
Study of Adsorption of Hydrogen on Al, Cu, Mg, Ti Surfaces in Al Alloy Melt via First Principles Calculation


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Surface Energy and Work Function of Al, Cu, Mg, and Ti
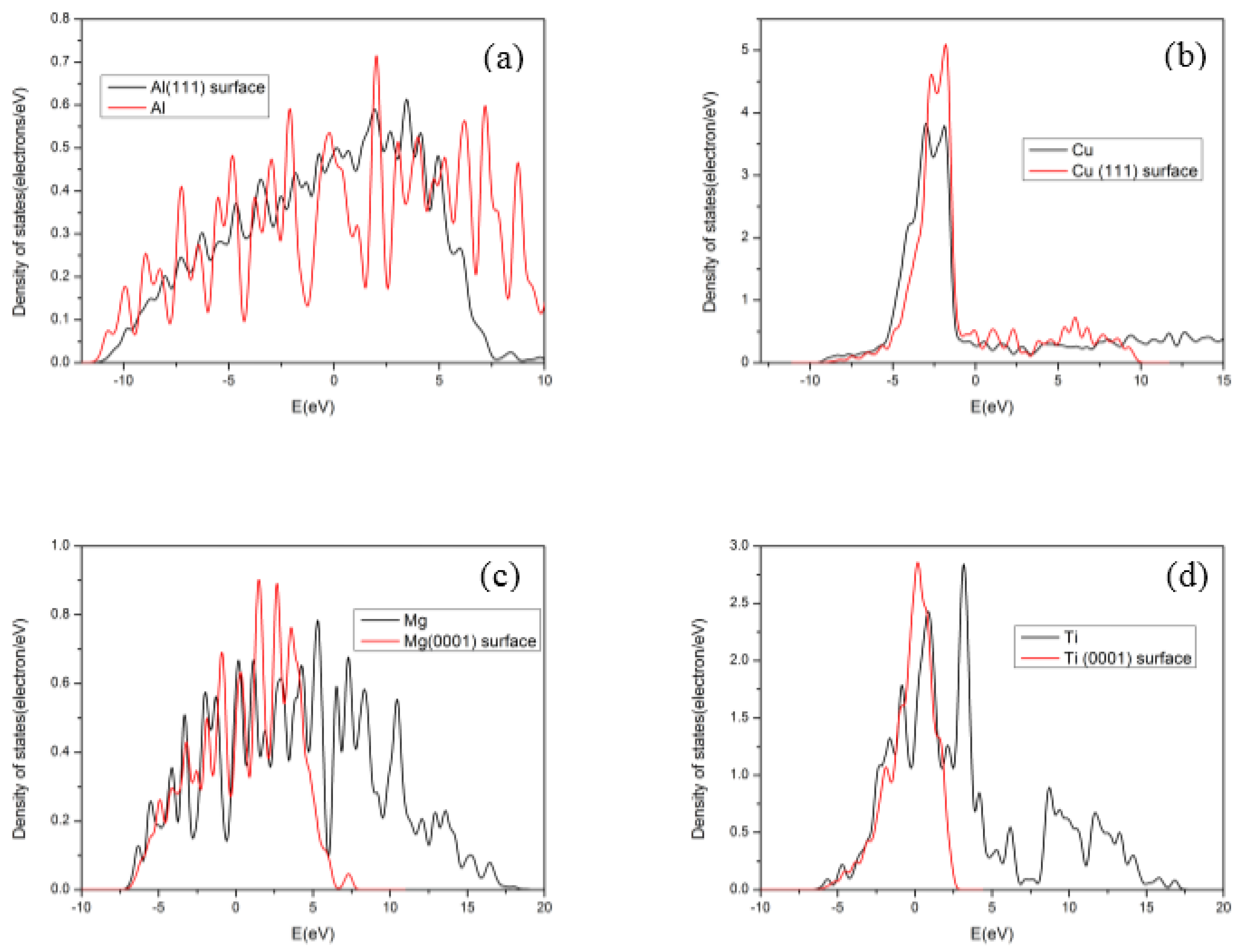
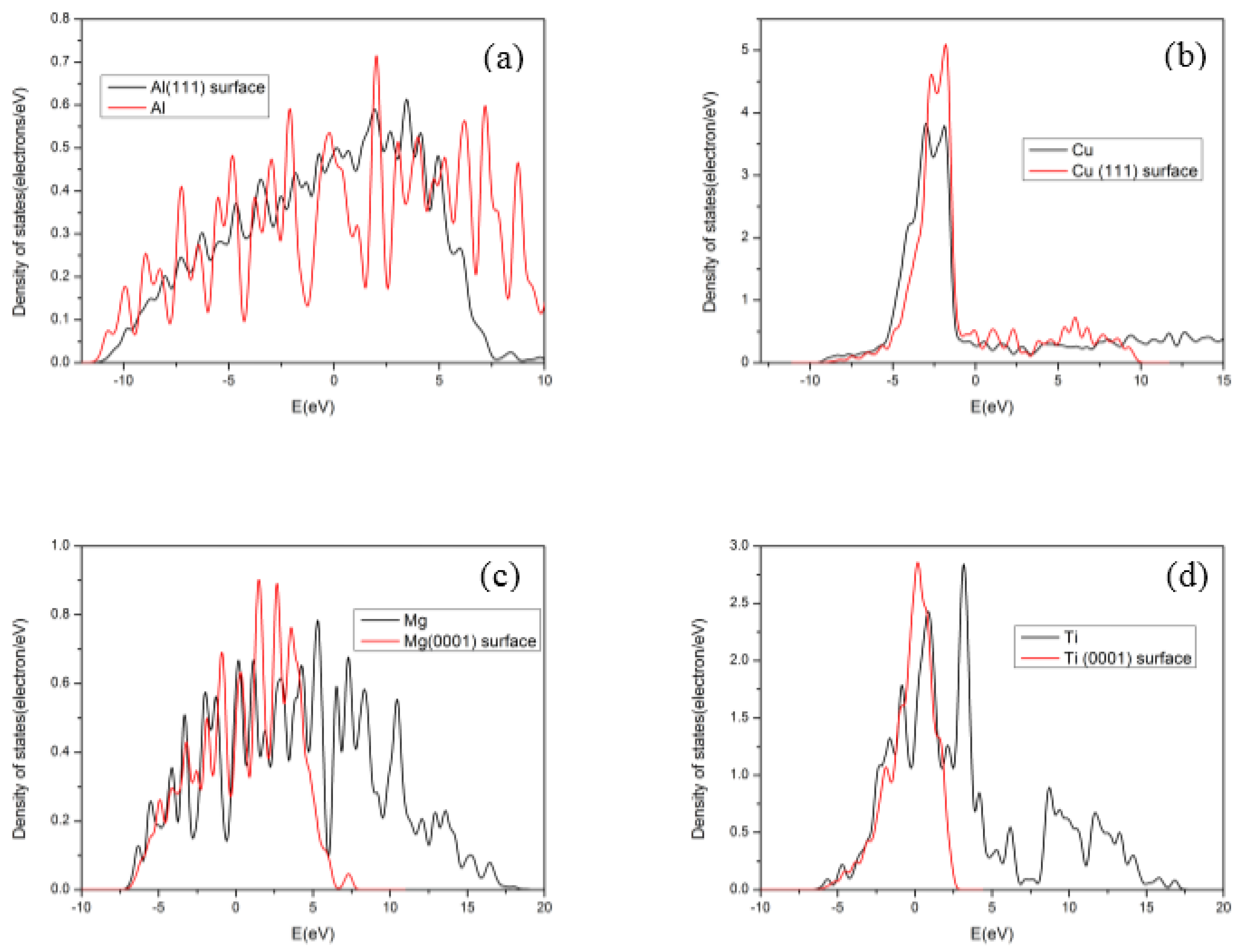
3.2. Electronic Structures of Slabs
3.3. H Adsorption on Al(111), Cu(111), Mg(0001), and Ti(0001) Surfaces
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wanhill, R.J.H.; Bray, G.H. Chapter 2—Aerostructural Design and Its Application to Aluminum-Lithium Alloys. Alum. Lithium Alloy. 2014, 14, 27–58. [Google Scholar]
- Li, Y.-P. Application and Prospect of Aluminum Alloys Automobile industry. Alum. Fabr. 2007, 173, 23–24. [Google Scholar]
- Shi, Q.; Xiong, W. Application and Development of Aluminium Alloys in Automobile Industry. New Technol. New Process 2006, 12, 55–58. [Google Scholar]
- Poirier, D.R.; Yeum, K.; Maples, A.L. A thermodynamic prediction for microporosity formation in aluminum-rich Al-Cu alloys. Metall. Trans. A 1987, 18, 1979–1987. [Google Scholar] [CrossRef]
- Han, Q.; Viswanathan, S. Hydrogen evolution during directional solidification and its effect on porosity formation in aluminum alloys. Metall. Mater. Trans. A 2002, 33, 2067–2072. [Google Scholar] [CrossRef]
- Felberbaum, M.; Désy, E.L.; Weber, L.; Rappaz, M. Effective hydrogen diffusion coefficient for solidifying aluminium alloys. Acta Mater. 2011, 59, 2302–2308. [Google Scholar] [CrossRef]
- Zhao, L.; Pan, Y.; Liao, H.; Wang, Q. Degassing of aluminum alloys during re-melting. Mater. Lett. 2012, 66, 328–331. [Google Scholar] [CrossRef]
- Xu, H.; Meek, T.T.; Han, Q. Effects of ultrasonic field and vacuum on degassing of molten aluminum alloy. Mater. Lett. 2007, 61, 1246–1250. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, D.; Tan, S. Improvement in the strength of reticulated porous ceramics by vacuum degassing. Mater. Lett. 2001, 51, 363–367. [Google Scholar] [CrossRef]
- Éskin, G.I. Prospects of ultrasonic (cavitational) treatment of the melt in the manufacture of aluminum alloy products. Metallurgist 1998, 42, 284–291. [Google Scholar] [CrossRef]
- Eskin, G.I. Principles of Ultrasonic Treatment: Application for Light Alloys Melts. Adv. Perform. Mater. 1997, 4, 223–232. [Google Scholar] [CrossRef]
- Wu, R.; Shu, D.; Sun, B.; Wang, J.; Li, F.; Chen, H.; Lu, Y. Theoretical analysis and experimental study of spray degassing method. Mater. Sci. Eng. A 2005, 408, 19–25. [Google Scholar] [CrossRef]
- Wu, R.; Qu, Z.; Sun, B.; Shu, D. Effects of spray degassing parameters on hydrogen content and properties of commercial purity aluminum. Mater. Sci. Eng. A 2007, 456, 386–390. [Google Scholar] [CrossRef]
- Warke, V.S.; Tryggvason, G.; Makhlouf, M.M. Mathematical modeling and computer simulation of molten metal cleansing by the rotating impeller degasser: Part I. Fluid flow. J. Mater. Process. Technol. 2005, 168, 112–118. [Google Scholar] [CrossRef]
- Warke, V.S.; Shankar, S.; Makhlouf, M.M. Mathematical modeling and computer simulation of molten aluminum cleansing by the rotating impeller degasser: Part II. Removal of hydrogen gas and solid particles. J. Mater. Process. Technol. 2005, 168, 119–126. [Google Scholar] [CrossRef]
- Wang, L.; Guo, E.; Huang, Y.; Lu, B. Rotary impeller refinement of 7075Al alloy. Rare Met. 2009, 28, 309–312. [Google Scholar] [CrossRef]
- Anyalebechi, P.N. Analysis of the effects of alloying elements on hydrogen solubility in liquid aluminum alloys. Scri. Metall. Mater. 1995, 33, 1209–1216. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Norm-conserving and ultrasoft pseudopotentials for first-row and transition elements. J. Phys. Condens. Matter 1994, 6, 8245. [Google Scholar] [CrossRef]
- Zope, R.R.; Mishin, Y. Interatomic potentials for atomistic simulations of the Ti-Al system. Phys. Rev. B 2003, 68, 024102. [Google Scholar] [CrossRef]
- Wang, J.W.; Gong, H.R. Adsorption and diffusion of hydrogen on Ti, Al, and TiAl surfaces. Int. J. Hydrog. Energy 2014, 39, 6068–6075. [Google Scholar] [CrossRef]
- Straumanis, M.E.; Yu, L.S. Lattice parameters, densities, expansion coefficients and perfection of structure of Cu and of Cu—In α phase. Acta Crystallogr. 1969, 25, 676–682. [Google Scholar] [CrossRef]
- Ganne, J.P.; Lebourgeois, R.; Paté, M.; Dubreuil, D; Pinier, L.; Pascard, H. The electromagnetic properties of Cu-substituted garnets with low sintering temperature. J. Eur. Ceram. Soc. 2007, 27, 2771–2777. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Addison-Wiley: New York, NY, USA, 2005. [Google Scholar]
- Wu, G.-X.; Zhang, J.Y.; Wu, Y.-Q.; Li, Q.; Chou, K.; Bao, X.H. First-Principle Calculations of the Adsorption, Dissociation and Diffusion of Hydrogen on the Mg(0001) Surface. Acta Phys. Chim. Sin. 2008, 24, 55–60. [Google Scholar] [CrossRef]
- Martin, A.S.; Manchester, F.D. The H-Ti (Hydrogen-Titanium) system. Bull. Alloy Phase Diagr. 1987, 8, 30–42. [Google Scholar] [CrossRef]
- Halas, S.; Durakiewicz, T.; Joyce, J.J. Surface energy calculation—Metals with 1 and 2 delocalized electrons per atom. Chem. Phys. 2002, 278, 111–117. [Google Scholar] [CrossRef]
- Tyson, W.R.; Miller, W.A. Surface free energies of solid metals: Estimation from liquid surface tension measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Polatoglou, H.M.; Methfessel, M.; Scheffler, M. Vacancy-formation energies at the (111) surface and in bulk Al, Cu, Ag, and Rh. Phys. Rev. B 1993, 48, 1877–1883. [Google Scholar] [CrossRef]
- Wright, A.F.; Feibelman, P.J.; Atlas, S.R. First-principles calculation of the Mg(0001) surface relaxation. Surf. Sci. 1994, 302, 215–222. [Google Scholar] [CrossRef]
- Murr, L.E. Interfacial Phenomena in Metals and Alloys; Addison-Wesley: Upper Saddle River, NJ, USA, 1974. [Google Scholar]
- Michaelson, H.B. The work function of the elements and its periodicity. J. Appl. Phys. 1977, 48, 4729–4733. [Google Scholar] [CrossRef]
- DeBoer, F.R.; Boom, R.; Mattens, W.C.M.; Miedema, A.R.; Niessen, A.K. Cohesion in Metals; HoIIand, N., Ed.; North Holland: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Skriver, H.L.; Rosengaard, N.M. Surface energy and work function of elemental metals. Phys. Rev. B 1992, 46, 7157–7168. [Google Scholar] [CrossRef] [Green Version]
- Ji, D.-P.; Zhu, Q.; Wang, S.-Q. Detailed first-principles studies on surface energy and work function of hexagonal metals. Surf. Sci. 2016, 651, 137–146. [Google Scholar] [CrossRef]
- Krumbein, A.D.; Malamud, H. Measurement of the effect of chlorine treatment on the work function of titanium and zirconium. J. Appl. Phys. 1954, 25, 591–592. [Google Scholar]
- Strömquist, J.; Bengtsson, L.; Persson, M.; Hammer, B. The dynamics of H absorption in and adsorption on Cu(111). Surf. Sci. 1998, 397, 382–394. [Google Scholar] [CrossRef]
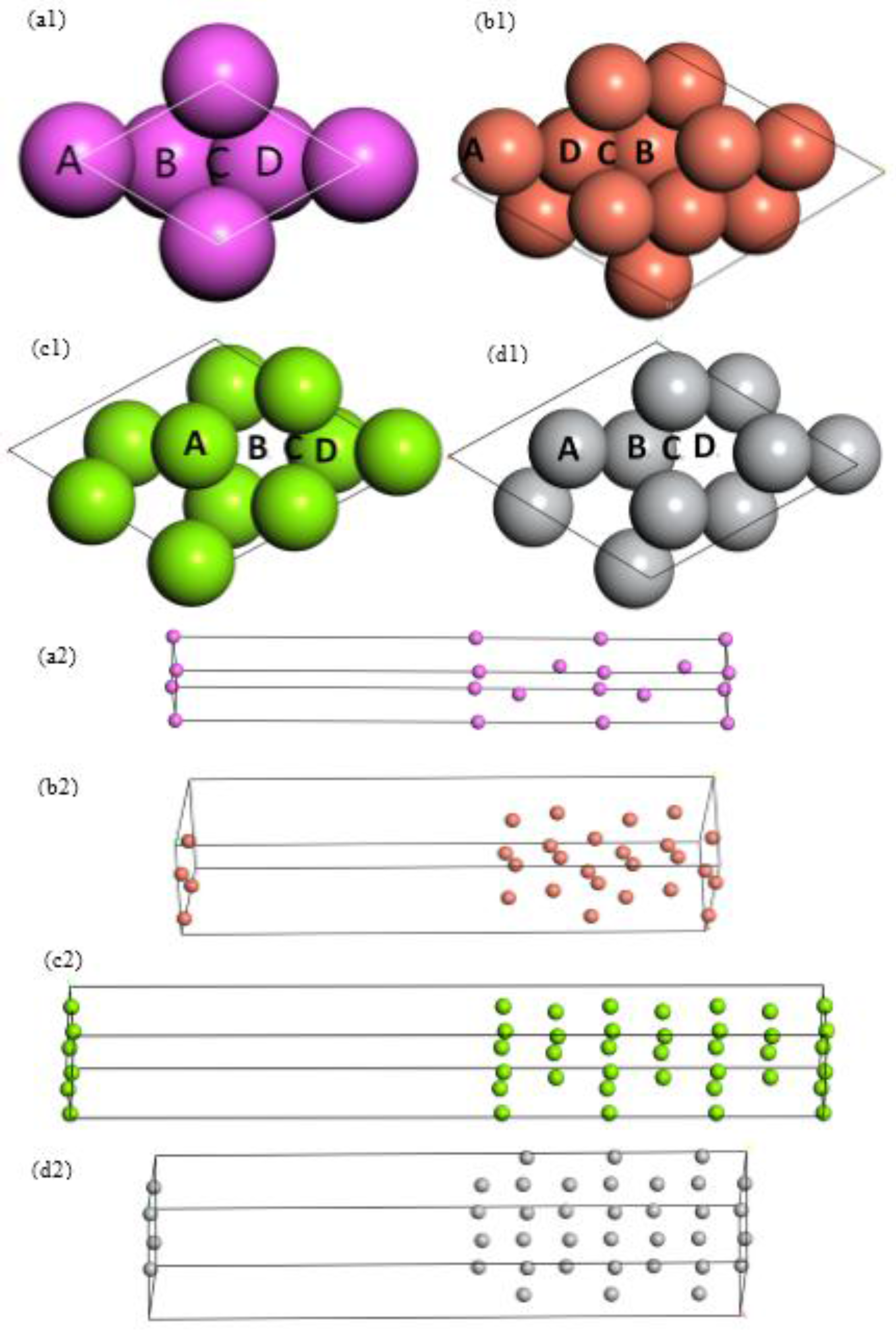

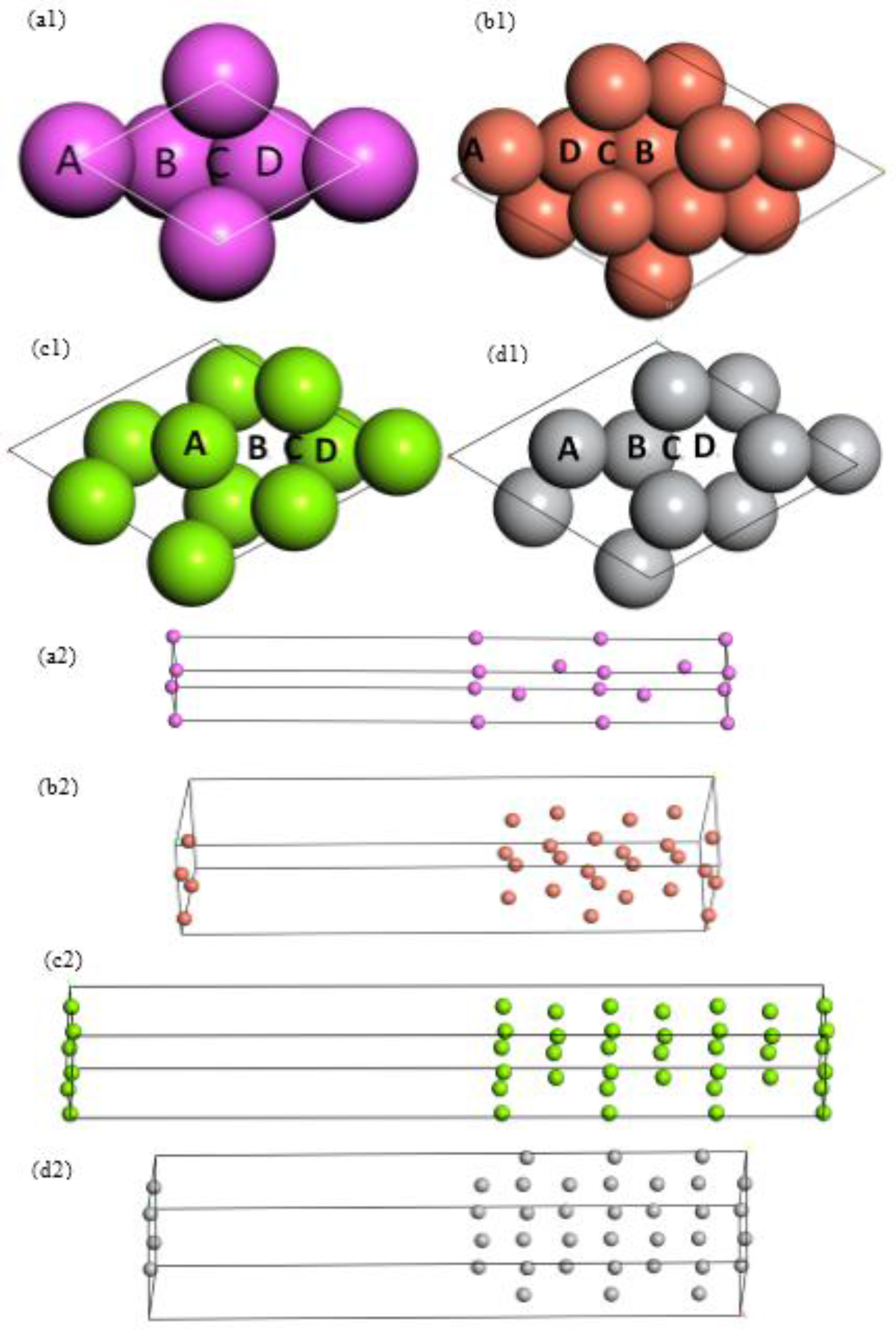{kind=link}
{kind=link}
| Elements | Lattice Constants (Å) | ||
|---|---|---|---|
| This Work | Experiments | Other Calculation | |
| Al | a = 4.021 | a = 4.05 [20] | a = 3.982 [21] |
| Cu | a = 3.641 | a = 3.61 [22] | a = 3.638 [23] |
| Mg | a = 3.192, c = 5.206 | a = 3.21, c = 5.213 [24] | a = 3.19, c = 5.17 [25] |
| Ti | a = 2.896, c = 4.626 | a = 2.904, c = 4.680 [26] | a = 2.864, c = 4.537 [21] |
| Surface | Surface Energy (J/m2) | ||
|---|---|---|---|
| This Work | Experiments | Other Calculation | |
| Al(111) | 0.864 | 1.14 [28] | 0.988 [21] |
| Cu(111) | 1.793 | 1.825 [28] | 1.94 [29] |
| Mg(0001) | 0.716 | 0.785 [28] | 0.641 [30] |
| Ti(0001) | 2.034 | 1.98 [31] | 2.235 [21] |
| Surfaces | Work Function (eV) | ||
|---|---|---|---|
| This Work | Experiments | Other Calculation | |
| Al(111) | 4.26 | 4.24 [32] | 4.22 [21] |
| Cu(111) | 4.98 | 4.94 [33] | 5.1 [29] |
| Mg(0001) | 3.80 | 3.86 [34] | 3.76 [35] |
| Ti(0001) | 4.58 | 4.45 [36] | 4.67 [21] |
| Surface | Sites | Eads (eV) | |
|---|---|---|---|
| This Work | Other Calculation | ||
| Al(111) | Top | 0.235 | 0.226 a |
| Fcc | 0.081 | 0.069 a | |
| Hcp | 0.132 | 0.122 a | |
| Cu(111) | Top | −1.667 | −1.83 b |
| Fcc | −2.385 | −2.37 b | |
| Hcp | −2.353 | −2.36 b | |
| Bridge | −2.178 | −2.22 b | |
| Mg(0001) | Top | −1.21 | −1.04 c |
| Fcc | −2.546 | −2.56 c | |
| Hcp | −2.471 | −2.52 c | |
| Bridge | −1.90 | −1.89 c | |
| Ti(0001) | Top | 0.610 | 0.400 a |
| Fcc | −1.486 | −1.342 a | |
| Hcp | −1.502 | −1.396 a | |
| Sites | Top | Fcc | Hcp | Bridge | |
|---|---|---|---|---|---|
| ∆Hf (kJ/mol) | Al8H | 5.04 | 1.74 | 2.83 | |
| Cu24H | −12.86 | −18.40 | −18.15 | −16.80 | |
| Mg29H | −7.78 | −16.37 | −15.89 | −12.22 | |
| Ti28H | 4.06 | −9.88 | −9.99 | ||
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Huang, Y.; Xiao, Z.; Reng, X. Study of Adsorption of Hydrogen on Al, Cu, Mg, Ti Surfaces in Al Alloy Melt via First Principles Calculation. Metals 2017, 7, 21. https://doi.org/10.3390/met7010021
Liu Y, Huang Y, Xiao Z, Reng X. Study of Adsorption of Hydrogen on Al, Cu, Mg, Ti Surfaces in Al Alloy Melt via First Principles Calculation. Metals. 2017; 7(1):21. https://doi.org/10.3390/met7010021
Chicago/Turabian StyleLiu, Yu, Yuanchun Huang, Zhengbing Xiao, and Xianwei Reng. 2017. "Study of Adsorption of Hydrogen on Al, Cu, Mg, Ti Surfaces in Al Alloy Melt via First Principles Calculation" Metals 7, no. 1: 21. https://doi.org/10.3390/met7010021