Comparative Genomics Identifies a Potential Marker of Human-Virulent Anaplasma phagocytophilum

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
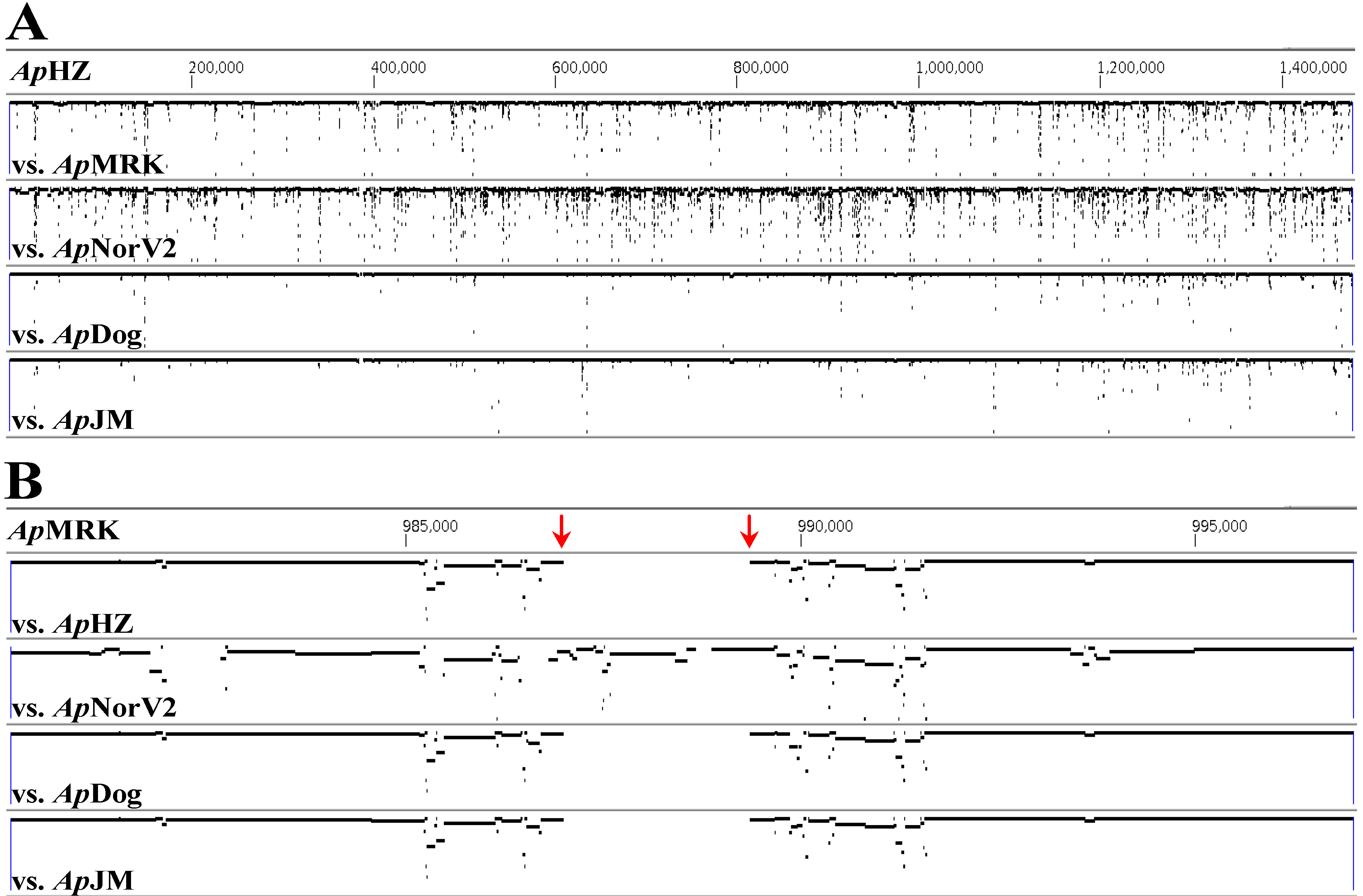
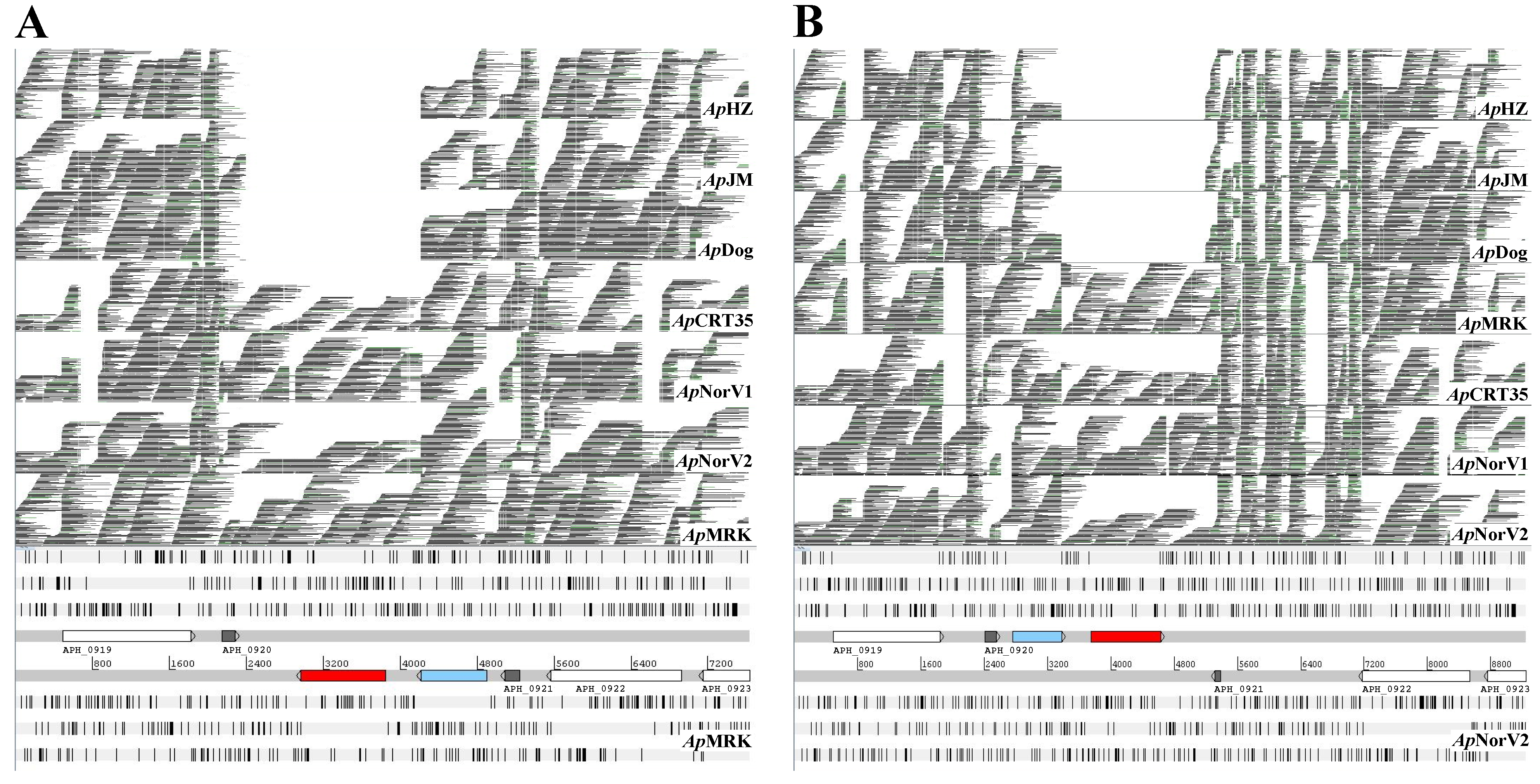
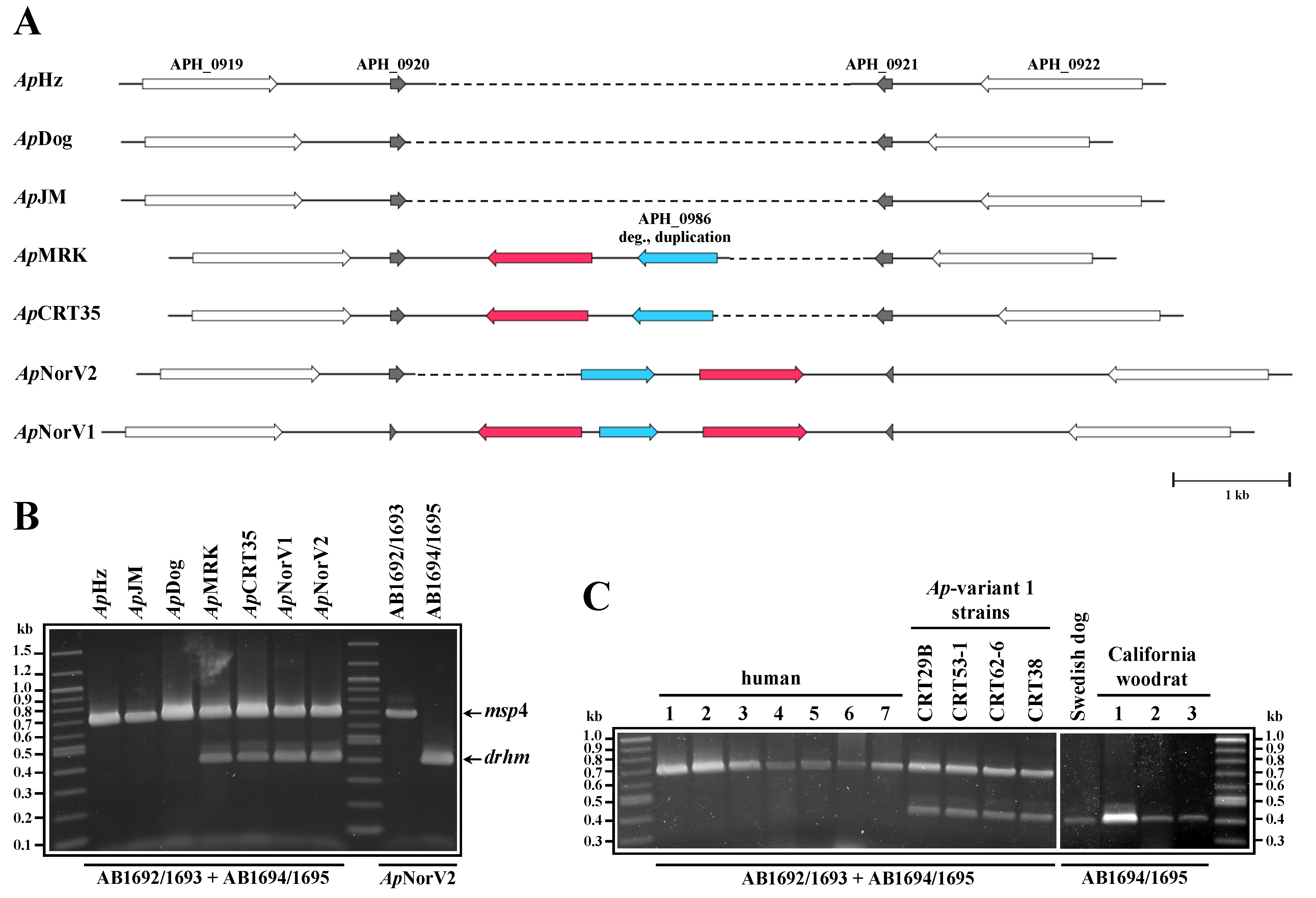
2. Results and Discussion




3. Experimental Section
3.1. A. phagocytophilum Strain Origins and Genomic DNA Isolation
3.2. Ethics Statement
3.3. 454 Genome Sequencing and Bioinformatics
3.4. PCR Amplification
3.5. GenBank Accession Numbers
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- CDC; Statistics and Epidemiology. Annual Cases of Anaplasmosis in the United States. Available online: http://www.cdc.gov/anaplasmosis/stats/ (accessed on 5 September 2013).
- Dahlgren, F.S.; Mandel, E.J.; Krebs, J.W.; Massung, R.F.; McQuiston, J.H. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000–2007. Am. J. Trop. Med. Hyg. 2011, 85, 124–131. [Google Scholar] [CrossRef]
- Annen, K.; Friedman, K.; Eshoa, C.; Horowitz, M.; Gottschall, J.; Straus, T. Two cases of transfusion-transmitted Anaplasma phagocytophilum. Am. J. Clin. Pathol. 2012, 137, 562–565. [Google Scholar] [CrossRef]
- Jereb, M.; Pecaver, B.; Tomazic, J.; Muzlovic, I.; Avsic-Zupanc, T.; Premru-Srsen, T.; Levicnik-Stezinar, S.; Karner, P.; Strle, F. Severe human granulocytic anaplasmosis transmitted by blood transfusion. Emerg. Infect. Dis. 2012, 18, 1354–1357. [Google Scholar] [CrossRef]
- Jin, H.; Wei, F.; Liu, Q.; Qian, J. Epidemiology and control of human granulocytic anaplasmosis: A systematic review. Vector Borne Zoonotic Dis. 2012, 12, 269–274. [Google Scholar] [CrossRef]
- Foley, J.E.; Nieto, N.C.; Massung, R.; Barbet, A.; Madigan, J.; Brown, R.N. Distinct ecologically relevant strains of Anaplasma phagocytophilum. Emerg. Infect. Dis. 2009, 15, 842–843. [Google Scholar] [CrossRef]
- Massung, R.F.; Priestley, R.A.; Miller, N.J.; Mather, T.N.; Levin, M.L. Inability of a variant strain of Anaplasma phagocytophilum to infect mice. J. Infect. Dis. 2003, 188, 1757–1763. [Google Scholar] [CrossRef]
- Massung, R.F.; Mather, T.N.; Priestley, R.A.; Levin, M.L. Transmission efficiency of the Ap-variant 1 strain of Anaplasma phagocytophila. Ann. N. Y. Acad. Sci. 2003, 990, 75–79. [Google Scholar] [CrossRef]
- Massung, R.F.; Mather, T.N.; Levin, M.L. Reservoir competency of goats for the Ap-variant 1 strain of Anaplasma phagocytophilum. Infect. Immun. 2006, 74, 1373–1375. [Google Scholar] [CrossRef]
- Rar, V.; Golovljova, I. Anaplasma, Ehrlichia, and “Candidatus neoehrlichia” Bacteria: Pathogenicity, biodiversity, and molecular genetic characteristics, a review. Infect. Genet. Evol. 2011, 11, 1842–1861. [Google Scholar] [CrossRef]
- Stuen, S.; van de Pol, I.; Bergström, K.; Schouls, L.M. Identification of Anaplasma phagocytophila (formerly Ehrlichia phagocytophila) variants in blood from sheep in Norway. J. Clin. Microbiol. 2002, 40, 3192–3197. [Google Scholar] [CrossRef]
- Stuen, S.; Nevland, S.; Moum, T. Fatal cases of tick-borne fever (tbf) in sheep caused by several 16S rRNA gene variants of Anaplasma phagocytophilum. Ann. N. Y. Acad. Sci. 2003, 990, 433–434. [Google Scholar] [CrossRef]
- Barbet, A.; Al-Khedery, B.; Stuen, S.; Granquist, E.; Felshein, R.; Munderloh, U. An emerging tick-borne disease of humans is caused by a subset of strains with conserved genome structure. Pathogens 2013, 2, 544–555. [Google Scholar] [CrossRef]
- Patz, J.A.; Olson, S.H.; Uejio, C.K.; Gibbs, H.K. Disease emergence from global climate and land use change. Med. Clin. North Am. 2008, 92, 1473–1491. [Google Scholar] [CrossRef]
- Morissette, E.; Massung, R.F.; Foley, J.E.; Alleman, A.R.; Foley, P.; Barbet, A.F. Diversity of Anaplasma phagocytophilum strains, USA. Emerg. Infect. Dis. 2009, 15, 928–931. [Google Scholar] [CrossRef]
- Rikihisa, Y.; Zhi, N.; Wormser, G.P.; Wen, B.; Horowitz, H.W.; Hechemy, K.E. Ultrastructural and antigenic characterization of a granulocytic ehrlichiosis agent directly isolated and stably cultivated from a patient in New York state. J. Infect. Dis. 1997, 175, 210–213. [Google Scholar] [CrossRef]
- Aguero-Rosenfeld, M.E.; Horowitz, H.W.; Wormser, G.P.; McKenna, D.F.; Nowakowski, J.; Muñoz, J.; Dumler, J.S. Human granulocytic ehrlichiosis: A case series from a medical center in New York state. Ann. Intern. Med. 1996, 125, 904–908. [Google Scholar] [CrossRef]
- Asanovich, K.M.; Bakken, J.S.; Madigan, J.E.; Aguero-Rosenfeld, M.; Wormser, G.P.; Dumler, J.S. Antigenic diversity of granulocytic ehrlichia isolates from humans in Wisconsin and New York and a horse in California. J. Infect. Dis. 1997, 176, 1029–1034. [Google Scholar]
- Gribble, D.H. Equine ehrlichiosis. J. Am. Vet. Med. Assoc. 1969, 155, 462–469. [Google Scholar]
- Madigan, J.E.; Gribble, D. Equine ehrlichiosis in northern California: 49 cases (1968–1981). J. Am. Vet. Med. Assoc. 1987, 190, 445–448. [Google Scholar]
- Massung, R.F.; Levin, M.L.; Munderloh, U.G.; Silverman, D.J.; Lynch, M.J.; Gaywee, J.K.; Kurtti, T.J. Isolation and propagation of the Ap-variant 1 strain of Anaplasma phagocytophilum in a tick cell line. J. Clin. Microbiol. 2007, 45, 2138–2143. [Google Scholar] [CrossRef]
- Al-Khedery, B.; Lundgren, A.M.; Stuen, S.; Granquist, E.G.; Munderloh, U.G.; Nelson, C.M.; Alleman, A.R.; Mahan, S.M.; Barbet, A.F. Structure of the Type IV secretion system in different strains of Anaplasma phagocytophilum. BMC Genomics 2012, 13, 678. [Google Scholar] [CrossRef]
- Barbet, A.F.; Meeus, P.F.; Bélanger, M.; Bowie, M.V.; Yi, J.; Lundgren, A.M.; Alleman, A.R.; Wong, S.J.; Chu, F.K.; Munderloh, U.G.; et al. Expression of multiple outer membrane protein sequence variants from a single genomic locus of Anaplasma phagocytophilum. Infect. Immun. 2003, 71, 1706–1718. [Google Scholar] [CrossRef]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef]
- Goecks, J.; Nekrutenko, A.; Taylor, J. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef]
- Blankenberg, D.; von Kuster, G.; Coraor, N.; Ananda, G.; Lazarus, R.; Mangan, M.; Nekrutenko, A.; Taylor, J. Galaxy: A web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010, Chapter 19, Unit 19.10.1–Unit 19.10.21. [Google Scholar]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef]
- Carver, T.; Harris, S.R.; Otto, T.D.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Bamview: Visualizing and interpretation of next-generation sequencing read alignments. Brief. Bioinform. 2013, 14, 203–212. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Recent developments in the mafft multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef]
- Goodstadt, L.; Ponting, C.P. Chroma: Consensus-based colouring of multiple alignments for publication. Bioinformatics 2001, 17, 845–846. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using targetp, signalp and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10, 1–6. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Nakai, K.; Horton, P. Psort: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999, 24, 34–36. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Khedery, B.; Barbet, A.F. Comparative Genomics Identifies a Potential Marker of Human-Virulent Anaplasma phagocytophilum. Pathogens 2014, 3, 25-35. https://doi.org/10.3390/pathogens3010025
Al-Khedery B, Barbet AF. Comparative Genomics Identifies a Potential Marker of Human-Virulent Anaplasma phagocytophilum. Pathogens. 2014; 3(1):25-35. https://doi.org/10.3390/pathogens3010025
Chicago/Turabian StyleAl-Khedery, Basima, and Anthony F. Barbet. 2014. "Comparative Genomics Identifies a Potential Marker of Human-Virulent Anaplasma phagocytophilum" Pathogens 3, no. 1: 25-35. https://doi.org/10.3390/pathogens3010025