Effect of Pyrite on Thiosulfate Leaching of Gold and the Role of Ammonium Alcohol Polyvinyl Phosphate (AAPP)
by
Xiaoliang Liu
1,2, 
Bin Xu
1,*,
Xin Min
1,
Qian Li
1,*,
Yongbin Yang
1,
Tao Jiang
1,
Yinghe He
2 and
Xi Zhang
1 1
School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China
2
College of Science and Engineering, James Cook University, Townsville 4814, Queensland, Australia
*
Authors to whom correspondence should be addressed.
Metals 2017, 7(7), 278; https://doi.org/10.3390/met7070278
Submission received: 18 June 2017
/
Revised: 13 July 2017
/
Accepted: 14 July 2017
/
Published: 19 July 2017
(This article belongs to the Special Issue Valuable Metal Recycling)
Abstract
:The effect of pyrite and the role of ammonium alcohol polyvinyl phosphate (AAPP) during gold leaching in ammoniacal thiosulfate solutions were investigated using pure gold foils. The results showed that pyrite catalyzed the decomposition and also significantly increased the consumption of thiosulfate. This detrimental effect became more severe with increasing pyrite content. Further, the presence of pyrite also substantially slowed the gold leaching kinetics and reduced the overall gold dissolution. The reduction in gold dissolution was found to be caused primarily by the surface passivation of the gold. The negative effects of pyrite, however, can be alleviated by the addition of AAPP. Comparison of zeta potentials of pyrite with and without AAPP suggests that AAPP had adsorbed on the surface of the pyrite and weakened the catalytic effect of pyrite on the thiosulfate decomposition by blocking the contact between the pyrite and thiosulfate anions. AAPP also competed with thiosulfate anions to complex with the cupric ion at the axial coordinate sites, and thus abated the oxidation of thiosulfate by cupric ions. Moreover, the indiscriminate adsorption of AAPP on the surfaces of gold and passivation species prevented the passivation of the gold surface by surface charge and electrostatic repulsion. Therefore, AAPP effectively stabilized the thiosulfate in the solution and facilitated the gold leaching in the presence of pyrite.
1. Introduction
Ammoniacal thiosulfate has been studied over the last three decades as a nontoxic and highly-efficient alternative lixiviant to cyanide for leaching gold from gold ores with copper(II) acting as a catalyst [1,2]. Much of the work has concentrated on copper-gold ores, carbonaceous gold ores and complex sulfidic gold ores where the recovery of gold by cyanidation is unsatisfactory or the consumption of cyanide is too high [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. The aerated thiosulfate leaching of gold is very slow in the absence of copper(II), but in the presence of copper(II) the rate of gold dissolution in ammoniacal thiosulfate solutions can be increased by 18–20 fold [1]. However, copper(II), together with the oxygen in the leach liquors, can also catalyze the degradation of thiosulfate, resulting in a high consumption of thiosulfate during gold leaching. The thiosulfate degradation not only leads to higher reagent cost, but also results in the generation of copper sulfide and sulfur layers which can passivate gold and hinder the leaching rate [19,20,21,22]. So the successful application of the thiosulfate leaching technology is obviously restricted by the high thiosulfate consumption and the gold surface passivation.
It has been shown that various associated minerals in gold ores present different influences on the stability of thiosulfate and the leaching behavior of gold [8,11,23,24,25,26]. Gold is commonly associated with sulfide minerals, of which pyrite is ubiquitous in sulfide orebodies [27]. The effect of pyrite on gold leaching in thiosulfate solutions has been studied mainly from the following four aspects:
- (i)
- Catalytic effect of pyrite on thiosulfate decomposition. The catalyzed decomposition of thiosulfate by pyrite has been reported by a number of researchers [8,23,28,29,30,31]. It is believed that the strong affinity of pyrite for aqueous sulfur species and the semiconducting properties of pyrite lead to the catalysis of pyrite in the degradation of thiosulfate in aqueous solutions of pH 2.9–8.6 [29]. An interfacial intermediate complex was considered to be formed by pyrite, thiosulfate and oxygen, of which thiosulfate is the aqueous electron donor on anodic sites and oxygen is the terminal electron acceptor on cathodic sites [24,29].
- (ii)
- Dissolution behavior of pyrite. It has been shown that iron-containing sulfide minerals such as pyrite, pyrrhotite, arsenopyrite, and chalcopyrite can be partially dissolved in ammoniacal thiosulfate liquors [8,28]. During the dissolution of pyrite, copper(II) and oxygen were found to be consumed and simultaneously a lower slurry potential was caused [24]. Scanning Electron Microscopy (SEM), Raman spectroscopy and X-ray Photoelectron Spectroscopy (XPS) analyses indicated that the leaching of pyrite largely took place at high-energy crystal boundaries and defect sites whilst iron oxide (hematite) was found to be formed at the pyrite surface after leaching [23,28].
- (iii)
- Gold adsorption and precipitation on pyrite surfaces. In thiosulfate-deficient solutions, dissolved gold can adsorb and precipitate on sulfide minerals, resulting in a lower gold recovery. The adsorption and precipitation of gold as the metal tended to occur at defect sites on the surface of sulfide mineral [31,32,33,34]. It was reported that gold precipitated on pyrite was difficult to be recovered unless undergoing the treatment with cyanide [31].
- (iv)
- Passivation of gold by iron species coating on gold surfaces. In the presence of pyrite, some iron-containing species can also be formed and may be responsible for the reduced dissolution of gold due to the surface passivation. The likely passivating products were iron hydroxides/oxides such as FeO·OH, Fe2O3 or Fe2O3·nH2O [8,23,24,28].
Hence, remedies to combat the detrimental effects of pyrite are crucial for thiosulfate leaching of gold. Besides the maintenance of appropriate reagent concentrations and leaching conditions, the pretreatment with an oxidative ammoniacal solution followed by thiosulfate leaching as an effective approach to diminish reagent consumption and enhance gold extraction from sulfide minerals has been researched by Feng and van Deventer [13]. More commonly, the usage of various additives has been attempted to optimize a copper-ammonia-thiosulfate leaching system. Sulfite was once added into the leach solution to stabilize thiosulfate [3,4], but sulfite significantly inhibited the thiosulfate leaching of gold by reducing copper(II) in solution [35]. The addition of sulfate could enhance gold leaching to a limited extent by inhibiting the pyrite dissolution in ammoniacal thiosulfate solutions [23]. In the absence of sulfide minerals, the effect of inorganic additives on the dissolution of gold colloids in copper(II) ammoniacal thiosulfate solutions was investigated by Senanayake and Zhang [36], and they found that cations such as Ag(I) and anions such as Cl−, SO42−, and CO32− had favorable effects on the gold dissolution mainly owning to enhancing the anodic reaction of gold with thiosulfate, increasing residual copper(II) concentration, or hindering the reaction of copper(II) with thiosulfate.
More recently, orthophosphate, polyphosphate, ethylenediaminetetraacetic acid (EDTA), ammonia triacetic acid (NTA), amino acids (glycine, dl-α-alanine, and l-histidine), guar type surfactant (Gempolym M47), carboxymethyl cellulose (CMC), sodium carboxymethyl starch (CMS), sodium humic acid (HA), etc. have also been adopted by researchers to stabilize thiosulfate and reduce gold passivation [8,15,25,37,38,39,40,41,42,43,44]. Organic additives, especially those containing plenty of functional groups such as hydroxyl, amino, carboxyl, phosphoryl, phosphate and so on are apt to present multiple beneficial effects such as (i) stabilizing thiosulfate; (ii) adjusting the residual copper(II) concentration and solution potential; (iii) improving the rheology of leach slurries; and (iv) preventing the formation of passivating layers or removing blocking products on gold surfaces to allow gold leaching over extended periods.
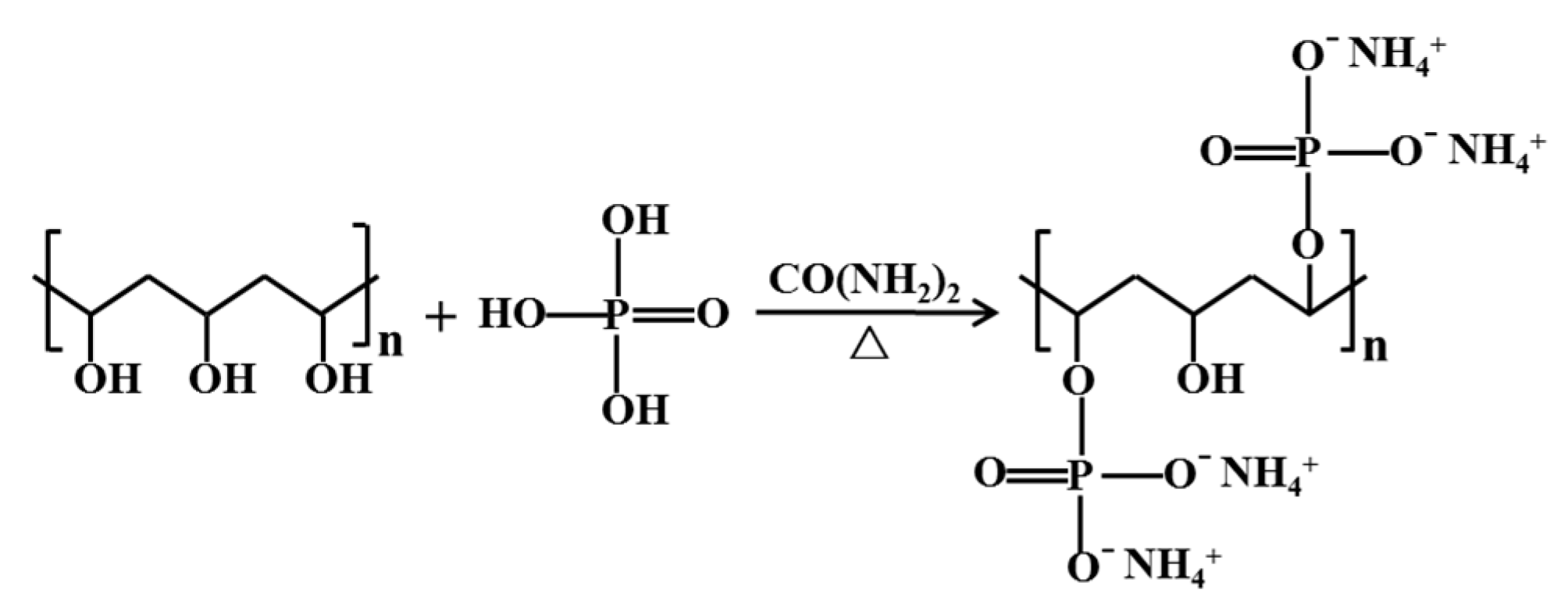
Ammonium alcohol polyvinyl phosphate (AAPP), as a synthetic polymer, has been used extensively in the materials industry for making nanocomposite, polyelectrolyte membrane [45,46], and so on, but rarely in the minerals and metallurgy industries. AAPP can be easily made from polyvinyl alcohol (PVA), phosphoric acid and urea. Similar to HA, CMC, CMS, and so on, AAPP possesses plentiful negatively charged functional groups of hydroxyl (see Figure 1). Also, AAPP has numerous phosphate groups, and it has been shown that orthophosphate, hexametaphosphate have beneficial effects on thiosulfate leaching of gold in the presence of sulfide minerals [8,25,40]. Hence, AAPP is expected to be an effective additive to relieve the unfavorable effect of pyrite.
This study aimed to investigate the effect of pyrite on thiosulfate leaching of gold and the role of AAPP in the presence of pyrite by leaching tests of gold foils suspended in ammoniacal thiosulfate solutions. Relevant thermodynamic calculations and the analyses of frontier orbital energy and XPS were conducted to illuminate the effect mechanism of pyrite. In the presence of pyrite, special emphasis was placed on the mechanism of AAPP in stabilizing thiosulfate and alleviating gold leaching passivation by the analyses of mixed slurry potential, zeta potential, SEM, XPS, and so on.
2. Experimental Section
2.1. Minerals and Reagents
The pure minerals of pyrite (Py) and quartz (Qz) were supplied from Mineral Specimen Supplies, Wuhan, China. Quantitative XRD (X-ray diffraction) results showed that the purities of pure minerals both exceed 99%. The minerals were first crushed, dry milled in a planetary ball mill and sieved into the size fraction of −44 µm over 95%. Then they were stored in air-tight plastic bags in a refrigerator in order to avoid any further oxidation. Gold foils (Au purity > 99.99%, thickness 0.2 mm) were used in the experiments with a surface area of 0.38 cm2. The chemical reagents of sodium thiosulfate pentahydrate, cupric sulfate, ammonia water (25%, mass fraction), urea, polyvinyl alcohol (PVA, average degree of polymerization 1750), phosphoric acid (85%, mass fraction) were all in analytically pure grade. De-ionized water was used throughout all experiments.
2.2. Preparation of Ammonium Alcohol Polyvinyl Phosphate (AAPP)
According to the typical synthetic steps, the AAPP was synthesized as follows [45,46]: Firstly, the PVA aqueous solution SA was obtained by dissolving PVA with deionized water in a beaker. Secondly, the phosphoric acid was blended with urea in a three-necked bottle and heated at 85 °C in a water bath for 2 h to acquire the aqueous solution SB. Thirdly, the solution SA was added into the solution SB, combining with agitating (by a magnetic stirrer, IKA EURO-STPCUS25, IKA, Staufen im Breisgau, Germany) and refluxing for 2 h. After the reaction was complete, a 95% ethanol aqueous solution was used to cool and precipitate the solution. A suction filter was used to obtain the precipitation product, and then de-ionized water was used to wash the product until the aqueous solution was neutral. Finally, AAPP was obtained after the wet product was dried under vacuum at 60 °C for 24 h. Figure 1 clearly depicts the simplified synthesis of AAPP.
2.3. Leaching Experiments
A 250 mL open-top reactor was used to perform the gold leaching tests at a rotating speed of 200 rpm using a magnetic stirrer (IKA EURO-STPCUS25). Desired amounts of the quartz and pyrite samples were added to 200 mL of solution containing certain quantities of reagents with and without the addition of AAPP. Before each test, the gold foils were polished by sand paper of 4000 grid, washed twice with acetone and then rinsed with deionized water. The gold foils were suspended in the middle of the solution to ensure no contact with the reactor wall during the leaching process. All experiments were conducted at 25 °C in a water bath and the pH of solutions was maintained in the range of 9.8–10.2. Samples were taken continuously at certain intervals from the slurry during a total retention time of 24 h. The obtained samples were centrifuged and filtered for the subsequent analyses. The gold dissolution rate was expressed in the mass of dissolved gold per square meter of the gold foil surface. All the experiments were performed with the reagent concentration of 0.1 M Na2S2O3·5H2O, 0.012 M CuSO4·5H2O and 0.5 M NH3·H2O.
2.4. Analytical Methods
The atomic absorption spectrometry (AA-6800, SHIMADZU, Kyoto, Japan) was adopted to assay the concentration of gold in solutions. The iodometric method was used to determine the thiosulfate concentration. Prior to the titration with the indicator Vitex, a certain amount of EDTA-2Na and formaldehyde were added to eliminate the impacts of cupric-ammonia complex and sulfite on the iodine titration. The mixed slurry potential was measured by a platinum electrode with a double junction reference electrode (Ag/AgCl, saturated KCl, Orion) to avoid the interference of thiosulfate with the reference electrode. All potentials were given with respect to standard hydrogen electrode (SHE). Zeta potential was measured by the micro electrophoretic method in a zeta potential analyzer (ZEN3690, Malvern Instruments Ltd., Malvern, UK). A 50 mg pyrite sample whose particle size was 100% less than 5 μm was dispersed in 50 mL of 0.01 M NaCl electrolyte solution for each measurement and the adjustment of pH was implemented with NaOH or HCl solution.
Morphological studies on leached gold foils were performed on SEM (Quanta 250 FEG, FEI, Hillsboro, OR, USA). The surface species on the leached gold foils were identified by XPS (ESCALAB 250Xi, Thermo Fisher, Waltham, MA, USA). XPS was carried out with a monochromatic Al Kα X-ray at 1486.6 eV operated at 200 W. Spectra of S 2p, Cu 2p and Fe 2p were collected using the pass energy of 20 eV and an energy step of 0.1 eV. The calibration of binding energy was based on C 1s at 284.6 eV. The leached gold foils were sufficiently rinsed with de-ionized water, dried under vacuum overnight and then were sealed in a special container filled with nitrogen before analyses of SEM and XPS.
3. Results
3.1. Thermodynamic Analysis
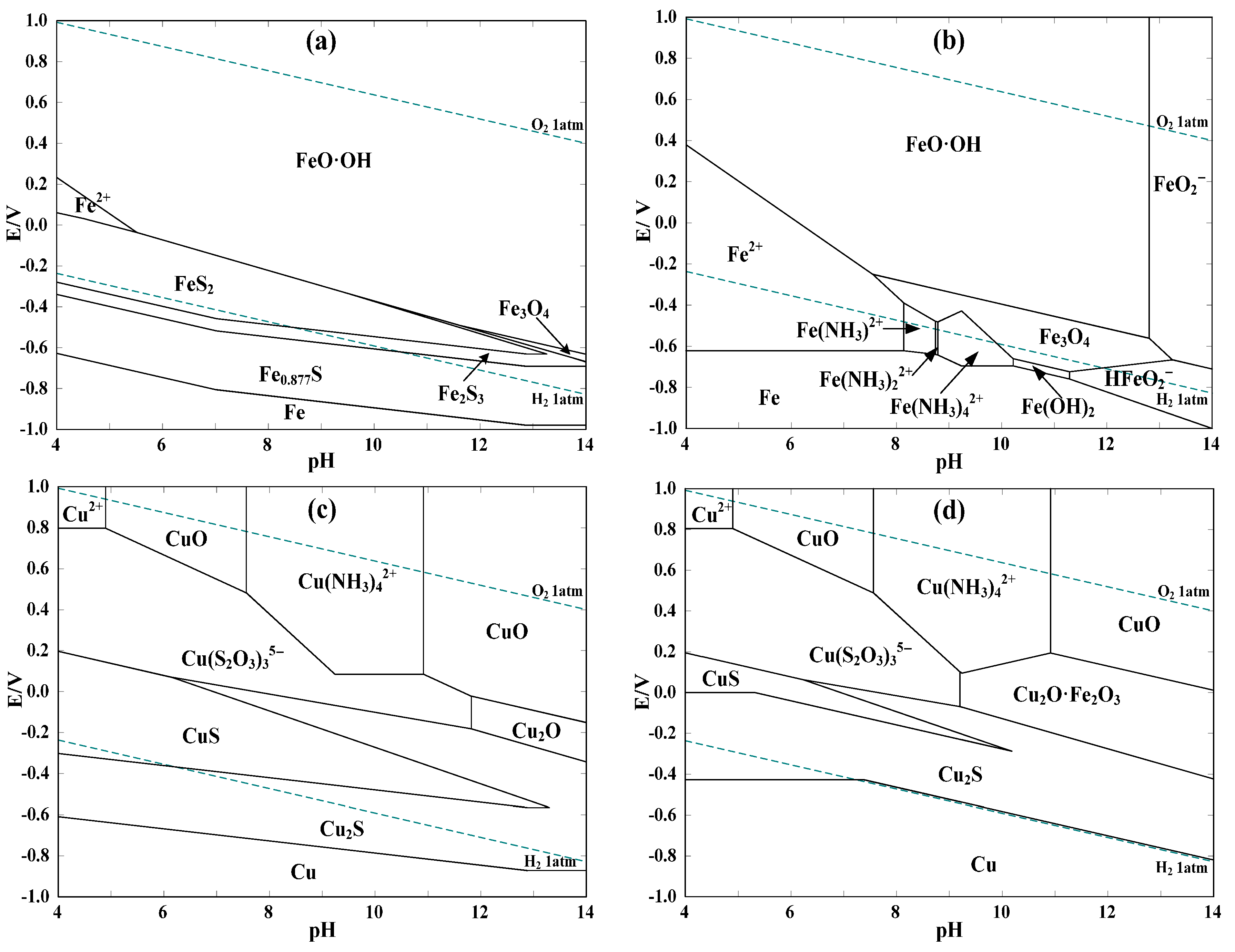
The relevant E-pH diagrams were drawn using the thermochemical software HSC Chemistry 6.0 (Outokumpu, Helsinki, Finland). The thermodynamic data are available from HSC database and are listed in Appendix A. To illustrate the dissolution and impacts of pyrite and possible intermediate products during the ammoniacal thiosulfate leaching process, the thermodynamic calculation was conducted by considering or neglecting some of the special species. The results of thermodynamic calculation for the systems of Fe-S-H2O, Fe-NH3-S2O3-H2O, Cu-NH3-S2O3-H2O and Cu-Fe-NH3-S2O3-H2O at 25 °C are presented in Figure 2.
As shown from Figure 2a, pyrite is rather unstable in the most relevant ranges of potential (0.25–0.3 V) and pH value (9–10) of thiosulfate leaching of gold [47]. The oxidation potential of pyrite is as low as −0.3–−0.4 V within pH 9–10. As a result, pyrite can be oxidized by dissolved oxygen and copper(II) during the ammoniacal thiosulfate leaching of gold, which is likely represented by the following simplified equation (ΔGo < 0, based on the HSC 6.0 database):
2 FeS2 + (13/2) O2 + 4 Cu(NH3)42+ + 7 H2O + 12 S2O32− = 2 FeO·OH + 4 (NH4)2SO4 +
4 Cu(S2O3)35− + 8 NH3 + 4 H+ (ΔGo = −616.1 kcal/mol)
4 Cu(S2O3)35− + 8 NH3 + 4 H+ (ΔGo = −616.1 kcal/mol)
Obviously, sulfur in pyrite can be dissolved as stable sulfate, whereas iron is much less soluble due to the formation of FeO·OH [28]. In order to investigate the possible impacts of dissolved iron species, the E-pH diagram of Fe-NH3-S2O3-H2O system at 25 °C (see Figure 2b) was constructed without considering the decomposition of thiosulfate. It is clear that soluble iron-ammonia complexes of Fe(NH3)x2+ (x = 1, 2, 4) can be formed in the pH range of 8–10. In terms of the potential, those complexes are thermodynamically unstable and are easily oxidized as Fe3O4 in the potential range of −0.6–−0.4 V which will be further oxidized as FeO·OH within potentials of −0.4–−0.3 V. In comparison with the much higher standard redox potentials of Cu(NH3)42+/Cu(S2O3)35− of 0.22 V [48] and Au(S2O3)23−/Au of 0.15 V [47], it may be that not only Cu(II) is consumed by Fe(NH3)x2+, but also Au(S2O3)23− is reduced and precipitated out as Au0 with the co-precipitation of iron in the form of Fe3O4 and FeO·OH. Moreover, the influence of pyrite on the copper species without regard for the oxidation of thiosulfate is shown by Figure 2c,d. In the presence of pyrite, the formation of Cu2O·Fe2O3 leads to the noticeable reduction of the predominance area of Cu(S2O3)35− in the pH range relevant to thiosulfate leaching of gold. This may lie in the reduction of Cu(NH3)42+ by dissolved iron species such as Fe(NH3)x2+, which causes the co-precipitation of copper with iron as Cu2O·Fe2O3 in aqueous solutions. A higher concentration of thiosulfate or ammonia is possibly beneficial for the re-dissolution of copper from Cu2O·Fe2O3 and thus renders Cu(S2O3)35− more stable.
So pyrite may be detrimental to gold leaching in copper ammoniacal thiosulfate solutions because of the following aspects: (i) oxidants of dissolved oxygen and copper(II) can be consumed due to the reducibility of pyrite; (ii) despite the rather limited dissolution of iron from pyrite [8,28], the dissolved iron species (e.g., ferrous ammine complexes) can reduce Au(S2O3)23− to Au0, which may be unbeneficial for gold leaching; (iii) the stability of redox couple of Cu(NH3)42+/Cu(S2O3)35− can be disrupted attributing to the generation of Cu2O·Fe2O3 which is more thermodynamically stable than Cu(S2O3)35−; and (iv) FeO·OH and Cu2O·Fe2O3 may be blocking products which can coat on gold surface and inhibit gold leaching.
3.2. Effect of Pyrite on Thiosulfate Leaching of Gold
Due to the inert properties of quartz, it can be used as the reference mineral to compare with pyrite [5,23,24]. During the gold leaching with thiosulfate, the effects of pyrite on thiosulfate stability and gold leaching were studied systematically.
3.2.1. Effect of Pyrite on Thiosulfate Decomposition
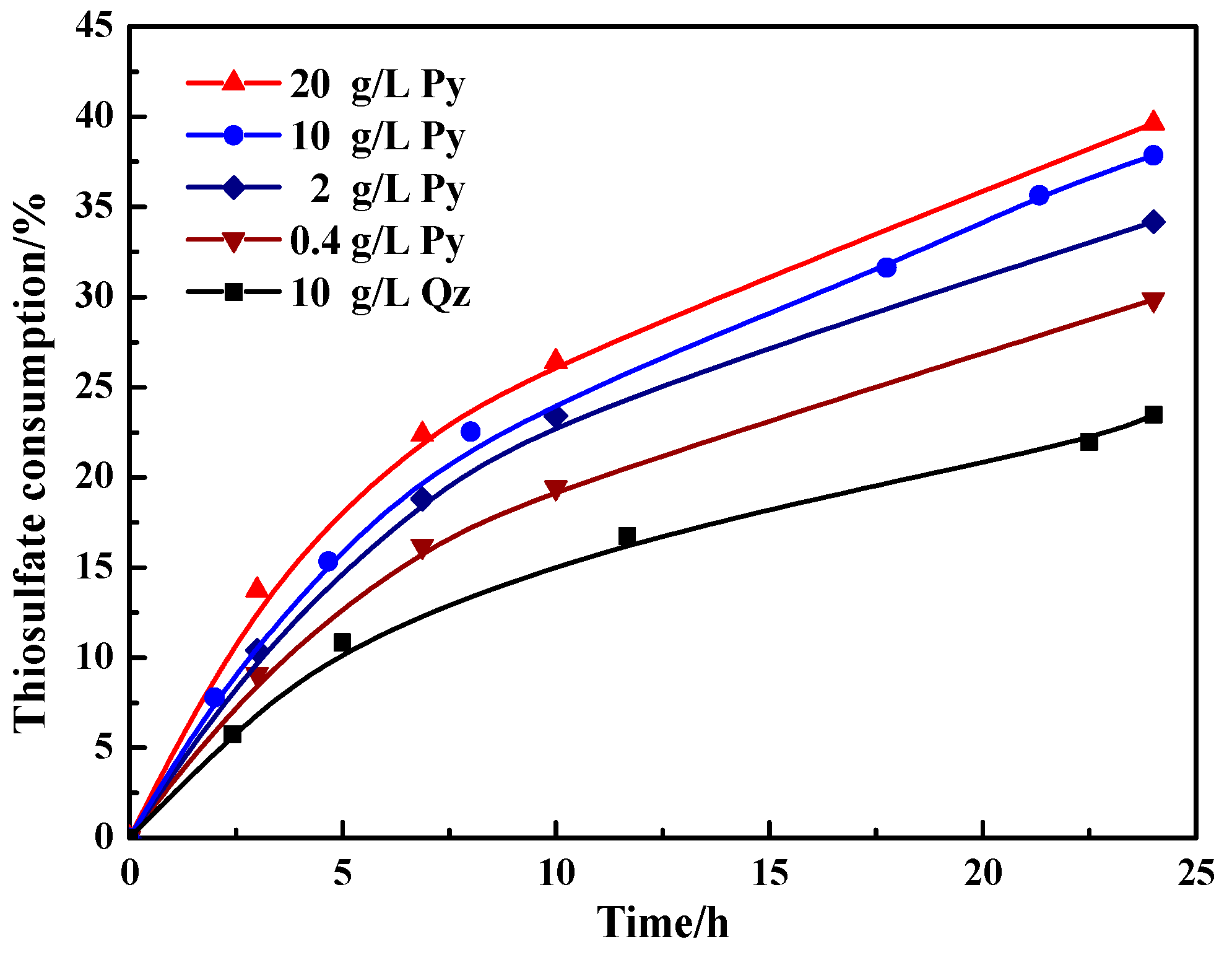
As shown in Figure 3, there was a marked growth of thiosulfate consumption both in the presence of pyrite and quartz with the increase of leaching time within 24 h. In the presence of quartz (10 g/L), 23.5% of thiosulfate degraded after leaching 24 h. However, when adding a small amount of pyrite, i.e., 0.4 g/L, the thiosulfate became unstable from the initial leaching (0–7 h), and after leaching 24 h, the consumption of thiosulfate increased to around 30%. With the increase of pyrite content, the consumption of thiosulfate further rose to about 34.2%, 37.9% and 39.6% in the presence of 2, 10 and 20 g/L pyrite, respectively. Therefore, pyrite significantly accelerated the decomposition of thiosulfate and the harmful effect could be aggravated with the increase of pyrite content.
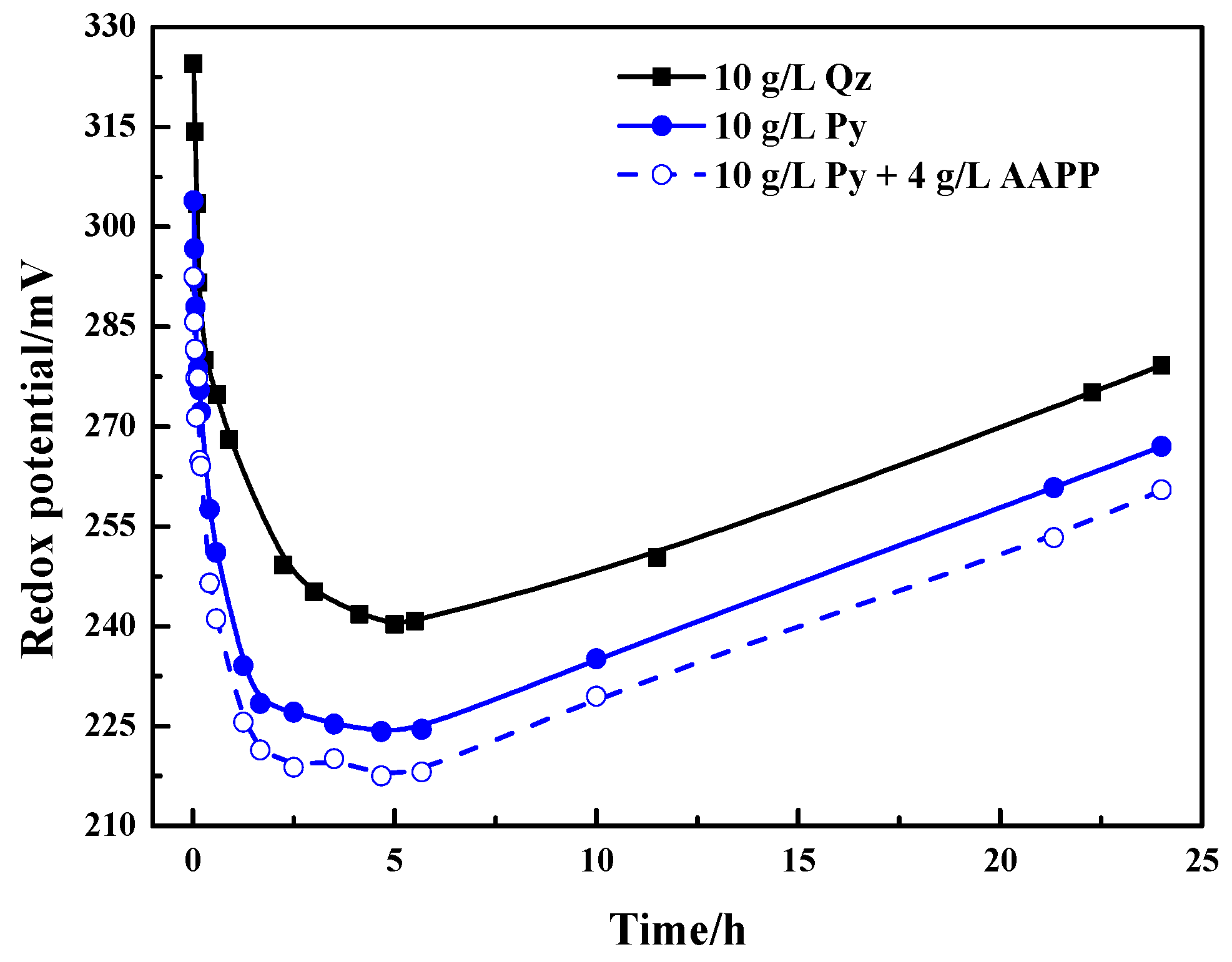
The variation of mixed slurry potential with time was also observed and recorded during the thiosulfate leaching of gold in the presence of quartz and pyrite with and without adding AAPP. The results are presented in Figure 4 and the situation in the absence of AAPP was only discussed in this section. Figure 4 indicates that the mixed slurry potential in the presence of 10 g/L quartz decreased rapidly from around 325 mV to 240–245 mV in the initial 5 h. Then the mixed potential started to increase again and rose relatively slowly to 280 mV after leaching 24 h. In the presence of 10 g/L pyrite, a similar trend of the variation of mixed potential is shown in comparison with the situation in the quartz slurry, but the potential levels were obviously lower. The mixed potential dramatically declined from about 300 to 225 mV, and then increased relatively slowly to nearly 265 mV. The potential level in the presence of pyrite was about 15–25 mV lower than that in the presence of quartz. As analyzed in Section 3.1, pyrite is easily oxidized by Cu(II) and dissolved O2 thermodynamically. According to the literature [12,36,38,42], the mixed slurry potential is determined by the Cu(II)/Cu(I) redox equilibrium potential. Hence, the mixed slurry potential in the presence of pyrite could decrease to lower levels attributing to the consumption of Cu(II) by pyrite. Lower mixed slurry potentials are more beneficial to the stability of thiosulfate, but the decomposition of thiosulfate was still aggravated by pyrite at lower mixed potentials. So during the leaching process, the mixed slurry potential is not the determinate factor of thiosulfate decomposition in the presence of pyrite. According to the above analyses, it is reasonable to believe that pyrite can catalyze the oxidative decomposing of thiosulfate.
It is thought that thiosulfates are apt to adsorb on the surface of pyrite and electrons are donated from thiosulfates to dissolved O2 via the conduction band of pyrite where electrons are highly reducing, and thereby catalyzing the oxidation of thiosulfate [29]. In an aerated copper ammoniacal thiosulfate system, Cu(II) also readily accepts electrons from the surface of pyrite and is reduced to Cu(I), which can be converted back to Cu(II) by the oxidation of dissolved O2. Cu(II) appears to be a more effective electron acceptor, likely owning to its much higher solubility (or activity) than that of dissolved O2, whose saturated solubility is only around 8 mg/L (or 0.25 mM) in water under ambient conditions of temperature and pressure [49]. Hence, the donation of electrons of thiosulfate to pyrite plays a crucial role in the catalysis process. In terms of the energy band theory, the electron in the highest occupied molecular orbital (HOMO) of the reductant will transfer to the lowest unoccupied molecular orbital (LUMO) of the oxidant in a redox reaction [29,50]. The frontier orbital energy (i.e., EHOMO and ELUMO) of pyrite (FeS2) [51] and thiosulfate anion (S2O32-) [52] are listed in Table 1 and the absolute values of difference between the frontier orbital energy of pyrite and thiosulfate anion are also shown. The closer the EHOMO and the ELUMO are, the more likely the redox reaction takes place. On the one hand, |ΔE1| of 1.780 eV is clearly less than |ΔE2| of 3.231 eV, meaning that pyrite is more likely to obtain the electron from thiosulfate anion; but on the other hand, the reaction between pyrite and thiosulfate anion alone cannot thermodynamically proceed towards the generation of tetrathionate, as seen from Equation (2) (ΔGo > 0, based on the HSC 6.0 database). In other words, the pyrite cannot figure as a terminal electron acceptor. This supports the conjecture that pyrite may act as a conduit, possibly because of its semiconducting property, where electrons from thiosulfate are delivered to a terminal electron acceptor (e.g., Cu(II) and dissolved O2) sorbed onto its surface. So in the presence of pyrite, the increased thiosulfate consumption mainly results from the pyrite-surface-catalyzed oxidation of thiosulfate. The detrimental catalysis was particularly severe in the starting stage of leaching (about 5–7 h) and seemed to be weakened over prolonged leaching periods likely due to the simultaneous oxidation of pyrite surfaces.
FeS2 + 2 S2O32− = S4O62− + FeS + S2− (ΔGo = 35.4 kcal/mol)
3.2.2. Effect of Pyrite on Gold Dissolution
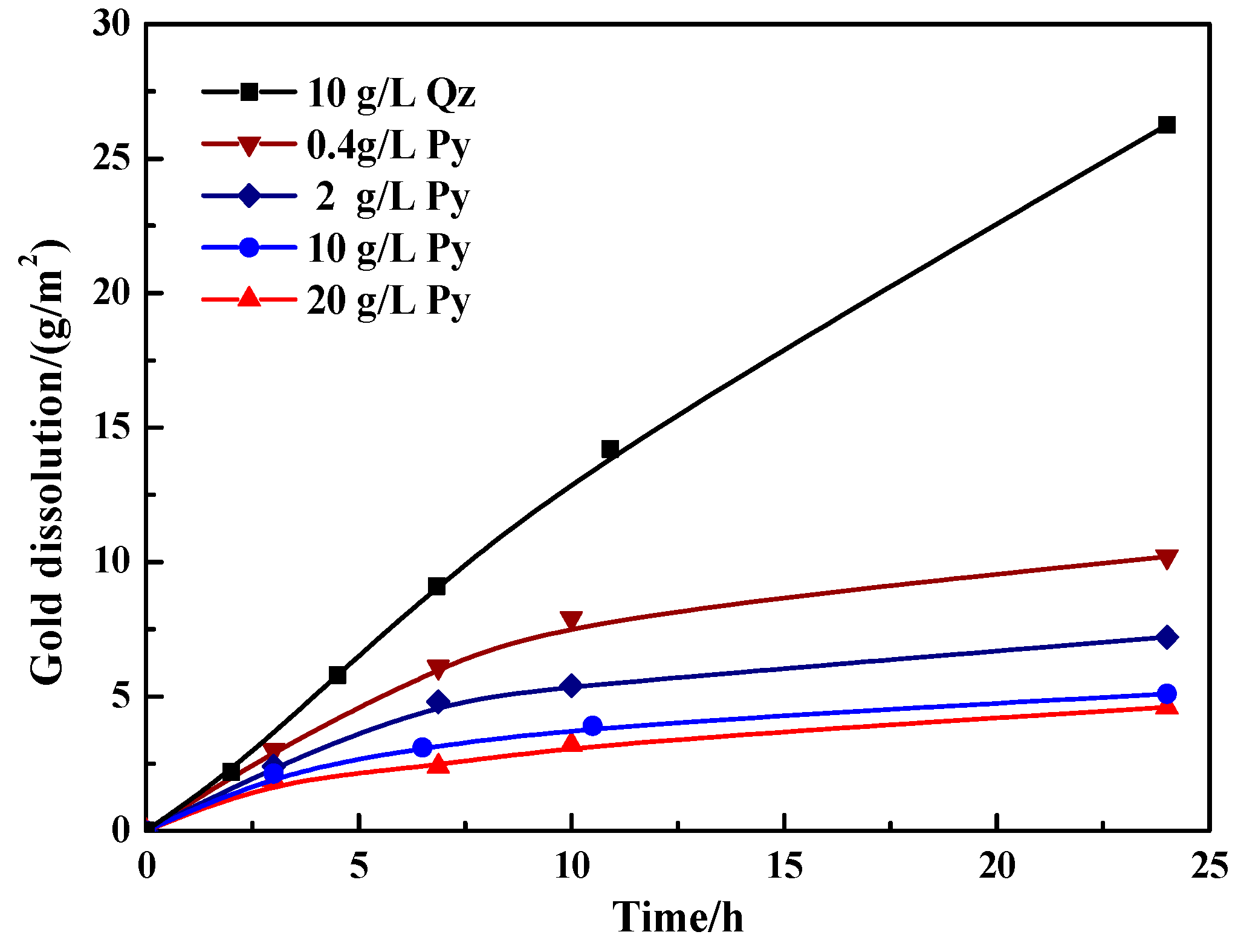
Figure 5 shows the gold dissolution in the presence of varied contents of pyrite. In the presence of quartz (10 g/L), the gold dissolution showed an approximately linear relationship with time. The dissolution of gold could increase from 2.2 g/m2 after leaching 2 h to 26.3 g/m2 after leaching 24 h in the quartz slurry. In contrast, the presence of 0.4 to 20 g/L pyrite significantly decreased the gold dissolution and this disadvantageous effect became more prominent with the increasing pyrite content (see Figure 5). After leaching 24 h, the gold dissolution was dramatically decreased to 10.2, 7.2, 5.1 and 4.6 g/m2 in the presence of 0.4, 2, 10 and 20 g/L pyrite, respectively. Particularly, the gold leaching kinetics was substantially restricted with over 10 g/L pyrite present.
Theoretically, the pyrite-catalyzed thiosulfate decomposition and the lowered mixed slurry potential are unfavorable for the leaching of gold. With respect to the mixed slurry potential, (i) it was lowered to about 225 mV in the initial leaching in the presence of 10 g/L pyrite, but it is still clearly higher than the redox couple potential of Au(S2O3)23−/Au, i.e., 150 mV; and (ii) even with the following rise of the mixed slurry potential, the gold leaching kinetics was still severely limited. Hence, the lowered mixed slurry potential may be not the primary cause for retarding gold leaching. As for the thiosulfate stability, the catalyzed decomposition of thiosulfate means the accelerated formation of tetrathionate, but tetrathionate is unstable in alkaline solutions [53]. Tetrathionate can further decompose to thiosulfate and sulfite ions together with some sulfide ion [54]. At the observed mixed slurry potential of 225–330 mV (see Figure 4) during gold leaching in the presence of pyrite, the generation of sulfide ions would not be thermodynamically expected [47]. However, the adsorption and decomposition of tetrathionate may occur on pyrite surfaces at lower mixed slurry potentials when the rate of Cu(II) and dissolved O2 consumption by the absorbed decomposing tetrathionate exceeds the rate of Cu(II) and dissolved O2 mass transfer from solution [31]. Consequently, sulfide ions may be generated on the surface of pyrite and diffuse to the solution, leading to the precipitation of copper as copper sulfides, which may coat on gold surfaces. It is possible that the steady accumulation of blocking products on the gold surface renders the gold leaching kinetics rather restricted.
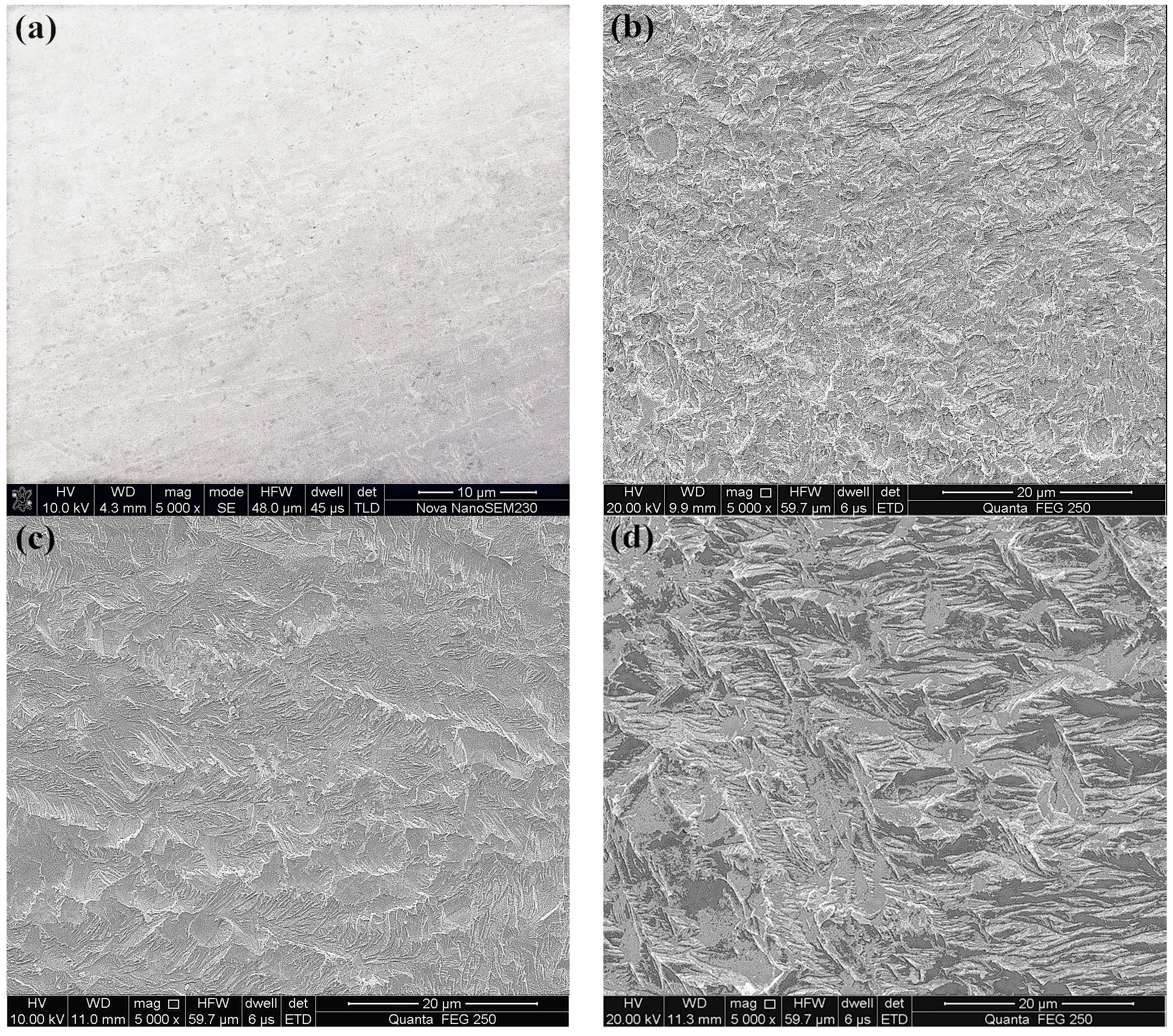
The surface morphology of the gold foils after leaching was subjected to SEM analysis. Figure 6 shows the SEM images of the original gold surface and the leached gold surfaces in the presence of quartz and pyrite without and with the addition of AAPP. The situation with adding AAPP would be discussed in detail in Section 3.3.
Clearly, different morphological structures and varied degrees of erosion were presented on the leached gold surfaces comparing with the original gold surface, such as in Figure 6a. In the quartz slurry, the leached gold surface presented obvious erosion characteristics of edges and pits as depicted in Figure 6b. Whereas in the presence of 10 g/L pyrite the leached gold surface was relatively smooth and only slightly wrinkled erosions were unevenly spotted on the gold surface, as shown in Figure 6c. A passivation film appeared to be formed on the gold surface.
A better understanding of the mechanism for pyrite passivating the leaching of gold can be achieved by the XPS analyses of leached gold surfaces. The XPS spectra were recorded for S 2p, Cu 2p and Fe 2p in the presence of 10 g/L quartz and 10 g/L pyrite without and with adding 4 g/L AAPP. Only the situations in the absence of AAPP were discussed in this section.
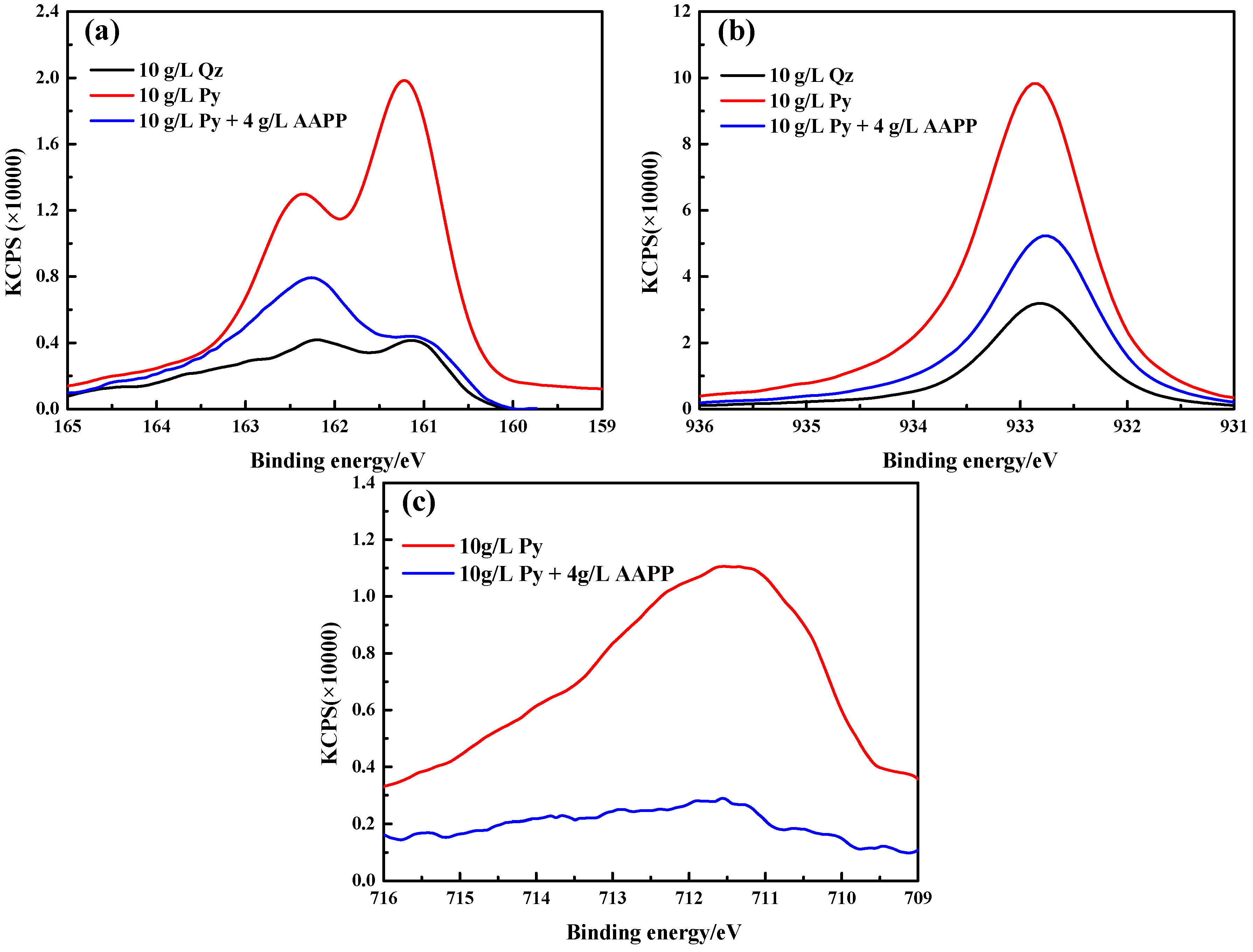
Figure 7a shows the S 2p spectra which were posited at 160–165 eV. There were mainly two peaks at approximately 161.2 and 162.4 eV. The lower binding energy peak is likely assigned to monosulfide S2− species of CuS, Cu2S, etc. [55,56]. The higher energy peak may have originated from S22− in disulfides such as CuS2 or FeS2 [57], of which FeS2 may be pyrite fines adsorbed on the gold surface whilst CuS2 may occur as an ammonia copper(I) polysulfide [21]. Obviously, the intensity and area of peak in the presence of pyrite was much stronger and larger than that in the presence of quartz, indicating that the increased quantity of sulfur element on gold surfaces. This possibly results from the pyrite-catalyzed decomposition of thiosulfate.
The XPS spectra of Cu 2p are presented in Figure 7b and all exhibited a peak at around 932.7 eV. It is likely derived from cuprous species of Cu2S/Cu2O [58,59] and cupric species of CuS/CuO/Cu(OH)2 [58,60,61]. Based on the intensity and area of peak, pyrite could also promote the precipitation of copper onto the gold surface.
As seen in Figure 7c, the Fe 2p spectra for the leached gold surface in the presence of pyrite presented an obvious peak at about 711.5 eV, which is likely originated from FeO·OH, Fe2O3/Cu2O·Fe2O3 [62,63]. This may stem from the oxidation of pyrite and its dissolved iron species.
So in the presence of pyrite, the likely composition of the passivation film included CuS, CuS2, Cu2S, CuO, Cu(OH)2, Cu2O, Cu2O·Fe2O3, FeO·OH, and so on. The copper species seemed to be dominant in the passivation film according to the intensity and area of peaks.
3.3. Role of AAPP Additive
In the copper ammoniacal thiosulfate solution with pyrite present, AAPP was added to investigate its possible role in stabilizing thiosulfate and enhancing gold leaching. The results of effect of AAPP on thiosulfate consumption and gold dissolution are presented in Figure 8.
3.3.1. Role of AAPP in Thiosulfate Decomposition
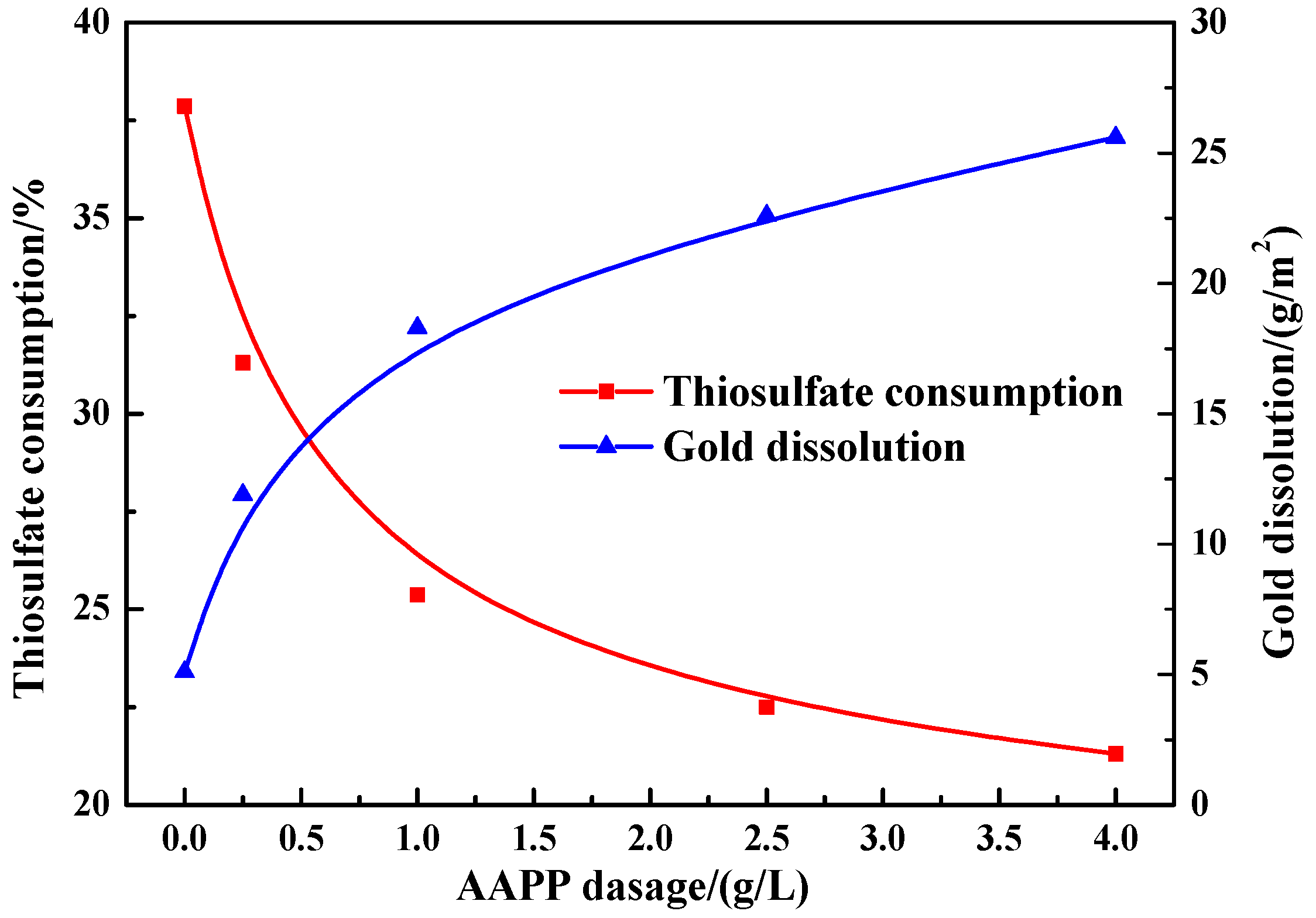
Figure 8 illustrates the consumption of thiosulfate after leaching 24 h in the presence of 10 g/L pyrite with adding varied dosages of AAPP. The presence of AAPP noticeably decreased the overall decomposition of thiosulfate, and this effect became more prominent at a higher dosage of AAPP. The thiosulfate consumption was significantly reduced from 37.9 (without adding AAPP) to 25.4% at a dosage of 1 g/L AAPP. When the dosage of AAPP exceeded 1 g/L, the decrease of thiosulfate consumption was relatively slow. The thiosulfate consumption was decreased to 21.3% at a higher dosage of 4 g/L AAPP, which is lower than that in the quartz slurry without adding AAPP (23.5%, see Figure 3).
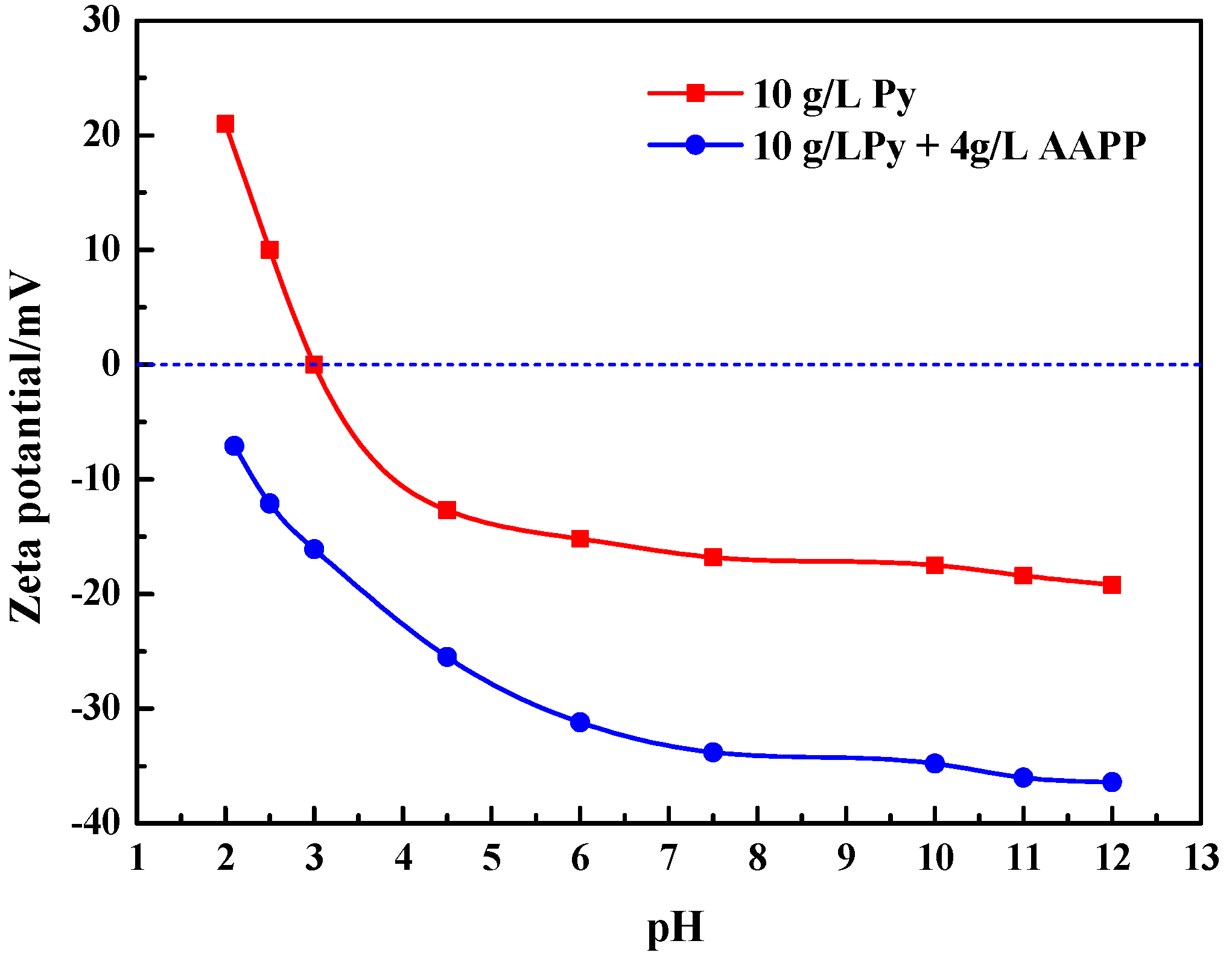
It has been concluded from Section 3.2.1 that the increased consumption of thiosulfate in the pyrite slurry possibly results from the decomposition of thiosulfate catalyzed by pyrite. AAPP possibly adsorbed on the pyrite surface especially where there were high-energy defect sites and crystal boundaries. Actually, Figure 9 exhibits the apparent zeta potential of pyrite particles as a function of pH in the absence and presence of 4 g/L AAPP. The zeta potential curve shows that the isoelectric point (IEP) of pyrite was around 3.0, which is close to the reported value [64]. It is clear that the presence of AAPP elevated the negativity of the surface charges of pyrite particles, demonstrating the adsorption of electronegative radical ions of AAPP on pyrite surfaces. This possibly results in the markedly decreasing affinity of pyrite surfaces for S2O32− because the surface active sites where S2O32− can easily adsorb had been occupied by AAPP radical ions, and thereby noticeably weakened the catalytic effect of pyrite. In addition, the adsorption of AAPP may also change the semiconducting properties of pyrite surfaces. Therefore, the pyrite-catalyzed decomposition of thiosulfate could be weakened with the addition of AAPP.
As described previously [44,65,66], in ammoniacal copper thiosulfate solution cupric ion primarily occurs as the complex of Cu(NH3)42+ with the remaining two axial coordinate sites occupied by water, and thiosulfate may replace water from a axial coordinate site. After adding AAPP, the hydroxyl and phosphate groups of AAPP in solution may enhance the competition with thiosulfate anions to complex with the cupric ion at the axial coordinate sites. Hence, AAPP could also lower the oxidation rate of S2O32− by Cu(II), and it was more favorable for the stabilization of thiosulfate at a higher AAPP dosage. It is depicted in Figure 1 that ammonium could also be ionized from AAPP in solution besides the radical ions of functional groups. Ammonium can not only maintain the solution pH by the buffering effect of NH4+/NH3, but also render cupric ions more stable owing to the increased total concentration of ammonia. As a result, the oxidant of Cu(II) became more stable in the presence of AAPP because of the coordination of functional groups and ammonia with cupric ions. It is predictable that the mixed slurry potential would be lowered since the mixed slurry potential is determined by the Cu(II)/Cu(I) redox equilibrium potential [12,36,38,42]. Figure 4 presents the variation of the mixed slurry potentials without and with the addition of 4 g/L AAPP. The variation trends of the mixed potential were alike, and as expected, the potential level was lowered after adding AAPP. A decrease of about 10 mV was achieved in the presence of AAPP. So it is reasonable to believe that AAPP can alleviate the oxidation of S2O32− by Cu(II).
3.3.2. Role of AAPP in Gold Dissolution
In the presence of 10 g/L pyrite, Figure 8 also indicates the dissolution of gold after leaching 24 h at varied dosages of AAPP. Clearly, with the addition of AAPP, the overall gold dissolution obtained a remarkable improvement, and a higher dosage of AAPP appeared to be more advantageous. In the presence of 1 g/L AAPP, the gold dissolution could substantially increase from 5.1 to 18.3 g/m2. As the addition of AAPP rose to 4 g/L, the gold dissolution constantly increased to 25.6 g/m2, which is comparable with that in the quartz slurry without adding AAPP (26.3 g/m2, see Figure 5).
As presented in Figure 4, the addition of AAPP reduced the mixed slurry potential in the presence of pyrite, but a pronounced enhancement of gold dissolution was still realized. This further suggests that the mixed slurry potential is not the primary cause of the hindered gold leaching. In association with results in Section 3.3.1, the significantly decreased thiosulfate consumption in the presence of AAPP could diminish the likelihood of forming excessive passivating products on the gold surface. So the improved dissolution of gold with the addition of AAPP is possibly owing to the reduction of leaching passivation, besides the stabilization of thiosulfate by AAPP.
Similar with other organic additives such as polyphosphate [40], CMC [42], HA [15,44], and so on, non-selective adsorption of AAPP on the surfaces of pyrite, gold and passivation species likely occurred owing to numerous negatively charged and hydrophilic functional groups of phosphate and hydroxyl. Besides the pyrite surface, the surfaces of gold and passivation species may also appear negatively charged and hydrophilic by AAPP adsorption. As a result, the precipitation of passivating products on the gold surface was likely prevented by electrostatic repulsion during the leaching process. It seems that a higher dosage of AAPP could achieve a higher coverage of the gold surface by AAPP, and thereby realize a greater extent of alleviation of the leaching passivation. Meanwhile, the adsorption of AAPP still enabled ammoniacal thiosulfate leach solutions readily accessible to the hydrophilic gold surface.
Typical SEM images of the leached gold surface after leaching 24 h in the presence of pyrite (10 g/L), with and without adding AAPP, (4 g/L) are shown in Figure 6. As clearly exhibited in Figure 6c,d, the wrinkles on the leached gold surface were still partial and uneven, but the slightly wrinkled erosion was distinctly enhanced in the presence of AAPP due to the serious erosion of matrix gold.
Figure 7 also illustrates the XPS spectra for S 2p, Cu 2p and Fe 2p of the leached gold surface after leaching 24 h with the addition of AAPP. In the presence of 10 g/L pyrite, the addition of AAPP (4 g/L) obviously decreased the peak intensities and areas of all these element spectra. This proves that AAPP effectively prevents the blocking products from forming and/or accumulating on the gold surface.
4. Conclusions
During gold leaching in copper ammoniacal thiosulfate solutions, pyrite could significantly accelerate the decomposition of thiosulfate and inhibit the leaching of gold. It was found that the mixed slurry potential was not the leading factor for both promoted thiosulfate decomposition and restricted gold leaching kinetics. The analysis of frontier orbital energy demonstrated that the catalysis of pyrite was the dominant cause for the increased thiosulfate consumption at lower mixed potentials. This detrimental effect presented as more severe at higher contents of pyrite. Meanwhile, it was obtained from the analyses of leaching tests, SEM and XPS that the passivation of the gold surface in the presence of pyrite caused the reduced gold dissolution. A higher content of pyrite was more unfavorable for the leaching of gold. The passivation film on the leached gold surface was mainly composed of copper species, including CuS, CuS2, Cu2S, CuO, Cu(OH)2, Cu2O, Cu2O·Fe2O3, etc.
Nevertheless, in the presence of pyrite, the addition of AAPP is beneficial for both the stability of thiosulfate and the leaching of gold from multiple aspects. This study showed that: (i) AAPP could effectively weaken the harmful effect of pyrite, catalyzing the decomposition of thiosulfate by blocking the contact of thiosulfate anions with pyrite surface; (ii) the phosphate and hydroxyl groups of AAPP also competed with thiosulfate anions to complex with the cupric ion at the axial coordinate sites, and thereby decreasing the oxidation of thiosulfate by cupric ions; simultaneously, (iii) the improved thiosulfate stability by AAPP reduced the quantity of possible passivation species; and (iv) AAPP could render both the gold surface and passivation species negatively charged due to the indiscriminate adsorption, and thus preventing passivation species from coating on the gold surface owing to surface charge and electrostatic repulsion.
Acknowledgments
Financial supports from the National Natural Science Foundation of China (Grant Nos. 51504293 and 51574284), the China Postdoctoral Science Foundation (Grant No. 2014M550422), the Hunan Provincial Natural Science Foundation of China (Grant No. 2015JJ3149), the Open-End Fund for the Valuable and Precision Instruments of Central South University (Grant No. CSUZC201704) and China Scholarship Council (Grant No. 201606370128) are all gratefully acknowledged.
Author Contributions
Bin Xu and Qian Li conceived and designed the study; Yongbin Yang and Tao Jiang contributed reagents/materials/analysis tools; Xin Min and Xi Zhang performed the experiments; Xiaoliang Liu and Yinghe He analyzed the data; Xiaoliang Liu wrote the paper with inputs from Yinghe He.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Free energies of formation (kcal/mol) for species of iron, copper, sulfur, etc.
| Species | ΔGo298/ (kcal/mol) | Species | ΔGo298/ (kcal/mol) | Species | ΔGo298/ (kcal/mol) | Species | ΔGo298/ (kcal/mol) |
|---|---|---|---|---|---|---|---|
| Fe | 0 | Fe(OH)O(aq) | −105.700 | Cu(NH3)42+(aq) | −28.011 | H2S(aq) | −6.607 |
| Fe0.877S | −25.533 | HFeO2(aq) | −105.700 | Cu(NH3)52+(aq) | −33.759 | HS−(aq) | 2.973 |
| FeS | −24.369 | HFeO2−(aq) | −95.353 | Cu(NH3)+(aq) | −2.516 | HSO3−(aq) | −126.103 |
| FeS2 | −38.247 | Fe(NH3)2+(aq) | −30.159 | Cu(NH3)2+(aq) | −15.614 | HSO4−(aq) | −180.524 |
| Fe2S3 | −67.03 | Fe(NH3)22+(aq) | −37.625 | Cu(S2O3)−(aq) | −126.885 | HS2O3−(aq) | −127.183 |
| FeO | −58.729 | Fe(NH3)42+(aq) | −52.421 | Cu(S2O3)23−(aq) | −254.368 | HS2O4−(aq) | −146.862 |
| Fe2O3 | −177.114 | Fe(S2O3)+(aq) | −131.632 | Cu(S2O3)35−(aq) | −381.404 | S2−(aq) | 20.548 |
| Fe3O4 | −241.956 | Fe(S2O3)(aq) | −147.928 | CuO22−(aq) | −41.220 | S22−(aq) | 19.055 |
| Fe(OH)2 | −117.578 | Cu | 0.000 | Cu(OH)+(aq) | −30.210 | S32−(aq) | 17.657 |
| Fe(OH)3 | −168.638 | CuS | −13.530 | Cu(OH)3−(aq) | −119.865 | S42−(aq) | 16.589 |
| Fe2O3·H2O | 5.776 | Cu2S | −20.607 | Cu(OH)42−(aq) | −156.970 | S52−(aq) | 15.821 |
| FeO·OH | −116.928 | CuO | −30.387 | Cu2(OH)3+(aq) | −16.424 | S62−(aq) | 15.827 |
| Fe3+(aq) | −4.107 | Cu2O | −35.335 | Cu2(OH)22+(aq) | −67.825 | SO32−(aq) | −116.287 |
| Fe2+(aq) | −21.875 | CuO·Fe2O3 | −205.969 | Cu3(OH)42+(aq) | −151.420 | SO42−(aq) | −177.907 |
| FeO(aq) | −50.715 | Cu2O·Fe2O3 | −220.015 | Cu(OH)O−(aq) | −60.096 | S2O32−(aq) | −124.825 |
| FeO+(aq) | −53.093 | Cu(OH)2 | −85.262 | H+(aq) | 0 | S2O42−(aq) | −143.539 |
| FeO2−(aq) | −92.642 | Cu2+(aq) | 15.545 | O2(aq) | 3.899 | S2O62−(aq) | −231.605 |
| FeOH2+(aq) | −57.830 | Cu+(aq) | 11.946 | H2O(aq) | −56.678 | S2O82−(aq) | −266.46 |
| FeOH+(aq) | −65.845 | Cu(NH3)2+(aq) | 3.400 | NH3(aq) | −6.375 | S3O62−(aq) | −228.815 |
| Fe(OH)2+(aq) | −108.077 | Cu(NH3)22+(aq) | −7.979 | NH4+(aq) | −18.977 | S4O62−(aq) | −248.627 |
| Fe2(OH)24+(aq) | −111.743 | Cu(NH3)32+(aq) | −18.500 | S | 0 | S5O62−(aq) | −228.331 |
References
- Aylmore, M.G.; Muir, D.M. Thiosulfate leaching of gold—A review. Miner. Eng. 2001, 14, 135–174. [Google Scholar] [CrossRef]
- Xu, B.; Kong, W.; Li, Q.; Yang, Y.; Jiang, T.; Liu, X. A review of thiosulfate leaching of gold: Focus on thiosulfate consumption and gold recovery from pregnant solution. Metals 2017, 7, 222. [Google Scholar] [CrossRef]
- Kerley, B.J. Recovery of Precious Metals from Difficult Ores. U.S. Patent 4,269,622, 26 May 1981. [Google Scholar]
- Kerley, B.J. Recovery of Precious Metals from Difficult Ores. U.S. Patent 4,369,061, 18 January 1983. [Google Scholar]
- Abbruzzese, C.; Fornari, P.; Massidda, R.; Vegliò, F.; Ubaldini, S. Thiosulphate leaching for gold hydrometallurgy. Hydrometallurgy 1995, 39, 265–276. [Google Scholar] [CrossRef]
- Marchbank, A.R.; Thomas, K.G.; Dreisinger, D.; Fleming, C. Gold Recovery from Refractory Carbonaceous Ores by Pressure Oxidation and Thiosulphate Leaching. U.S. Patent 5,536,297, 16 July 1996. [Google Scholar]
- Wan, R.Y.; Brierley, J.A. Thiosulfate leaching following biooxidation pretreatment for gold recovery from refractory carbonaceous-sulfidic ore. Miner. Eng. 1997, 76, 76–80. [Google Scholar]
- Xia, C. Associated Sulfide Minerals in Thiosulfate Leaching of Gold: Problems and Solutions. Ph.D. Thesis, Department of Mining Engineering, Queen’s University, Kingston, ON, Canada, 2008. [Google Scholar]
- Feng, D.; van Deventer, J.S.J. The effect of sulphur species on thiosulphate leaching of gold. Miner. Eng. 2007, 20, 273–281. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. The role of oxygen in thiosulphate leaching of gold. Hydrometallurgy 2007, 85, 193–202. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Effect of hematite on thiosulphate leaching of gold. Int. J. Miner. Process. 2007, 82, 138–147. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Effect of thiosulphate salts on ammoniacal thiosulphate leaching of gold. Hydrometallurgy 2010, 105, 120–126. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Oxidative pre-treatment in thiosulphate leaching of sulphide gold ores. Int. J. Miner. Process. 2010, 94, 28–34. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Liu, X.; Xu, B.; Yang, Y.; Jiang, T. Improvement of Gold Leaching from a Refractory Gold Concentrate Calcine by Separate Pretreatment of Coarse and Fine Size Fractions. Minerals 2017, 7, 80. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Y.; Jiang, T.; Li, Q.; Zhang, X.; Wang, D. Improved thiosulfate leaching of a refractory gold concentrate calcine with additives. Hydrometallurgy 2015, 152, 214–222. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Y.; Li, Q.; Jiang, T.; Li, G. Stage leaching of a complex polymetallic sulfide concentrate: Focus on the extraction of Ag and Au. Hydrometallurgy 2015, 159, 87–94. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Y.; Li, Q.; Yin, W.; Jiang, T.; Li, G. Thiosulfate leaching of Au, Ag and Pd from a high Sn, Pb and Sb bearing decopperized anode slime. Hydrometallurgy 2016, 164, 278–287. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Y.; Li, Q.; Jiang, T.; Liu, S.; Li, G. The development of an environmentally friendly leaching process of a high C, As and Sb bearing sulfide gold concentrate. Miner. Eng. 2016, 89, 138–147. [Google Scholar] [CrossRef]
- Pedraza, A.M.; Villegas, I.; Freund, P.L.; Chornik, B. Electro-oxidation of thiosulphate ion on gold: Study by means of cyclic voltammetry and Auger electron spectroscopy. J. Electroanal. Chem. 1988, 250, 443–449. [Google Scholar] [CrossRef]
- Wan, R.-Y.; LeVier, K.M. Solution chemistry factors for gold thiosulfate heap leaching. Int. J. Miner. Process. 2003, 72, 311–322. [Google Scholar] [CrossRef]
- Jeffrey, M.I.; Watling, K.; Hope, G.A.; Woods, R. Identification of surface species that inhibit and passivate thiosulfate leaching of gold. Miner. Eng. 2008, 21, 443–452. [Google Scholar] [CrossRef]
- Mirza, J.; Smith, S.R.; Baron, J.Y.; Choi, Y.; Lipkowski, J. A SERS characterization of the stability of polythionates at the gold-electrolyte interface. Surf. Sci. 2015, 631, 196–206. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Ammoniacal thiosulphate leaching of gold in the presence of pyrite. Hydrometallurgy 2006, 82, 126–132. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Effect of sulfides on gold dissolution in ammoniacal thiosulfate medium. Metall. Mater. Trans. B 2003, 34, 5–13. [Google Scholar] [CrossRef]
- Yang, Y.B.; Zhang, X.; Xu, B.; Li, Q.; Jiang, T.; Wang, Y.X. Effect of arsenopyrite on thiosulfate leaching of gold. Trans. Nonferr. Met. Soc. 2015, 25, 3454–3460. [Google Scholar] [CrossRef]
- Liu, X.; Xu, B.; Yang, Y.; Li, Q.; Jiang, T.; Zhang, X.; Zhang, Y. Effect of galena on thiosulfate leaching of gold. Hydrometallurgy 2017, 171, 157–164. [Google Scholar] [CrossRef]
- Marsden, J.O.; House, C.I. The Chemistry of Gold Extraction, 2nd ed.; Society for Mining, Metallurgy, and Exploration: Littleton, CO, USA, 2006. [Google Scholar]
- Feng, D.; van Deventer, J.S.J. Leaching behaviour of sulfides in ammoniacal thiosulphate systems. Hydrometallurgy 2002, 63, 189–200. [Google Scholar] [CrossRef]
- Xu, Y.; Schoonen, M.A.A. The stability of thiosulfate in the presence of pyrite in low-temperature aqueous solutions. Geochim. Cosmochim. Acta 1995, 59, 4605–4622. [Google Scholar] [CrossRef]
- Zhang, H.; Jeffrey, M.I. A study of pyrite catalysed oxidation of thiosulfate. In Proceedings of the Sixth International Symposium Hydrometallurgy 2008, Phoenix, AZ, USA, 17–20 August 2008; Young, C.A., Talor, P.R., Anderson, C.G., Choi, Y., Eds.; Society for Mining, Metallurgy, and Exploration: Littleton, CO, USA, 2008; pp. 769–778. [Google Scholar]
- Aylmore, M.G.; Muir, D.M.; Staunton, W.P. Effect of minerals on the stability of gold in copper ammoniacal thiosulfate solutions—The role of copper, silver and polythionates. Hydrometallurgy 2014, 143, 12–22. [Google Scholar] [CrossRef]
- Jean, G.E.; Michael, B.G. An XPS and SEM study of gold deposition at low temperatures on sulphide mineral surfaces: Concentration of gold by adsorption/reduction. Geochim. Cosmochim. Acta 1985, 49, 979–987. [Google Scholar] [CrossRef]
- Schoonen, M.A.A.; Fisher, N.S.; Wente, M. Gold sorption onto pyrite and goethite: A radiotracer study. Geochim. Cosmochim. Acta 1992, 56, 1801–1814. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Preg-robbing phenomena in the thiosulphate leaching of gold ores. Miner. Eng. 2001, 14, 1387–1402. [Google Scholar] [CrossRef]
- Zhang, X.M.; Senanayake, G. A review of ammoniacal thiosulfate leaching of gold: An update useful for further research in non-cyanide gold lixiviants. Miner. Process. Extr. Metall. Rev. 2016, 37, 385–411. [Google Scholar] [CrossRef]
- Senanayake, G.; Zhang, X.M. Gold leaching by copper(II) in ammoniacal thiosulphate solutions in the presence of additives. Part II: Effect of residual Cu(II), pH and redox potentials on reactivity of colloidal gold. Hydrometallurgy 2012, 115–116, 21–29. [Google Scholar] [CrossRef]
- Senanayake, G. Analysis of reaction kinetics, speciation and mechanism of gold leaching and thiosulfate oxidation by ammoniacal copper(II) solutions. Hydrometallurgy 2004, 75, 55–75. [Google Scholar] [CrossRef]
- Senanayake, G. Gold leaching by copper(II) in ammoniacal thiosulphate solutions in the presence of additives. Part I: A review of the effect of hard–soft and Lewis acid-base properties and interactions of ions. Hydrometallurgy 2012, 115–116, 1–20. [Google Scholar] [CrossRef]
- Xia, C.; Yen, W.T.; Deschenes, G. Improvement of thiosulfate stability in gold leaching. Miner. Metall. Process. 2003, 20, 68–72. [Google Scholar]
- Feng, D.; van Deventer, J.S.J. Thiosulphate leaching of gold in the presence of orthophosphate and polyphosphate. Hydrometallurgy 2011, 106, 38–45. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Thiosulphate leaching of gold in the presence of ethylenediaminetetraacetic acid (EDTA). Miner. Eng. 2010, 23, 143–150. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. Thiosulphate leaching of gold in the presence of carboxymethyl cellulose (CMC). Miner. Eng. 2011, 24, 115–121. [Google Scholar] [CrossRef]
- Feng, D.; van Deventer, J.S.J. The role of amino acids in the thiosulphate leaching of gold. Miner. Eng. 2011, 24, 1022–1024. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Y.; Li, Q.; Jiang, T.; Zhang, X.; Li, G. Effect of common associated sulfide minerals on thiosulfate leaching of gold and the role of humic acid additive. Hydrometallurgy 2017, 171, 44–52. [Google Scholar] [CrossRef]
- Dong, Y.; Zhu, Y.; Dai, X.; Zhao, D.; Zhou, X.; Qi, Y.; Koo, J.H. Ammonium alcohol polyvinyl phosphate intercalated LDHs/epoxy nanocomposites. J. Therm. Anal. Calorim. 2015, 122, 135–144. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Zou, J.; Duan, H.; Li, J.; Chen, C.; Meng, P. Preparation and membrane separation performances of quarternized ammonium cationic polyvinyl alcohol. J. Appl. Polym. Sci. 2011, 119, 2584–2594. [Google Scholar] [CrossRef]
- Aylmore, M.G.; Muir, D.M. Thermodynamic analysis of gold leaching by ammoniacal thiosulfate using Eh/pH and speciation diagrams. Miner. Metall. Proc. 2001, 18, 221–227. [Google Scholar]
- Hiskey, B.J.; Atluri, V.P. Dissolution chemistry of gold and silver in different lixiviants. Miner. Process. Extr. Met. Rev. 1988, 4, 95–134. [Google Scholar] [CrossRef]
- Weiss, R.F. The solubility of nitrogen, oxygen and argon in water and seawater. Deep-Sea Res. 1970, 17, 721–735. [Google Scholar] [CrossRef]
- Luther, G.W. The frontier-molecular-orbital theory approach in geochemical process. In Aquatic Chemical Kinetics; Stumm, W., Ed.; John Wiley & Sons: Toronto, ON, Canada, 1990; Chapter 6; pp. 173–198. [Google Scholar]
- Zhang, X. Research on the Effect of Sulfide Minerals on Thiosulfate Leaching of Gold and the Intensification Method. Master’s Thesis, School of Minerals Processing and Bioengineering, Central South University, Changsha, China, 2015. [Google Scholar]
- Ouyang, S. Research on Quantum Chemistry of Effects of Impurities and Flotation Separation Process of Pb-Zn Sulfide Containing High Sulfur in Huanjiang Guangxi. Master’s Thesis, Department of Mining Engineering, Guangxi University, Nanning, China, 2010. [Google Scholar]
- Zhang, H.; Dreisinger, D.B. The adsorption of gold and copper onto ion-exchange resins from ammoniacal thiosulphate solutions. Hydrometallurgy 2002, 66, 67–76. [Google Scholar] [CrossRef]
- Miura, Y.; Koh, T. Spectroscopic determination of tetrathionate by means of its alkaline decomposition. Nippon Kagaku Kaishi 1983, 11, 1597–1601. [Google Scholar] [CrossRef]
- Langer, D.W.; Vesely, C.J. Electronic core levels of zinc chalcogenides. Phys. Rev. B 1970, 2, 4885–4892. [Google Scholar] [CrossRef]
- Manocha, A.S.; Park, R.L. Flotation related ESCA studies on PbS surfaces. Appl. Surf. Sci. 1977, 1, 129–141. [Google Scholar] [CrossRef]
- Laajalehto, K.; Kartio, I.; Nowak, P. XPS study of clean metal sulfide surfaces. Appl. Surf. Sci. 1994, 81, 11–15. [Google Scholar] [CrossRef]
- Perry, D.L.; Taylor, J.A. X-ray photoelectron and Auger spectroscopic studies of Cu2S and CuS. J. Mater. Sci. Lett. 1986, 5, 384–386. [Google Scholar] [CrossRef]
- Jolley, J.G.; Geesey, G.G.; Hankins, M.R.; Wright, R.B.; Wichlacz, P.L. Auger electron and X-ray photoelectron spectroscopic study of the biocorrosion of copper by alginic acid polysaccharide. Appl. Surf. Sci. 1989, 37, 469–480. [Google Scholar] [CrossRef]
- Deroubaix, G.; Marcus, P. X-ray photoelectron spectroscopy analysis of copper and zinc oxides and sulphides. Surf. Interface Anal. 1992, 18, 39–46. [Google Scholar] [CrossRef]
- Losev, A.; Rostov, K.; Tyuliev, G. Electron beam induced reduction of CuO in the presence of a surface carbonaceous layer: An XPS/HREELS study. Surf. Sci. 1989, 213, 564–579. [Google Scholar] [CrossRef]
- Tan, B.J.; Klabunde, K.J.; Sherwood, P.M.A. X-ray photoelectron spectroscopy studies of solvated metal atom dispersed catalysts. Monometallic iron and bimetallic iron-cobalt particles on alumina. Chem. Mater. 2002, 2, 186–191. [Google Scholar] [CrossRef]
- Oscarson, D.W.; Huang, P.M.; Defosse, C.; Herbillon, A. Oxidative power of Mn(IV) and Fe(III) oxides with respect to As(III) in terrestrial and aquatic environments. Nature 1981, 291, 50–51. [Google Scholar] [CrossRef]
- Reyes-Bozo, L.; Escudey, M.; Vyhmeister, E.; Higueras, P.; Godoy-Faúndez, A.; Salazar, J.L.; Valdés-González, H.; Wolf-Sepúlveda, G.; Herrera-Urbina, R. Adsorption of biosolids and their main components on chalcopyrite, molybdenite and pyrite: Zeta potential and FTIR spectroscopy studies. Miner. Eng. 2015, 78, 128–135. [Google Scholar] [CrossRef]
- Byerley, J.J.; Fouda, S.A.; Rempel, G.L. Kinetics and mechanism of the oxidation of thiosulphate ions by copper(II) ions in aqueous ammonia solution. J. Chem. Soc. Dalton Trans. 1973, 8, 889–893. [Google Scholar] [CrossRef]
- Breuer, P.L.; Jeffrey, M.I. The reduction of copper(II) and the oxidation of thiosulfate and oxysulfur anions in gold leaching solutions. Hydrometallurgy 2003, 70, 163–173. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of the synthesis of ammonium alcohol polyvinyl phosphate (AAPP).
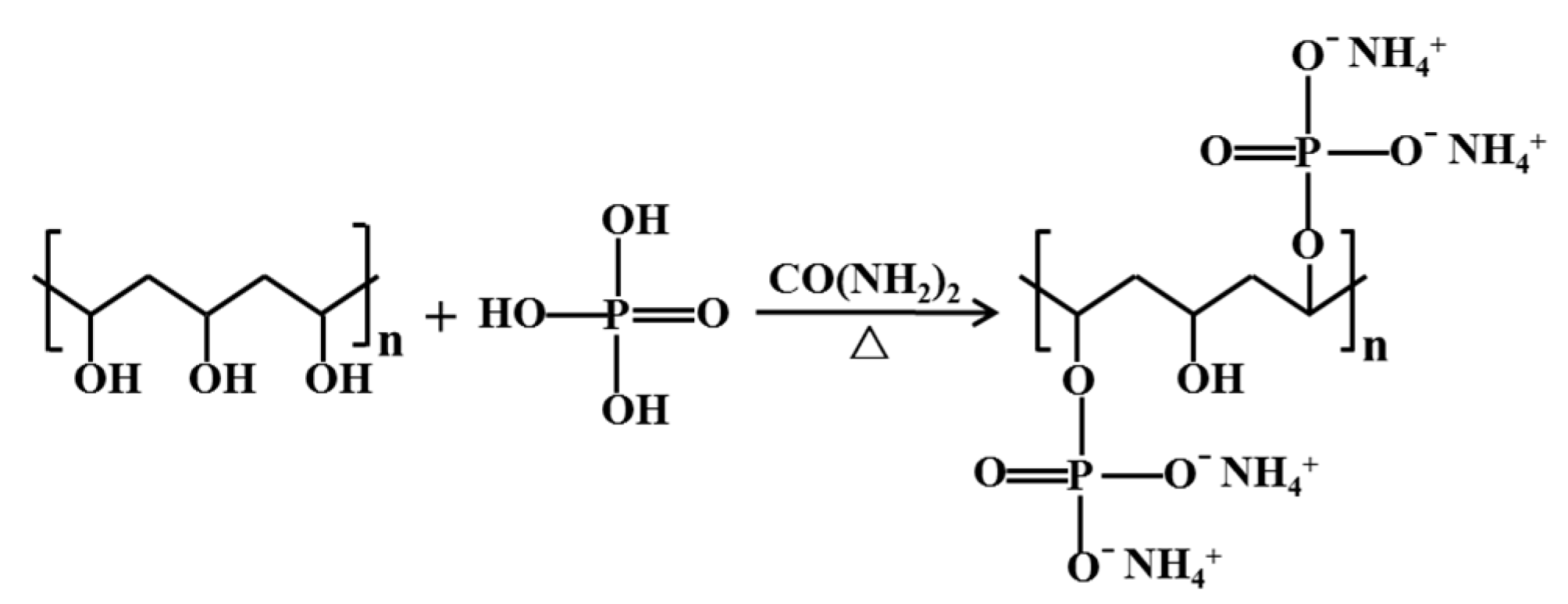
Figure 2.
E-pH diagrams for systems of (a) Fe-S-H2O; (b) Fe-NH3-S2O3-H2O; (c) Cu-NH3-S2O3-H2O; and (d) Cu-Fe-NH3-S2O3-H2O. Conditions: (a) Fe 3 × 10−3 M, S 0.22 M; (b) Fe 1 × 10−5 M, NH3/NH4+ 0.5 M, S2O32− 0.1 M; (c) Cu 0.012 M, NH3/NH4+ 0.5 M, S2O32− 0.1 M; (d) Cu 0.012 M, Fe 3 × 10−3 M, NH3/NH4+ 0.5 M, S2O32− 0.1 M.
Figure 2.
E-pH diagrams for systems of (a) Fe-S-H2O; (b) Fe-NH3-S2O3-H2O; (c) Cu-NH3-S2O3-H2O; and (d) Cu-Fe-NH3-S2O3-H2O. Conditions: (a) Fe 3 × 10−3 M, S 0.22 M; (b) Fe 1 × 10−5 M, NH3/NH4+ 0.5 M, S2O32− 0.1 M; (c) Cu 0.012 M, NH3/NH4+ 0.5 M, S2O32− 0.1 M; (d) Cu 0.012 M, Fe 3 × 10−3 M, NH3/NH4+ 0.5 M, S2O32− 0.1 M.
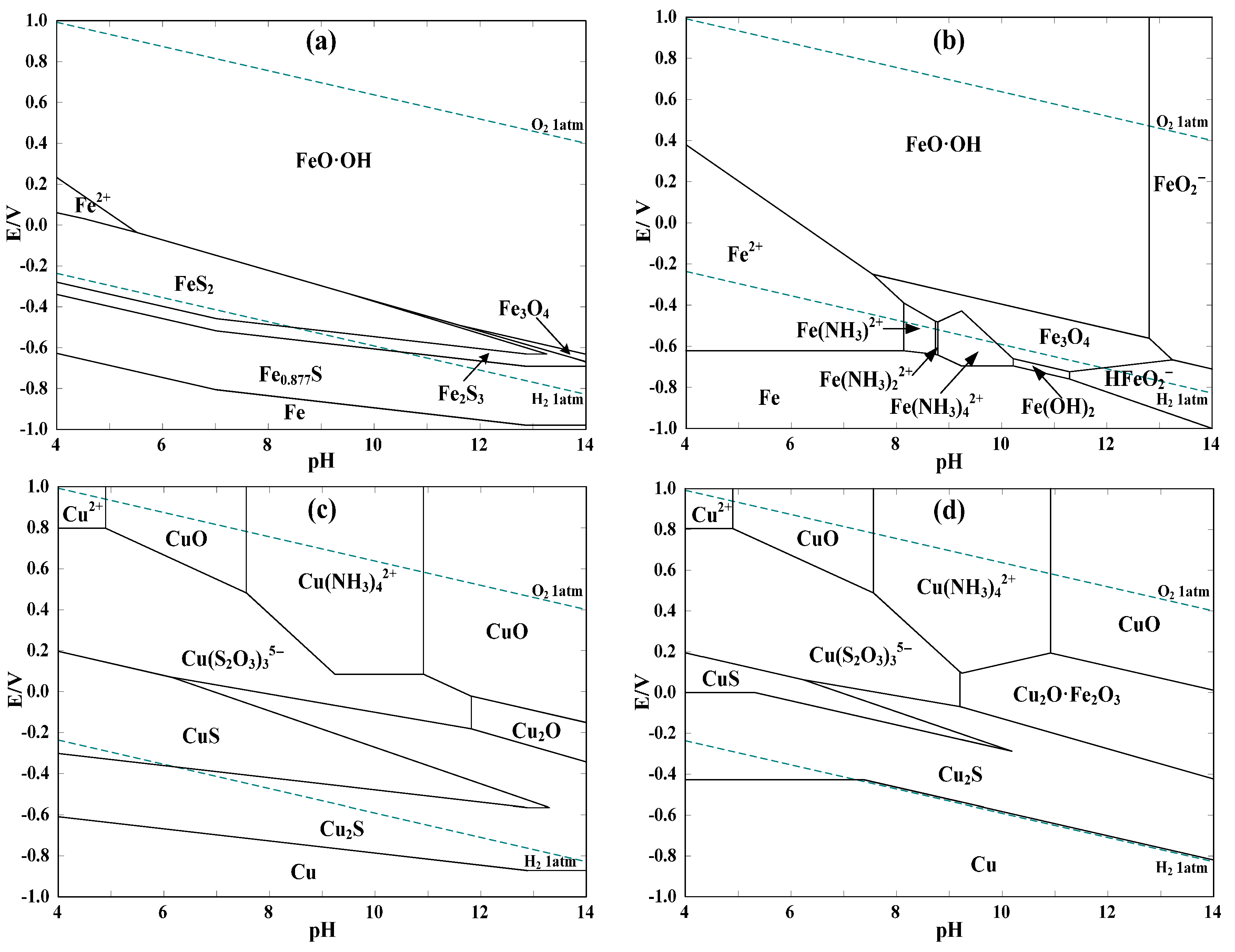
Figure 3.
Variation of the thiosulfate consumption with time during thiosulfate leaching of gold in the presence of pyrite and quartz. Mineral content: 0.4, 2, 10 and 20 g/L pyrite; 10 g/L quartz.
Figure 3.
Variation of the thiosulfate consumption with time during thiosulfate leaching of gold in the presence of pyrite and quartz. Mineral content: 0.4, 2, 10 and 20 g/L pyrite; 10 g/L quartz.
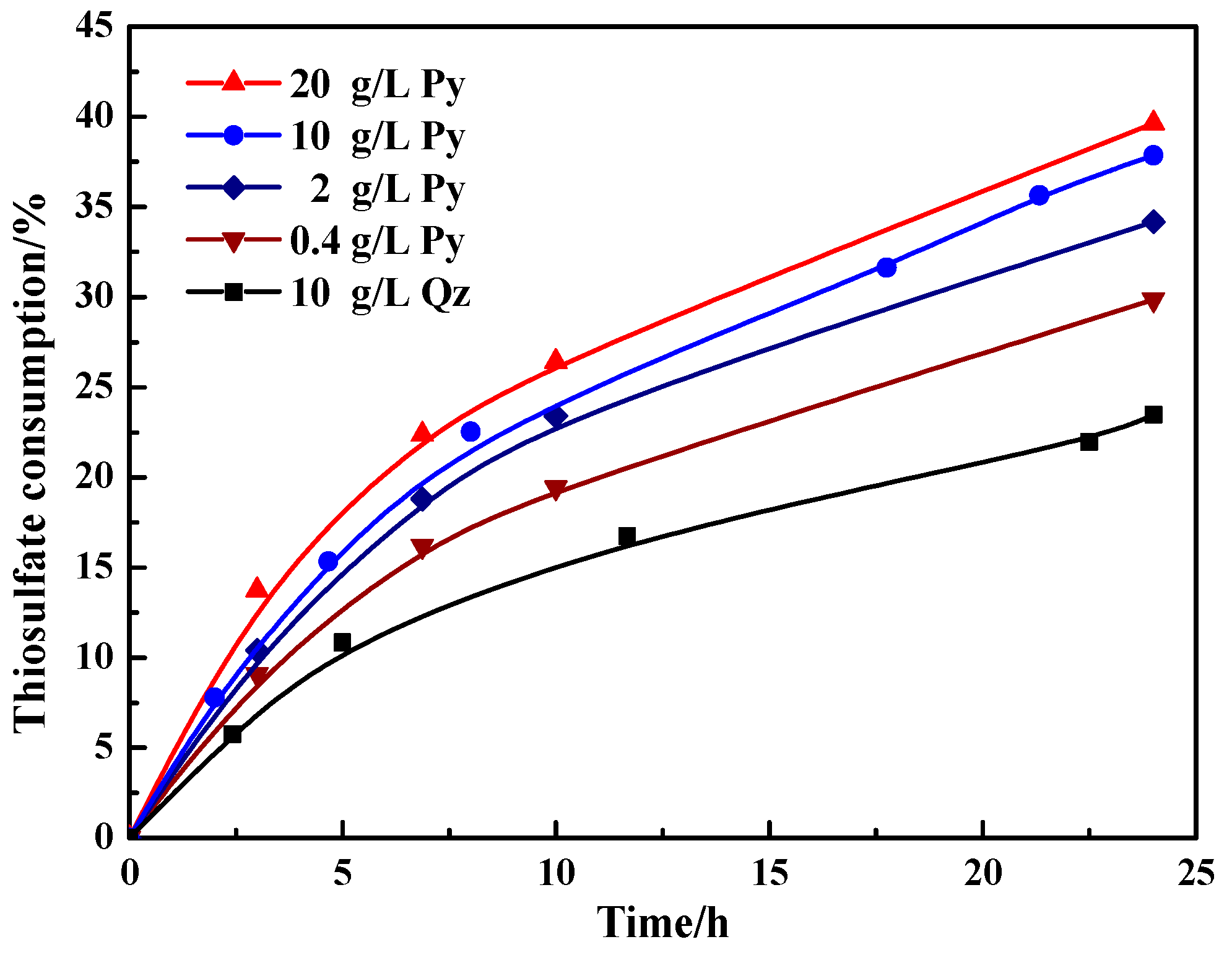
Figure 4.
Variation of the mixed slurry potential with time during thiosulfate leaching of gold in the presence of 10 g/L pyrite and 10 g/L quartz without and with the addition of 4 g/L AAPP.
Figure 4.
Variation of the mixed slurry potential with time during thiosulfate leaching of gold in the presence of 10 g/L pyrite and 10 g/L quartz without and with the addition of 4 g/L AAPP.
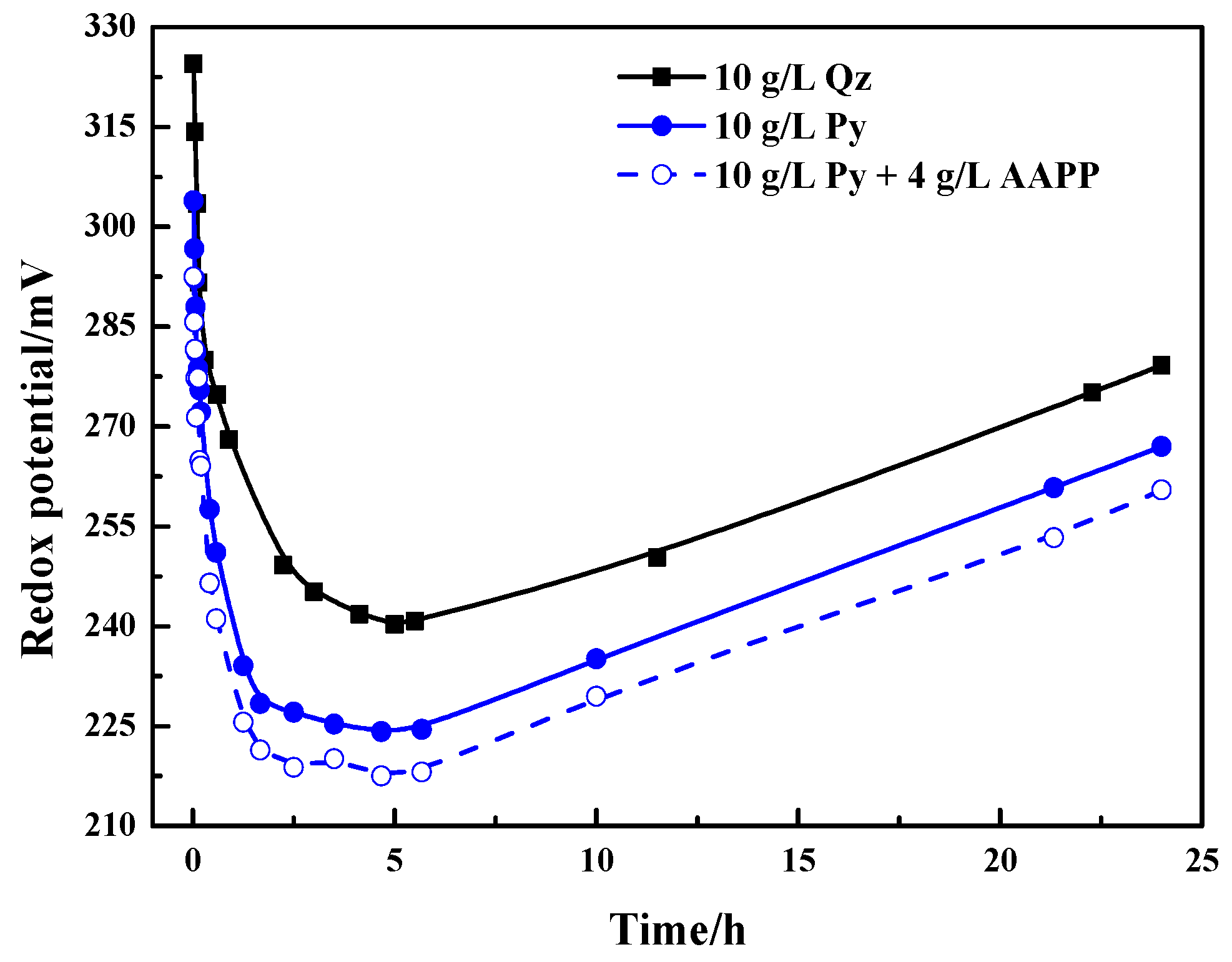
Figure 5.
Variation of the gold dissolution with time during thiosulfate leaching of gold in the presence of pyrite and quartz. Mineral content: 0.4, 2, 10 and 20 g/L pyrite; 10 g/L quartz.
Figure 5.
Variation of the gold dissolution with time during thiosulfate leaching of gold in the presence of pyrite and quartz. Mineral content: 0.4, 2, 10 and 20 g/L pyrite; 10 g/L quartz.
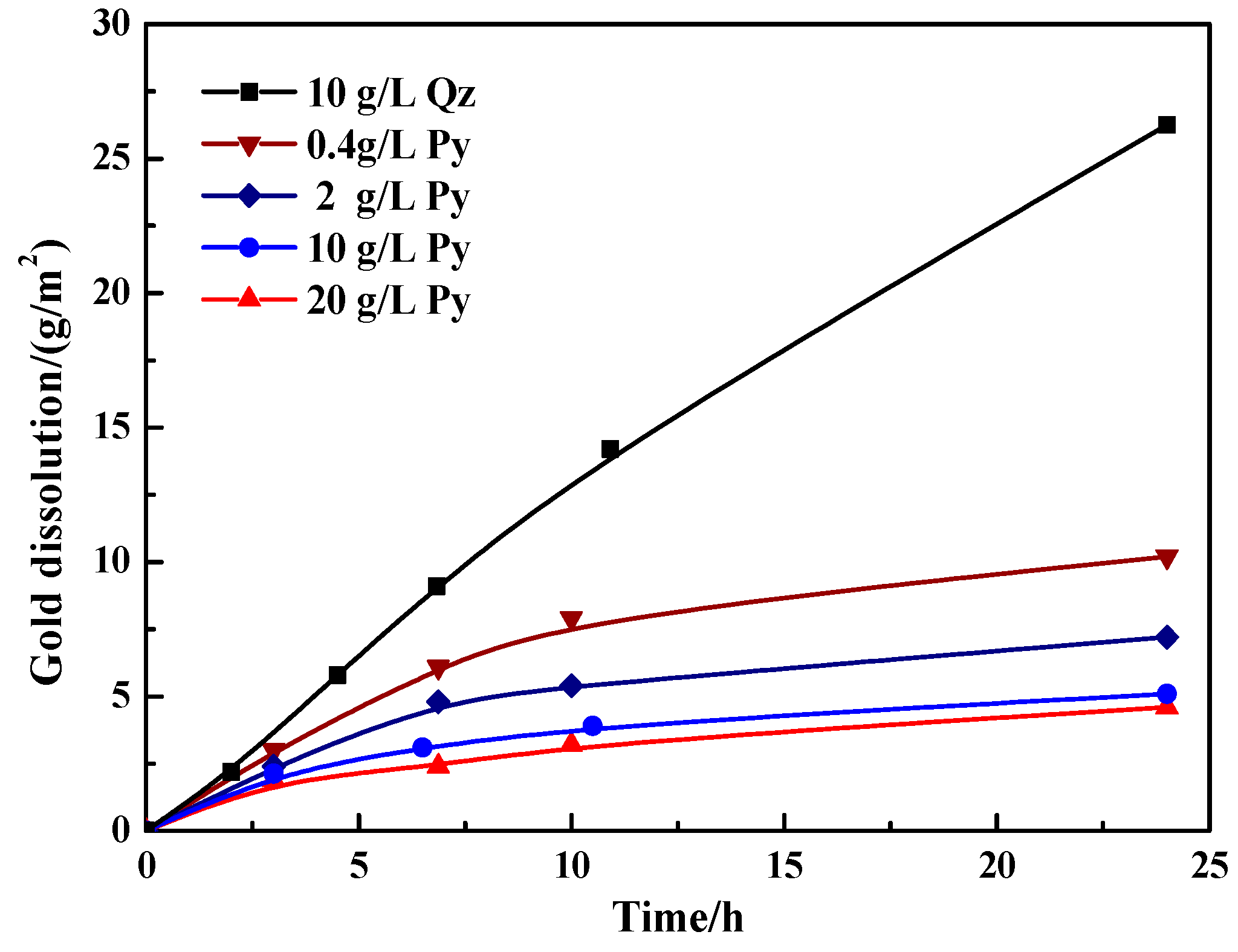
Figure 6.
Scanning electron microscopy (SEM) images of gold foils before and after thiosulfate leaching 24 h. (a) The original gold foil surface; the leached gold surface of (b) in the presence of 10 g/L quartz, (c) in the presence of 10 g/L pyrite and (d) in the presence of 10 g/L pyrite with the addition of 4 g/L AAPP.
Figure 6.
Scanning electron microscopy (SEM) images of gold foils before and after thiosulfate leaching 24 h. (a) The original gold foil surface; the leached gold surface of (b) in the presence of 10 g/L quartz, (c) in the presence of 10 g/L pyrite and (d) in the presence of 10 g/L pyrite with the addition of 4 g/L AAPP.

Figure 7.
X-ray photoelectron spectroscopy (XPS) spectra of (a) S 2p; (b) Cu 2p and (c) Fe 2p for leached gold foil surfaces in the presence of 10 g/L quartz, 10 g/L pyrite and 10 g/L pyrite with the addition of 4 g/L AAPP.
Figure 7.
X-ray photoelectron spectroscopy (XPS) spectra of (a) S 2p; (b) Cu 2p and (c) Fe 2p for leached gold foil surfaces in the presence of 10 g/L quartz, 10 g/L pyrite and 10 g/L pyrite with the addition of 4 g/L AAPP.

Figure 8.
Effect of AAPP on thiosulfate consumption and gold dissolution in thiosulfate leaching of gold in the presence of 10 g/L pyrite.
Figure 8.
Effect of AAPP on thiosulfate consumption and gold dissolution in thiosulfate leaching of gold in the presence of 10 g/L pyrite.
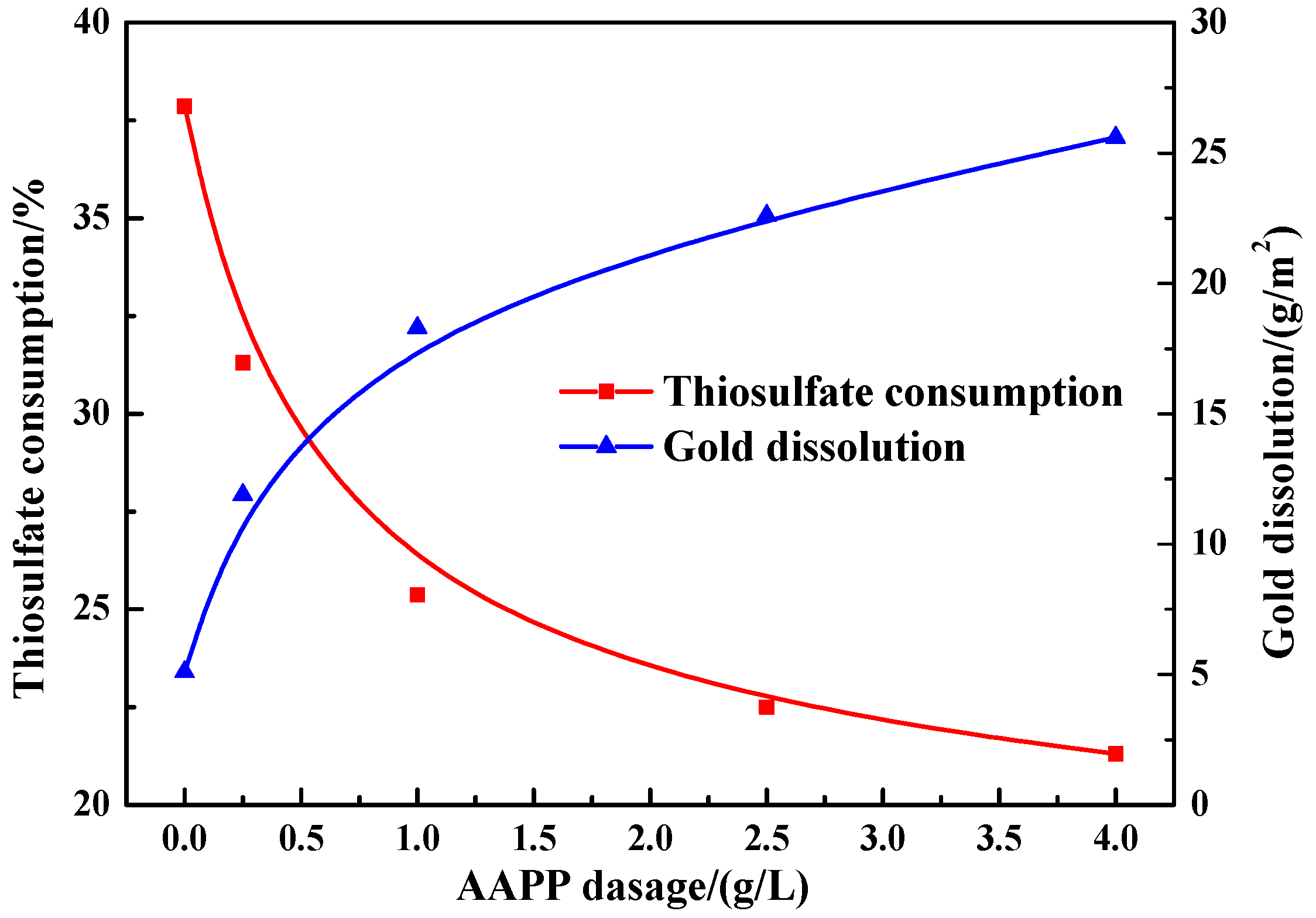
Figure 9.
Zeta potential versus pH for pyrite in the absence and presence of 4 g/L AAPP.

Table 1.
The absolute values of difference between the frontier orbital energy of pyrite and thiosulfate anion.
Table 1.
The absolute values of difference between the frontier orbital energy of pyrite and thiosulfate anion.
| Reactant | EHOMO/eV | ELUMO/eV | |ΔE1|/eV a | |ΔE2|/eV b |
|---|---|---|---|---|
| FeS2 | −6.295 [51] | −5.732 [51] | 1.780 | 3.231 |
| S2O32− | −7.703 [52] | −3.064 [52] |
a |ΔE1|=|E(HOMO, thiosulfate)–E(LUMO, pyrite)|; b |ΔE2|=|E(HOMO, pyrite)–E(LUMO, thiosulfate)|.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, X.; Xu, B.; Min, X.; Li, Q.; Yang, Y.; Jiang, T.; He, Y.; Zhang, X. Effect of Pyrite on Thiosulfate Leaching of Gold and the Role of Ammonium Alcohol Polyvinyl Phosphate (AAPP). Metals 2017, 7, 278. https://doi.org/10.3390/met7070278
AMA Style
Liu X, Xu B, Min X, Li Q, Yang Y, Jiang T, He Y, Zhang X. Effect of Pyrite on Thiosulfate Leaching of Gold and the Role of Ammonium Alcohol Polyvinyl Phosphate (AAPP). Metals. 2017; 7(7):278. https://doi.org/10.3390/met7070278
Chicago/Turabian StyleLiu, Xiaoliang, Bin Xu, Xin Min, Qian Li, Yongbin Yang, Tao Jiang, Yinghe He, and Xi Zhang. 2017. "Effect of Pyrite on Thiosulfate Leaching of Gold and the Role of Ammonium Alcohol Polyvinyl Phosphate (AAPP)" Metals 7, no. 7: 278. https://doi.org/10.3390/met7070278
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.