
Molecular Analysis of SARS-CoV-2 Genetic Lineages in Jordan: Tracking the Introduction and Spread of COVID-19 UK Variant of Concern at a Country Level



Abstract
:1. Introduction
2. Results
2.1. Characteristics of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Jordanian Dataset
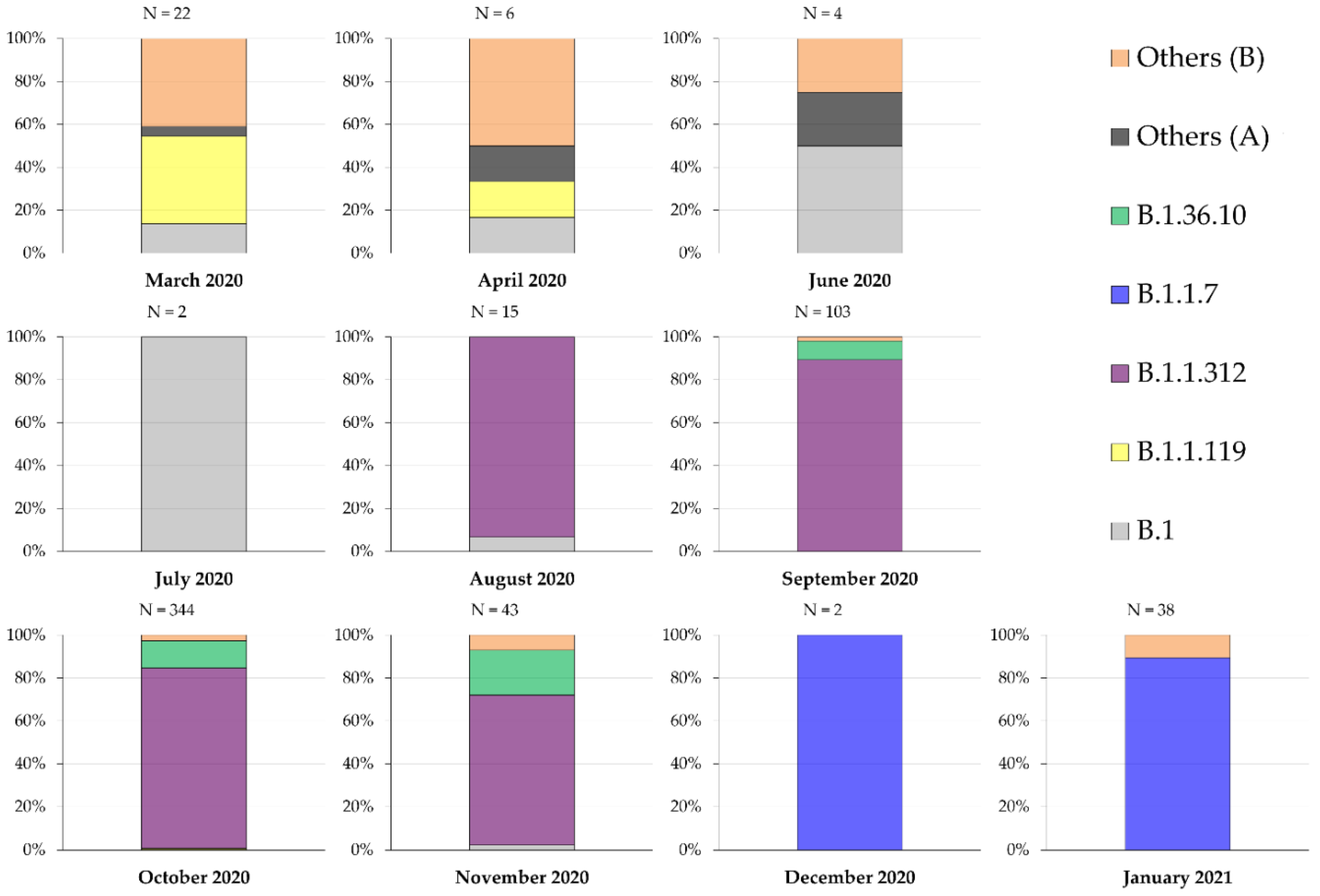
2.2. Description of the Genetic Lineages of SARS-CoV-2 in Jordan Using the “Pango” System
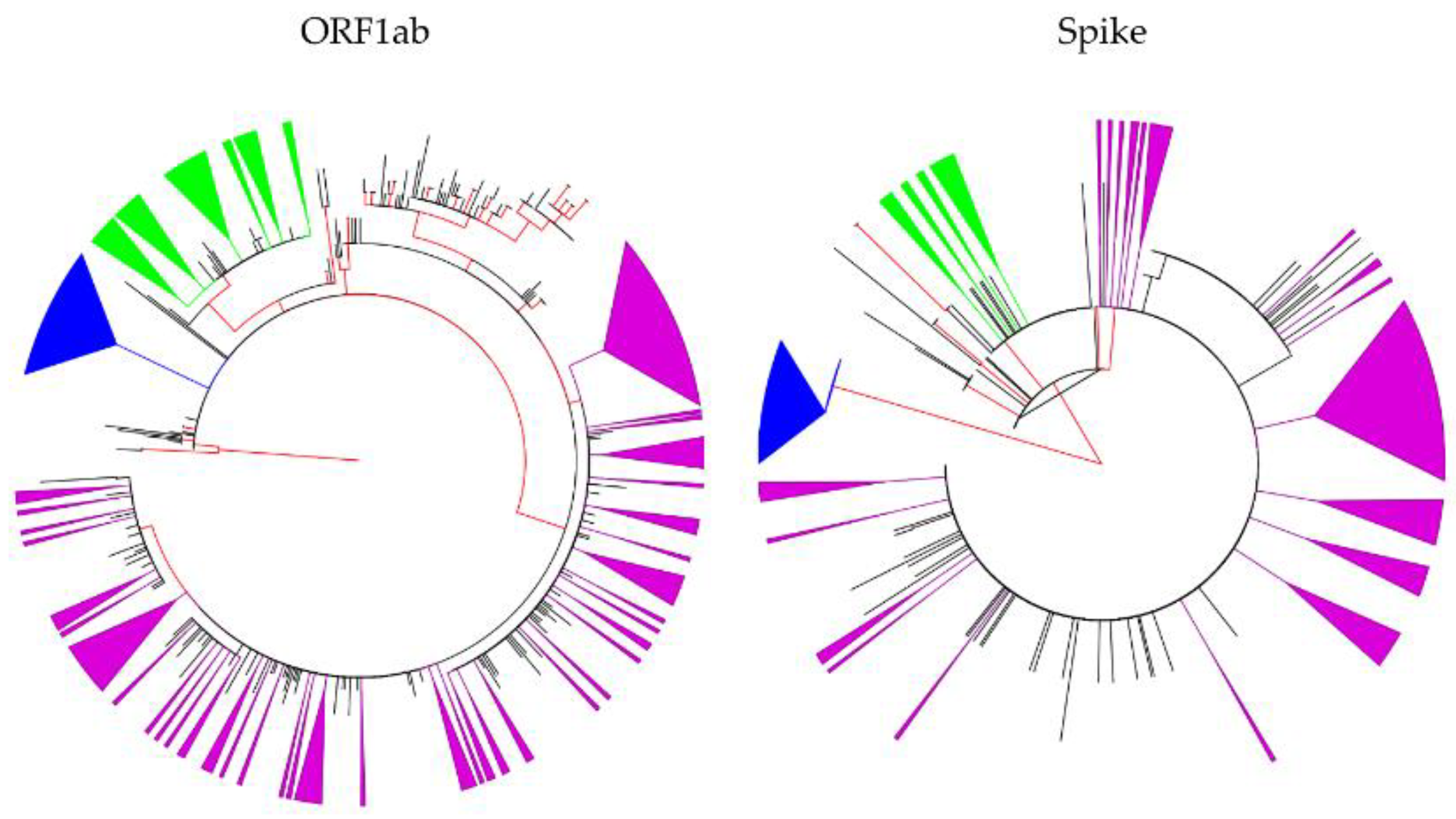
2.3. The Proportion of Phylogenetic Clustering among the Three Most Common Lineages in Jordan
2.4. Amino Acid Substitutions in the Surface Glycoprotein of the Three Major Genetic Lineages in Jordan
2.5. The UK Variant of Concern was Introduced into Jordan in Late November 2020
3. Discussion
Study Strengths and Limitations
4. Materials and Methods
4.1. Compilation of SARS-CoV-2 Jordanian Dataset and Epidemiologic Data
4.2. SARS-CoV-2 Lineage Assignment
4.3. Assessment Spike Protein of the Major Lineages in Jordan
4.4. Maximum Likelihood Phylogenetic Analysis
4.5. Timing of B.1.1.7 Lineage (UK Variant) Introduction into Jordan
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIC | Akaike Information Criterion |
| aLRT-SH | Approximate likelihood Shimodaira-Hasegawa statistical support |
| BEAST | Bayesian evolutionary analysis by sampling trees |
| COVID-19 | Coronavirus disease 2019 |
| D614G | Replacement of aspartic acid by glycine at position 614 of the spike glycoprotein |
| del | Deletion |
| E484K | Replacement of glutamic acid by lysine at position 484 of the spike glycoprotein |
| ESS | Effective sample size |
| GISAID | The global science initiative and primary source for genomic data of influenza viruses |
| K417N | Replacement of lysine by asparagine at position 417 of the spike glycoprotein |
| MCC | Maximum clade credibility |
| MCMC | Markov chain Monte Carlo |
| ML | Maximum likelihood |
| N501Y | Replacement of asparagine by tyrosine at position 501 of the spike glycoprotein |
| Ne | Effective population size |
| ORF | Open reading frame |
| P681H | Replacement of proline by histidine at position 681 of the spike glycoprotein |
| PROVEAN | Protein Variation Effect Analyzer tool |
| RNA | Ribonucleic acid |
| S | Spike gene of SARS-CoV-2 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| tMRCA | Time to the most recent common ancestor |
| UK | United Kingdom |
References
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Sevajol, M.; Subissi, L.; Decroly, E.; Canard, B.; Imbert, I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014, 194, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Worldometer. COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 31 January 2021).
- Sironi, M.; Hasnain, S.E.; Rosenthal, B.; Phan, T.; Luciani, F.; Shaw, M.A.; Sallum, M.A.; Mirhashemi, M.E.; Morand, S.; Gonzalez-Candelas, F.; et al. SARS-CoV-2 and COVID-19: A genetic, epidemiological, and evolutionary perspective. Infect. Genet. Evol. 2020, 84, 104384. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Tatem, A.J.; Lemey, P. Virus evolution and transmission in an ever more connected world. Proc. Biol. Sci. 2015, 282, 20142878. [Google Scholar] [CrossRef] [Green Version]
- Nie, Q.; Li, X.; Chen, W.; Liu, D.; Chen, Y.; Li, H.; Li, D.; Tian, M.; Tan, W.; Zai, J. Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res. 2020, 287, 198098. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. Addendum: A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2021, 6, 415. [Google Scholar] [CrossRef]
- Wise, J. Covid-19: New coronavirus variant is identified in UK. BMJ 2020, 371, m4857. [Google Scholar] [CrossRef]
- Kupferschmidt, K. New mutations raise specter of ‘immune escape’. Am. Assoc. Adv. Sci. 2021, 371, 329–330. [Google Scholar] [CrossRef]
- Faria, N.R.; Morales Claro, I.; Candido, D.; Moyses Franco, L.A.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.M.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S. Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 3 February 2021).
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Molecular Mechanism of the N501Y Mutation for Enhanced Binding between SARS-CoV-2’s Spike Protein and Human ACE2 Receptor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020. [Google Scholar] [CrossRef]
- van Dorp, L.; Richard, D.; Tan, C.C.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Robson, B. COVID-19 Coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed achilles’ heel conserved region to minimize probability of escape mutations and drug resistance. Comput. Biol. Med. 2020, 121, 103749. [Google Scholar] [CrossRef] [PubMed]
- Sallam, M.; Ababneh, N.A.; Dababseh, D.; Bakri, F.G.; Mahafzah, A. Temporal increase in D614G mutation of SARS-CoV-2 in the Middle East and North Africa. Heliyon 2021, 7, e06035. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; Silva, F.R.D.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.d.C.; Mariani, D.; Costa, R.M.d.; Ferreira, O.C.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv 2020. [Google Scholar] [CrossRef]
- Sallam, M.; Dababseh, D.; Yaseen, A.; Al-Haidar, A.; Taim, D.; Eid, H.; Ababneh, N.A.; Bakri, F.G.; Mahafzah, A. COVID-19 misinformation: Mere harmless delusions or much more? A knowledge and attitude cross-sectional study among the general public residing in Jordan. PLoS ONE 2020, 15, e0243264. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Health, The Hashemite Kingdom of Jordan. COVID-19 Updates in Jordan. Available online: https://corona.moh.gov.jo/en (accessed on 3 February 2021).
- Reuters. Jordan to Close Border with Syria after Spike in COVID-19 Cases. Available online: https://www.reuters.com/article/idUSL1N2FE0IK (accessed on 3 February 2021).
- Sallam, M.; Dababseh, D.; Eid, H.; Al-Mahzoum, K.; Al-Haidar, A.; Taim, D.; Yaseen, A.; Ababneh, N.A.; Bakri, F.G.; Mahafzah, A. High Rates of COVID-19 Vaccine Hesitancy and Its Association with Conspiracy Beliefs: A Study in Jordan and Kuwait among Other Arab Countries. Vaccines 2021, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Sallam, M.; Dababseh, D.; Yaseen, A.; Al-Haidar, A.; Ababneh, N.A.; Bakri, F.G.; Mahafzah, A. Conspiracy Beliefs Are Associated with Lower Knowledge and Higher Anxiety Levels Regarding COVID-19 among Students at the University of Jordan. Int. J. Environ. Res. Public Health 2020, 17, 4915. [Google Scholar] [CrossRef]
- Lam, T.T.; Hon, C.C.; Tang, J.W. Use of phylogenetics in the molecular epidemiology and evolutionary studies of viral infections. Crit. Rev. Clin. Lab. Sci. 2010, 47, 5–49. [Google Scholar] [CrossRef]
- Scudiero, O.; Lombardo, B.; Brancaccio, M.; Mennitti, C.; Cesaro, A.; Fimiani, F.; Gentile, L.; Moscarella, E.; Amodio, F.; Ranieri, A.; et al. Exercise, Immune System, Nutrition, Respiratory and Cardiovascular Diseases during COVID-19: A Complex Combination. Int. J. Environ. Res. Public Health 2021, 18, 904. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Cook, A.R.; Lim, J.T.; Sun, Y.; Dickens, B.L. A Systematic Review of COVID-19 Epidemiology Based on Current Evidence. J. Clin. Med. 2020, 9, 967. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Pierson, E.; Koh, P.W.; Gerardin, J.; Redbird, B.; Grusky, D.; Leskovec, J. Mobility network models of COVID-19 explain inequities and inform reopening. Nature 2021, 589, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, N.; Siddique, A.B.; Andalibi, A. Assessment of SARS-CoV-2 transmission among attendees of live concert events in Japan using contact-tracing data. J. Travel Med. 2020, 27, taaa096. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Graham, M.S.; Sudre, C.H.; May, A.; Antonelli, M.; Murray, B.; Varsavsky, T.; Kläser, K.; Canas, L.S.; Molteni, E.; Modat, M.; et al. The effect of SARS-CoV-2 variant B.1.1.7 on symptomatology, re-infection and transmissibility. medRxiv 2021. [Google Scholar] [CrossRef]
- Makowski, L.; Olson-Sidford, W.; W-Weisel, J. Biological and Clinical Consequences of Integrin Binding via a Rogue RGD Motif in the SARS CoV-2 Spike Protein. Viruses 2021, 13, 146. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Callaway, E. The coronavirus is mutating-does it matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Diez-Fuertes, F.; Iglesias-Caballero, M.; Garcia-Perez, J.; Monzon, S.; Jimenez, P.; Varona, S.; Cuesta, I.; Zaballos, A.; Jimenez, M.; Checa, L.; et al. A Founder Effect Led Early SARS-CoV-2 Transmission in Spain. J. Virol. 2021, 95, e01583-20. [Google Scholar] [CrossRef]
- Ruan, Y.; Luo, Z.; Tang, X.; Li, G.; Wen, H.; He, X.; Lu, X.; Lu, J.; Wu, C.-I. On the founder effect in COVID-19 outbreaks: How many infected travelers may have started them all? Natl. Sci. Rev. 2020, 8, nwaa246. [Google Scholar] [CrossRef]
- Farkas, C.; Fuentes-Villalobos, F.; Garrido, J.L.; Haigh, J.; Barria, M.I. Insights on early mutational events in SARS-CoV-2 virus reveal founder effects across geographical regions. PeerJ 2020, 8, e9255. [Google Scholar] [CrossRef]
- DemIr, A.B.; Benvenuto, D.; AbacioGlu, H.; Angeletti, S.; Ciccozzi, M. Identification of the nucleotide substitutions in 62 SARS-CoV-2 sequences from Turkey. Turk. J. Biol. 2020, 44, 178–184. [Google Scholar] [CrossRef]
- Roya News. Updated List of Green, Yellow and Red Countries Eligible to Travel to and from Jordan. Available online: https://en.royanews.tv/news/22149/Updated-list-of-green-yellow-and-red-countries-eligible-to-travel-to-and-from-Jordan (accessed on 3 February 2021).
- Maitra, A.; Sarkar, M.C.; Raheja, H.; Biswas, N.K.; Chakraborti, S.; Singh, A.K.; Ghosh, S.; Sarkar, S.; Patra, S.; Mondal, R.K.; et al. Mutations in SARS-CoV-2 viral RNA identified in Eastern India: Possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J. BioSci. 2020, 45, 76. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun 2020, 11, 6013. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Ritchie, H. Our World in Data: Coronavirus Source Data. Available online: https://ourworldindata.org/coronavirus-source-data (accessed on 30 January 2021).
- O’Toole, Á.; McCrone, J.T.; Scher, E. Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin COVID-19 Lineage Assigner). Available online: https://pangolin.cog-uk.io/ (accessed on 30 January 2021).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1. 4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 24 August 2017).
- Parker, E.; Anderson, C.; Zeller, M.; Havens, J.; Tibi, A.; Hussein, L.; Ali, S.; Saddedin, B.; Robles-Sikisaka, R.; Kurzban, E.; et al. The emergence of the B.1.1.7 lineage in Jordan. Available online: https://virological.org/t/the-emergence-of-the-b-1-1-7-lineage-in-jordan/634 (accessed on 17 March 2021).



{kind=link}
{kind=link}
{kind=link}
| Month | Newly Diagnosed COVID-19 1 Cases | COVID-19 Related Deaths | Number of SARS-CoV-2 2 Sequences | Percentage of Sequences Compared to New Cases |
|---|---|---|---|---|
| March 2020 | 274 | 5 | 22 | 8.0292% |
| April 2020 | 179 | 3 | 6 | 3.3520% |
| May 2020 | 286 | 1 | 0 | 0 |
| June 2020 | 393 | 0 | 4 | 1.0178% |
| July 2020 | 61 | 2 | 2 | 3.2787% |
| August 2020 | 841 | 4 | 15 | 1.7836% |
| September 2020 | 9791 | 46 | 103 | 1.0520% |
| October 2020 | 60,782 | 768 | 344 | 0.5660% |
| November 2020 | 146,823 | 1922 | 43 | 0.0293% |
| December 2020 | 75,064 | 1083 | 2 | 0.0027% |
| January 2021 | 30,539 | 447 | 38 | 0.1244% |
| SARS-CoV-2 Lineage | Amino Acid Substitution | PROVEAN 1 Score | Prediction (Cutoff = −2.5) |
|---|---|---|---|
| UK variant of concern (B.1.1.7) | H69_V70del | 0.808 | Neutral |
| V143_Y144del | 1.318 | Neutral | |
| N501Y | −0.090 | Neutral | |
| A570D | −0.682 | Neutral | |
| D614G | 0.598 | Neutral | |
| P681H | 0.060 | Neutral | |
| T716I | −3.293 | Deleterious | |
| S982A | −1.505 | Neutral | |
| D1118H | −1.142 | Neutral | |
| First Jordan lineage (B.1.1.312) | D614G | 0.598 | Neutral |
| Q957L | −2.929 | Deleterious | |
| Second Jordan Lineage (B.1.36.10) | D614G | 0.598 | Neutral |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sallam, M.; Mahafzah, A. Molecular Analysis of SARS-CoV-2 Genetic Lineages in Jordan: Tracking the Introduction and Spread of COVID-19 UK Variant of Concern at a Country Level. Pathogens 2021, 10, 302. https://doi.org/10.3390/pathogens10030302
Sallam M, Mahafzah A. Molecular Analysis of SARS-CoV-2 Genetic Lineages in Jordan: Tracking the Introduction and Spread of COVID-19 UK Variant of Concern at a Country Level. Pathogens. 2021; 10(3):302. https://doi.org/10.3390/pathogens10030302
Chicago/Turabian StyleSallam, Malik, and Azmi Mahafzah. 2021. "Molecular Analysis of SARS-CoV-2 Genetic Lineages in Jordan: Tracking the Introduction and Spread of COVID-19 UK Variant of Concern at a Country Level" Pathogens 10, no. 3: 302. https://doi.org/10.3390/pathogens10030302