Genome Sequencing of Xanthomonas vasicola Pathovar vasculorum Reveals Variation in Plasmids and Genes Encoding Lipopolysaccharide Synthesis, Type-IV Pilus and Type-III Secretion Effectors


Abstract
:
1. Introduction
2. Results and Discussion

2.1. Overview of Sequence Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate (NCPPB number) | Source and date of isolation | Source |
|---|---|---|
| Xvv 206 a | South Africa 1948 | Zea mays |
| Xvv 702 b | Zimbabwe 1959 | Saccharum officinarum |
| Xvv 890 c | South Africa 1960 | S. officinarum |
| Xvv 895 c | Malagasy Republic 1960 | S. officinarum |
| Xvv 1326 a | Zimbabwe 1962 | S. officinarum |
| Xvv 1381 a | Zimbabwe 1962 | S. officinarum |
| Xcm 2005 a | Ethiopia 1967 | Ensete ventricosum |
| Xcm 2251 a | Ethiopia 1969 | Musa sp. |
| Xcm 4379 a | Uganda 2007 | Musa sp. |
| Xcm 4380 a | Uganda 2007 | Musa sp. |
| Xcm 4381 b | Uganda 2007 | Musa sp. |
| Xcm 4383 a | Uganda 2007 | Musa sp. |
| Xcm 4384 a | Uganda 2007 | Musa sp. |
| Xcm 4387 a | D. R. Congo 2007 | Musa sp. |
| Xcm 4389 a | Rwanda 2007 | Musa sp. |
| Xcm 4392 a | Tanzania 2007 | Musa sp. |
| Xcm 4394 a | Tanzania 2007 | Musa sp. |
| Xcm 4395 a | Tanzania 2007 | Musa sp. |
| Xcm “Kenyan” d | Kenya (year not known) | Musa sp. |
| Isolate (NCPPB number) | Number of read pairs | Read length | Coverage | SRA accession |
|---|---|---|---|---|
| Xvv 206 a | 2,579,404 | 76 | 70 x | SRR494500.3 |
| Xvv 702 b | 2,913,785 | 36 | 35 x | SRR020202.3 |
| Xvv 890 c | 4,843,028 | 67 | 58 x | SRR1045340 |
| Xvv 895 c | 2,867,513 | 67 | 50 x | SRR1045341 |
| Xvv 1326 a | 2,365,912 | 76 | 63 x | SRR494491.5 |
| Xvv 1381 a | 2,450,234 | 76 | 66 x | SRR494499.3 |
| Xcm 2005 a | 2,536,030 | 76 | 72 x | SRR489154.7 |
| Xcm 4379 a | 3,652,875 | 76 | 102 x | SRR494484.2 |
| Xcm 4380 a | 4,069,509 | 76 | 113 x | SRR494485.2 |
| Xcm 4381 b | 5,052,905 | 36 | 56 x | SRR020203.3 |
| Xcm 4384 a | 1,976,797 | 76 | 55 x | SRR494488.2 |
| Xcm 4392 a | 2,554,161 | 76 | 72 x | SRR494498.3 |
| Xcm 4394 a | 3,295,956 | 76 | 92 x | SRR494489.1 |
| Xcm 4395 a | 4,117,662 | 76 | 117 x | SRR494490.2 |
| Isolate (NCPPB number) | Number of scaffolds | Scaffolds N50 | Total length (b.p.) | GenBank accession |
|---|---|---|---|---|
| Xvv 206 a | 177 | 103,376 | 4,825,935 | AKBM00000000.1 |
| Xvv 702 b | 97 | 205,000 | 5,478,002 | ACHS00000000.1 |
| Xvv 890 c | 2,565 | 3,627 | 4,951,053 | AKBN00000000.1 |
| Xvv 895 c | 1,229 | 8,362 | 4,803,807 | AKBO00000000.1 |
| Xvv 1326 a | 253 | 74,455 | 4,951,570 | AKBK00000000.1 |
| Xvv 1381 a | 137 | 108,088 | 4,958,101 | AKBL00000000.1 |
| Xcm 2005 a | 156 | 61,233 | 4,692,764 | AKBE00000000.1 |
| Xcm 4379 a | 95 | 147,554 | 4,758,198 | AKBF00000000.1 |
| Xcm 4380 a | 87 | 149,435 | 4,751,644 | AKBG00000000.1 |
| Xcm 4381 b | 115 | 143,688 | 4,793,900 | ACHT00000000.1 |
| Xcm 4384 a | 84 | 157,780 | 4,741,777 | AKBH00000000.1 |
| Xcm 4392 a | 162 | 90,974 | 4,728,564 | AKBI01000000.1 |
| Xcm 4394 a | 85 | 151,917 | 4,792,617 | AKBJ00000000.1 |
| Xcm “Kenyan” d | 510 | 36,401 | 4,907,936 | AGFQ01000000.1 |




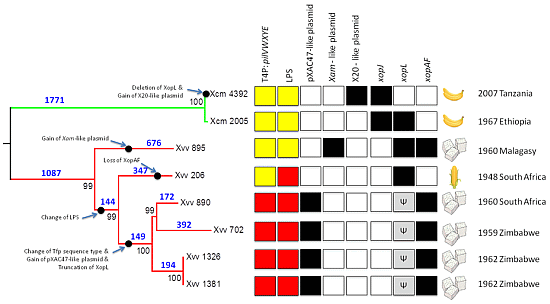
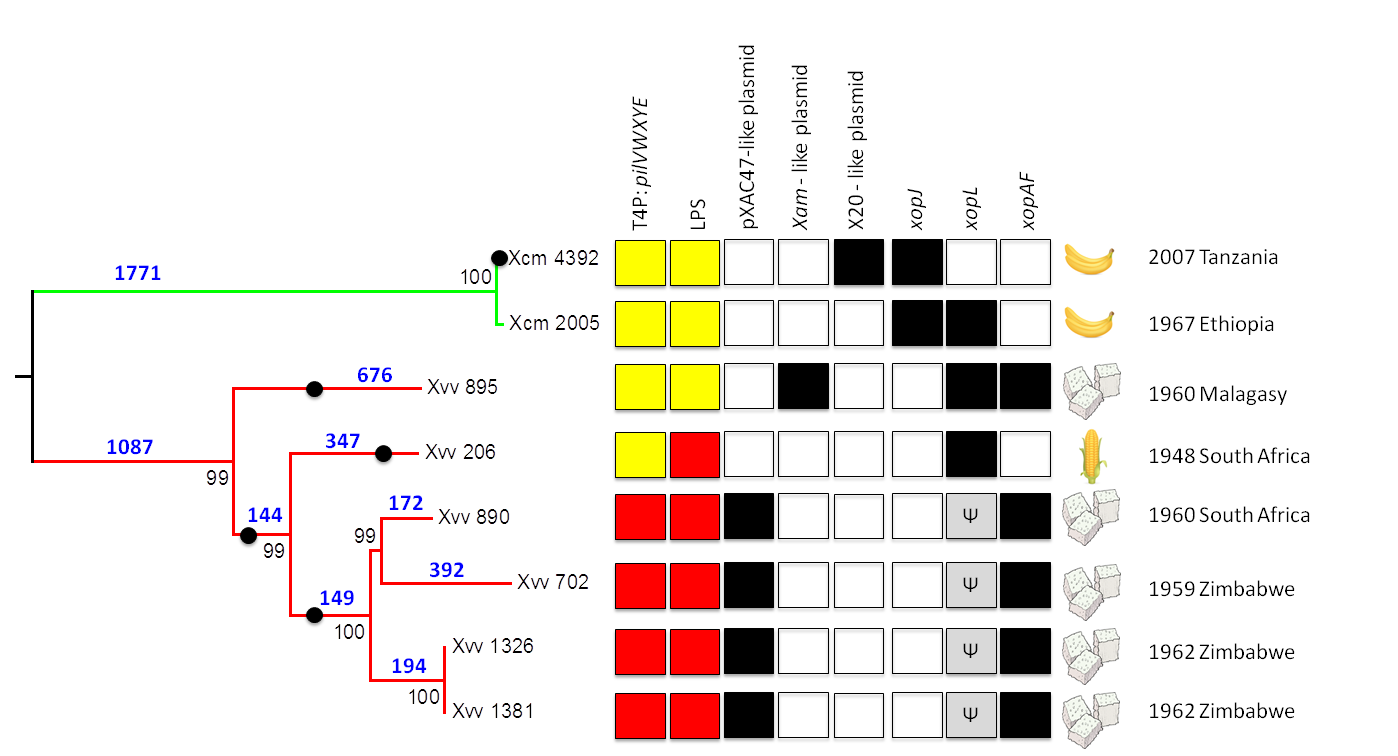
2.2. Phylogenetic Relationships among Xvv Strains
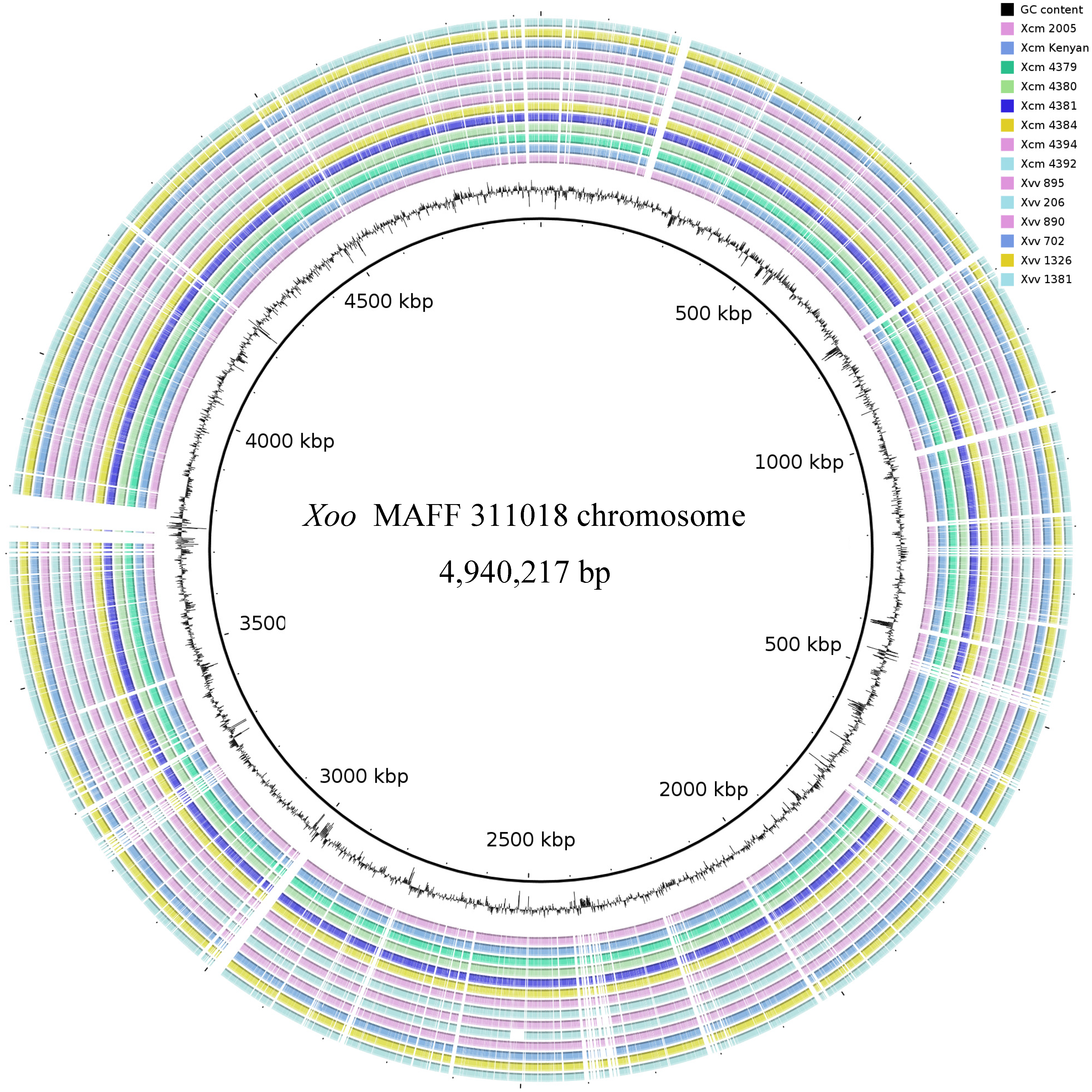
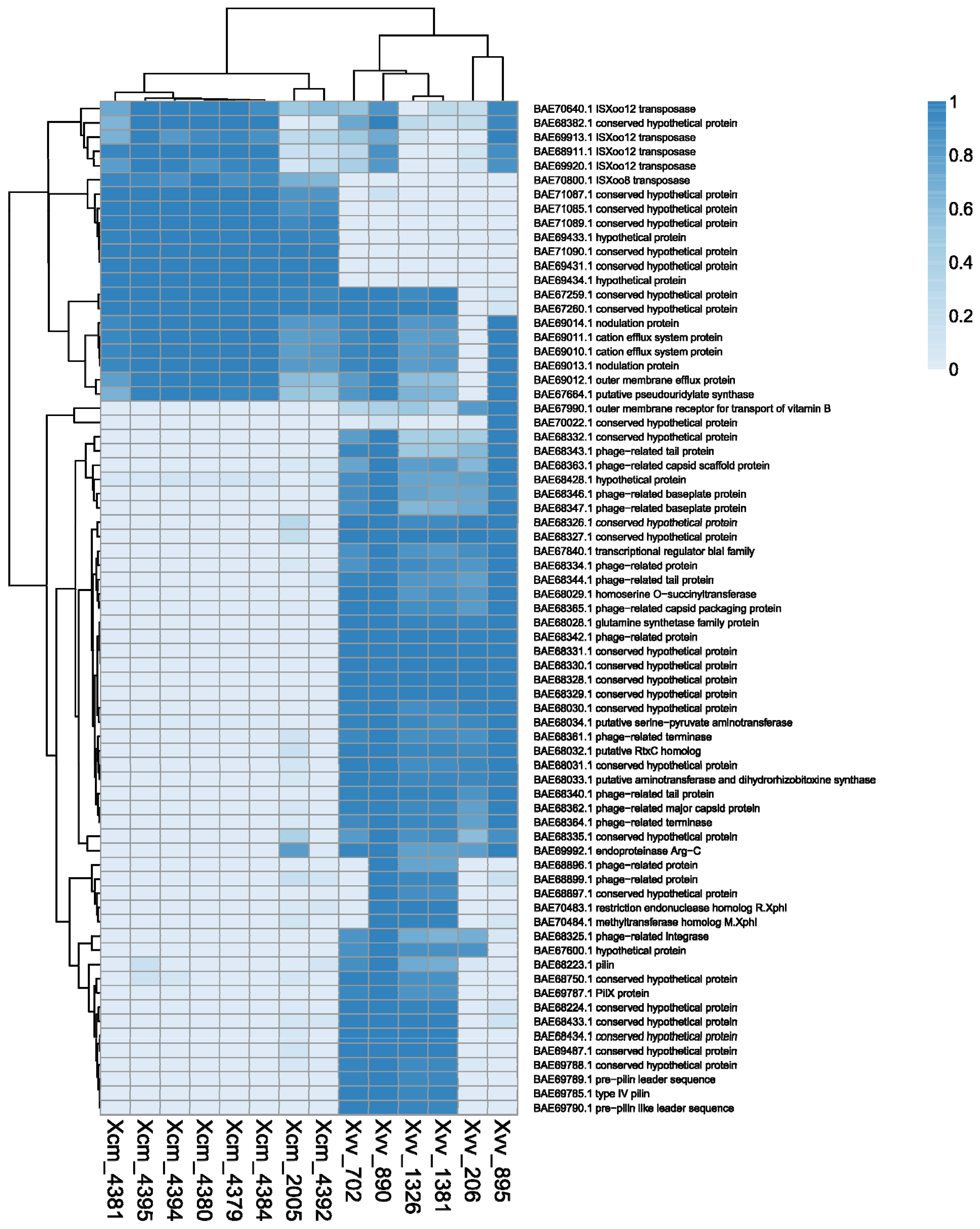
2.3. Global Genomic Comparison of Genomes of Xvv and Xcm Isolates
2.4. Xvv Isolates NCPPB 890, 702, 1326 and 1381 Contain Sequences Similar to Plasmid pXAC47
2.5. Xvv Isolate NCPPB 895 Contains Sequences Similar to a Plasmid from X. axonopodis pv. manihotis (Xam)
2.6. Xcm Isolates NCPPB 4379, 4380, 4383, 4384, 4392 and 4395 Contain Sequences Similar to a Plasmid from X. citri pv. malvacearum Strain X20
2.7. Genetic Variation in the Type-IV Pilus (T4P) Among Isolates of Xvv
2.7.1. The pilVWXYE Gene Cluster
2.7.2. The pilCABRS Gene Cluster
2.7.3. Concluding Remarks about Variation in T4P Genes
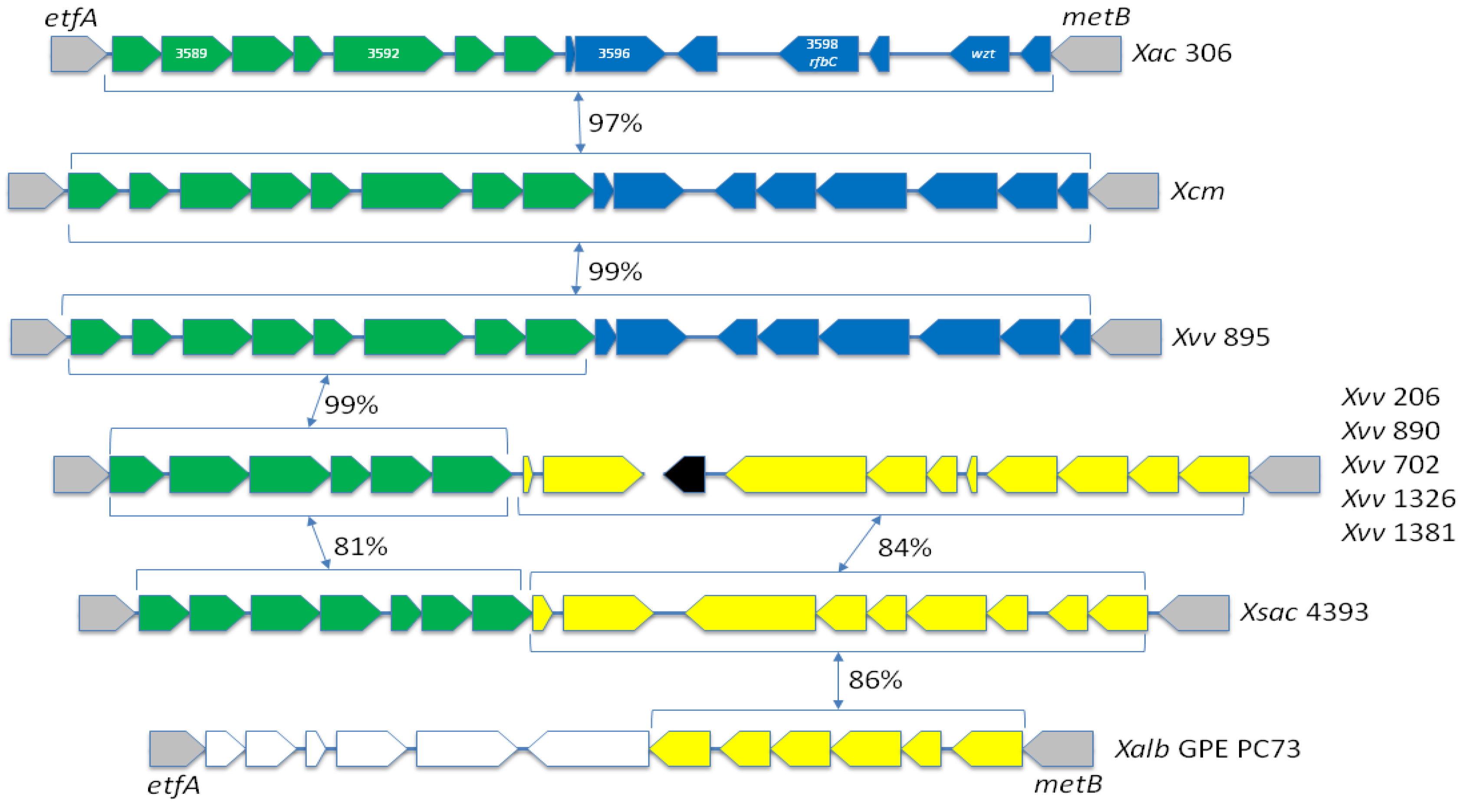
2.8. There Are Two Distinct Sequence Types of LPS Biosynthesis Clusters in Xvv

2.9. Xvv Isolates Differ in Their Repertoires of T3SS Effector Genes
2.9.1. Gene for XopAF is Absent from Xvv 206, Isolated from Maize
2.9.2. Gene Encoding Homologue of XopL Underwent Truncation in Common Ancestor of Xvv 890, 702, 1326 and 1381
2.9.3. Absence of Genes Encoding Homologues of XopJ Distinguishes Xcm from Xvv
3. Experimental Section
3.1. Sources of Bacterial Strains
3.2. Preparation of Genomic DNA
3.3. Genome Sequencing
3.4. Alignment of Sequence Reads Against Reference Genome Sequences
3.5. SNP Calling and Phylogenetic Analysis
3.6. Genome Assembly
3.7. Identification of Presence and Absence of Genes
3.8. Visualisation of Genome-Wide Patterns of Sequence Conservation
3.9. Identification of Potential PIP Boxes
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hayward, A.C. The hosts of Xanthomonas. In Xanthomonas; Springer: Dordrecht, The Netherlands, 1993; pp. 1–119. [Google Scholar]
- Ryan, R.P.; Vorhölter, F.-J.; Potnis, N.; Jones, J.B.; van Sluys, M.-A.; Bogdanove, A.J.; Dow, J.M. Pathogenomics of Xanthomonas: understanding bacterium-plant interactions. Nat. Rev. Microbiol. 2011, 9, 344–355. [Google Scholar] [CrossRef]
- Dookun, A.; Stead, D.E.; Autrey, L.J. Variation among strains of Xanthomonas campestris pv. vasculorum from Mauritius and other countries based on fatty acid analysis. Syst. Appl. Microbiol. 2000, 23, 148–155. [Google Scholar] [CrossRef]
- Aritua, V.; Parkinson, N.; Thwaites, R.; Heeney, J.V.; Jones, D.R.; Tushemereirwe, W.; Crozier, J.; Reeder, R.; Stead, D.E.; Smith, J. Characterization of the Xanthomonas sp causing wilt of enset and banana and its proposed reclassification as a strain of X. vasicola. Plant Pathol. 2008, 57, 170–177. [Google Scholar]
- Odipio, J.; Tusiime, G.; Tripathi, L. Genetic homogeneity among Ugandan isolates of Xanthomonas campestris pv. musacearum revealed by randomly amplified polymorphic DNA analysis. Afr. J. Biotechnol. 2009. [Google Scholar] [CrossRef]
- Aritua, V.; Nanyonjo, A.; Kumakech, F.; Tushemereirwe, W. Rep-PCR reveals a high genetic homogeneity among Ugandan isolates of Xanthomonas campestris pv musacearum. Afr J Biotechnol. 2007, 6, 179–183. [Google Scholar]
- Wasukira, A.; Tayebwa, J.; Thwaites, R.; Paszkiewicz, K.; Aritua, V.; Kubiriba, J.; Smith, J.; Grant, M.; Studholme, D.J. Genome-wide sequencing reveals two major sub-lineages in the genetically monomorphic pathogen Xanthomonas campestris pathovar musacearum. Genes. 2012, 3, 361–377. [Google Scholar] [CrossRef]
- Studholme, D.J.; Kemen, E.; MacLean, D.; Schornack, S.; Aritua, V.; Thwaites, R.; Grant, M.; Smith, J.; Jones, J.D.G. Genome-wide sequencing data reveals virulence factors implicated in banana Xanthomonas wilt. FEMS Microbiol. Lett. 2010, 310, 182–192. [Google Scholar] [CrossRef]
- Ivey, M.L.L.; Tusiime, G.; Miller, S.A. A Polymerase Chain Reaction Assay for the Detection of Xanthomonas campestris pv. musacearum in Banana. Plant Dis. 2010, 94, 109–114. [Google Scholar] [CrossRef]
- Qhobela, M.; Claflin, L.E. Eastern and southern African strains of Xanthomonas campestris pv. vasculorum are distinguishable by restriction fragment length polymorphism of DNA and polyacrylamide gel electrophoresis of membrane proteins. Plant Pathol. 1992, 41, 113–121. [Google Scholar] [CrossRef]
- Vauterin, L.; Hoste, B.; Kersters, K.; Swings, J. Reclassification of Xanthomonas. Int. J. Syst. Bacteriol. 1995, 472–489. [Google Scholar]
- Rademaker, J.L.W.; Louws, F.J.; Schultz, M.H.; Rossbach, U.; Vauterin, L.; Swings, J.; de Bruijn, F.J. A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology 2005, 95, 1098–1111. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Sommer, D.D.; Schatz, M.C.; Phillippy, A.M.; Rabinowicz, P.D.; Tsuge, S.; Furutani, A.; Ochiai, H.; Delcher, A.L.; Kelley, D.; et al. Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genom. 2008, 9, 204. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Bart, R.; Cohn, M.; Kassen, A.; McCallum, E.J.; Shybut, M.; Petriello, A.; Krasileva, K.; Dahlbeck, D.; Medina, C.; Alicai, T.; et al. High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc. Natl. Acad. Sci. USA. 2012, 109, E1972–E1979. [Google Scholar] [CrossRef]
- Cunnac, S.; Bolot, S.; Serna, N.F.; Ortiz, E.; Szurek, B.; Noël, L.D.; Arlat, M.; Jacques, M.-A.; Gagnevin, L.; Carrere, S.; et al. High-quality draft genome sequences of two Xanthomonas citri pv. malvacearum strains. Genome Announc. 2013. [Google Scholar] [CrossRef]
- Potnis, N.; Krasileva, K.; Chow, V.; Almeida, N.F.; Patil, P.B.; Ryan, R.P.; Sharlach, M.; Behlau, F.; Dow, J.M.; Momol, M.T.; et al. Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genom. 2011, 12, 146. [Google Scholar] [CrossRef]
- Taguchi, F.; Ichinose, Y. Role of type IV pili in virulence of Pseudomonas syringae pv. tabaci 6605: Correlation of motility, multidrug resistance, and HR-inducing activity on a nonhost plant. Mol. Plant. Microbe. Interact. 2011, 24, 1001–1011. [Google Scholar] [CrossRef]
- Liu, H.; Kang, Y.; Genin, S.; Schell, M.A.; Denny, T.P. Twitching motility of Ralstonia solanacearum requires a type IV pilus system. Microbiology 2001, 147, 3215–3229. [Google Scholar]
- Mattick, J.S. Type IV pili and twitching motility. Annu. Rev. Microbiol. 2002, 56, 289–314. [Google Scholar] [CrossRef]
- De La Fuente, L.; Burr, T.J.; Hoch, H.C. Mutations in type I and type IV pilus biosynthetic genes affect twitching motility rates in Xylella fastidiosa. J. Bacteriol. 2007, 189, 7507–7510. [Google Scholar] [CrossRef]
- Li, Y.; Hao, G.; Galvani, C.D.; Meng, Y.; De La Fuente, L.; Hoch, H.C.; Burr, T.J. Type I and type IV pili of Xylella fastidiosa affect twitching motility, biofilm formation and cell-cell aggregation. Microbiology 2007, 153, 719–726. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Wan, D.-S.; Huang, S.-S.; Leng, F.-F.; Yan, L.; Ni, Y.-Q.; Li, H.-Y. Type IV pili of Acidithiobacillus ferrooxidans are necessary for sliding, twitching motility, and adherence. Curr. Microbiol. 2010, 60, 17–24. [Google Scholar] [CrossRef]
- Pelicic, V. Type IV pili: E pluribus unum? Mol. Microbiol. 2008, 68, 827–837. [Google Scholar] [CrossRef]
- Burdman, S.; Bahar, O.; Parker, J.K.; De La Fuente, L. Involvement of type IV Pili in pathogenicity of plant pathogenic bacteria. Genes. 2011, 2, 706–735. [Google Scholar] [CrossRef]
- Jenkins, A.T.A.; Buckling, A.; McGhee, M.; ffrench-Constant, R.H. Surface plasmon resonance shows that type IV pili are important in surface attachment by Pseudomonas aeruginosa. J. R. Soc. Interface 2005, 2, 255–259. [Google Scholar] [CrossRef]
- Heijstra, B.D.; Pichler, F.B.; Liang, Q.; Blaza, R.G.; Turner, S.J. Extracellular DNA and Type IV pili mediate surface attachment by Acidovorax temperans. Antonie Van Leeuwenhoek 2009, 95, 343–349. [Google Scholar] [CrossRef]
- Roine, E.; Raineri, D.M.; Romantschuk, M.; Wilson, M.; Nunn, D.N. Characterization of type IV pilus genes in Pseudomonas syringae pv. tomato DC3000. Mol. Plant. Microbe. Interact. 1998, 11, 1048–1056. [Google Scholar] [CrossRef]
- Shime-Hattori, A.; Iida, T.; Arita, M.; Park, K.-S.; Kodama, T.; Honda, T. Two type IV pili of Vibrio parahaemolyticus play different roles in biofilm formation. FEMS Microbiol. Lett. 2006, 264, 89–97. [Google Scholar] [CrossRef]
- Darsonval, A.; Darrasse, A.; Meyer, D.; Demarty, M.; Durand, K.; Bureau, C.; Manceau, C.; Jacques, M. The Type III secretion system of Xanthomonas fuscans subsp. fuscans is involved in the phyllosphere colonization process and in transmission to seeds of susceptible beans. Appl. Environ. Microbiol. 2008, 74, 2669–2678. [Google Scholar] [CrossRef]
- Varga, J.J.; Therit, B.; Melville, S.B. Type IV pili and the CcpA protein are needed for maximal biofilm formation by the gram-positive anaerobic pathogen Clostridium perfringens. Infect. Immun. 2008, 76, 4944–4951. [Google Scholar] [CrossRef]
- Mhedbi-Hajri, N.; Darrasse, A.; Pigné, S.; Durand, K.; Fouteau, S.; Barbe, V.; Manceau, C.; Lemaire, C.; Jacques, M.-A. Sensing and adhesion are adaptive functions in the plant pathogenic xanthomonads. BMC Evol. Biol. 2011, 11, 67. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.-J.; Chung, I.-Y.; Choi, K.B.; Lau, G.W.; Cho, Y.-H. Genome sequence comparison and superinfection between two related Pseudomonas aeruginosa phages, D3112 and MP22. Microbiology 2007, 153, 2885–2895. [Google Scholar] [CrossRef]
- Lerouge, I.; Vanderleyden, J. O-antigen structural variation: Mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol. Rev. 2002, 26, 17–47. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Hendrick, C.A.; Sequeira, L. Lipopolysaccharide-DEFECTIVE Mutants of the Wilt Pathogen Pseudomonas solanacearum. Appl. Environ. Microbiol. 1984, 48, 94–101. [Google Scholar]
- Drigues, P.; Demery-Lafforgue, D.; Trigalet, A.; Dupin, P.; Samain, D.; Asselineau, J. Comparative studies of lipopolysaccharide and exopolysaccharide from a virulent strain of Pseudomonas solanacearum and from three avirulent mutants. J. Bacteriol. 1985, 162, 504–509. [Google Scholar]
- Jayaswal, R.K.; Bressan, R.A.; Handa, A.K. Effects of a mutation that eliminates UDP glucose-pyrophosphorylase on the pathogenicity of Erwinia carotovora subsp. carotovora. J. Bacteriol. 1985, 164, 473–476. [Google Scholar]
- Schoonejans, E.; Expert, D.; Toussaint, A. Characterization and virulence properties of Erwinia chrysanthemi lipopolysaccharide-defective, phi EC2-resistant mutants. J. Bacteriol. 1987, 169, 4011–4017. [Google Scholar]
- Kingsley, M.T.; Gabriel, D.W.; Marlow, G.C.; Roberts, P.D. The opsX locus of Xanthomonas campestris affects host range and biosynthesis of lipopolysaccharide and extracellular polysaccharide. J. Bacteriol. 1993, 175, 5839–5850. [Google Scholar]
- Dow, J.M.; Osbourn, A.E.; Wilson, T.J.; Daniels, M.J. A locus determining pathogenicity of Xanthomonas campestris is involved in lipopolysaccharide biosynthesis. Mol. Plant. Microbe. Interact. 1995, 8, 768–777. [Google Scholar]
- Titarenko, E.; López-Solanilla, E.; García-Olmedo, F.; Rodríguez-Palenzuela, P. Mutants of Ralstonia (Pseudomonas) solanacearum sensitive to antimicrobial peptides are altered in their lipopolysaccharide structure and are avirulent in tobacco. J. Bacteriol. 1997, 179, 6699–6704. [Google Scholar]
- Li, J.; Wang, N. The gpsX gene encoding a glycosyltransferase is important for polysaccharide production and required for full virulence in Xanthomonas citri subsp. citri. BMC Microbiol. 2012, 12, 31. [Google Scholar] [CrossRef]
- Yan, Q.; Hu, X.; Wang, N. The novel virulence-related gene nlxA in the lipopolysaccharide cluster of Xanthomonas citri ssp. citri is involved in the production of lipopolysaccharide and extracellular polysaccharide, motility, biofilm formation and stress resistance. Mol. Plant Pathol. 2012, 13, 923–934. [Google Scholar] [CrossRef]
- Li, J.; Wang, N. The wxacO gene of Xanthomonas citri ssp. citri encodes a protein with a role in lipopolysaccharide biosynthesis, biofilm formation, stress tolerance and virulence. Mol. Plant Pathol. 2011, 12, 381–396. [Google Scholar] [CrossRef]
- Patil, P.B.; Bogdanove, A.J.; Sonti, R.V. The role of horizontal transfer in the evolution of a highly variable lipopolysaccharide biosynthesis locus in xanthomonads that infect rice, citrus and crucifers. BMC Evol Biol 2007, 7, 243. [Google Scholar] [CrossRef]
- Patil, P.; Sonti, R. Variation suggestive of horizontal gene transfer at a lipopolysaccharide (lps) biosynthetic locus in Xanthomonas oryzae pv. oryzae, the bacterial leaf blight pathogen of rice. BMC Microbiol. 2004, 4, 40. [Google Scholar] [CrossRef]
- Lu, H.; Patil, P.; van Sluys, M.-A.; White, F.F.; Ryan, R.P.; Dow, J.M.; Rabinowicz, P.; Salzberg, S.L.; Leach, J.E.; Sonti, R.; et al. Acquisition and evolution of plant pathogenesis-associated gene clusters and candidate determinants of tissue-specificity in Xanthomonas. PLoS One 2008, 3, e3828. [Google Scholar] [CrossRef]
- Da Silva, A.C.R.; Ferro, J.A.; Reinach, F.C.; Farah, C.S.; Furlan, L.R.; Quaggio, R.B.; Monteiro-Vitorello, C.B.; van Sluys, M.A.; Almeida, N.F.; Alves, L.M.C.; et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 2002, 417, 459–463. [Google Scholar] [CrossRef]
- Pieretti, I.; Royer, M.; Barbe, V.; Carrere, S.; Koebnik, R.; Cociancich, S.; Couloux, A.; Darrasse, A.; Gouzy, J.; Jacques, M.-A. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genom. 2009, 10, 616. [Google Scholar] [CrossRef]
- Pieretti, I.; Royer, M.; Barbe, V.; Carrere, S.; Koebnik, R.; Couloux, A.; Darrasse, A.; Gouzy, J.; Jacques, M.-A.; Lauber, E.; et al. Genomic insights into strategies used by Xanthomonas albilineans with its reduced artillery to spread within sugarcane xylem vessels. BMC Genom. 2012, 13, 658. [Google Scholar] [CrossRef] [Green Version]
- Studholme, D.J.; Wasukira, A.; Paszkiewicz, K.; Aritua, V.; Thwaites, R.; Smith, J.; Grant, M. Correction: Studholme et al. Draft Genome Sequences of Xanthomonas sacchari and Two Banana-Associated Xanthomonads Reveal Insights into the Xanthomonas Group 1 clade. Genes 2011, 2, 1050–1065. [Google Scholar] [CrossRef]
- Dow, M.; Newman, M.-A.; von Roepenack, E. The induction and modulation of plant defense responses by bacterial lipopolysaccharides. Annu. Rev. Phytopathol. 2000, 38, 241–261. [Google Scholar] [CrossRef]
- Meyer, A.; Pühler, A.; Niehaus, K. The lipopolysaccharides of the phytopathogen Xanthomonas campestris pv. campestris induce an oxidative burst reaction in cell cultures of Nicotiana tabacum. Planta 2001, 213, 214–222. [Google Scholar] [CrossRef]
- Keshavarzi, M.; Soylu, S.; Brown, I.; Bonas, U.; Nicole, M.; Rossiter, J.; Mansfield, J. Basal defenses induced in pepper by lipopolysaccharides are suppressed by Xanthomonas campestris pv. vesicatoria. Mol. Plant. Microbe. Interact. 2004, 17, 805–815. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Chou, C.-P.; Kuo, T.-T.; Lin, S.-H.; Yang, M.-K. PilR enhances the sensitivity of Xanthomonas axonopodis pv. citri to the infection of filamentous bacteriophage Cf. Curr. Microbiol. 2004, 48, 251–261. [Google Scholar] [CrossRef]
- Pal, S.; Wu, L.P. Pattern recognition receptors in the fly: Lessons we can learn from the Drosophila melanogaster immune system. Fly 2009, 3, 121–129. [Google Scholar]
- Sarkar, S.F.; Gordon, J.S.; Martin, G.B.; Guttman, D.S. Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics 2006, 174, 1041–1056. [Google Scholar] [CrossRef]
- Hajri, A.; Brin, C.; Hunault, G.; Lardeux, F.; Lemaire, C.; Manceau, C.; Boureau, T.; Poussier, S. A “repertoire for repertoire” hypothesis: Repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS One 2009, 4, e6632. [Google Scholar]
- Hajri, A.; Pothier, J.F.; Fischer-Le Saux, M.; Bonneau, S.; Poussier, S.; Boureau, T.; Duffy, B.; Manceau, C. Type three effector genes distribution and sequence analysis provides new insights into pathogenicity of plant pathogenic Xanthomonas arboricola. Appl. Environ. Microbiol. 2012, 78, 371–384. [Google Scholar] [CrossRef]
- Hajri, A.; Brin, C.; Zhao, S.; David, P.; Feng, J.-X.; Koebnik, R.; Szurek, B.; Verdier, V.; Boureau, T.; Poussier, S. Multilocus sequence analysis and type III effector repertoire mining provide new insights into the evolutionary history and virulence of Xanthomonas oryzae. Mol. Plant Pathol. 2012, 13, 288–302. [Google Scholar] [CrossRef] [Green Version]
- Astua-Monge, G.; Minsavage, G.V.; Stall, R.E.; Davis, M.J.; Bonas, U.; Jones, J.B. Resistance of tomato and pepper to T3 strains of Xanthomonas campestris pv. vesicatoria is specified by a plant-inducible avirulence gene. Mol. Plant. Microbe. Interact. 2000, 13, 911–921. [Google Scholar] [CrossRef]
- Jalan, N.; Kumar, D.; Andrade, M.O.; Yu, F.; Jones, J.B.; Graham, J.H.; White, F.F.; Setubal, J.C.; Wang, N. Comparative genomic and transcriptome analyses of pathotypes of Xanthomonas citri subsp. citri provide insights into mechanisms of bacterial virulence and host range. BMC Genom. 2013, 14, 551. [Google Scholar] [CrossRef] [Green Version]
- Thieme, F.; Koebnik, R.; Bekel, T.; Berger, C.; Boch, J.; Büttner, D.; Caldana, C.; Gaigalat, L.; Goesmann, A.; Kay, S.; et al. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J. Bacteriol. 2005, 187, 7254–7266. [Google Scholar] [CrossRef]
- Singer, A.U.; Schulze, S.; Skarina, T.; Xu, X.; Cui, H.; Eschen-Lippold, L.; Egler, M.; Srikumar, T.; Raught, B.; Lee, J.; Scheel, D.; Savchenko, A.; Bonas, U. A pathogen type III effector with a novel E3 ubiquitin ligase architecture. PLoS Pathog. 2013, 9, e1003121. [Google Scholar] [CrossRef]
- Jiang, W.; Jiang, B.-L.; Xu, R.-Q.; Huang, J.-D.; Wei, H.-Y.; Jiang, G.-F.; Cen, W.-J.; Liu, J.; Ge, Y.-Y.; Li, G.-H.; et al. Identification of six type III effector genes with the PIP box in Xanthomonas campestris pv. campestris and five of them contribute individually to full pathogenicity. Mol. Plant. Microbe. Interact. 2009, 22, 1401–1411. [Google Scholar] [CrossRef]
- Büttner, D.; Bonas, U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 2010, 34, 107–133. [Google Scholar] [CrossRef]
- Koebnik, R.; Krüger, A.; Thieme, F.; Urban, A.; Bonas, U. Specific binding of the Xanthomonas campestris pv. vesicatoria AraC-type transcriptional activator HrpX to plant-inducible promoter boxes. J. Bacteriol. 2006, 188, 7652–7660. [Google Scholar] [CrossRef]
- Wengelnik, K.; Bonas, U. HrpXv, an AraC-type regulator, activates expression of five of the six loci in the hrp cluster of Xanthomonas campestris pv. vesicatoria. J. Bacteriol. 1996, 178, 3462–3469. [Google Scholar]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2013, 1, 409. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.-A.; Barrell, B.G.; Parkhill, J. ACT: The artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile Hidden Markov Models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- stün, S.; Bartetzko, V.; Börnke, F. The Xanthomonas campestris Type III Effector XopJ Targets the Host Cell Proteasome to Suppress Salicylic-Acid Mediated Plant Defence. PLoS Pathog. 2013, 9, e1003427. [Google Scholar] [CrossRef]
- Qhobela, M.; Leach, J.E. Characterisation of strains of Xanthomonas campestris pv. holcicola by PAGE of membrane proteins and by REA and RFLP analysis of genomic DNA. Plant Dis. 1991, 75, 32–36. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wasukira, A.; Coulter, M.; Al-Sowayeh, N.; Thwaites, R.; Paszkiewicz, K.; Kubiriba, J.; Smith, J.; Grant, M.; Studholme, D.J. Genome Sequencing of Xanthomonas vasicola Pathovar vasculorum Reveals Variation in Plasmids and Genes Encoding Lipopolysaccharide Synthesis, Type-IV Pilus and Type-III Secretion Effectors. Pathogens 2014, 3, 211-237. https://doi.org/10.3390/pathogens3010211
Wasukira A, Coulter M, Al-Sowayeh N, Thwaites R, Paszkiewicz K, Kubiriba J, Smith J, Grant M, Studholme DJ. Genome Sequencing of Xanthomonas vasicola Pathovar vasculorum Reveals Variation in Plasmids and Genes Encoding Lipopolysaccharide Synthesis, Type-IV Pilus and Type-III Secretion Effectors. Pathogens. 2014; 3(1):211-237. https://doi.org/10.3390/pathogens3010211
Chicago/Turabian StyleWasukira, Arthur, Max Coulter, Noorah Al-Sowayeh, Richard Thwaites, Konrad Paszkiewicz, Jerome Kubiriba, Julian Smith, Murray Grant, and David J. Studholme. 2014. "Genome Sequencing of Xanthomonas vasicola Pathovar vasculorum Reveals Variation in Plasmids and Genes Encoding Lipopolysaccharide Synthesis, Type-IV Pilus and Type-III Secretion Effectors" Pathogens 3, no. 1: 211-237. https://doi.org/10.3390/pathogens3010211