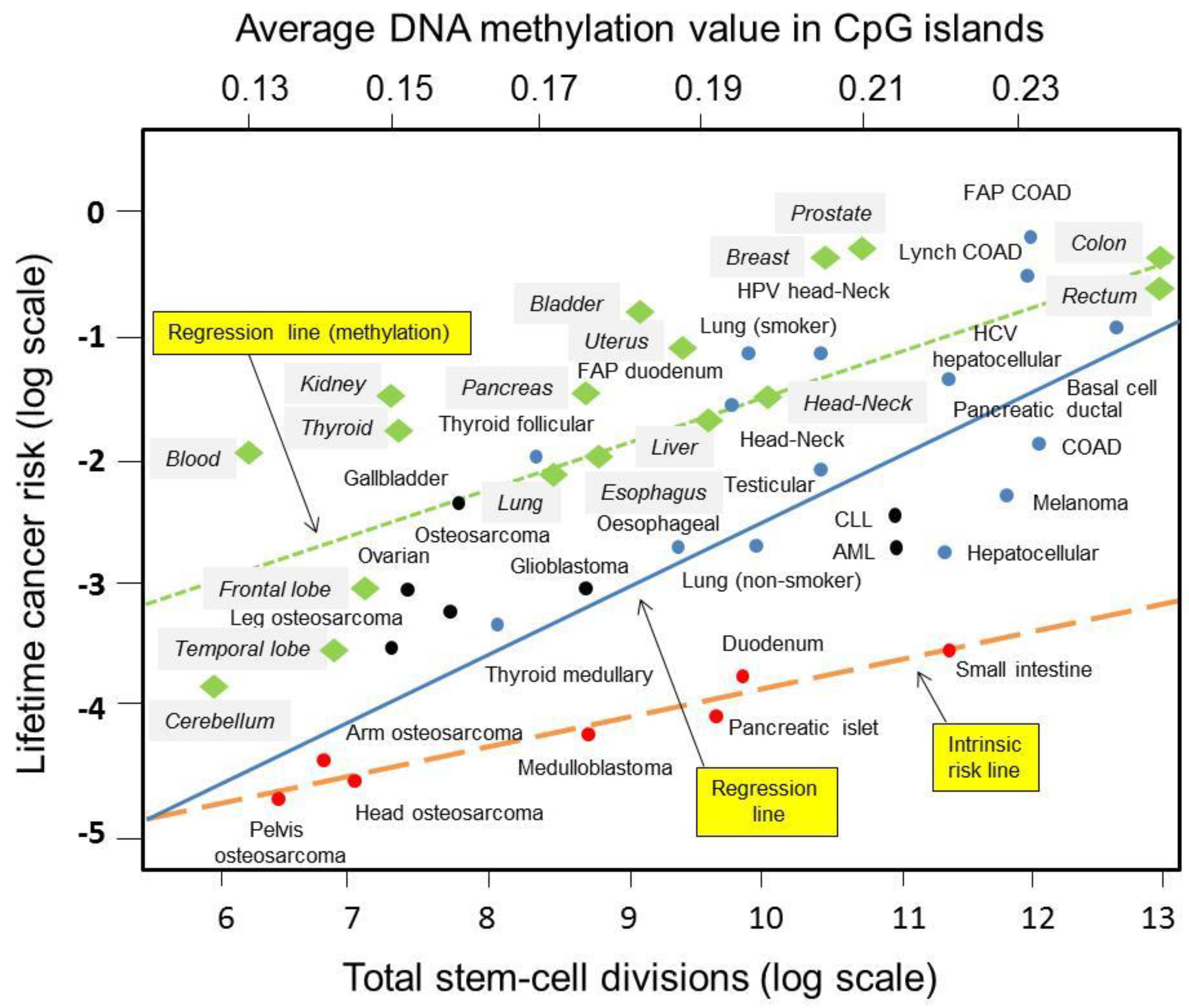
Chemical-induced epimutations and not genetic mutations could be a cause of many types of cancers [42]. It is well-known that aberrant de novo methylation takes place preferentially with PRC2 Polycomb-bound CpG islands [39]. The level of CpG-island methylation varies over a relatively wide range in continually renewed normal tissues through stem cell divisions. Remarkably, Klutstein and colleagues found the strongest correlation between DNA methylation and lifetime cancer risk (R = 0.8, p < 0.0001) in normal tissues and organs of 70-year-old people [43]. It was noticeable that tissues and organs with higher cancer incidence had increased epigenetic marks [43]. Figure 1 shows the regression line for epigenetic factors’ contribution to lifetime cancer risk as compared to intrinsic and extrinsic factors. Overall, these data show that cross-talk between DNA repair and DNA methylation pathways exists and may be complementary during DNA damage response [11,18,44]. Systemic interrogations would tell us whether programmed, accidental, or environmentally induced patterns correlate with genomic signatures induced by intrinsic and extrinsic factors involved in carcinogenesis as described in recent studies [4,20].
6. Discussion and Perspectives
Public knowledge of the real risk factors producing any type of cancer is critical for taking decisions and establishing preventive guidelines for environmental factor exposure across a lifetime. Doll and Peto’s quantitative probabilities of human cancer risk in the population of the USA, reported in 1981, were based on lifestyle and exposure to environmental factors, and did not provide a genetic mechanism at that time [46]. The theory of “bad luck” proposed by Tomasetti and Vogelstein indicates one molecular mechanism that could lead to an underestimation of the absolute contribution of these environmental factors in cancer risk. “Bad luck” cannot explain changes in cancer incidences; for example: Melanomas, which continue to increase; and, lung, cervix, and uterine cancers, which have declined during the past decades. Nor can this variation be explained by tissue-stem cell divisions. Many authors have questioned and provided further evidence for divergent results in the literature [45,47,48,49,50]. Wu and colleagues examined the same cohort of cancer patients and applied diverse mathematic model approaches, taking into account the data of epidemiological studies and 30 cancer mutational signatures as described [36]. First, they consider a hypothetical scenario, caused by a nuclear bomb in which radiation quadruples the lifetime risks for all cancers, and the hypothesized cancer risk per stem cell division, and concluded that a simple regression analysis does not distinguish the contribution of extrinsic versus intrinsic factors to cancer risk. Next, they constructed one plot (Figure 1) to compare the two regression lines according to the mathematical approaches used in the Tomasetti and Volgelstein study and in their own study, considering the smallest cancer risks for a given cancer. They found that cancer risk due to intrinsic stem-cell mutation errors alone was low for almost all cancers that require over two mutations [45]. When considering three or more mutations, the contribution of extrinsic factors to the lifetime risk of cancer was greater than 70–90% for most common cancers. In another study, Noble and colleagues concluded that the total number of stem cells and the lifetime number of divisions per stem cell each significantly, and independently of one another, explained the variation in the lifetime risk of cancer [50]. Their results gave further support for the hypothesis that tissues and organs have evolved differential mechanisms for cancer protection [50]. Tissues of the breast, prostate, lung, and colon undergo a continuous renewal of cells supported by an active division of stem cells that originate basal and luminal cells in their compartments. Luminal cells in such tissues are of great cancer risk, which may be explained by use of the error-prone NHEJ pathway to both endogenous and exogenous DNA damage [9,32,34]. The self-renewal pathway of stem cells is unique to hematological cells as compared to epithelial cells in solid tissues. Despite the co-localization at the same anatomical site and high proliferative activity, small intestine stem cells have evolved mechanisms that prevent cancer incidence as compared to colorectal stem cells. Thus, it is proposed that cancer suppression is an adaptive trait under evolving natural selection, which fits into Darwinian evolution, and this mechanism could be linked to either genetic or epigenetic control of cell division and DNA repair mechanisms.
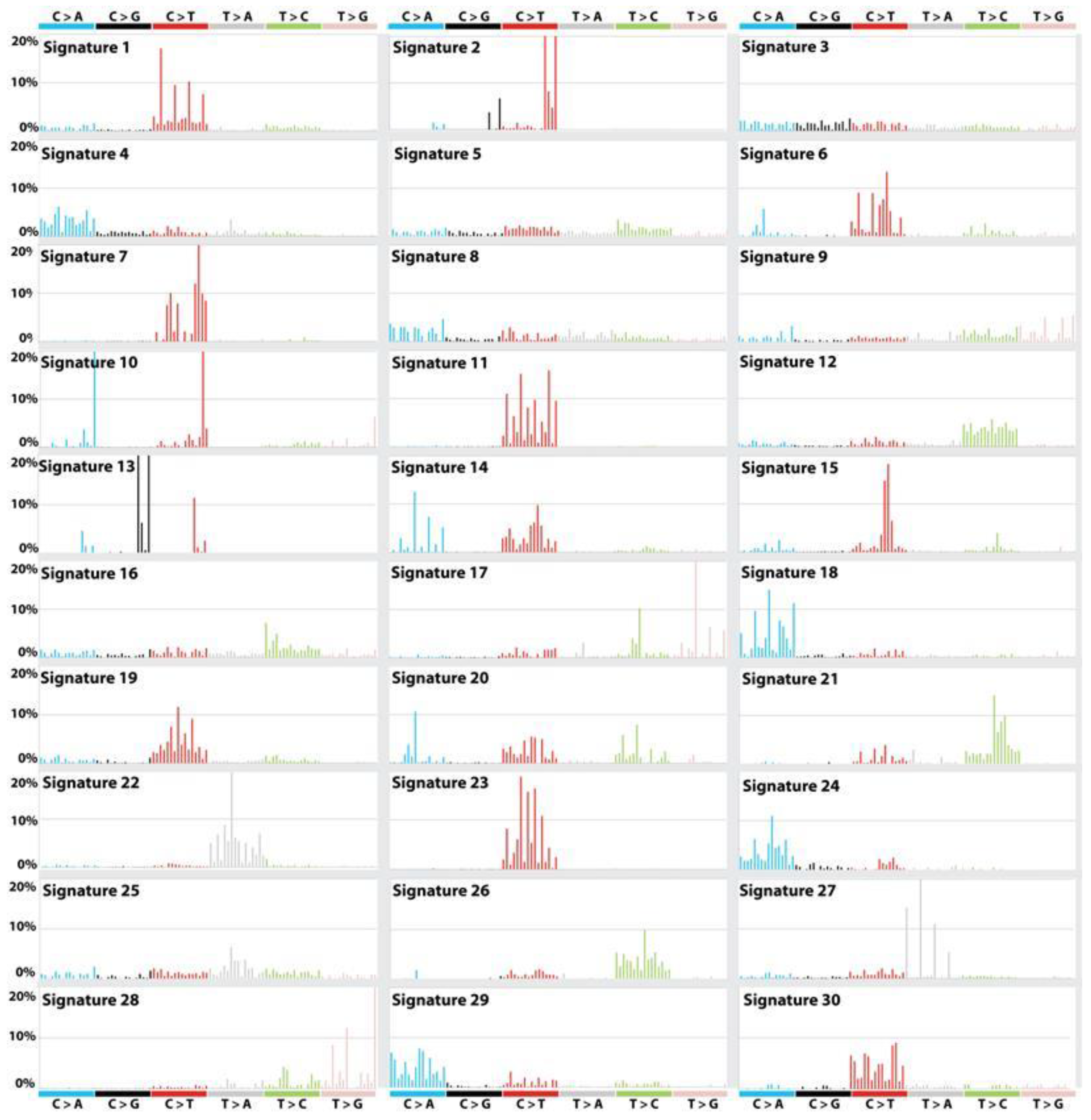
The complex mechanisms in the etiologies of cancers across populations have been explored through next-generation DNA sequencing technologies. More than 30 distinct mutational signatures across 36 cancer types were found to contribute to cancer phenotypes [35,36,51]. They are available in the COSMIC database and are shown in Figure 2. The APOBEC-associated signature is the second most common, and is likely involved in somatic mutagenesis in the lagging single DNA strand, as compared with the leading strand, during the process of DNA replication [35,36,51]. It is remarkable that APOBEC proteins are also involved in anti-virus defense, as well as genomic surveillance of the retro-transposition of the endogenous long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs) and long terminal repeat (LTR) retrotransposons [52,53]. These nuclear elements are epigenetically silenced by DNA methylation. Interestingly, pharmaceutical and natural environmental factors increase their transcription in adulthood and/or senescence, which coincides with cancer rising [53]. High-throughput sequencing technologies have revealed diverse transcriptional changes in response to many types of genomic and epigenomic alterations. Thus, transcriptome signatures can also provide insights not only into DNA repair deficiencies within a mutational process, but also into cancer etiology.
We know that cancer and stem cells share some essential signaling pathways that switch between the cell type-specific cellular states and the tridimensional (3D) genome organization [14,21,30]. The mutation, aberrant expression, activation, or deletion, of oncogenes and tumor suppressor together with epigenetic modulators and modifiers may contribute to stem cell reprogramming into cancer stem cells [18,21]. Signaling and metabolic pathways activated by chemical insults from the environment, injury, inflammation, and other forms of stress, may increase normal cell epigenetic reprogramming to more stem-like states [14,21,30]. Nutrition deficiencies such as in folate, methionine, betaine, choline, tryptophan, and vitamin B12, impact on global DNA hypomethylation, histone modification, and chromatin organization [3,42]. Transgenerational epigenetic inheritance is increasingly implicated in genome instability and predisposition to childhood and adulthood diseases [38,54]. Somatic cells, and likely stem cells, gradually acquire mutations in their non-replicating DNA [43]. These mutations commonly arise at CpG sites and appear to accumulate in an essentially replication-independent manner. Thus, we need to know if “specific epigenetic patterns” can be inheritable across generations and if they may work as an evolutionary determinant of organ-specific resistance or susceptibility to cancer growth as described in nutritional epidemiology studies [54].
The cell of origin of a cancer and the mathematical equation to predict its evolution, prognosis (including, for example, patient survival time), and response to treatment, varies from cancer to cancer [34]. More studies are needed to delineate the stochastic processes that arise and progress in most cancer cells, as well as to understand the co-evolution of tumor cells and their complex and non-linear interaction with their microenvironment [14]. A more complex pattern of mutational cancer signatures, which are connected via underlying biological mechanisms and processes, may emerge across cancer genomic and epigenomic maps [36]. The large oncogenomic databases now available express inter- and intra-patient heterogeneity, and should be used to advance cancer research into accurately modeling signaling networks and clinical outcomes from new therapeutics [55,56]. In summary, Tomasetti and Volgelstein’s discoveries will help us in the development of new approaches to approximate the probability of relative cancer risk variation between organs and tissues due to heredity or mutagenic factors, and in the compilation of evidence of random copy errors in stem cells as origin of cancers.
Funding
The J.E.B.’s studies are supported by “Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq proc 486048/2011 and 312206/2016-0), and Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP proc 2012/02497-7 and 2014/20847-0)”.
Conflicts of Interest
The author declares no conflict of interest. The views expressed herein are those of the author only and these may not necessarily be the views of the institution/organization that the author is associated with.
References
- Egeblad, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [Green Version]
- Navin, N.E.; Hicks, J. Tracing the tumor lineage. Mol. Oncol. 2010, 4, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F.; et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peto, R.; Roe, F.J.C.; Lee, P.N.; Levy, L.; Clack, J. Cancer and ageing in mice and men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunney, L.; Muir, B. Peto’s paradox and the hallmarks of cancer: Constructing an evolutionary framework for understanding the incidence of cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150161. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, A.S. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum. Mutat. 2013, 21, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pazhanisamy, S.K. Stem cells, DNA damage, ageing and cancer. Hematol. Oncol. Stem Cell Ther. 2009, 2, 375–384. [Google Scholar] [CrossRef]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2008, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, P.; Shibata, D. A simple algebraic cancer equation: Calculating how cancers may arise with normal mutation rates. BMC Cancer 2010, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrret, R.; Moseley, S. Evolution of resistance and progression to disease during clonal expansion of cancer. Theor. Popul. Biol. 2010, 77, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddel, R.R. The role of senescence and immortalization in carcinogenesis. Carcinogenesis 2000, 21, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Furth, J.; Kahn, M. The transmission of leukaemia of mice with a single cell. Am. J. Cancer 1937, 31, 276–282. [Google Scholar]
- Wang, J.C.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 2005, 15, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Arock, M.; Block, C.; Georg, T.I.; Galli, S.J.; Gotlib, J.; Haferlach, T.; Hoermann, G.; Hermine, O.; et al. Proposed terminology and classification of pre-malignant neoplastic conditions: A consensus proposal. EBioMedicine 2017, 16, 17–24. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Smith, B.D. Eliminating cancer stem cells by targeting embryonic signaling pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Weeden, C.E.; Asselin-Labat, M.L. Mechanisms of DNA damage repair in adult stem cells and implications for cancer formation. Biochim. Biophys. Acta 2018, 1864, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.; Lerner, L.K.; Okamoto, O.K.; Marchetto, M.C.; Menck, C.F. The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat. Res. 2013, 752, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Altrock, P.M.; Liu, L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2015, 12, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Rabhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Reddington, J.P.; Sproul, D.; Meehan, R.R. DNA methylation reprogramming in cancer: Does it act by re-configuring the binding landscape of Polycomb repressive complexes? Bioessays 2014, 36, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.P.; Rao, A. DNA methylation and methylcytosine oxidation in cell fate decisions. Curr. Opin. Cell Biol. 2013, 25, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klutstein, M.; Moss, J.; Kaplan, M.; Cedar, H. Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc. Natl. Acad. Sci. USA 2017, 114, 2230–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Peto, R. The causes of cancer: Quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 1981, 166, 1191–1308. [Google Scholar] [CrossRef]
- Blot, W.L.; Tarone, R.E. Doll and Peto’s quantitative estimates of cancer risks: Holding generally true for 35 years. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.V. Cancer: Bad luck or punishment? Biochemistry (Moscow) 2017, 82, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Waclaw, B. Genes, environment, and ‘‘bad luck’’. Science 2017, 355, 1266–1267. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.; Kaltz, O.; Hochberg, M.E. Peto’s paradox and human cancers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150104. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Magiorkinis, G.; Belshaw, R.; Katzourakis, A. “There and back again”: Revisiting the pathophysiological roles of human endogenous retroviruses in the post-genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120504. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Jacobs, D.R., Jr.; Porta, M. Hypothesis: A unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ. Health Perspect. 2009, 117, 1799–1802. [Google Scholar] [CrossRef] [PubMed]
- Belizário, J.E.; Sangiuliano, B.A.; Perez-Sosa, M.; Neyra, J.M.; Moreira, D.F. Using pharmacogenomic databases for discovering patient-target genes and small molecule candidates to cancer therapy. Front. Pharmacol. 2016, 7, 312. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Regression lines for intrinsic, extrinsic and epigenetic factors’ contribution to lifetime cancer risk. Green diamond dots are estimated methylation in Polycomb-bound CpG islands in normal tissue and organs. The green dashed line is the regression line for lifetime cancer risk versus DNA methylation according to Klutstein et al., 2017 [43]. The solid blue line is the regression line for total stem-cell division versus lifetime cancer risk according to Tomasetti and Vogelstein, 2015 [4]. Red dots are cancer types used to determine the regression line for the intrinsic risk (painted as dashed red line) according to Wu and colleagues, 2015 [45]. Blue dots are cancers with substantial extrinsic risk as estimated by epidemiology studies. Figure abbreviations: COAD, colon adenomatous disease; FAP, familial adenomatous polyposis; HCV, hepatitis C virus; HPV, human papillomavirus; CLL, chronic lymphoid leukemia; AML, acute myeloid leukemia. Adapted from [4,43,45].
Figure 1.
Regression lines for intrinsic, extrinsic and epigenetic factors’ contribution to lifetime cancer risk. Green diamond dots are estimated methylation in Polycomb-bound CpG islands in normal tissue and organs. The green dashed line is the regression line for lifetime cancer risk versus DNA methylation according to Klutstein et al., 2017 [43]. The solid blue line is the regression line for total stem-cell division versus lifetime cancer risk according to Tomasetti and Vogelstein, 2015 [4]. Red dots are cancer types used to determine the regression line for the intrinsic risk (painted as dashed red line) according to Wu and colleagues, 2015 [45]. Blue dots are cancers with substantial extrinsic risk as estimated by epidemiology studies. Figure abbreviations: COAD, colon adenomatous disease; FAP, familial adenomatous polyposis; HCV, hepatitis C virus; HPV, human papillomavirus; CLL, chronic lymphoid leukemia; AML, acute myeloid leukemia. Adapted from [4,43,45].

Figure 2.
Mutational signatures across the spectrum of human cancer types. The profile of 30 mutational signatures displayed using the six substitution subtypes (i.e., C>A, C>G, C>T, T>A, T>C, and T>G), based on the actual trinucleotide frequencies of the reference human genome version GRCh37. The probability bars for each of the six types of substitutions and the mutated bases are displayed in different colors. The mutation types are displayed on the horizontal axes, while vertical axes depict the percentage of mutations attributed to a specific mutation type. Signature 2 found in 22 cancer types is attributed to activity of the AID/APOBEC family of cytidine deaminases. Signature 4 (found in head and neck cancer; liver cancer; lung adenocarcinoma; lung squamous carcinoma; small cell lung carcinoma; and, esophageal cancer), is attributed to tobacco carcinogens. Signature 3 (found in breast, pancreatic, and ovarian cancer), is strongly associated with germline and somatic BRCA1 and BRCA2 mutations. Fourteen mutational signatures are of an unknown etiology. Further details on each of 30 cancer signatures are described in COSMIC (https://cancer.sanger.ac.uk/cosmic/signatures).
Figure 2.
Mutational signatures across the spectrum of human cancer types. The profile of 30 mutational signatures displayed using the six substitution subtypes (i.e., C>A, C>G, C>T, T>A, T>C, and T>G), based on the actual trinucleotide frequencies of the reference human genome version GRCh37. The probability bars for each of the six types of substitutions and the mutated bases are displayed in different colors. The mutation types are displayed on the horizontal axes, while vertical axes depict the percentage of mutations attributed to a specific mutation type. Signature 2 found in 22 cancer types is attributed to activity of the AID/APOBEC family of cytidine deaminases. Signature 4 (found in head and neck cancer; liver cancer; lung adenocarcinoma; lung squamous carcinoma; small cell lung carcinoma; and, esophageal cancer), is attributed to tobacco carcinogens. Signature 3 (found in breast, pancreatic, and ovarian cancer), is strongly associated with germline and somatic BRCA1 and BRCA2 mutations. Fourteen mutational signatures are of an unknown etiology. Further details on each of 30 cancer signatures are described in COSMIC (https://cancer.sanger.ac.uk/cosmic/signatures).

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Belizário, J.E. Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures. Epigenomes 2018, 2, 13. https://doi.org/10.3390/epigenomes2030013
AMA Style
Belizário JE. Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures. Epigenomes. 2018; 2(3):13. https://doi.org/10.3390/epigenomes2030013
Chicago/Turabian StyleBelizário, José E. 2018. "Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures" Epigenomes 2, no. 3: 13. https://doi.org/10.3390/epigenomes2030013