Genomic and Global Approaches to Unravelling How Hypermutable Sequences Influence Bacterial Pathogenesis

Abstract
:1. Introduction
2. Localised Hypermutation, Phase Variation and the Mechanisms of Hypermutation
3. Detection of Hypermutable Sequences in Genomes
3.1. A Definition of Functional SSR Classes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of SSR | Mutation Rate (Mutations/Division) | Predicted Range of Repeat Numbers (References 1) | |
|---|---|---|---|
| Proto-mutable | 10−9 to 10−7 | <6G [19] | |
| 2-4CA [20] | |||
| 2-4TAAA | |||
| 2-4AGTC | |||
| GACGAGAAGA | |||
| Mutable | 10−7 to 10−5 | 6G-8G [21] | |
| 4-10CA [20] | |||
| 3-9TAAA | |||
| 3-12AGTC [15] | |||
| 2GACGA | |||
| Hypermutable | 10−5 to 10−2 | >7G [21,22,23] | |
| >10CA[20] | |||
| >9 TAAA[24] | |||
| >10 AGTC [15] | |||
| 3+GACGA [25] | |||
3.2. Detection of Multiple SSR in the Bacterial Genomes
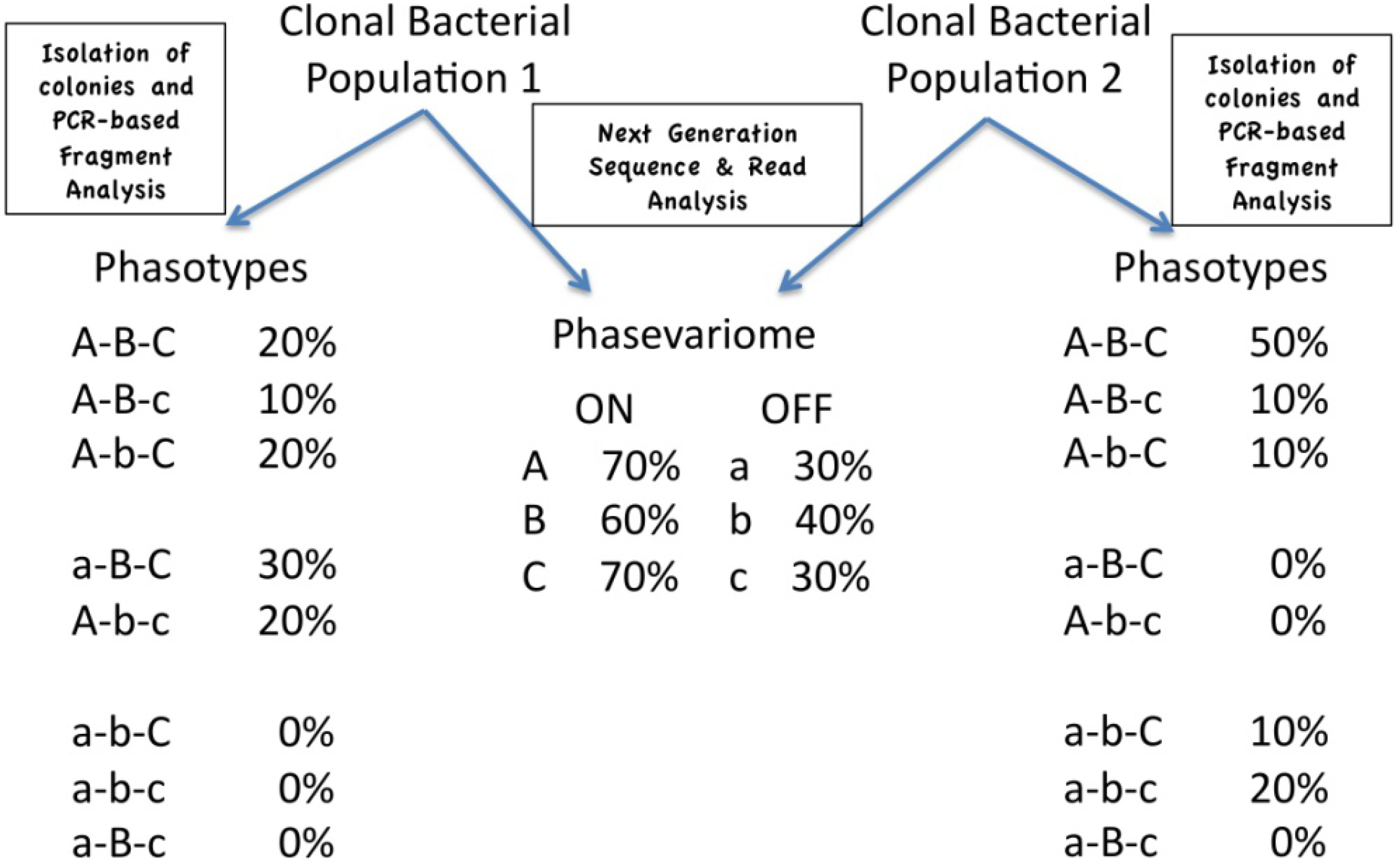
4. Expression States, Phasotypes and the Phasevariome
4.1. Analysis of Expression States of Phase Variable Genes
4.2. Phasotypes
| Gene | fetA 1 | porA 1 | opc 1 | nadA 1 | hpuA 2 | nalP 2 |
|---|---|---|---|---|---|---|
| Tract Length | 9C | 12G | 11C | 9TAAA | 10G | 11C |
| Expression State1 | 2 | 2 | 1 | 0 | 2 | 2 |
| Phasotype | 2-2-1-0-2-2 | |||||
4.3. Phasotypes and the Phasevariome

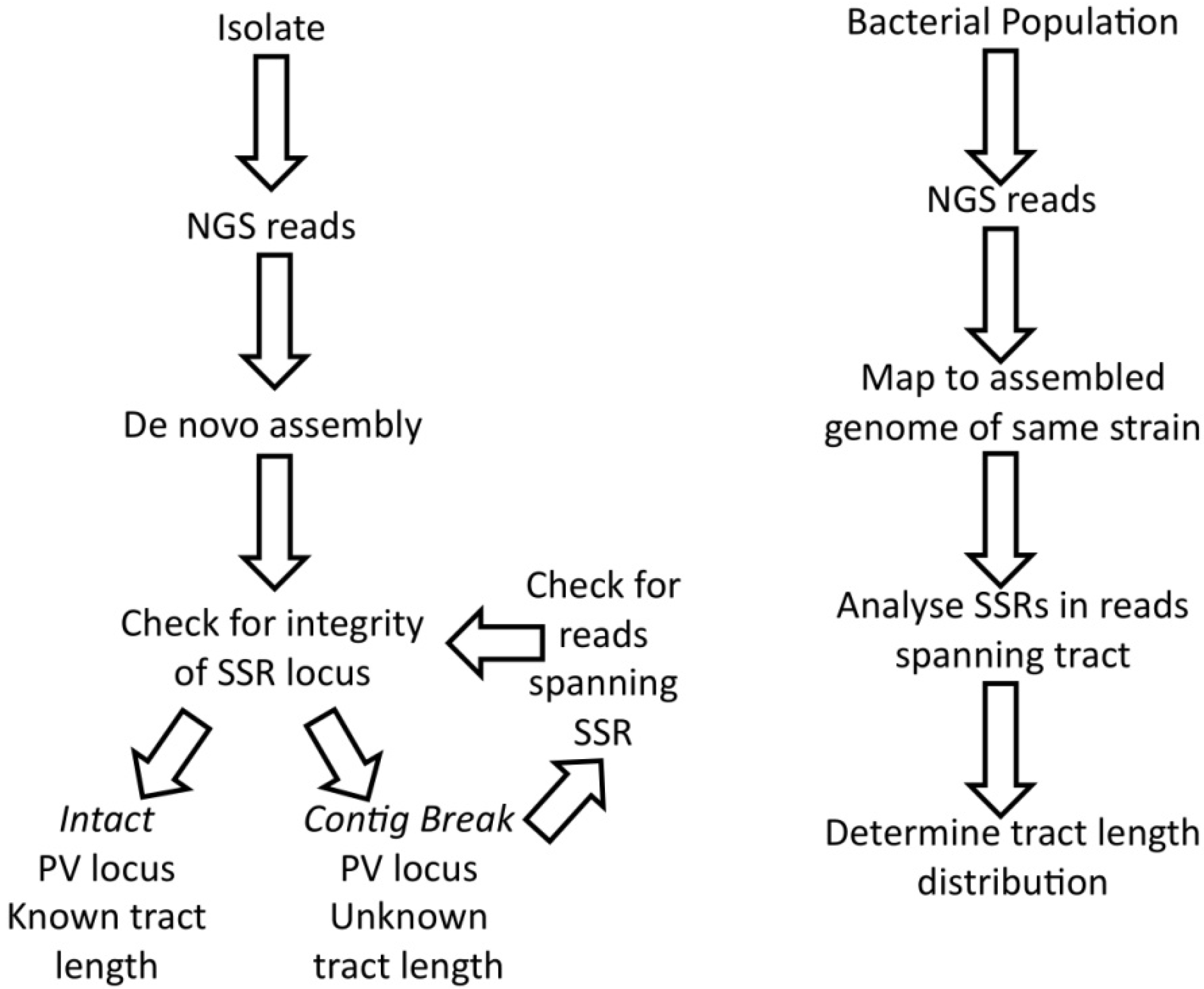
5. Analysis of SSR Diversity by Next Generation Sequencing
5.1. Utility of NGS for Comparison of SSR Prevalence in Bacterial Genomes
5.2. Methodological Impact of NGS on the Determination of Tract Length
5.3. Methodological Impact of Assembly on Analysis of Multiple-Copy Loci
6. Analysis of SSRs by NGS versus PCR-Based Approaches
6.1. NGS Approach

6.2. PCR-Based Approach
6.3. Use of NGS versus PCR to Analyse C. jejuni Phase Variable Genes
7. Case Studies Illustrating Global Approaches to SSR Analysis in Clinical Isolates
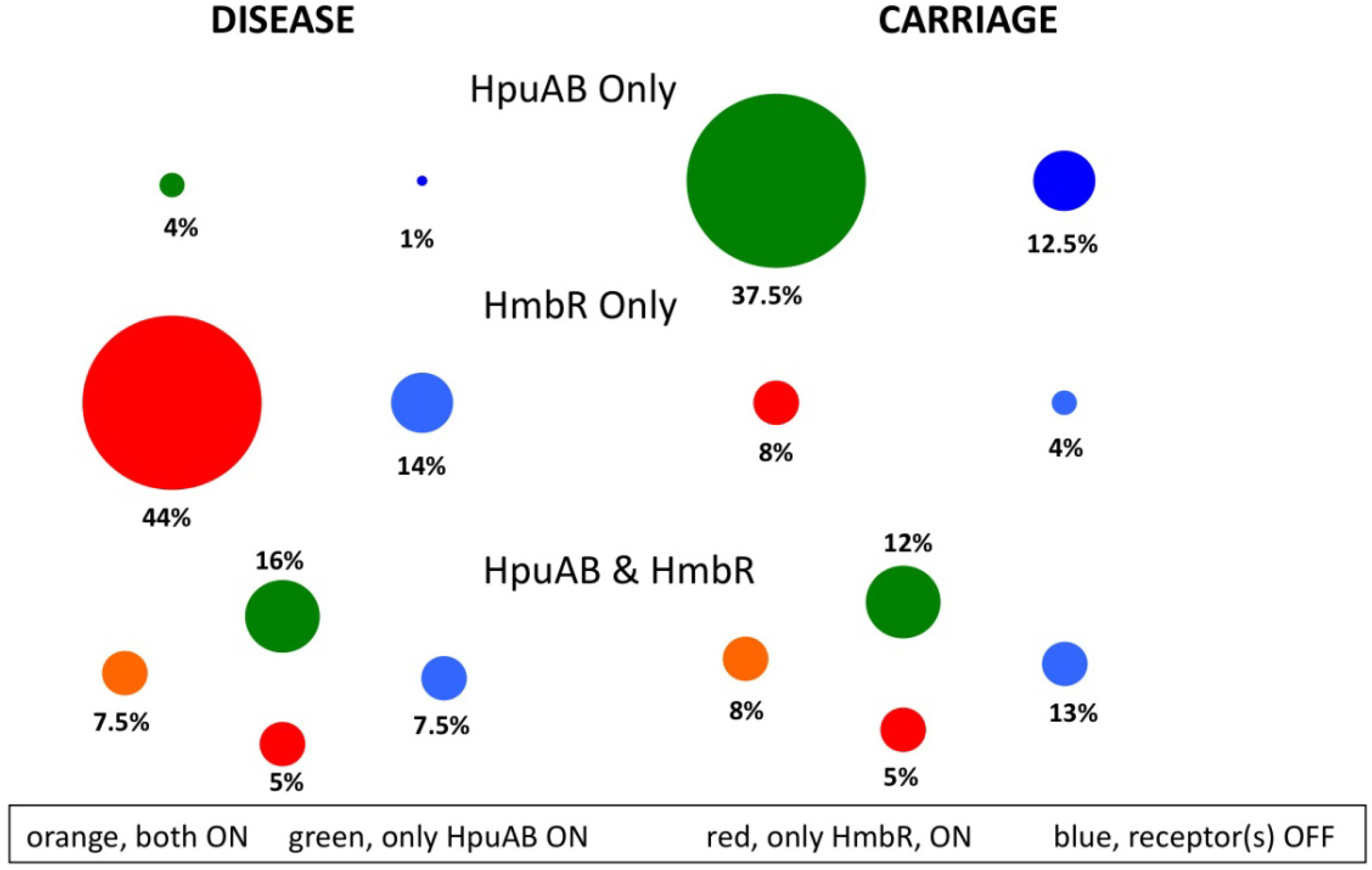
7.1. Comparison of Hb-Receptor Prevalence and Expression in Meningococcal Isolate Collections

7.2. Analysis of Clinical Samples from Meningococcal Patients for Hb-Receptor Expression Status
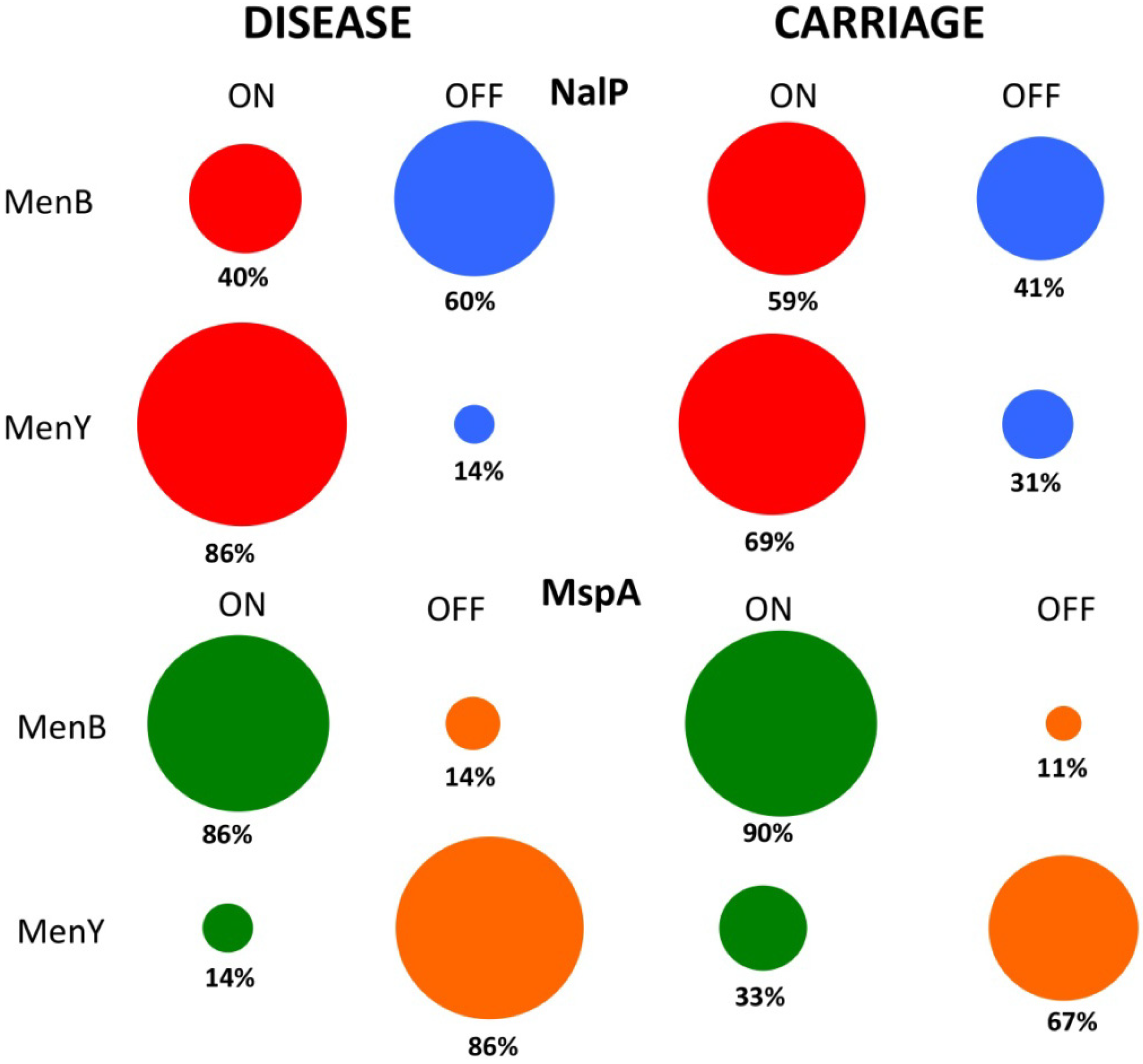
7.3. Phase Variable Autotransporters in Clinical Meningococcal Isolates

7.4. Alterations in Outer Membrane Protein of Neisseria meningitidis and Haemophilus influenzae
8. Summary
Author Contributions
Conflicts of Interest
References and Notes
- Deitsch, K.W.; Lukehart, S.A.; Stringer, J.R. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 2009, 7, 493–503. [Google Scholar] [CrossRef]
- Moxon, E.R.; Bayliss, C.D.; Hood, D.W. Bacterial contingency loci: The role of simple sequence DNA repeats in bacterial adaptation. Ann. Rev. Genet. 2007, 40, 307–333. [Google Scholar] [CrossRef]
- Van der Woude, M.W.; Baumler, A.J. Phase and antigenic variation in bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611. [Google Scholar] [CrossRef]
- Bayliss, C.D. Determinants of phase variation rate and the fitness implications of differing rates for bacterial pathogens and commensals. FEMS Microbiol. Rev. 2009, 33, 504–520. [Google Scholar] [CrossRef]
- Hall, L.M.; Henderson-Begg, S.K. Hypermutable bacteria isolated from humans—a critical analysis. Microbiology 2006, 152, 2505–2514. [Google Scholar] [CrossRef]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef]
- Bayliss, C.D.; Moxon, E.R. Repeats and variation in pathogen selection. In The Implicit Genome; Caporale, L.H., Ed.; Oxford University Press: Oxford, UK, 2005; pp. 54–76. [Google Scholar]
- Moxon, E.R.; Rainey, P.B.; Nowak, M.A.; Lenski, R. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 1994, 4, 24–33. [Google Scholar] [CrossRef]
- Cahoon, L.A.; Seifert, H.S. Transcription of a cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog. 2013, 9, e1003074. [Google Scholar] [CrossRef]
- Yogev, D.; Browning, G.; Wise, K.S. Mechanisms of surface variation. In Molecular biology and pathogenicity of mycoplasma; Razin, S., Herrmann, R., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2002; pp. 417–443. [Google Scholar]
- Cahoon, L.A.; Manthei, K.A.; Rotman, E.; Keck, J.L.; Seifert, H.S. Neisseria gonorrhoeae RecQ helicase HRDC domains are essential for efficient binding and unwinding of the pilE guanine quartet structure required for pilin antigenic variation. J. Bacteriol. 2013, 195, 2255–2261. [Google Scholar] [CrossRef]
- Criss, A.K.; Kline, K.A.; Seifert, H.S. The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Mol. Microbiol. 2005, 58, 510–519. [Google Scholar] [CrossRef]
- Andrewes, F.W. Studies in group-agglutination I. The salmonella group and its antigenic structure. J. Path. Bacteriol. 1922, 25, 505–521. [Google Scholar] [CrossRef]
- Henderson, I.R.; Owen, P.; Nataro, J.P. Molecular switches—the ON and OFF of bacterial phase variation. Mol. Micro. 1999, 33, 919–932. [Google Scholar] [CrossRef]
- De Bolle, X.; Bayliss, C.D.; Field, D.; van de Ven, T.; Saunders, N.J.; Hood, D.W.; Moxon, E.R. The length of a tetranucleotide repeat tract in Haemophilus influenzae determines the phase variation rate of a gene with homology to type III DNA methyltransferases. Mol. Microbiol. 2000, 35, 211–222. [Google Scholar] [CrossRef]
- Hendrixson, D.R. Restoration of flagellar biosynthesis by varied mutational events in Campylobacter jejuni. Mol. Microbiol. 2008, 70, 519–536. [Google Scholar] [CrossRef]
- Tran, H.T.; Keen, J.D.; Kricker, M.; Resnick, M.A.; Gordenin, D.A. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997, 17, 2859–2865. [Google Scholar]
- Lin, W.H.; Kussell, E. Evolutionary pressures on simple sequence repeats in prokaryotic coding regions. Nucleic Acids Res. 2012, 40, 2399–2413. [Google Scholar]
- Gawel, D.; Jonczyk, P.; Bialoskorska, M.; Schaaper, R.M.; Fijalkowska, I.J. Asymmetry of frameshift mutagenesis during leading and lagging-strand replication in Escherichia coli. Mutat. Res. 2002, 501, 129–136. [Google Scholar] [CrossRef]
- Strauss, B.S.; Sagher, D.; Acharya, S. Role of proofreading and mismatch repair in maintaining the stability of nucleotide repeats in DNA. Nucleic Acids Res. 1997, 25, 806–813. [Google Scholar] [CrossRef]
- Martin, P.; Sun, L.; Hood, D.W.; Moxon, E.R. Involvement of genes of genome maintenance in the regulation of phase variation frequencies in Neisseria meningitidis. Microbiology 2004, 150, 3001–3012. [Google Scholar] [CrossRef]
- Bayliss, C.D.; Bidmos, F.A.; Anjum, A.; Manchev, V.T.; Richards, R.L.; Grossier, J.P.; Wooldridge, K.G.; Ketley, J.M.; Barrow, P.A.; Jones, M.A.; et al. Phase variable genes of Campylobacter jejuni exhibit high mutation rates and specific mutational patterns but mutability is not the major determinant of population structure during host colonization. Nuc. Acids Res. 2012, 5876–5889. [Google Scholar]
- Richardson, A.R.; Yu, Z.; Popovic, T.; Stojiljkovic, I. Mutator clones of Neisseria meningitidis in epidemic serogroup A disease. Proc. Natl. Acad. Sci. USA 2002, 99, 6103–6107. [Google Scholar] [CrossRef]
- Martin, P.; Makepeace, K.; Hill, S.A.; Hood, D.W.; Moxon, E.R. Microsatellite instability regulates transcription factor binding and gene expression. Proc. Natl. Acad. Sci. USA 2005, 102, 3800–3804. [Google Scholar] [CrossRef]
- Zaleski, P.; Wojciechowski, M.; Piekarowicz, A. The role of Dam methylation in phase variation of Haemophilus influenzae genes involved in defence against phage infection. Microbiology 2005, 151, 3361–3369. [Google Scholar] [CrossRef]
- Sreenu, V.B.; Ranjitkumar, G.; Swaminathan, S.; Priya, S.; Bose, B.; Pavan, M.N.; Thanu, G.; Nagaraju, J.; Nagarajaram, H.A. MICAS: A fully automated web server for microsatellite extraction and analysis from prokaryote and viral genomic sequences. Appl. Bioinformatics 2003, 2, 165–168. [Google Scholar]
- Hood, D.W.; Deadman, M.E.; Allen, T.; Masoud, H.; Martin, A.; Brisson, J.R.; Fleischmann, R.; Venter, J.C.; Richards, J.C.; Moxon, E.R. Use of the complete genome sequence information of Haemophilus influenzae strain Rd to investigate lipopolysaccharide biosynthesis. Mol. Microbiol. 1996, 22, 951–965. [Google Scholar]
- Parkhill, J.; Wren, B.W.; Mungall, K.; Ketley, J.M.; Churcher, C.; Basham, D.; Chillingworth, T.; Davies, R.M.; Feltwell, T.; Holroyd, S.; et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 2000, 403, 665–668. [Google Scholar] [CrossRef]
- Saunders, N.J.; Jeffries, A.C.; Peden, J.F.; Hood, D.W.; Tettelin, H.; Rappouli, R.; Moxon, E.R. Repeat-associated phase variable genes in the complete genome sequence of Neisseria. meningitidis strain MC58. Mol. Micro. 2000, 37, 207–215. [Google Scholar] [CrossRef]
- Saunders, N.J.; Peden, J.; Hood, D.; Moxon, E.R. Simple sequence repeats in the Helicobacter pylori genome. Mol. Microbiol. 1998, 27, 1091–1098. [Google Scholar] [CrossRef]
- Martin, P.; van de Ven, T.; Mouchel, N.; Jeffries, A.C.; Hood, D.W.; Moxon, E.R. Experimentally revised repertoire of putative contingency loci in Neisseria. meningitidis strain MC58: Evidence for a novel mechanism of phase variation. Mol. Microbiol. 2003, 50, 245–257. [Google Scholar] [CrossRef]
- Power, P.M.; Sweetman, W.A.; Gallacher, N.J.; Woodhall, M.R.; Kumar, G.A.; Moxon, E.R.; Hood, D.W. Simple sequence repeats in Haemophilus influenzae. Infect. Genet. Evol. 2009, 9, 216–228. [Google Scholar] [CrossRef]
- Snyder, L.A.S.; Butcher, S.A.; Saunders, N.J. Comparative whole-genome analyses reveal over 100 putative phase-variable genes in the pathogenic Neisseria spp. Microbiology 2001, 147, 2321–2332. [Google Scholar]
- Dixon, K.; Bayliss, C.D.; Makepeace, K.; Moxon, E.R.; Hood, D.W. Identification of the functional initiation codons of a phase-variable gene of Haemophilus influenzae, lic2A, with the potential for differential expression. J. Bacteriol. 2007, 189, 511–521. [Google Scholar] [CrossRef]
- Snyder, L.A.; Loman, N.J.; Linton, J.D.; Langdon, R.R.; Weinstock, G.M.; Wren, B.W.; Pallen, M.J. Simple sequence repeats in Helicobacter canadensis and their role in phase variable expression and C-terminal sequence switching. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Van der Ende, A.; Hopman, C.T.; Dankert, J. Multiple mechanisms of phase variation of PorA in Neisseria meningitidis. Infect. Immun. 2000, 68, 6685–6690. [Google Scholar] [CrossRef]
- Van Ham, S.M.; van Alphen, L.; Mooi, F.R.; van Putten, J.P. Phase variation of H. influenzae fimbriae: Transcriptional control of two divergent genes through a variable combined promoter region. Cell 1993, 73, 1187–1196. [Google Scholar] [CrossRef]
- Metruccio, M.M.; Pigozzi, E.; Roncarati, D.; Berlanda Scorza, F.; Norais, N.; Hill, S.A.; Scarlato, V.; Delany, I. A novel phase variation mechanism in the meningococcus driven by a ligand-responsive repressor and differential spacing of distal promoter elements. PLoS Pathog. 2009, 5, e1000710. [Google Scholar] [CrossRef]
- Carson, S.D.; Stone, B.; Beucher, M.; Fu, J.; Sparling, P.F. Phase variation of the gonococcal siderophore receptor FetA. Mol Microbiol 2000, 36, 585–593. [Google Scholar]
- Tauseef, I.; Harrison, O.B.; Wooldridge, K.G.; Feavers, I.M.; Neal, K.R.; Gray, S.J.; Kriz, P.; Turner, D.P.; Ala'Aldeen, D.A.; Maiden, M.C.; et al. Influence of the combination and phase variation status of the haemoglobin receptors HmbR and HpuAB on meningococcal virulence. Microbiology 2011, 157, 1446–1456. [Google Scholar] [CrossRef]
- Power, P.M.; Roddam, L.F.; Rutter, K.; Fitzpatrick, S.Z.; Srikhanta, Y.N.; Jennings, M.P. Genetic characterization of pilin glycosylation and phase variation in Neisseria meningitidis. Mol. Microbiol. 2003, 49, 833–847. [Google Scholar]
- Aas, F.E.; Vik, A.; Vedde, J.; Koomey, M.; Egge-Jacobsen, W. Neisseria gonorrhoeae O-linked pilin glycosylation: functional analyses define both the biosynthetic pathway and glycan structure. Mol. Microbiol. 2007, 65, 607–624. [Google Scholar] [CrossRef]
- Churbanov, A.; Ryan, R.; Hasan, N.; Bailey, D.; Chen, H.; Milligan, B.; Houde, P. HighSSR: high-throughput SSR characterization and locus development from next-gen sequencing data. Bioinformatics 2012, 28, 2797–2803. [Google Scholar] [CrossRef]
- Fondon, J.W., III; Martin, A.; Richards, S.; Gibbs, R.A.; Mittelman, D. Analysis of microsatellite variation in Drosophila melanogaster with population-scale genome sequencing. PLoS One 2012, 7, e33036. [Google Scholar]
- Bayliss, C.D. A preliminary analysis of genome assemblies of meningococcal carriage isolates. 2014; in preparion. [Google Scholar]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
- Bilek, N.; Ison, C.A.; Spratt, B.G. Relative contributions of recombination and mutation to the diversification of the opa gene repertoire of Neisseria gonorrhoeae. J. Bacteriol. 2009, 191, 1878–1890. [Google Scholar] [CrossRef]
- Lucidarme, J.; Findlow, J.; Chan, H.; Feavers, I.M.; Gray, S.J.; Kaczmarski, E.B.; Parkhill, J.; Bai, X.; Borrow, R.; Bayliss, C.D. The distribution and “in vivo” phase variation status of haemoglobin receptors in invasive meningococcal serogroup B disease: Genotypic and phenotypic analysis. PLoS One 2013, 8, e76932. [Google Scholar] [CrossRef]
- Jerome, J.P.; Bell, J.A.; Plovanich-Jones, A.E.; Barrick, J.E.; Brown, C.T.; Mansfield, L.S. Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLoS One 2011, 6, e16399. [Google Scholar]
- Caldwell, M.B.; Guerry, P.; Lee, E.C.; Burans, J.P.; Walker, R.I. Reversible expression of flagella in Campylobacter jejuni. Infect. Immun. 1985, 50, 941–943. [Google Scholar]
- Linton, D.; Gilbert, M.; Hitchen, P.G.; Dell, A.; Morris, H.R.; Wakarchuk, W.W.; Gregson, N.A.; Wren, B.W. Phase variation of a beta-1,3 galactosyltransferase involved in generation of the ganglioside GM1-like lipo-oligosaccharide of Campylobacter jejuni. Mol. Microbiol. 2000, 37, 501–514. [Google Scholar]
- Bacon, D.J.; Szymanski, C.M.; Burr, D.H.; Silver, R.P.; Alm, R.A.; Guerry, P. A phase-variable capsule is involved in virulence of Campylobacter jejuni 81–176. Mol. Microbiol. 2001, 40, 769–777. [Google Scholar] [CrossRef]
- Guerry, P.; Szymanski, C.M.; Prendergast, M.M.; Hickey, T.E.; Ewing, C.P.; Pattarini, D.L.; Moran, A.P. Phase variation of Campylobacter jejuni 81–176 lipooligosaccharide affects ganglioside mimicry and invasiveness in vitro. Infect. Immun. 2002, 70, 787–793. [Google Scholar] [CrossRef]
- Karlyshev, A.V.; Champion, O.L.; Churcher, C.; Brisson, J.R.; Jarrell, H.C.; Gilbert, M.; Brochu, D.; St. Michael, F.; Li, J.; Wakarchuk, W.W.; et al. Analysis of Campylobacter jejuni capsular loci reveals multiple mechanisms for the generation of structural diversity and the ability to form complex heptoses. Mol. Microbiol. 2005, 55, 90–103. [Google Scholar]
- Szymanski, C.M.; Michael, F.S.; Jarrell, H.C.; Li, J.; Gilbert, M.; Larocque, S.; Vinogradov, E.; Brisson, J.R. Detection of conserved N-linked glycans and phase-variable lipooligosaccharides and capsules from campylobacter cells by mass spectrometry and high resolution magic angle spinning NMR spectroscopy. J. Biol. Chem. 2003, 278, 24509–24520. [Google Scholar] [CrossRef]
- Prendergast, M.M.; Tribble, D.R.; Baqar, S.; Scott, D.A.; Ferris, J.A.; Walker, R.I.; Moran, A.P. In vivo phase variation and serologic response to lipooligosaccharide of Campylobacter jejuni in experimental human infection. Infect. Immun. 2004, 72, 916–922. [Google Scholar] [CrossRef]
- Ashgar, S.S.; Oldfield, N.J.; Wooldridge, K.G.; Jones, M.A.; Irving, G.J.; Turner, D.P.; Ala’Aldeen, D.A. CapA, an autotransporter protein of Campylobacter jejuni, mediates association with human epithelial cells and colonization of the chicken gut. J. Bacteriol. 2007, 189, 1856–1865. [Google Scholar] [CrossRef]
- Lango-Scholey, L.; Aidley, J.; Akinyemi, N.M.; Woodacre, A.; Jones, M.A.; Bayliss, C.D. Development of a multiplex GeneScan assay for analysis of the phase variable genes of Campylobacter jejuni strain NCTC11168. 2014; In preparation. [Google Scholar]
- Schryvers, A.B.; Gonzalez, G.C. Receptors for transferrin in pathogenic bacteria are specific for the host’s protein. Can. J. Microbiol. 1990, 36, 145–147. [Google Scholar] [CrossRef]
- Ala'Aldeen, D.A.; Borriello, S.P. The meningococcal transferrin-binding proteins 1 and 2 are both surface exposed and generate bactericidal antibodies capable of killing homologous and heterologous strains. Vaccine 1996, 14, 49–53. [Google Scholar] [CrossRef]
- Lewis, L.A.; Gray, E.; Wang, Y.P.; Roe, B.A.; Dyer, D.W. Molecular characterization of hpuAB, the haemoglobin-haptoglobin-utilization operon of Neisseria meningitidis. Mol. Microbiol. 1997, 23, 737–749. [Google Scholar]
- Stojiljkovic, I.; Larson, J.; Hwa, V.; Anic, S.; So, M. HmbR outer membrane receptors of pathogenic Neisseria spp.: Iron-regulated, hemoglobin-binding proteins with a high level of primary structure conservation. J. Bacteriol. 1996, 178, 4670–4678. [Google Scholar]
- Lewis, L.A.; Gipson, M.; Hartman, K.; Ownbey, T.; Vaughn, J.; Dyer, D.W. Phase variation of HpuAB and HmbR, two distinct haemoglobin receptors of Neisseria meningitidis DNM2. Mol. Microbiol. 1999, 32, 977–989. [Google Scholar] [CrossRef]
- Harrison, O.B.; Evans, N.J.; Blair, J.M.; Grimes, H.S.; Tinsley, C.R.; Nassif, X.; Kriz, P.; Ure, R.; Gray, S.J.; Derrick, J.P.; et al. Epidemiological evidence for the role of the hemoglobin receptor, hmbR, in meningococcal virulence. J. Infect. Dis. 2009, 200, 94–98. [Google Scholar] [CrossRef]
- Harrison, O.B.; Bennett, J.S.; Derrick, J.P.; Maiden, M.C.; Bayliss, C.D. Distribution and diversity of the haemoglobin-haptoglobin iron-acquisition systems in pathogenic and non-pathogenic Neisseria. Microbiology 2013, 159, 1920–1930. [Google Scholar] [CrossRef]
- Omer, H.; Rose, G.; Jolley, K.A.; Frapy, E.; Zahar, J.R.; Maiden, M.C.; Bentley, S.D.; Tinsley, C.R.; Nassif, X.; Bille, E. Genotypic and phenotypic modifications of Neisseria meningitidis after an accidental human passage. PLoS One 2011, 6, e17145. [Google Scholar] [CrossRef]
- Richardson, A.R.; Stojiljkovic, I. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol. Micro. 2001, 40, 645–655. [Google Scholar] [CrossRef]
- Perkins-Balding, D.; Ratliff-Griffin, M.; Stojiljkovic, I. Iron transport systems in Neisseria meningitidis. Microbiol. Mol. Biol. Rev. 2004, 68, 154–171. [Google Scholar] [CrossRef]
- Oldfield, N.J.; Matar, S.; Bidmos, F.A.; Alamro, M.; Neal, K.R.; Turner, D.P.; Bayliss, C.D.; Ala’Aldeen, A.A. Prevalence and Phase Variable Expression Status of Two Autotransporters, NalP and MspA, in Carriage and Disease Isolates of Neisseria meningitidis. PLoS One 2013, 8, e69746. [Google Scholar] [CrossRef]
- Turner, D.P.; Marietou, A.G.; Johnston, L.; Ho, K.K.; Rogers, A.J.; Wooldridge, K.G.; Ala’Aldeen, D.A. Characterization of MspA, an immunogenic autotransporter protein that mediates adhesion to epithelial and endothelial cells in Neisseria meningitidis. Infect. Immun. 2006, 74, 2957–2964. [Google Scholar] [CrossRef]
- Van Ulsen, P.; Adler, B.; Fassler, P.; Gilbert, M.; van Schilfgaarde, M.; van der Ley, P.; van Alphen, L.; Tommassen, J. A novel phase-variable autotransporter serine protease, AusI, of Neisseria meningitidis. Microbes Infect. 2006, 8, 2088–2097. [Google Scholar] [CrossRef]
- Serruto, D.; Spadafina, T.; Ciucchi, L.; Lewis, L.A.; Ram, S.; Tontini, M.; Santini, L.; Biolchi, A.; Seib, K.L.; Giuliani, M.M.; et al. Neisseria meningitidis GNA2132, a heparin-binding protein that induces protective immunity in humans. Proc. Natl. Acad. Sci. USA 2010, 107, 3770–3775. [Google Scholar] [CrossRef]
- Arenas, J.; Nijland, R.; Rodriguez, F.J.; Bosma, T.N.; Tommassen, J. Involvement of three meningococcal surface-exposed proteins, the heparin-binding protein NhbA, the alpha-peptide of IgA protease and the autotransporter protease NalP, in initiation of biofilm formation. Mol. Microbiol. 2013, 87, 254–268. [Google Scholar] [CrossRef]
- Del Tordello, E.; Vacca, I.; Ram, S.; Rappuoli, R.; Serruto, D. Neisseria meningitidis NalP cleaves human complement C3, facilitating degradation of C3b and survival in human serum. Proc. Natl. Acad. Sci. USA 2014, 111, 427–432. [Google Scholar]
- Dawid, S.; Barenkamp, S.J.; St. Geme, J.W., III. Variation in expression of the Haemophilus influenzae HMW adhesins: a prokaryotic system reminiscent of eukaryotes. Proc. Natl. Acad. Sci. USA 1999, 96, 1077–1082. [Google Scholar] [CrossRef]
- Cholon, D.M.; Cutter, D.; Richardson, S.K.; Sethi, S.; Murphy, T.F.; Look, D.C.; St. Geme, J.W., III. Serial isolates of persistent Haemophilus influenzae in patients with chronic obstructive pulmonary disease express diminishing quantities of the HMW1 and HMW2 adhesins. Infect. Immun. 2008, 76, 4463–4468. [Google Scholar] [CrossRef]
- Bidmos, F.A.; Neal, K.R.; Oldfield, N.J.; Turner, D.P.; Ala'Aldeen, D.A.; Bayliss, C.D. Persistence, replacement, and rapid clonal expansion of meningococcal carriage isolates in a 2008 university student cohort. J. Clin. Microbiol. 2011, 49, 506–512. [Google Scholar] [CrossRef]
- Alamro, M.; Bidmos, F.A.; Chan, H.; Oldfield, N.J.; Newton, E.; Bai, X.; Aidley, J.; Care, R.; Mattick, C.; Turner, D.P.J.; et al. Phase variation mediates reductions in expression of surface proteins during persistent meningococcal carriage. Infect. Immun. 2014. Submitted. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bidmos, F.A.; Bayliss, C.D. Genomic and Global Approaches to Unravelling How Hypermutable Sequences Influence Bacterial Pathogenesis. Pathogens 2014, 3, 164-184. https://doi.org/10.3390/pathogens3010164
Bidmos FA, Bayliss CD. Genomic and Global Approaches to Unravelling How Hypermutable Sequences Influence Bacterial Pathogenesis. Pathogens. 2014; 3(1):164-184. https://doi.org/10.3390/pathogens3010164
Chicago/Turabian StyleBidmos, Fadil A., and Christopher D. Bayliss. 2014. "Genomic and Global Approaches to Unravelling How Hypermutable Sequences Influence Bacterial Pathogenesis" Pathogens 3, no. 1: 164-184. https://doi.org/10.3390/pathogens3010164